

Abstract
GFP and the red fluorescent protein, DsRed, have been combined to design a protease assay that allows not only for fluorescence resonance energy transfer (FRET) studies but also for dual-color crosscorrelation analysis, a single-molecule-based method that selectively probes the concomitant movement of two distinct tags. The measurement principle is based on a spectrally resolved detection of single molecules diffusing in and out of a diffraction-limited laser focus. Double-labeled substrate molecules are separated into two single-labeled products by specific cleavage at a protease cleavage site between the two flanking tags, DsRed and GFP, thus disrupting joint fluctuations in the two detection channels and terminating FRET between the two labels. In contrast to enzyme assays based solely on FRET, this method of dual-color crosscorrelation is not limited to a certain range of distances between the fluorophores and is much more versatile with respect to possible substrate design. To simplify the measurement setup, two-photon excitation was used, allowing for simultaneous excitation of both tags with a single infrared laser wavelength. The general concept was experimentally verified with a GFP–peptide–DsRed construct containing the cleavage site for tobacco etch virus protease. Two-photon excitation in the infrared and the use of cloneable tags make this assay easily adaptable to intracellular applications. Moreover, the combination of FRET and crosscorrelation analysis in a single-molecule-based approach promises exciting perspectives for miniaturized high-throughput screening based on fluorescence spectroscopy.
Protein–protein interactions have emerged as a critical theme in biochemistry in the postgenomic era. With an increasing number of novel proteins revealed by genomic research and the subsequent task of characterizing their functions, new technologies have been developed for in vivo applications and high-throughput screening. Confocal detection coupled with analysis of sparse or single fluorescent molecules in an aqueous environment (1–3) is currently considered one of the most promising tools because of its high sensitivity, relative noninvasiveness, and low sample consumption. On the basis of fluorescent reporter molecules, generally proteins or nucleic acids tagged with synthetic dyes, these tools allow diverse biological processes to become amenable to sensitive biophysical techniques. Fluorescence correlation spectroscopy (FCS) (1, 4, 5), one of the most prominent of these techniques, analyzes minute spontaneous fluctuations in the emission behavior of small molecular ensembles, which reflect underlying inter- and intramolecular dynamics. Because FCS observes fluorescent molecules in the nanomolar range, it can yield information closely mimicking actual physiological conditions about a diverse number of processes, such as association and dissociation reactions, transport, enzymatic turnovers of biochemical substrates, and intramolecular (e.g., structural) dynamics, over a wide resolution range in space and time.
One of the most promising spectroscopic tools for studying protein–protein interactions in the living cell, fluorescence resonance energy transfer (FRET), has recently been used for the development of efficient intracellular ligand-binding (6) and protease assays (7). Here, cellular processes can be evidenced by spectral changes of the emission signal because of distance-dependent energy transfer from an excited donor dye to a long-wavelength acceptor. Because of its high specificity, FRET has proven useful to investigate molecular interactions and conformational changes even on a single-molecule scale (2, 8). However, because FRET requires spatial distances between donor and acceptor dyes of typically 20–60 Å (8), this imposes a fundamental limit on probing molecular interactions or the formation or cleavage of linkages (i.e., to indicate enzyme activity).
Dual-color fluorescence crosscorrelation spectroscopy (dcFCCS) analysis (9) evades this limitation by recording concomitant signal fluctuations from a confocal detection volume in two spectrally distinct emission channels and allows for probing of extremely low fluorophore concentrations with a temporal resolution down to tens of nanoseconds. In the single-molecule regime, coincident signal fluctuations in both detection channels unambiguously indicate the presence of tight chemical or physical linkages between the fluorophores. Previously, this has been successfully used to quantitatively study endonucleolytic cleavage of double-labeled double-stranded DNA (dsDNA) in real time (10, 11). By limiting readout parameters to either crosscorrelation amplitudes or coincidence measurements, the speed of the assay could be improved to meet high-throughput screening standards, enabling reliable yes-or-no assessments within subsecond sampling times (12). Furthermore, dcFCCS has recently been applied to study intracellular phenomena, such as endocytosis (13).
Two-photon excitation (TPE) coupled with dcFCCS confers particular advantages for intracellular applications, such as limitation of cell damage and sample consumption by photobleaching, similar to analogous in vivo two-photon FCS experiments (14, 15). TPE occurs only in the immediate vicinity of the focal spot by quasisimultaneous absorption of two near-infrared photons. Because different parity selection rules apply for the transition probabilities between the ground and excited states, two-photon excitation spectra can differ considerably from their respective one-photon spectra. Therefore, another substantial advantage of two-photon excitation is the ability to simultaneously excite spectrally distinct dyes with the same infrared wavelength.
