

Abstract
The restriction endonuclease EcoRII requires the cooperative interaction with two copies of the sequence 5′CCWGG for DNA cleavage. We found by limited proteolysis that EcoRII has a two-domain structure that enables this particular mode of protein–DNA interaction. The C-terminal domain is a new restriction endonuclease, EcoRII-C. In contrast to the wild-type enzyme, EcoRII-C cleaves DNA specifically at single 5′CCWGG sites. Moreover, substrates containing two or more cooperative 5′CCWGG sites are cleaved much more efficiently by EcoRII-C than by EcoRII. The N-terminal domain binds DNA specifically and attenuates the activity of EcoRII by making the enzyme dependent on a second 5′CCWGG site. Therefore, we suggest that a precursor EcoRII endonuclease acquired an additional DNA-binding domain to enable the interaction with two 5′CCWGG sites. The current EcoRII molecule could be an evolutionary intermediate between a site-specific endonuclease and a protein that functions specifically with two DNA sites such as recombinases and transposases. The combination of these functions may enable EcoRII to accomplish its own propagation similarly to transposons.
Keywords: DNA recombination/DNA restriction/EcoRII evolution/type IIE restriction endonuclease
Introduction
In the last years, a structural relationship between sequence-specific type II restriction endonucleases and enzymes that are involved in site-specific DNA recombination, transposition and repair has been revealed by a number of crystal structure analyses (Ban and Yang, 1998; Kovall and Matthews, 1998; Hickman et al., 2000; Hadden et al., 2001; Pingoud and Jeltsch, 2001; Tsutakawa and Morikawa, 2001). Currently, 14 crystal structures of ∼3400 discovered restriction endonucleases are solved. These structurally solved restriction endonucleases share a similar three-dimensional fold in the catalytic core, but, in general, do not share similarities in their primary amino acid sequences (Roberts and Macelis, 2001; Pingoud and Jeltsch, 2001; Grazulis et al., 2002; Horton et al., 2002). The majority of all restriction endonucleases are homodimeric type II restriction endonucleases that recognize short (4–8 bp) palindromic DNA sequences. These endonucleases cleave the DNA within or near the palindromic sequence (Pingoud and Jeltsch, 2001). In contrast to orthodox type II restriction endonucleases, subtype IIE restriction endonucleases interact stochiometrically and cooperatively with two copies of a defined DNA sequence. One of these DNA sites functions as an allosteric effector to activate DNA cleavage (Krüger et al., 1988, 1995; Conrad and Topal, 1989; Gabbara and Bhagwat, 1992).
EcoRII is a homodimeric type IIE restriction endonuclease. It recognizes the DNA sequence 5′CCWGG-(N)x-CCWGG. The unspecific spacer (N)x should not exceed 1000 bp to allow cooperative interaction (Pein et al., 1991). Similarly to bacterial and viral repressors, transcription factors, site-specific recombinases or replication proteins, EcoRII forms intermediate DNA loops on a linear DNA substrate that contains two 5′CCWGG sites (Reuter et al., 1998; Mücke et al., 2000). Based on a stretch of conserved and functional amino acids, an evolutionary relationship between EcoRII and the integrase family of site-specific recombinases may exist (Topal and Conrad, 1993; Nunes-Duby et al., 1998). Moreover, an evolutionary connection between DNA endonucleases and topoisomerases is suggested by the crystal structure and biochemical data of NaeI, another type IIE restriction endonuclease (Jo and Topal, 1995; Huai et al., 2000, 2001).
Specific regions and individual amino acids of EcoRII that are involved in DNA recognition, catalysis and protein–protein interactions have been identified despite the absence of a crystal structure (Reuter et al., 1999; Mücke et al., 2000, 2002). Although these specific DNA-binding regions and amino acids suggested a two-domain structure, functional domains of EcoRII have remained unclear.
Therefore, we subjected EcoRII to limited proteolysis by trypsin or chymotrypsin. We identified two functional domains for EcoRII. Both protease-resistant domains were cloned and expressed together with an N-terminal His6 tag. We found that the N-terminal domain binds sequence specifically, but cannot cleave the DNA substrate. In contrast, the C-terminal domain cleaves DNA sequence specifically and as effectively as a type II restriction endonuclease. Thus, the C-terminal domain of EcoRII is the first described domain of a restriction endonuclease that is a fully active endonuclease itself. We believe that an ancient precursor EcoRII endonuclease acquired the N-terminal DNA-binding domain by, for example, genomic rearrangements, thus resulting in the essential binding of a second 5′CCWGG site and in the reduced cleavage efficiency of full-length EcoRII. Domain acquisitions such as this may explain the evolution of new protein functions. Many other proteins in genomes reflect this evolutionary strategy as well, because they are often the result of the recombination of two or more domains (Babbitt and Gerlt, 2000). Because restriction–modification systems are thought to be mobile genetic elements that could be associated with genome rearrangements (Arber, 2000; Kobayashi, 2001), EcoRII could benefit from interacting with two sites with respect to its more specific transposition within a genome or between genomes.
Results
Limited proteolysis of EcoRII
To search for functional domains of EcoRII, we subjected EcoRII to limited proteolysis by either trypsin or chymotrypsin in the presence or absence of DNA. This limited proteolysis can provide information about protein folding, because regions of the protein that are tightly folded are less accessible to proteases than linker regions or loops between tightly folded domains. Therefore, tightly folded regions of the protein can occur as stable intermediates during proteolysis and can be isolated and identified by N-terminal sequencing.
