

Abstract
Single molecule fluorescence resonance energy transfer (FRET) and fluorescence correlation spectroscopy were used to investigate DNA looping by NgoMIV restriction endonuclease. Using a linear double-stranded DNA (dsDNA) molecule labeled with a fluorescence donor molecule, Cy3, and fluorescence acceptor molecule, Cy5, and by varying the concentration of NgoMIV endonuclease from 0 to 3 × 10−6 M, it was possible to detect and determine diffusion properties of looped DNA/protein complexes. FRET efficiency distributions revealed a subpopulation of complexes with an energy transfer efficiency of 30%, which appeared upon addition of enzyme in the picomolar to nanomolar concentration range (using 10−11 M dsDNA). The concentration dependence, fluorescence burst size analysis, and fluorescence correlation analysis were all consistent with this subpopulation arising from a sequence specific interaction between an individual enzyme and a DNA molecule. A 30% FRET efficiency corresponds to a distance of ∼65 Å, which correlates well with the distance between the ends of the dsDNA molecule when bound to NgoMIV according to the crystal structure of this complex. Formation of the looped complexes was also evident in measurements of the diffusion times of freely diffusing DNA molecules with and without NgoMIV. At very high protein concentrations compared to the DNA concentration, FRET and fluorescence correlation spectroscopy results revealed the formation of larger DNA/protein complexes.
INTRODUCTION
A number of proteins that participate in gene expression, DNA replication, DNA modification, and recombination perform their functions by forming DNA loops (Schleif, 1992). For example, it is well established that the gal operon repression in Escherichia coli (E. coli) is mediated by GalR repressor-induced formation of DNA loops. Other examples in E. coli include DNA looping-mediated regulation of lac, ara, or deo operons (Schleif, 1992; Voet and Voet, 1995).
Some of the type II restriction endonucleases, such as SfiI, Cfr10I, and NgoMIV, also function by making DNA loops. These enzymes bind to two recognition sites on a double-stranded DNA (dsDNA) molecule and cleave it (Deibert et al., 2000; Embleton et al., 2001; Halford et al., 2000; Nobbs and Halford, 1995; Siksnys et al., 1999; Watson et al., 2000). Cleavage occurs much more rapidly when these enzymes simultaneously bind at two restriction sites instead of one; for example SfiI activity increases up to 40 times when the DNA substrate has two recognition sequences (Wentzell et al., 1995). The binding and cleaving reaction also depends on the distance between the sites. If the restriction sites are very close (less then 30 basepairs (bp) from one another), the enzyme is unable to generate a loop in the DNA molecule due to the rigidity of the short DNA strand and this inhibits the cleavage reaction. However, if the sites are too far away, cleavage is also inhibited or prevented (Bickle and Kruger, 1993; Siksnys et al., 1999). The formation of dsDNA loops by SfiI and Cfr10I restriction endonucleases has been visualized by electron microscopy (Friedhoff et al., 2001; Siksnys et al., 1999). The DNA looping kinetics of SfiI and Cfr10I endonucleases were investigated using inhibition of the Tn21 resolvase reaction (Milsom et al., 2001; Oram et al., 1997). This assay is based on two competing reactions: DNA looping by restriction enzyme and recombination by Tn21 resolvase. Tn21 resolvase can complete its reaction only when the restriction enzyme-DNA complex is dissociated. It was determined that for Cfr10I the looped complexes existed as long as 90 s, whereas with the SfiI enzyme, DNA looped complexes were stable for more than 7 h (Milsom et al., 2001).
Another type II restriction enzyme that forms DNA loops is NgoMIV. The crystal structure of NgoMIV bound to two short dsDNA fragments was solved to 1.6 Å resolution (Deibert et al., 2000). This protein consists of four identical subunits of ∼32 kDa each. It simultaneously binds two copies of the DNA recognition sequence 5′-GCCGGC-3′ and cleaves after the 5′ G leaving a four base overhang. The protein has dimensions of 60 × 70 × 80 Å, whereas the helices of DNA molecules are separated by ∼55 Å from each other and have a 60° angle between their helical axes (see Fig. 1). This enzyme apparently binds to two recognition sites on the double-stranded DNA, forming a DNA loop, and then cleaves the DNA at the two recognition sites (Deibert et al., 2000; Milsom et al., 2001). However, the DNA looping kinetics by NgoMIV endonuclease were too fast to be resolved in bulk measurements using Tn21 resolvase inhibition (Milsom et al., 2001).