Since the introduction of cloneable fluorescent tags such as GFP, the most attractive assays, particularly with respect to intracellular applicability, are certainly those that can be fully adapted to such “living color” reporter systems, because their interference with the organism can be minimized. Thus far, the development of an all-protein-based enzyme or protein-binding assay for possible use with dcFCCS or other kinds of single-molecule analysis in living cells has, however, been limited by the absence of suitable pairs of fluorescent proteins that fulfill the necessary criteria for the assay, i.e., largely different emission spectra but similar photostabilities. In this study, we provide proof of the principle that state-of-the-art dual-color FCCS technology combined with TPE can indeed be applied to genetically encoded fluorescent tags to perform kinetic enzyme analysis on the single molecule level. To achieve this goal, a combined FRET/crosscorrelation enzyme assay was created based on red-shifted GFP (rsGFP) (16) and DsRed (17) connected by a short protein linker of 32-aa length (see Fig. 1a), containing the target site of rTEV, a recombinant fragment from tobacco etch virus (TEV) protease (18). On protease cleavage, the separation of the fluorescent proteins from the reporter construct was monitored quantitatively by using dual-color crosscorrelation, which allows for the discrimination of different enzyme concentrations within the nanomolar range (see Fig. 1b). FRET efficiency of the substrate was simultaneously observed during the protease digest by recording the molecular photon yield per second, providing an alternative means to monitor the cleavage reaction. Although in theory a FRET efficiency of 100% would seriously hamper crosscorrelation analysis, we show that transfer efficiencies of between 30% and 60% can be tolerated if properly corrected for in the data analysis.
Fig 1.

Schematic explanations of the STEV-ST and the fluorescent protease assay. (a) Domain structure of STEV-ST. Composition and linear arrangement of STEV-ST are illustrated (not drawn to scale). The rsGFP and DsRed domains of STEV-ST are joined by a 32-aa protein linker containing the protease recognition sequence from the TEV protease. A C-terminal Strep-Tag II was added for protein purification. The vertical numbers indicate the amino acid position within the protein. (b) A schematic representation depicting the protease assay based on STEV-ST. Cleavage in the peptide linker region of the reporter construct by TEV protease terminates both FRET and crosscorrelation because of the separation of the fluorescent proteins.
These promising results, although obtained in a controlled in vitro system, clearly demonstrate that the future use of this or related reporter constructs in vivo is well conceivable. The crucial advantage of this assay, in contrast to previous FRET- or FCS-based single-molecule assays, is that it does not rely on tedious fluorescence labeling and cell-loading strategies.
Materials and Methods
Theoretical Concept.
A detailed discussion of the theory and implications of crosscorrelation analysis can be found elsewhere (9, 19). The normalized fluorescence correlation function is generally defined by:
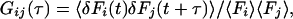 |
where Fi(j)(t) are fluorescence signals. For i = j, Eq. 1 defines the autocorrelation function for one molecular species in a single detection channel. If fluorescence fluctuations of different fluorescent species (i ≠ j) are simultaneously recorded in two emission channels and crosscorrelated, Gij or Gx defines the crosscorrelation function. Under ideal conditions with minimal spectral crosstalk between the dyes and in the absence of FRET, the crosscorrelation amplitude Gx(0) is directly proportional to the relative concentration of double-labeled species (i.e., the uncleaved substrate in our protease assay). The absolute concentration of double-labeled substrate can be derived from the corresponding autocorrelation amplitudes according to:
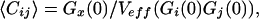 |
with Veff being the effective detection volume. In the absence of double-labeled species, Gx(0) equals zero.
Again under idealized conditions, the temporal decay of the crosscorrelation curves can be evaluated by a three-parameter model for a single diffusing species (19):
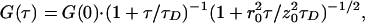 |
where τD = r /8D is defined as the average lateral diffusion time for a single molecule of double-labeled species in the case of two-photon excitation. Here, r0 and z0 are the characteristic lateral and axial dimensions of the effective focal volume element Veff = (π/2)3/2r
/8D is defined as the average lateral diffusion time for a single molecule of double-labeled species in the case of two-photon excitation. Here, r0 and z0 are the characteristic lateral and axial dimensions of the effective focal volume element Veff = (π/2)3/2r z0. With known Veff, the local concentration of fluorescent molecules 〈Ci〉 can thus be determined by consideration of autocorrelation amplitudes, according to 〈Ci〉 = G
z0. With known Veff, the local concentration of fluorescent molecules 〈Ci〉 can thus be determined by consideration of autocorrelation amplitudes, according to 〈Ci〉 = G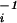 (0)⋅V
(0)⋅V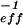 . The size of Veff, on the other hand, can be determined by calibration measurements with pure dye of a well-known diffusion coefficient.