The digestion of EcoRII with trypsin without specific DNA released an intermediate fragment cluster of ∼33–34 kDa (fragments A) (Figure 1A). The apparent molecular masses of the proteolytic fragments were estimated by SDS–PAGE and compared with the theoretically expected fragment pattern. The presence of specific DNA changed the tryptic cleavage pattern. We found a stable fragment with a molecular mass of ∼23 kDa (fragment B) and transient fragments of ∼30 kDa (fragments C).
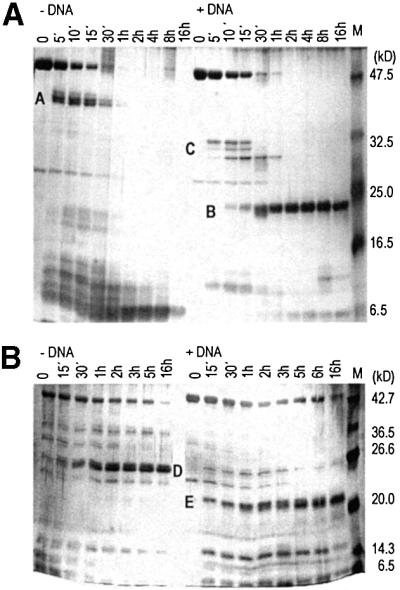
Fig. 1. (A) Digestion of EcoRII by trypsin in the presence or absence of specific DNA. A, B and C are proteolytic fragments: A, 33–34 kDa; B, 23 kDa; and C, ∼30 kDa. Digestion times are given at the top of each lane. M, pre-stained protein molecular weight marker (New England Biolabs). Molecular weights were estimated using the Broad Range molecular weight marker (New England Biolabs) not shown here. (B) Digestion of EcoRII by chymotrypsin in the presence or absence of specific DNA. D and E are proteolytic fragments: D, 26–27 kDa; and E, 21–22 kDa. Digestion times are given at the top of each lane. M, Broad Range protein molecular weight marker (New England Biolabs).
The digestion of EcoRII with chymotrypsin without specific DNA released a stable fragment of 26–27 kDa (fragment D) (Figure 1B). In the presence of specific DNA, we observed a stable fragment of ∼21–22 kDa (fragment E). In the presence of non-specific DNA, the proteolytic fragment patterns corresponded to those obtained in the absence of DNA (data not shown). Whereas the presence of specific DNA changed the fragment patterns, the presence of DNA per se did not interfere with proteolysis.
The N-terminal sequence of the proteolytic fragments B, C and E, which occurred in the presence of specific DNA, was determined by Edman degradation of the first 10 amino acids. Thus, these fragments we identified to be N-terminal fragments of EcoRII (Figure 2). The fragments A and D, obtained in the absence of specific DNA, could be assigned to the C-terminus of EcoRII based on their N-terminal amino acid sequence and on their apparent molecular mass (Figure 2). The N-terminal half of EcoRII was resistant to proteolysis in the presence of specific DNA, whereas the C-terminal half of EcoRII was more accessible to proteases in the presence than in the absence of specific DNA. We conclude from these data that EcoRII might consist of two domains that correspond to the N- and C-terminal halves of the protein. Furthermore, because specific DNA protected particularly the N-terminal half of EcoRII against proteolysis, the N-terminal half appears to be involved in specific DNA binding.

Fig. 2. Top: identification of the proteolytic cleavage fragments by Edman degradation. Possible cleavage positions in the EcoRII primary sequence were determined from the molecular weight of the fragments in SDS–polyacrylamide gels. Bottom: assignment of the proteolytic fragments to the EcoRII sequence. The amino acid sequences of the N-terminal fragments B and E start with the His6 tag of the protein. The length of the bars represents the length of the EcoRII sequence (404 amino acids) and of the proteolytic fragments, respectively.
Cloning of the protease-resistant domains of EcoRII and purification
To test if the identified N- and C-terminal proteolytic fragments correspond to stable functional domains of EcoRII, we molecularly cloned the sequence encoding the N-terminal amino acid residues 4–192 and that encoding the C-terminal amino acid residues 173–404 of EcoRII (cf. Figure 2). Both truncated proteins, termed EcoRII-N and EcoRII-C, respectively, were expressed separately and purified with an N-terminal His6 tag according to Reuter et al. (1998).
EcoRII-N migrated as two bands of ∼26 and 22 kDa in a denaturing SDS–polyacrylamide gel (not shown). The 22 kDa band corresponded to the theoretical molecular mass of 23 kDa of EcoRII-N as calculated from the amino acid sequence. The 26 kDa band did not agree with the theoretically expected molecular mass, but may be due to hydrophobic regions on the surface of EcoRII-N that could lead to abnormal running behavior. The C-terminal protein domain EcoRII-C migrated as a single 27 kDa band, which could correspond to the theoretical molecular mass of 28 kDa of EcoRII-C as calculated from its amino acid sequence. Western blot analysis with anti-EcoRII and anti-His6 antibodies verified that the two protein species of EcoRII-N and the one observed for EcoRII-C were specific for EcoRII and the His6 tag (not shown).