FIGURE 1.

Structure of NgoMIV with two bound fragments of DNA molecules: (a) side view and (b) top view. α-helices of the protein are shown in light green, β-strands in yellow, other regions in dark green, DNA molecules are shown in purple. The figure was created using PDB entry 1FIU (Deibert et al., 2000).
One of the fundamental problems in investigating DNA looping and especially its dynamics is the stochastic nature of this process. It is practically impossible to synchronize this kind of process between large numbers of molecules, and thus the dynamic behavior remains largely uninvestigated. However, single molecule techniques provide a possible avenue for the study of dynamic process like DNA looping. Single molecule measurements can be used to resolve the time trajectories of processes that are inherently not correlated between different molecules, as well as to characterize the dynamic populations of intermediates during the course of a reaction (Weiss, 1999, 2000). One powerful technique that can be applied at the single molecule level is fluorescence resonance energy transfer (FRET). This allows one to look at single molecule dynamics in terms of millisecond time scale variations in the distances between fluorophores bound to biomolecules (Deniz et al., 1999; Dietrich et al., 2002; Ha et al., 1999a,b; Kim et al., 2002; Norman et al., 2000; Widengren et al., 2001; Ying et al., 2000; Zhuang et al., 2000).
Here, single molecule fluorescence resonance energy transfer methodologies are applied to study the DNA/protein looped complexes as a function of the concentration of NgoMIV restriction endonuclease. FRET occurring between the donor (Cy3) and acceptor (Cy5) molecules, which are attached to the ends of a DNA fragment with appropriate binding sites, allows the measurement of the distance between the DNA ends. In this study, relative populations of different DNA/protein geometries are measured by observing NgoMIV/DNA complexes at equilibrium in solution. This work sets the stage for future studies of the dynamics of this process using immobilized DNA molecules. Such measurements should lend insight into the conformational motion of the DNA molecule caused by its interaction with NgoMIV endonuclease.
MATERIALS AND METHODS
Enzyme and DNA substrate preparation
NgoMIV was purchased from Promega (Madison, WI) and used without further purification. The approximate enzyme concentration was determined spectroscopically as previously described (Pace and Schmid, 1997). A 172-bp dsDNA molecule containing two NgoMIV recognition sequences was generated using polymerase chain reaction (PCR) techniques as follows. Two NgoMIV restriction sites at the positions 769 and 929 of the plasmid pBR322 were used for the design of the dsDNA molecule. A forward primer 5′-AGGTGCCGGCAGGCTCT-3′ was labeled with a Cy3 dye molecule at the 5′ end phosphate group through a C3 linker (NgoMIV binding site is underlined) and a reverse primer 5′-CCATGCCGGCGATAATGG-3′ was labeled with a Cy5 dye molecule at the 5′ end. The pBR322 region between the primers, from position 765 to 937, was amplified using PCR. A dsDNA molecule without endonuclease restriction sites was also made, as control, for testing the specific binding of NgoMIV enzyme to DNA. To generate that DNA fragment, a forward primer 5′-AGGTACGGACAGGCTCT-3′ with Cy3 at the 5′ end and a reverse primer 5′-CCATACGGACGATAATGG-3′ with Cy5 at the 5′ end were used in the PCR reaction, again to amplify the pBR322 plasmid's region between the positions 765 and 937, but this time with three changes in each of the restriction sites. All primers were purchased from Sigma-Genosys (Woodland, Texas). Primers similar to the NgoMIV specific primers (5′-TAGGACAGGTGCCGGC-3′ forward and 5′-CGGCCGCCATGCCGGC-3′ reverse) without the dyes were used to synthesize a 184-bp dsDNA molecule to verify that NgoMIV cuts a linear DNA molecule with 160-bp distance between the restriction sites (this 184-bp dsDNA molecule was the same as the 172-bp dsDNA molecule used for the FRET measurement except the ends were extended to resolve the cut from the uncut DNA on a gel). The reaction between NgoMIV and the dsDNA substrate was performed according to the manufacturer's instructions and the resulting fragments were separated on 3% agarose gel (data not shown).