. The size of Veff, on the other hand, can be determined by calibration measurements with pure dye of a well-known diffusion coefficient.
If dual-color analysis is combined with FRET between the distinct fluorophores, the evaluation of both auto- and crosscorrelation amplitudes must be modified. In this case, the molecular brightness η, measured in photons per molecule and second, must be taken into account. For a certain dye, η can be generally determined in standard FCS measurements by multiplication of the respective autocorrelation amplitude, being inversely proportional to the number of observed molecules, with the average fluorescence count rate (9). If ηi,F and ηj,F denote the brightness values in the presence of FRET (in our assay, of the uncleaved crosscorrelating substrate), and ηi and ηj describe the brightness values of the dyes unaffected by FRET (in our assay, of the cleaved fragments), the auto- and crosscorrelation amplitudes change to:
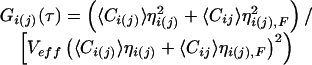 |
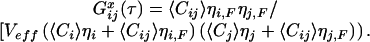 |
To rate the overall effect of FRET to the correlation amplitudes, we introduce the parameters fi = ηi,F/ηi and fj = ηj,F/ηj. The relative amplitudes in the presence and absence of FRET can then be written as:
Autocorrelation:
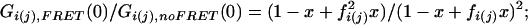 |
Crosscorrelation:
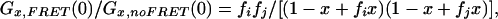 |
where x = Cij/C0 is the actual relative fraction of crosscorrelating (i.e., uncleaved) substrate with respect to the initial substrate concentration C0. The correction factor for a proper evaluation of the amplitudes is hence a dynamic one, depending on the relative concentrations and on the stage of the reaction.
It can be verified that for many fi/fj combinations, in particular if a dramatic quenching of the donor or enhancement of the acceptor occurs, the relationship between the two amplitudes Gx(0) with and without FRET is highly nonlinear. Consequently, if absolute concentrations of double-labeled species ought to be derived, proper correction requires experimental control of both fi and fj. The quantitative relationship between Gx,FRET(0) and the absolute concentration of the dual-color species Cij is given by:
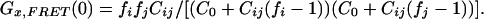 |
Materials.
Proteolytic assays were performed by using rTEV protease [29 kDa (18)], purchased from GIBCO/BRL with an activity of 10 units/μl (1 unit corresponds to 100 ng or 3.5 pmol of enzyme, as defined by the manufacturer). PCR amplifications were performed by using the Expand High Fidelity PCR System (Roche Applied Science, Mannheim, Germany). For cloning purposes, Escherichia coli XL1-blue was used; protein expression performed with E. coli BL21(DE3) (Stratagene). Strep-Tactin Sepharose and oligonucleotides were obtained from IBA (Göttingen, Germany). PCR purification and gel extraction kits were purchased from Qiagen (Hilden, Germany).
Plasmid Constructions.
A multistep cloning strategy was used to create a fusion protein of rsGFP and DsRed linked by the rTEV target site (from the native sequence of the TEV polyprotein) (amino acids 2781–2797, National Center for Biotechnology Information accession no. M11458). The coding sequence of rsGFP was amplified from pQBI63 (Q-BIOgene, San Diego) by PCR and cloned into the pDsRed plasmid (CLONTECH) upstream of the DsRed coding sequence. A DNA fragment encoding the TEV polyprotein linker region (amino acid sequence, QSTLEDPDIPTTENLYFQSGTVDADPRVPVAT) was inserted into the BamHI restriction site between rsGFP and DsRed. The generated construct was cloned into the bacterial expression vector pASK-IBA3 (IBA), 21 bp upstream of a C-terminal Strep-Tag II. The resulting gene (1,554 bp) was termed STEV-ST (S for the short 32-aa linker region and -ST referring to Strep-Tag II).
Protein Purification.