DNA binding and catalytic properties of the protease-resistant domains
To determine if the protease-resistant domains would bind specifically to DNA, we performed electrophoretic mobility shift assays (EMSAs). In these assays, EcoRII-N formed two complexes with a specific 191 bp DNA substrate. These complexes had a higher electrophoretic mobility than wild-type EcoRII–DNA complexes. The higher electrophoretic mobility of the EcoRII-N–DNA complexes results from the lower molecular mass of EcoRII-N compared with EcoRII (Figure 3). The two bands for EcoRII-N–DNA complexes could be due to either nonhomogeneity of the enzyme preparation or dimerization of EcoRII-N in the presence of specific DNA. The DNA–substrate affinity of EcoRII-N differed from that of the wild-type EcoRII by only one order of magnitude; the apparent KD was ∼26 nM for EcoRII-N and ∼1 nM for the wild-type EcoRII. The specificity of DNA binding was verified by competition experiments using a 5000-fold molar excess of unlabeled specific or unspecific oligonucleotide duplexes over the 32P-labeled DNA substrate (not shown). The EcoRII-N-DNA complexes disappeared in the presence of an excess of unlabeled specific over labeled specific DNA, but were maintained in the presence of an excess of unlabeled unspecific over labeled specific DNA. Therefore, EcoRII-N bound specifically to the DNA substrate. Furthermore, these data confirmed that the N-terminal domain of EcoRII contributes importantly to DNA binding of EcoRII. In contrast to the EcoRII-N–DNA complexes found, we could not demonstrate specific DNA binding of EcoRII-C by our EMSA, because the portion of these complexes was beyond the detection limit (not shown).
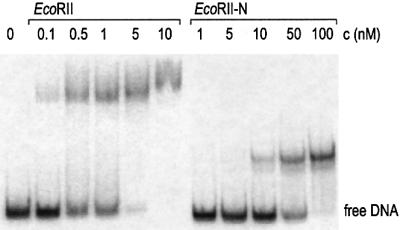
Fig. 3. Electrophoretic mobility shift assay with wild-type EcoRII and EcoRII-N in the presence of a 191 bp DNA molecule (0.6 nM) containing a single 5′CCWGG site. Enzyme concentrations c (nM) are indicated at the top of each lane.
Although EcoRII-N bound specifically to the DNA, it was not able to cleave DNA substrates (not shown). In contrast to EcoRII-N, however, EcoRII-C cleaved linearized pBR322 DNA specifically and much more efficiently than wild-type EcoRII. The cleavage efficiency was determined by analyzing the time dependence of DNA cleavage (Figure 4A). Whereas EcoRII-C cleaved pBR322 DNA completely after 1 min, the wild-type enzyme did not cleave the DNA completely, not even after 1 h. The high cleavage efficiency of EcoRII-C is equivalent to that of type II restriction endonucleases such as the EcoRII isoschizomer BstNI (Figure 4A). Based on these data, it appears that EcoRII-C possesses all structural and functional components corresponding to type II restriction endonucleases that cleave specifically at single DNA recognition sequences.

Fig. 4. (A) Kinetics of the cleavage reactions of wild-type EcoRII and EcoRII-C with linearized pBR322 Dcm– DNA. The reaction times are given at the top of each lane. BstNI, an isoschizomer of EcoRII (positive control); M, molecular weight marker. (B) Cleavage of T3 DNA with EcoRII-C and wild-type EcoRII. Enzyme amounts are given at the top of each lane. Left lane, T3 DNA without enzyme; BstNI, positive control. Molecular weight markers are given on the right.
To test this theory, we chose bacteriophage T3 DNA for a cleavage assay. It is known that wild-type EcoRII cannot cleave the DNA of phage T3 (Krüger et al., 1988). The inability of EcoRII to cleave T3 DNA is due to the required simultaneous interaction of wild-type EcoRII with two copies of 5′CCWGG. In the phage genome, however, this sequence occurs at low frequency (three times in 38 740 bp). The long distances (>1000 bp) between these sites do not permit EcoRII-mediated site cooperation and DNA cleavage.
In the cleavage assay, we found that EcoRII-C cleaved T3 DNA as efficiently as the type II restriction endonuclease BstNI, whereas wild-type EcoRII did not cleave T3 DNA under the same conditions (Figure 4B). We infer from this result that the C-terminal domain of EcoRII corresponds to an endonuclease-like domain. The fact that EcoRII-C cleaved DNA specifically implies that it can bind specifically to DNA as well. Therefore, EcoRII must consist of two domains that can interact specifically and independently with DNA. Typical of dimeric type II restriction endonucleases, EcoRII-C cleaved both strands of the DNA. Thus it is possible that EcoRII-C acts as a dimer upon DNA cleavage.
Oligomeric states of the protease-resistant domains EcoRII-N and EcoRII-C
We analyzed the oligomeric states of both protease-resistant domains, EcoRII-N and EcoRII-C, in solution under equilibrium conditions by analytical ultracentrifugation. Assuming a monomer–dimer equilibrium, we determined the concentration dependence of the oligomeric states of EcoRII-N and EcoRII-C (Figure 5). In the concentration range examined, the apparent molecular mass of EcoRII-N was found to be 37–40 kDa. These values lie between the theoretical molecular masses for the monomer (23 kDa) and the dimer (46 kDa) and, therefore, support a monomer–dimer equilibrium. EcoRII-C showed an apparent molecular mass of ∼50 kDa over the examined concentration range. This value also lies between the theoretical molecular masses for the monomer (28 kDa) and the dimer (56 kDa).