The single molecule measurements were performed in a 10-μl sample volume placed on a 0.13-mm thick glass cover slip (VWR, West Chester, PA) by detecting fluorescence from freely diffusing molecules or complexes. For these measurements, the DNA generated from the PCR reaction was diluted to 10−11 M final concentration in sterilized ultrapure water. NgoMIV restriction endonuclease concentration in the sample was varied from 0 to 3 × 10−6 M first by diluting the enzyme stock solution in the storage buffer (10 mM Tris-HCl (pH 7.3), 30 mM NaCl, 0.1 mM EDTA, 1 mM DTT, 0.5 mg/ml BSA) and then diluting 10 times by adding it to the DNA sample. These sample preparation conditions were chosen to maintain the enzyme under conditions in which no cleavage could occur (there was no magnesium in the enzyme storage buffer), while both maintaining appropriate conditions for binding and minimizing sources of fluorescent contaminants. Use of the Multi-Core buffer containing magnesium (Promega) instead, which is recommended for the optimal enzyme activity, resulted in very fast cleavage of DNA molecules even at room temperature (see below). Data were also obtained using Multi-Core buffer in which magnesium ions were chelated by EDTA. However, the addition of EDTA to the buffer resulted in increase of the background fluorescence. The ultrapure water used for the samples was deoxygenated using argon gas before each measurement to improve the photostability of the Cy3 and Cy5 molecules. A 15-bp dsDNA molecule labeled with Cy3 and Cy5 dye molecules at the 5′ ends of the complimentary strands was created as a control for calibration of single molecule FRET between Cy3 and Cy5 dyes. In this 15-bp dsDNA molecule, the donor and acceptor FRET pair are expected to be 51 Å apart. This construct should show an energy transfer efficiency of 55% assuming an R0 (distance at which energy transfer is 50%) of 53 Å, as reported previously for this dye pair (Ishii et al., 1999). For these control measurements using the 15-bp dsDNA, 50% (v/v) glycerol was added to the samples to slow the diffusion of the DNA molecules in solution.
Apparatus for single molecule detection
Excitation light at 514.5 nm from a CW argon ion laser (Lexel Lasers, Fremont, CA) was used to excite the Cy3 molecules. Single molecule fluorescence was detected using an inverted Nikon microscope (Nikon TE200) with a high numerical aperture Nikon objective (Nikon 100×, 1.4 NA, oil immersion). The fluorescence light collected by the same objective was transmitted through a dichroic mirror (part No. 525DRLP, Omega Optical, Brattleboro, VT) and focused onto a 50-μm diameter pinhole. The fluorescence was then passed through a dichroic mirror (630DRLP, Omega Optical, Brattleboro, VT) to split it into two different wavelength detection channels. Interference band pass filters centered at 570 nm and at 670 nm with 40 nm spectral bandwidths were used to spectrally select the fluorescence from Cy3 and Cy5 molecules, respectively (570 DF 40 and 670 DF 40 from Omega Optical, Brattleboro, VT). Fluorescence in each channel was detected using an avalanche photodiode (SPCM-AQ 151, EG&G, Vaudreuil, Canada). Data collection was performed with two multichannel scaler cards (MCS-plus, EG&G Ortec, Canada), one for each wavelength channel. Fluorescence from both channels was collected at the same time with an integration time of either 0.2 or 1 ms.