For protein expression, pSTEV-ST was transformed into E. coli BL21(DE3) by using electroporation. Liquid cultures were inoculated with 500 μl of an overnight culture, grown at 37°C to an A550 of 0.5, and induced with 0.2 μg/ml of anhydrotetracyclin (IBA). After 5 hr at 25°C, cells were harvested at 4,000 × g for 15 min and resuspended in 8 ml of buffer W (100 mM Tris⋅HCl, pH 8.0/150 mM NaCl/1 mM MgCl2). The cell pellet was lysed by using a French press and clarified by centrifugation at 23,420 × g for 45 min. The supernatant was applied to a Strep-Tactin Sepharose column (2 ml of bed volume; gravity flow; IBA) equilibrated in buffer W. The column was washed with 5 ml of buffer W, and the fusion protein was eluted with buffer W plus 2.5 mM desthiobiotin (IBA). Fractions (0.5 ml) containing both green and red fluorescence were pooled, concentrated by Vivaspin 50,000 molecular weight cutoff concentrators (Sartorius), and incubated at room temperature for 48 hr to allow for complete fluorophore maturation. The concentration of STEV-ST was determined by using ɛDsRed = 75,000/M⋅cm at 558 nm (19) and ɛrsGFP = 42,933/M⋅cm at 490 nm (CLONTECH) along with a BCA protein assay and SDS/PAGE.
The final product consists of 80% completely functional STEV-ST and its derivatives, resulting from partial degradation or incomplete fluorophore maturation, whereas a nonspecific impurity of approximately 20% was observed after SDS/PAGE. Above protein concentration measurements and FCS analysis (see below) showed a 50% excess of unpaired (only red) DsRed molecules and that 50% of the purified protein was functional STEV-ST. Because of the C-terminal affinity tag, no unpaired rsGFP protein fragments were purified.
Proteolytic Assay.
Protease reactions were carried out in 15-μl volumes in eight-well chambers (Nunc) under conditions specified by the manufacturer. Both the fluorescent substrate and protease were diluted to the appropriate concentrations immediately before analysis: 10 units of rTEV and 4.5 μM STEV-ST for spectral analysis and 0.4, 0.6, 1.3, and 2.5 units of rTEV (69, 118, 236, 437 nM rTEV) and 110 nM STEV-ST for FCCS measurements. Reactions were accomplished at room temperature and were initiated by the addition of rTEV. Fig. 1a illustrates the composition and arrangement of the various components of STEV-ST. The rTEV cleavage reaction produces a 29.0-kDa rsGFP and a 29.2-kDa DsRed fragment from the 58.2-kDa fusion protein.
Experimental Setup.
Two-photon crosscorrelation spectroscopy measurements were carried out on an inverted Olympus (Hamburg, Germany) IX-50 microscope (Fig. 2, ref. 11). Two-photon excitation was achieved by using an 80-MHz 100-fs pulsed mode-locked tunable titanium/sapphire laser (Spectra-Physics). The back aperture of the objective (UPlanApo 60×, 1.2 NA, Olympus) was slightly overfilled to create a diffraction-limited focal spot. The size of the effective volume element in this setup is ≈0.25 fl (11). The emitted light of around 550–700 nm from DsRed and 490–550 nm from rsGFP (single-emission spectra not shown) was collimated, split by a dichroic mirror (530 DCLP, AHF, Tübingen, Germany), and detected by two optical fiber-coupled avalanche photodiodes (SPCM-200, EG&G, Vaudreuil, PQ, Canada) after passing a D620/100 (AHF) bandpass filter in the DsRed and a HQ525/50M (AHF) bandpass filter in the rsGFP detection channel. For single-color calibration measurements, an emission filter 600DF200 (AHF) was sufficient for suppression of infrared excitation light. Confocal spectra were taken with a fiber-coupled spectrometer (Ocean Optics, Dunedin, FL) by using an entrance fiber slit of 100 μm. The digital pulses of the avalanche photodiodes were processed and time was correlated by an ALV-5000 multiple-τ correlator card (ALV, Langen, Germany).
Fig 2.

Optical setup as realized in an inverted Olympus IX-50 microscope. A single Titanium-Sapphire laser line with tuneable wavelength is used for illumination. The emitted light is split by a dichroic mirror behind the collimating lens and imaged onto two different optical fibers. Both fibers can be alternatively coupled into a spectrometer.
Results
Spectral Analysis and Calibration Measurements of the Assay Under TPE.
The selection of suitable dye combinations for two-photon crosscorrelation spectroscopy was based on quantum yields, two-photon absorption spectra, and photostability, because excitation intensity cannot be tuned independently for both dyes. To test the suitability of rsGFP and DsRed for simultaneous TPE, their emission signals were first recorded in a single-color setup with excitation at different wavelengths from 800 to 1,000 nm (Fig. 3a). Under optimal excitation conditions, close to saturation but still safely below photobleaching, emissions peaked at 940 nm (rsGFP) and 960 nm (DsRed) excitation. The maximum photon yields were 20 kHz/molecule for rsGFP and 12 kHz per molecule for DsRed. Because rsGFP was used as a FRET donor in the described protease assay, the excitation conditions were tuned in favor of this dye. Thus, the optimal wavelength for joint excitation of both fluorophores in the FRET/crosscorrelation measurements described below was 940 nm. A maximum intensity of 20 mW at this wavelength yielded sufficient excitation for both dyes.