Fig. 5. Fraction (%) of the dimeric form of EcoRII-N and EcoRII-C determined by analytical ultracentrifugation. Open circles, EcoRII-N; filled circles, EcoRII-C; graph without data points, wild-type EcoRII for comparison as determined previously (Behlke et al., 1997).
The dimer dissociation constants (KDs) were calculated from the concentration dependence of the molecular mass for each of the protease-resistant domains (Behlke et al., 1997). The KD values for protein dimerization of EcoRII-N and EcoRII-C were 1.85 ± 0.19 µM and 75.4 ± 9.1 nM, respectively. For comparison, wild-type EcoRII forms a stable dimer in solution with a KD of 2.89 ± 1.08 nM (Mücke et al., 2000). Based on the KD of EcoRII-C, we believe that EcoRII-C preferentially forms a dimer as suggested by its specific cleavage of double-stranded DNA. The EcoRII-C dimer, however, is 26-fold less stable than that of wild-type EcoRII. In contrast to EcoRII-C and based on the KD of EcoRII-N, this domain exists primarily as a monomer and was 640-fold destabilized compared with wild-type EcoRII. Because EcoRII-N is predominantly monomeric and EcoRII-C is predominantly dimeric, we suggest that within the EcoRII dimer, the C-terminal domain (amino acids 173–404) mainly mediates the protein–protein contacts of EcoRII. Preliminary studies using peptide libraries to determine protein–protein contacts of EcoRII further support that the EcoRII dimer is formed vitally by contacts in the C-terminal domain (C.Petter and M.Reuter, unpublished data). Nevertheless, it is also conceivable that the N-terminal domain dimerizes upon DNA binding, because we observed two bands of protein–DNA complexes in EMSAs (Figure 3).
Discussion
In this study, we have engineered an endonuclease EcoRII-C that cleaves DNA at single 5′CCWGG sites. This is in contrast to the full-length EcoRII, which requires two copies of this sequence for DNA cleavage (Krüger et al., 1995). Thus, we have generated a new restriction endonuclease with a new specificity by deleting the N-terminal domain of the full-length EcoRII restriction endonuclease. Furthermore, the EcoRII-C restriction endonuclease cleaves DNA much faster than the full-length EcoRII. Therefore, we propose that truncation of restriction endonucleases such as EcoRII could be a strategy to promote their endonucleolytic activity.
In addition, because EcoRII-C cleaved much more efficiently than full-length EcoRII, the presence of the N-terminal domain of EcoRII obviously slows down the cleavage efficiency of the C-terminally encoded restriction endonuclease in the full-length enzyme and necessitates the simultaneous binding of two 5′CCWGG sites for DNA cleavage.
Nonetheless, the N-terminal domain contributes significant components to the substrate binding capacity of the EcoRII enzyme. The affinity of EcoRII-N is only about one order of magnitude lower than that of the wild-type enzyme. The C-terminal catalytic domain has a lower DNA binding affinity than the N-terminal domain. However, the DNA binding affinity of the C-terminal domain is sufficient to enable its specific and efficient endonucleolytic function. Therefore, we propose that EcoRII is composed of an N-terminal DNA-binding domain and a C-terminal catalytic domain.
The fact that both EcoRII domains interact specifically and independently with DNA confirms the two separate DNA-binding regions of EcoRII, which had been narrowed down previously by membrane-bound peptide libraries and mutational analysis (Reuter et al., 1999; Mücke et al., 2000, 2002).
The domain organization of EcoRII is similar to the two-domain structure of NaeI, another type IIE restriction endonuclease (Colandene and Topal, 1998; Huai et al., 2000, 2001). The N-terminal domain of EcoRII appears to correspond to the Topo domain, and the C-terminal domain of EcoRII to the Endo domain of NaeI. The domain organization of EcoRII is also similar to that of FokI that consists of two separable domains (Li et al., 1992). Because of the marked similarities of type IIS (FokI) to type IIE restriction endonucleases (EcoRII, NaeI) with respect to structure and interaction with two sites, both subtypes, IIS and IIE, might be more related than anticipated so far (Wah et al., 1997, 1998; Bitinaite et al., 1998; Huai et al., 2000; Vanamee et al., 2001). Although the presence of a second DNA-binding domain in particular seems to be a feature of both subtypes, to the best of our knowledge, this is the first report on the dissection of a restriction endonuclease into functional domains that uncovered a still functionally active restriction endonuclease unit.
Based on these results, we suggest that in evolution, the C-terminal domain of EcoRII, which is an active restriction endonuclease, acquired an additional DNA-binding domain and thus evolved a new protein function. Based on recent sequence homology studies of restriction endonucleases of different subtypes, this acquisition of an additional domain for DNA binding has also been suggested for EcoRII (Pingoud et al., 2002). The newly acquired function of EcoRII consisted of the simultaneous recognition of two identical DNA sequences which also resulted in a reduction in the endonucleolytic activity of the enzyme. One consequence of the requirement for two unmodified DNA sites for DNA cleavage is a protection against suicidal restriction of the rare unmodified sites in the cellular DNA. Such unmethylated sites may arise by DNA repair or by incomplete DNA methylation (Bickle and Krüger, 1993).