Fluorescence correlation spectroscopy (FCS) was used to investigate single molecule diffusion times. For this purpose, a Digital Correlator card (Correlator.com, Bridgewater, NJ) was used. Cross-correlation and autocorrelation curves from both detection channels were measured with either 30 or 60 s integration time. The errors for the diffusion time were determined by measuring and fitting the results five times.
RESULTS AND DISCUSSION
Predicting distances using single molecule FRET
Fluorescence time traces of a 15-bp DNA molecule labeled with the Cy3 on one end and Cy5 on the other were measured using a signal binning time of 1 ms. Cross-correlation of fluorescence bursts in the Cy3 and Cy5 channels was observed (raw data not shown). Fluorescence resonance energy transfer efficiency was calculated for each bin according to the equation,
 |
Here IA stands for fluorescence intensity from the acceptor channel, ID is the intensity of the donor channel, and γ corrects for the bleedthrough of Cy3 fluorescence into the Cy5 detection channel (Ha et al., 1999a). The bleedthrough parameter was calculated as 0.15 from measurements of a DNA sample with only the Cy3 fluorophore attached. There was essentially no cross talk in the other direction (Cy5 fluorescence in the Cy3 channel) and thus no correction for this was necessary. The 0.15 value of γ was used in all FRET efficiency calculations. For these calculations, fluorescence intensity per bin was required to have a minimum of 30 counts after summing the fluorescence from both the Cy3 and the Cy5 channels, or it was not counted. It should also be noted that in the FRET efficiency calculations, there was no background subtraction performed from either channel, as the average background was less than 1 count in each 1-ms bin. The FRET efficiency calculations also depend on any difference between the detection efficiencies of the two detector channels (Dahan et al., 1999; Ha et al., 1999a). Based on the control results using the 15-bp DNA molecule labeled with either Cy3, Cy5, or both dyes, the detection efficiencies in two channels was estimated to be ∼1. This is consistent with assumptions made in previous studies using the same or similar dyes and similar detection techniques (Dahan et al., 1999; Deniz et al., 1999; Ha et al., 1999a).
Fig. 2 shows the distribution of FRET efficiencies for single molecule measurements using the 15-bp DNA molecule labeled on one end with Cy3 and the other end with Cy5. A primary peak with a maximum around 0.5 and small peak with a maximal efficiency of 0 is observed. The R0 for the Cy3/Cy5 pair has been determined previously to be 53 Å (Ishii et al., 1999). Thus, using a value of 2/3 for the orientation factor κ2, the 50% efficiency peak should correspond to a distance between the dye molecules of about R0 or 50–55 Å. The distance between the donor and acceptor based on the DNA structure results in about a 51 Å separation of the dye molecules, in excellent agreement with the FRET results. The peak at 0% results from the fact that sometimes only Cy3 fluorescence is detected. This could not represent a situation in which the donor and acceptor were far apart because the distance between the two fluorophores is necessarily limited by the DNA structure. Instead, it is likely that there is occasionally photochemical bleaching of the Cy5 acceptor in some molecules, as previously observed in various single molecule FRET measurements (Deniz et al., 1999; Ying et al., 2000). In any case, the values estimated for FRET efficiency from the main distribution correspond well to what would be expected from the structure, supporting the assumptions regarding the orientation factor, κ2, and the relative detection efficiencies of the two detection channels. The same calculations and assumptions were used for distance determination in the DNA/endonuclease system as well (described below).
FIGURE 2.

FRET efficiency distribution of 15-bp double-stranded DNA molecule labeled with a Cy3 dye molecule at one end and a Cy5 dye molecule at the other (see Methods).