Fig 3.

Characterizing the STEV-ST protease assay. (a) Wavelength dependence of the fluorescence emission yield η for rsGFP and DsRed with two-photon excitation. Photon yields per molecule were measured in a single-color setup (no chromatic beam splitting) under conditions close to saturation but well below photobleaching. Both fluorophores were efficiently excited by TPE with peak emissions at 940 nm for rsGFP (20 kHz per molecule) and 960 nm for DsRed (12 kHz per molecule). The optimal wavelength for joint excitation of both fluorophores was 940 nm with a maximum intensity of 20 mW. (b) Proteolytic changes in the STEV-ST emission spectrum during the digestion process. The emission spectrum of the fusion protein under TPE at 940 nm (black line) corresponds to ≈40% FRET. The spectrum continuously changes on initiation of the proteolytic digestion with addition of rTEV (1.7 μM) to the substrate STEV-ST (4.5 μM), as indicated by the gray lines. Measurements were taken for 10 s over a 5-min duration.
To analyze the spectral properties of the fusion protein substrate under actual measurement conditions for TPE at 940 nm, the emission spectrum was recorded during a proteolytic digestion of STEV-ST in the absence of all emission filters with the entire focal volume directly coupled to the entrance slit of a fiber-coupled spectrometer (Fig. 3b). The time course of this protease digest was monitored as an ensemble measurement in a 4.5-μM STEV-ST solution after addition of 10 units of rTEV/15 μl (1.7 μM rTEV) in single measurements of 10-s duration over 5 min. At all stages of the reaction, the spectrum exhibited two distinctive peaks around 510 and 580 nm, corresponding to rsGFP and DsRed emission maxima, respectively. Proteolytic cleavage of STEV-ST resulted in a shift of the emission spectrum and revealed FRET between rsGFP and DsRed in the system. In comparison to the spectrum of uncleaved substrate at the beginning of the reaction, rsGFP showed an almost 50% increase, whereas DsRed fluorescence decreased accordingly. Quantitatively analyzing the spectra of the fusion protein and dyes alone, the FRET efficiency for the intact substrate was estimated to be almost 40%. On the basis of the number of amino acids (32 aa), the length of the protein linker between the fluorophores was estimated to range between 58 Å for an α-helical conformation to approximately 100 Å for a relaxed conformation.
Real-Time Single-Molecule-Based FCS Analysis of Cleavage Reaction
Autocorrelation/FRET Analysis.
With the suitability of the rsGFP/DsRed dye combination for two-photon crosscorrelation spectroscopy and the biological functionality of the all-protein-based substrate demonstrated, the fluorescent protein-based protease assay was subject to FCS measurements on a single-molecule scale. In a reaction mix containing 236 nM rTEV and 110 nM STEV-ST, the protease digest was monitored over 10 min at a rate of two measurements per minute by recording continuous photon count rates corresponding to rsGFP and DsRed emission along with the corresponding autocorrelation functions for both detection channels. A large increase in the green and a decrease in the red count rate were observed, whereas the corresponding autocorrelation functions remained constant (Fig. 4). Because the average molecular brightness per molecule η at any given time is easily determined by multiplying the autocorrelation amplitude by the photon count rate, FCS allows for very sensitive monitoring of the emission properties in the presence and absence of FRET (20). As plotted in Fig. 4, the photon emission rate per single molecule increases for rsGFP by more than 50% from 4.6 kHz (ηGFP,FRET) at the beginning of the enzymatic reaction to 7 kHz (ηGFP,noFRET) at the end. This increase corresponds to approximately 35% of energy transfer in the intact substrate, which agrees reasonably well with the value of 40% determined from the emission spectra analysis at higher substrate and enzyme concentrations.
Fig 4.

FRET on a single molecule scale. Autocorrelation functions and photon counts per molecule in kHz (Inset) for rsGFP and DsRed were determined in parallel during an incubation of 110 nM STEV-ST with 197 nM rTEV. Changes in fluorescence intensity of the FRET donor (rsGFP) and acceptor (DsRed) were detected immediately after enzyme addition, whereas autocorrelation G(0) values and fluorescent particle numbers remained constant. The monitored increase of rsGFP fluorescence corresponds to approximately 35% FRET in the intact substrate.