The requirement for two DNA sites is also seen for enzymes that accomplish cellular processes such as site-specific DNA recombination and transposition. Evidence for a connection between EcoRII and these enzymes came from conserved amino acids for EcoRII and the Int family of site-specific recombinases (Topal and Conrad, 1993; Nunes-Duby et al., 1998). Furthermore, because proteins with an endonuclease-like protein fold include DNA transposases, recombinases and DNA repair enzymes in prokaryotes, eukaryotes and archaea (Ban and Yang, 1998; Kovall and Matthews, 1998; Tsutakawa et al., 1999a,b; Yang et al., 1999; Daiyasu et al., 2000; Hickman et al., 2000), the evolution of those DNA-processing enzymes may be based on a very distant ancestral nuclease. This hypothesis is supported by an evolutionary tree of the restriction endonuclease-like superfamily (Bujnicki, 2000). Based on this phylogenetic relationship, it has been proposed that the non-specific DNA cleavage domain of the type IIS restriction endonuclease FokI is evolutionarily older than other endonucleases of its branch. Furthermore, FokI might have acquired a separate domain for specific DNA recognition (Bujnicki, 2000). Because of the evolutionary relationship between restriction endonucleases and DNA-processing enzymes, we suppose that acquiring an additional DNA-binding domain could be the first step on the way to protein functions beyond DNA phosphodiester bond cleavage for a restriction endonuclease.
The structural homology of protein folds described above unequivocally links enzymes involved in DNA recombination and transposition with restriction endonucleases. The biological role of restriction–modification systems could explain this connection. In addition to the well known biological role of restriction–modification systems as protectors against foreign molecular parasites (Bickle and Krüger, 1993), two other hypothe ses exist to explain the development and maintenance of the impressively high number and diversity of restriction–modification genes during evolution (Arber, 2000; Kobayashi, 2001). First, because genetic variation is a pre-condition for biological evolution, the existence of evolution genes is postulated, which benefit biological evolution (Arber, 2000). According to this hypothesis, restriction–modification enzymes, together with DNA repair enzymes, are regarded as modulators of the frequency of genetic variation. Restriction enzymes, on the one hand, are thought to reduce the uptake of foreign DNA into a cell to a low level. On the other hand, restricted foreign DNA fragments, which are potentially recombinogenic DNA molecules, can be incorporated into the bacterial genome (Arber, 2000). The functional cooperation of restriction–modification and DNA repair enzymes as modulators could be a driving force to connect endonucleolytic activity and additional DNA binding capacity.
An alternative hypothesis considers restriction– modification genes to be selfish mobile genetic elements, like viruses or transposons (Kobayashi, 2001). Therefore, restriction–modification systems might be molecular invaders themselves. They ensure that they are maintained in a population to the cost of the host cell as they kill cells that have eliminated them. Moreover, genome comparisons suggest that restriction–modification systems can move to different positions within a genome as well as between genomes, and are associated with genome rearrangements (Arber, 2000; Kobayashi, 2001). Possibly, the newly acquired recognition of two identical DNA sites could promote the mobility of restriction– modification genes within a genome or between genomes.
The acquisition of new protein domains by either the predicted frequent horizontal gene transfer of restriction– modification systems associated with genomic rearrangements or by acquisition of DNA fragments after foreign DNA restriction could be an efficient evolutionary strategy that results in new proteins and protein functions/domains. Several protein superfamilies prove this evolutionary strategy, because their structures are often the result of a combination of two or more domains (Babbitt and Gerlt, 2000). In the case of EcoRII, the new protein function is the dependence of the EcoRII enzymatic activity on two 5′CCWGG sites. This feature is an essential prerequisite for enzymes that play a role in DNA transposition and DNA recombination; it might enable the EcoRII restriction–modification genes to provide its own propagation into new habitats as transposons do.
Materials and methods
Limited proteolytic digestion of EcoRII
A 30 µg aliquot of EcoRII (325 pmol dimer) was digested with trypsin at a ratio of wEcoRII/wtrypsin of 100/1. EcoRII was digested in the absence or presence of 1.3 nmol of a specific 20 bp oligonucleotide (5′GCTGCCAACCTGGCTCTAAC, EcoRII-specific sequence in bold letters) at 37°C. Digestion of 48.3 µg of EcoRII (520 pmol dimer) by chymotrypsin was performed at a ratio of wEcoRII/wchymotrypsin of 80/1 in the absence or presence of 1.05 nmol specific 20 bp oligonucleotide at 25°C. All digestions were done in 10 mM Tris–HCl pH 8.5 and in a total volume of 200 µl. Samples of 18 µl were removed over a time period of 16 h (Figure 1A and B). The digestions were stopped with SDS gel loading buffer [62.5 mM Tris–HCl pH 6.8, 2% (w/v) SDS, 10% glycerol, 0.01% Bromophenol Blue, 40 mM dithiothreitol (DTT)]. After digestion, samples were denatured immediately at 95°C for 5 min. Proteolytic fragments were separated on a 12% SDS–polyacrylamide gel and bands were visualized by silver staining.