The evaluation of ηDsRed was slightly more complicated because of a ≈50% excess of unpaired DsRed fragments in our sample. The presence of proteolyzed DsRed fragments was verified by the 2-fold lower autocorrelation amplitude in the red compared with the green channel. Therefore, the value ηDsRed,FRET is not equivalent to ηDsRed,0 at the beginning of the cleavage process. If n fluorescent species of different brightness values ηn are represented in a single FCS autocorrelation curve, the average brightness 〈η〉 is given by 〈η〉 = ∑n Cnη /∑n Cnηn, where Cn are the relative concentrations. Thus, in a mixture of FRET-active paired and FRET-inactive unpaired DsRed particles, with ηDsRed,noFRET = 6.7 kHz/molecule known from endpoint measurements, ηDsRed,FRET = 8.1 kHz/molecule was derived from an average count rate of ηDsRed,0 = 7.5 kHz/molecule at time 0. That the decrease of the DsRed fluorescence after termination of FRET is only 20%, compared with the 35% increase for GFP, can be explained by direct acceptor excitation and the complex photophysical nature of DsRed (21), suggesting that energy is dissipated by transfer from GFP to a transiently nonfluorescent state of the acceptor.
/∑n Cnηn, where Cn are the relative concentrations. Thus, in a mixture of FRET-active paired and FRET-inactive unpaired DsRed particles, with ηDsRed,noFRET = 6.7 kHz/molecule known from endpoint measurements, ηDsRed,FRET = 8.1 kHz/molecule was derived from an average count rate of ηDsRed,0 = 7.5 kHz/molecule at time 0. That the decrease of the DsRed fluorescence after termination of FRET is only 20%, compared with the 35% increase for GFP, can be explained by direct acceptor excitation and the complex photophysical nature of DsRed (21), suggesting that energy is dissipated by transfer from GFP to a transiently nonfluorescent state of the acceptor.
A remarkable consistency of the autocorrelation functions' amplitudes throughout the proteolytic cleavage shows that there was essentially no loss of fluorescent molecules because of adsorption or photodegradation. One of the most important implications of this constancy is the exclusion of significant oligomerization of DsRed in the fusion protein. If the DsRed–peptide–GFP substrate contained DsRed as a dimer or tetramer, as presumed in previous studies (22), proteolytic cleavage would most likely increase the number of released GFP particles. The stability of the GFP autocorrelation amplitude thus indicates a complete absence of multimers among the observed (fluorescent) substrate molecules.
Crosscorrelation Analysis in the Presence of FRET.
Cleavage of STEV-ST results in dissociation of the two fluorophores (Fig. 1b) and, consequently, in a decay of the crosscorrelation amplitude. Fig. 5 shows the time course of crosscorrelation curves recorded over 380 s from a sample containing 110 nM STEV-ST and 437 nM rTEV. Green and red fluorescence were measured continuously, and crosscorrelation analysis was carried out in intervals of 40 s (low enzyme concentrations) and 20 s (high enzyme concentrations).
Fig 5.

Crosscorrelation curves measured during a proteolytic cleavage reaction. 110 nM STEV-ST was incubated with 437 nM rTEV at room temperature. The curves were averaged from two successive 20 s measurements each and fitted with Eq. 3. During the course of the reaction, the amplitudes of G(0) gradually decreased whereas the corresponding diffusion times remained constant, assuring the identity of the substrate.
The decay of crosscorrelation amplitudes Gx(0) closely resembles the decrease of the intact substrate concentration Cij. However, in the presence of FRET, there exists no simple linear relationship between Gx(0) and Cij. Rather, the determination of absolute substrate concentrations requires both the autocorrelation amplitudes and fluorescence count rates (i.e., brightness parameters η as indicated in Theory). Eq. 6 describes the magnitude of this effect, which depends on the parameters fi = ηi,F/ηi and fj = ηj,F/ηj to compare the emission properties of both fluorophores in the presence (F) and absence of FRET. From the values determined above, ηGFP,FRET = 4.6 kHz, ηGFP,noFRET = 7 kHz, ηDsRed,FRET = 8.1 kHz, and ηDsRed,noFRET = 6.7 kHz, we assessed frsGFP = 0.66 and fDsRed = 1.2 for our system.
Fig. 6 visualizes the general effect of different theoretical cases of FRET (expressed in terms of fDsRed and fGFP) to the relative crosscorrelation amplitudes Gx,FRET(0)/Gx,noFRET(0). The experimental conditions for the GFP/DsRed assay described here are highlighted. It can be seen that the existence of FRET in our case leads to an underestimation of relative substrate fraction that gets the more severe the less substrate is left. For all fi/fj combinations below the 1/1 line, the presence of FRET generally emphasizes changes in the regime of high relative concentrations of double-labeled species. For combinations above the 1/1 line, relative changes in the low concentration regime of crosscorrelating species are more pronounced. For the autocorrelation amplitudes, a slight effect induced by FRET can also be observed, as indicated by the inset of Fig. 6. However, for the combination frsGFP = 0.66 and fDsRed = 1.2 as derived for our assay, no significant changes (less than 5% for rsGFP, less than 1% for DsRed) can be expected. The effect is too small to be visible beyond the noise on the correlation functions in Fig. 4, which explains their apparent constancy.