Amino acid sequencing
Proteolytic fragments of EcoRII were semi-dry blotted on ProBlott™ membranes (Applied Biosystems) for 1 h using anode buffer 1 (0.3 M trishydroxymethane, 15% CH3OH), anode buffer 2 (25 mM trishydroxymethane, 15% CH3OH) and cathode buffer (25 mM trishydroxymethane, 40 mM ε-aminocapronic acid, 15% CH3OH). Bands were visualized by staining with 0.1% Coomassie Blue R250, 1% CH3COOH, 40% CH3OH in H2O. Bands of EcoRII fragments were cut out of the membrane and sequenced using a PE/ABI model 492.
Generation of the protease-resistant domains
DNA encoding the protease-resistant domains was amplified by PCR using the pQE-30 (Qiagen GmbH)-derived expression plasmid pQE-RII, which encodes the EcoRII restriction endonuclease as template (Reuter et al., 1998). For the N-terminal domain EcoRII-N, the primers 5′AGGCGTATCA CGAGGCCCTT TCGTCTTCAC and 5′GCGCA GGTGC CAGTCTTCAG GTAGAATATA were used for PCR. The pQE-30 vector was linearized by EcoRI and SmaI and dephosphorylated using calf intestinal phosphatase (CIP) (Roche). The 633 bp PCR product was cleaved with EcoRI, phosphorylated with T4 polynucleotide kinase (New England Biolabs) and ATP, and ligated into the pQE-30 vector using the Ready-To-Go-Ligase Kit (Amersham Pharmacia Biotech).
For amplifying the C-terminal domain EcoRII-C, the primers 5′CGCGGATCCT CTCTACAGCA AGCGCCAGTA AATCATAAA and 5′GTACCTATGG AATATCTGCG TAAAGCCCTG T were used in PCR with plasmid pQE-RII as template. The pQE-30 vector DNA was cleaved sequentially by SmaI and BamHI, and dephosphorylated using CIP (Roche). The 703 bp PCR product was cleaved with BamHI, phosphorylated with T4 polynucleotide kinase and ligated into the pQE-30 vector using T4 DNA ligase (New England Biolabs). Competent Escherichia coli JM109 (pDK1r–m+) cells were transformed with the resulting recombinant plasmids and the DNA sequence was verified by sequencing with the ThermoSequenase Kit (Amersham Pharmacia Biotech). All amino acid sequence positions were related to the EcoRII sequence AJ224995 which starts with amino acid Met3 of EcoRII. The pQE-30 plasmid also encodes the N-terminal His tag of both mutants (amino acids MRGSHHHHHHGS).
Enzyme preparations
N-terminally His6-tagged wild-type EcoRII and truncated proteins were expressed in E.coli JM109 (pDK1r–m+) and purified as described (Reuter et al., 1998). The proteins were analyzed by western blotting using the primary antibodies polyclonal rabbit EcoRII antiserum and monoclonal mouse anti-His antibodies (No. 34610, Qiagen) as described (Reuter et al., 1999).
Gel shift and cleavage assays
Gel shift assays and estimation of the apparent KD of the truncated proteins were performed as described (Reuter et al., 1999). For cleavage assays, 300 ng of T3 DNA (35.1 fmol 5′CCWGG sites) were incubated with EcoRII or EcoRII-C at enzyme to site ratios of 0.5 and 500 in 1× universal buffer (Stratagene) at 37°C for 30 min. The total reaction volume was 20 µl. DNA fragments were separated on a 0.6% agarose gel and stained with ethidium bromide. To determine the kinetics of the cleavage reaction, 3 µg of HindIII-linearized pBR322 Dcm– DNA that contains six 5′CCWGG sites per 4361 bp (6.22 pmol sites) were incubated with 3 pmol of wild-type EcoRII or EcoRII-C at 37°C. The reaction mixture contained 10 mM MgCl2 and 0.5 mg/ml bovine serum albumin (BSA). The total reaction volume was 200 µl. Aliquots of 20 µl were removed over a time period of 60 min (see legend to Figure 4). Reactions were stopped with 10× Bromophenol Blue solution [final concentrations: 0.042% (w/v) Bromophenol Blue, 20 mM EDTA, 1.5% Ficoll]. DNA fragments were separated on a 0.8% agarose gel and bands were stained with ethidium bromide.
Analytical ultracentrifugation
Mr analyses were carried out in an XL-A-type analytical ultracentrifuge (Beckman) with UV absorbance scanner optics. The sedimentation equilibirium was analyzed using externally loaded six-channel centerpieces of 12 mm optical path length usually filled with 70 µl of protein solution. Three of these cells were used to analyze different samples in one run simultaneously. Sedimentation equilibrium was reached after 2 h of overspeed at 24 000 r.p.m., followed by an equilibrium speed of 20 000 r.p.m. at 10°C for 24–30 h. The radial absorbancies of each compartment were scanned at 275, 280 and 285 nm, or 230, 233 and 236 nm depending on concentration and extinction of the samples. We used the molecular absorption coefficients determined with EcoRII wild-type. Mr calculations were done as described (Behlke et al., 1997). The molecular mass values of EcoRII-N and EcoRII-C, respectively, depended on protein concentration typically for a monomer–dimer equilibrium. Therefore, the association constants (K2) were obtained by fitting the radial concentration distribution (cr) at sedimentation equilibrium to the following multi-exponential equation
cr = crm exp[MF(r2 – rm2)] + 2K2crm2 exp[2MF(r2 – rm2)]
with F = [(1 –ρ̄)ω2/2RT]. Here, M is the theoretical molecular mass derived from the amino acid composition of EcoRII-N and EcoRII-C, respectively, rm is the meniscus radius, ρ̄ the partial specific volume, ρ is the solvent density, ω is the angular velocity, R the gas constant, and T the absolute temperature. For the fitting procedure, we used our computer program POLYMOLE (Behlke et al., 1997). The hyperbolic curves are the theoretical ones obtained from the averaged equilibrium constants. EcoRII-N was dissolved in 20 mM Tris–HCl pH 7.6, 200 mM KCl. The solution of EcoRII-C contained additionally 2 mM EDTA to protect against proteases.