Fig 6.

Simulation of relative amplitudes of Gx,FRET(0)/Gx,NoFRET(0) for different concentrations x of double-labeled FRET-substrate during a proteolytic digest. At the beginning of the reaction, x = 1. The relative crosscorrelation amplitude, determining the effect of FRET on FCS data analysis, changes in the time course of a digest and depends on the individual ratios f = ηintact/ηcleaved for both fluorophores, respectively. From our FRET measurements, we derived fGFP = 0.66 and fDsRed = 1.2 for STEV-ST (▪). (Inset) Effect of FRET on autocorrelation amplitudes, assuming the same fi.
For a quantitative determination of enzymatic cleavage, STEV-ST cleavage reactions were carried out at 69, 118, 236, and 437 nM rTEV concentrations and 110 nM STEV-ST, 3 orders of magnitude below the KM = 69 μM proposed by Parks et al. (18). Gx (0) values were obtained at a rate of 2–3 measurements per min over a 6- to 10-min reaction. Absolute substrate concentrations at given times (Fig. 7) were determined according to Eqs. 2–6. Because of the complete overlap of detection volumes and minimal spectral crosstalk of less than 10% (data not shown), the experimental error was determined solely by the uncertainty of specific labeling. This was estimated to be less than 5% based on the GFP-labeled, FRET-active substrate molecules.
Fig 7.

Kinetics of the proteolytic cleavage reaction at different enzyme concentrations. Proteolytic reactions were carried out at room temperature by using 110 nM STEV-ST. Enzyme concentrations ranging from 69 to 473 nM were easily discriminable, particularly from their initial reaction rates (see Inset). Initial reaction rates were derived from data points in the first 60–180 s of the reaction by linear regression after FRET correction of the amplitudes as described.
Enzyme concentrations differing by less than a factor of 2 could be easily distinguished over the time course of the reaction and more quantitatively from the initial reaction rates (Fig. 7 Inset). No decrease of the crosscorrelation signal was detected for a negative control (without enzyme) during a 10-min measurement. As seen in Fig. 5, the amplitude of the crosscorrelation function did not decrease below ≈20% of its initial value even for longer reaction times, indicating that a residual amount of crosscorrelating substrate was not affected by the cleavage process. Because the protease target site is flanked by two rather large globular domains, the cleavage site might not have been fully accessible in this residual fraction.
Summary and Conclusion
A protease assay suitable for dual-color two-photon fluorescence crosscorrelation spectroscopy on a single-molecule scale was developed solely on the basis of genetically encoded tags. This specifically designed fusion protein substrate, STEV-ST, consists of rsGFP-peptide-DsRed with a cleavage site for TEV protease within the peptide region and shows a decay of crosscorrelation as well as FRET between the fluorophores during the enzymatic cleavage while being monitored in real time. There exists a tremendous need for alternative and universally applicable systems for measuring intracellular molecular interactions and determining reaction kinetics of proteins and enzymes, particularly with regard to the elucidation of protein function and high-throughput screening applications.
The combination of TPE, FCS/FCCS, and FRET coupled with an all-protein fluorescent substrate assay for monitoring proteolytic degradation results in an extremely sensitive and selective real-time method for following dynamic processes under physiological conditions Furthermore, this type of assay is amenable to adaptation for cellular applications. The limits imposed by alternative techniques, such as standard FRET, with regard to the distances between the fluorophores, can be overcome by the use of dual-color crosscorrelation, which takes into account the dynamic concomitant movement of dye molecules independent of their size and proximity. In comparison to previous work using two-photon FCCS measurements with synthetic dyes (11), the combination of the fluorescent proteins, DsRed and rsGFP, appears to be well suited for simultaneous excitation with TPE. DsRed, despite its slow maturation and tendency to aggregate as a native protein, worked well within the fusion construct being linked to protein fragments at both its N- and C-terminal end. In conclusion, this study provides proof of principle for the extension of two-photon crosscorrelation spectroscopy applications to protein-based reporter molecules with a bright outlook of applying this technology to diagnostics, screening applications, and cell biological research.