Acknowledgments
Acknowledgements
We would like to thank Dr Angelika Hofmann for critical reading of the manuscript. We gratefully acknowledge Petra Mackeldanz, Ulrike Marzahn and Ursula Scherneck for skilful technical assistance. This work was supported by Deutsche Forschungsgemeinschaft (Re 879/2), Fonds der chemischen Industrie, Universitäre Forschungsförderung of the Humboldt University and Sonnenfeld-Stiftung.
References
- Arber W. (2000) Genetic variation: molecular mechanisms and impact on microbial evolution. FEMS Microbiol. Rev., 24, 1–7. [DOI] [PubMed] [Google Scholar]
- Babbitt P.C. and Gerlt,J.A. (2000) New functions from old scaffolds: how nature reengineers enzymes for new functions. Adv. Protein Chem., 55, 1–28. [DOI] [PubMed] [Google Scholar]
- Ban C. and Yang,W. (1998) Structural basis for MutH activation in E.coli mismatch repair and relationship of MutH to restriction endonucleases. EMBO J., 17, 1526–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behlke J., Ristau,O. and Schonfeld,H.J. (1997) Nucleotide-dependent complex formation between the Escherichia coli chaperonins GroEL and GroES studied under equilibrium conditions. Biochemistry, 36, 5149–5156. [DOI] [PubMed] [Google Scholar]
- Bickle T.A. and Krüger,D.H. (1993) Biology of DNA restriction. Microbiol. Rev., 57, 434–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitinaite J., Wah,D.A., Aggarwal,A.K. and Schildkraut,I. (1998) FokI dimerization is required for DNA cleavage. Proc. Natl Acad. Sci. USA, 95, 10570–10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujnicki J.M. (2000) Phylogeny of the restriction endonuclease-like superfamily inferred from comparison of protein structures. J. Mol. Evol., 50, 39–44. [DOI] [PubMed] [Google Scholar]
- Colandene J.D. and Topal,M.D. (1998) The domain organization of NaeI endonuclease: separation of binding and catalysis. Proc. Natl Acad. Sci. USA, 95, 3531–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad M. and Topal,M.D. (1989) DNA and spermidine provide a switch mechanism to regulate the activity of restriction enzyme NaeI. Proc. Natl Acad. Sci. USA, 86, 9707–9711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiyasu H., Komori,K., Sakae,S., Ishino,Y. and Toh,H. (2000) Hjc resolvase is a distantly related member of the type II restriction endonuclease family. Nucleic Acids Res., 28, 4540–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbara S. and Bhagwat,A.S. (1992) Interaction of EcoRII endonuclease with DNA substrates containing single recognition sites. J. Biol. Chem., 267, 18623–18630. [PubMed] [Google Scholar]
- Grazulis S. et al. (2002) Crystal structure of the Bse634I restriction endonuclease: comparison of two enzymes recognizing the same DNA sequence. Nucleic Acids Res., 30, 876–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadden J.M., Convery,M.A., Declais,A.C., Lilley,D.M. and Phillips,S.E. (2001) Crystal structure of the Holliday junction resolving enzyme T7 endonuclease I. Nat. Struct. Biol., 8, 62–67. [DOI] [PubMed] [Google Scholar]
- Hickman A.B., Li,Y., Mathew,S.V., May,E.W., Craig,N.L. and Dyda,F. (2000) Unexpected structural diversity in DNA recombination: the restriction endonuclease connection. Mol. Cell, 5, 1025–1034. [DOI] [PubMed] [Google Scholar]
- Horton N.C., Dorner,L.F. and Perona,J.J. (2002) Sequence selectivity and degeneracy of a restriction endonuclease mediated by DNA intercalation. Nat. Struct. Biol., 9, 42–47. [DOI] [PubMed] [Google Scholar]
- Huai Q., Colandene,J.D., Chen,Y., Luo,F., Zhao,Y., Topal,M.D. and Ke,H. (2000) Crystal structure of NaeI—an evolutionary bridge between DNA endonuclease and topoisomerase. EMBO J., 19, 3110–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huai Q., Colandene,J.D., Topal,M.D. and Ke,H. (2001) Structure of NaeI–DNA complex reveals dual-mode DNA recognition and complete dimer rearrangement. Nat. Struct. Biol., 8, 665–669. [DOI] [PubMed] [Google Scholar]
- Jo K. and Topal,M.D. (1995) DNA topoisomerase and recombinase activities in NaeI restriction endonuclease. Science, 267, 1817–1820. [DOI] [PubMed] [Google Scholar]
- Kobayashi I. (2001) Behavior of restriction–modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res., 29, 3742–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovall R.A. and Matthews,B.W. (1998) Structural, functional and evolutionary relationships between λ-exonuclease and the type II restriction endonucleases. Proc. Natl Acad. Sci. USA, 95, 7893–7897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger D.H., Barcak,G.J., Reuter,M. and Smith,H.O. (1988) EcoRII can be activated to cleave refractory DNA recognition sites. Nucleic Acids Res., 16, 3997–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger D.H., Kupper,D., Meisel,A., Reuter,M. and Schroeder,C. (1995) The significance of distance and orientation of restriction endonuclease recognition sites in viral DNA genomes. FEMS Microbiol. Rev., 17, 177–184. [DOI] [PubMed] [Google Scholar]