Acknowledgments
We thank Michael Jahnz, Elke Haustein, and Petra Dittrich for helpful discussions; Karin Birkenfeld for assistance with sample preparation; and Sally Kim for critical proofreading. Financial support by the German Ministry for Education and Research (Biofuture Grants 0311845 and 16SV1257) and Evotec BioSystems AG (Hamburg, Germany) is gratefully acknowledged. Structural data for Fig. 1b were obtained from the Research Collaboratory for Structural Bioinformatics (www.rcsb.org/pdb/; IDs: 1G7K; 1EMF) and were visualized with rasmol (www.umass.edu/microbio/rasmol/).
Abbreviations
rsGFP, red-shifted GFP
TPE, two-photon excitation
FRET, fluorescence resonance energy transfer
dcFCCS, dual-color fluorescence crosscorrelation spectroscopy
dsDNA, double-stranded DNA
FCS, fluorescence correlation spectroscopy
TEV, tobacco etch virus
References
- 1.Eigen M. & Rigler, R. (1994) Proc. Natl. Acad. Sci. USA 91, 5740-5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weiss S. (1999) Science 283, 1676-1683. [DOI] [PubMed] [Google Scholar]
- 3.Schwille P. & Kettling, U. (2001) Curr. Opin. Biotechnol. 12, 382-386. [DOI] [PubMed] [Google Scholar]
- 4.Magde D., Elson, E. L. & Webb, W. W. (1972) Phys. Rev. Lett. 29, 705-708. [Google Scholar]
- 5.Schwille P. (2001) Cell. Biochem. Biophys. 34, 383-408. [DOI] [PubMed] [Google Scholar]
- 6.Miyawaki A., Llopis, J., Heim, R., Mccaffery, J. M., Adams, J. A., Ikura, M. & Tsien, R. Y. (1997) Nature (London) 388, 882-887. [DOI] [PubMed] [Google Scholar]
- 7.Tawa P., Tam, J., Cassady, R., Nicholson, D. W. & Xanthoudakis, S. (2001) Cell Death Differ. 8, 30-37. [DOI] [PubMed] [Google Scholar]
- 8.Selvin P. R. (2000) Nat. Struct. Biol. 7, 730-734. [DOI] [PubMed] [Google Scholar]
- 9.Schwille P. (2000) in Fluorescence Correlation Spectroscopy—Theory and Applications, eds. Rigler, R. & Elson, E. (Springer, Heidelberg), pp. 360–378.
- 10.Kettling U., Koltermann, A., Schwille, P. & Eigen, M. (1998) Proc. Natl. Acad. Sci. USA 95, 1416-1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heinze K. G., Koltermann, A. & Schwille, P. (2000) Proc. Natl. Acad. Sci. USA 97, 10377-10382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koltermann A., Kettling, U., Bieschke, J., Winkler, T. & Eigen, M. (1998) Proc. Natl. Acad. Sci. USA 95, 1421-1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bacia K., Majoul, I. V. & Schwille, P. (2002) Biophys. J. 83, 1184-1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Denk W., Strickler, J. H. & Webb, W. W. (1990) Science 248, 73-76. [DOI] [PubMed] [Google Scholar]
- 15.Schwille P., Haupts, U., Maiti, S. & Webb, W. W. (1999) Biophys. J. 77, 2251-2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Delagrave S., Hawtin, R. E., Silva, C. M., Yang, M. M. & Youvan, D. C. (1995) BioTechnology 13, 151-154. [DOI] [PubMed] [Google Scholar]
- 17.Matz M. V., Fradkov, A. F., Labas, Y. A., Savitsky, A. P., Zaraisky, A. G., Markelov, M. L. & Lukyanov, S. A. (1999) Nat. Biotechnol. 17, 969-973. [DOI] [PubMed] [Google Scholar]
- 18.Parks T. D., Howard, E. D., Wolpert, T. J., Arp, D. J. & Dougherty, W. G. (1995) Virology 210, 194-201. [DOI] [PubMed] [Google Scholar]
- 19.Schwille P., Meyer-Almes, F. J. & Rigler, R. (1997) Biophys. J. 72, 1878-1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Widengren J., Schweinberger, E., Berger, S. & Seidel, C. A. M. (2001) J. Phys. Chem. 105, 6851-6866. [Google Scholar]
- 21.Malvezzi-Campeggi F., Jahnz, M., Heinze, K. G., Dittrich, P. & Schwille, P. (2001) Biophys. J. 81, 1776-1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baird G. S., Zacharias, D. A. & Tsien, R. Y. (2000) Proc. Natl. Acad. Sci. USA 97, 11984-11989. [DOI] [PMC free article] [PubMed] [Google Scholar]