- Li L., Wu,L.P. and Chandrasegaran,S. (1992) Functional domains in FokI restriction endonuclease. Proc. Natl Acad. Sci. USA, 89, 4275–4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mücke M., Lurz,R., Mackeldanz,P., Behlke,J., Krüger,D.H. and Reuter,M. (2000) Imaging DNA loops induced by restriction endonuclease EcoRII. A single amino acid substitution uncouples target recognition from cooperative DNA interaction and cleavage. J. Biol. Chem., 275, 30631–30637. [DOI] [PubMed] [Google Scholar]
- Mücke M., Pingoud,V., Grelle,G., Kraft,R., Krüger,D.H. and Reuter,M. (2002) Asymmetric photocross-linking pattern of restriction endonuclease EcoRII to the DNA recognition sequence. J. Biol. Chem., 277, 14288–14293. [DOI] [PubMed] [Google Scholar]
- Nunes-Duby S.E., Kwon,H.J., Tirumalai,R.S., Ellenberger,T. and Landy,A. (1998) Similarities and differences among 105 members of the Int family of site-specific recombinases. Nucleic Acids Res., 26, 391–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pein C.-D., Reuter,M., Meisel,A., Cech,D. and Krüger,D.H. (1991) Activation of restriction endonuclease EcoRII does not depend on the cleavage of stimulator DNA. Nucleic Acids Res., 19, 5139–5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pingoud A. and Jeltsch,A. (2001) Structure and function of type II restriction endonucleases. Nucleic Acids Res., 29, 3705–3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pingoud V., Kubareva,E., Stengel,G., Friedhoff,P., Bujnicki,J.M., Urbanke,C., Sudina,A. and Pingoud,A. (2002) Evolutionary relationship between different subgroups of restriction endonucleases. J. Biol. Chem., 277, 14306–14314. [DOI] [PubMed] [Google Scholar]
- Reuter M., Kupper,D., Meisel,A., Schroeder,C. and Krüger,D.H. (1998) Cooperative binding properties of restriction endonuclease EcoRII with DNA recognition sites. J. Biol. Chem., 273, 8294–8300. [DOI] [PubMed] [Google Scholar]
- Reuter M., Schneider-Mergener,J., Kupper,D., Meisel,A., Mackeldanz,P., Krüger,D.H. and Schroeder,C. (1999) Regions of endonuclease EcoRII involved in DNA target recognition identified by membrane-bound peptide repertoires. J. Biol. Chem., 274, 5213–5221. [DOI] [PubMed] [Google Scholar]
- Roberts R.J. and Macelis,D. (2001) REBASE—restriction enzymes and methylases. Nucleic Acids Res., 29, 268–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topal M.D. and Conrad,M. (1993) Changing endonuclease EcoRII Tyr308 to Phe abolishes cleavage but not recognition: possible homology with the Int-family of recombinases. Nucleic Acids Res., 21, 2599–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutakawa S.E. and Morikawa,K. (2001) The structural basis of damaged DNA recognition and endonucleolytic cleavage for very short patch repair endonuclease. Nucleic Acids Res., 29, 3775–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutakawa S.E., Jingami,H. and Morikawa,K. (1999a) Recognition of a TG mismatch: the crystal structure of very short patch repair endonuclease in complex with a DNA duplex. Cell, 99, 615–623. [DOI] [PubMed] [Google Scholar]
- Tsutakawa S.E., Muto,T., Kawate,T., Jingami,H., Kunishima,N., Ariyoshi,M., Kohda,D., Nakagawa,M. and Morikawa,K. (1999b) Crystallographic and functional studies of very short patch repair endonuclease. Mol. Cell, 3, 621–628. [DOI] [PubMed] [Google Scholar]
- Vanamee E.S., Santagata,S. and Aggarwal,A.K. (2001) FokI requires two specific DNA sites for cleavage. J. Mol. Biol., 309, 69–78. [DOI] [PubMed] [Google Scholar]
- Wah D.A., Hirsch,J.A., Dorner,L.F., Schildkraut,I. and Aggarwal,A.K. (1997) Structure of the multimodular endonuclease FokI bound to DNA. Nature, 388, 97–100. [DOI] [PubMed] [Google Scholar]
- Wah D.A., Bitinaite,J., Schildkraut,I. and Aggarwal,A.K. (1998) Structure of FokI has implications for DNA cleavage. Proc. Natl Acad. Sci. USA, 95, 10564–10569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Malik,H.S. and Eickbush,T.H. (1999) Identification of the endonuclease domain encoded by R2 and other site-specific, non-long terminal repeat retrotransposable elements. Proc. Natl Acad. Sci. USA, 96, 7847–7852. [DOI] [PMC free article] [PubMed] [Google Scholar]