At least two distinct NgoMIV/DNA complexes are formed at different enzyme concentrations
Single molecule fluorescence time traces of 172-bp DNA molecules labeled with Cy3 and Cy5 (at 10−11 M concentration; see Methods for details of DNA design and generation) were recorded at various NgoMIV concentrations from 0 to 3 × 10−6 M. DNA cleavage is prevented by the absence of magnesium ions in the solution (see Methods). The same measurements were performed on two different DNA molecules, one of which contained two NgoMIV restriction sites separated by 160 bp and one that did not contain restriction sites. In both cases, fluorescence time traces were recorded with both 0.2 and 1 ms binning times, and FRET efficiency distributions were calculated as described above (see Fig. 3). Fig. 3 a shows the case in which the enzyme NgoMIV is absent in the system. In this case, no fluorescence energy transfer is detected. Fluorescence is almost exclusively observed from the Cy3 donor resulting in an efficiency peak near 0% (so called “zero peak”). When 10−12 M NgoMIV endonuclease is present in the sample with the DNA molecules containing restriction sites, two populations in the FRET efficiency distribution can be resolved. The first population corresponds to a zero peak, which is present due to DNA molecules with no NgoMIV bound or when the ends of the DNA strand are still separated by a large distance (there could also be a small population in which the FRET acceptor is photobleached as described for the double-labeled DNA control above). The second population ranges from 20% to 40% efficiency with the maximum at around 30% (see Fig. 3 b). This second peak is due to the energy transfer from the donor Cy3 to the acceptor Cy5 molecule as a result of the NgoMIV binding to both recognition sites on the DNA (for distance calculations and comparison to the structure, see below).
FIGURE 3.

FRET efficiency histograms of a 10−11 M Cy3 and Cy5 end-labeled dsDNA fragment containing two NgoMIV restriction endonuclease sites 160 bp apart (see Methods) at four different NgoMIV concentrations: (a) 0 M NgoMIV, (b) 10−12 M NgoMIV, (c) 10−7 M NgoMIV, (d) 3 × 10−6 M NgoMIV.
It is more difficult to interpret the relative size of the peaks in the FRET efficiency distribution. At low concentration of enzyme (10−12 M), it is very likely that one NgoMIV enzyme binds to only one DNA molecule and most of the DNA should be detected in the unbound (zero FRET efficiency) form. This is because the DNA molecules outnumber the enzyme molecules by a factor of 10. However, Fig. 3 b shows similar amounts of the zero and 30% peaks. Taken alone, this would imply that ∼50% of the DNA molecules have the bound enzyme, rather than the maximum of 10% predicted by the stoichiometry. This discrepancy arises because of the way in which fluorescence bins were selected. As mentioned above, for FRET efficiency calculations only the bins with a total Cy3 plus Cy5 intensity of more that 30 counts were used. This results in the preferential selection of the FRET events over the free DNA detection events, as many of the Cy3 only bursts (corresponding to free DNA) are less than 30 counts (see burst size distributions, Fig. 5). The problems associated with the effect of burst thresholds on the ability to quantitatively compare the amounts different subpopulations in a heterogeneous mixture have been discussed previously (Dahan et al., 1999). Thus, these measurements demonstrate that there are two distinct species present in the DNA/NgoMIV system, but it is not possible to compare directly the concentrations of the different species in the sample using these methods.
FIGURE 5.

Burst size distributions calculated from the data measured at 10−12 M (▵) and 10−7 M (○) NgoMIV concentration. See text for details.
Essentially the same FRET efficiency distributions are observed at all NgoMIV concentrations from 10−12 to 10−8 M (data not shown). Fitting the FRET efficiency distributions obtained for this range of enzyme concentrations several times (using data taken on different days) and assuming a Gaussian distribution of efficiencies, the center value obtained is 28 ± 2%. The error value of 2% reflects both the variance of the center value with the change in the enzyme concentration as well as the reproducibility of the results during various days of measurements. The width of the distribution, of course, is considerably larger than 2% of the mean value, because individual measurements involve limited numbers of photons. The center value of the distribution, however, is quite reproducible. At an even higher concentration of NgoMIV, 10−7 M, a population with ∼10%–15% energy transfer efficiency can be resolved (see Fig. 3 c). This may correspond to NgoMIV/DNA complexes that are geometrically distinct from those formed at lower concentrations or could be a consequence of aggregation (discussed in more detail in terms of the structure below). At NgoMIV concentrations of 10−6 to 3 × 10−6 M the 10% efficiency population becomes dominant (Fig. 3 d).
To insure that the enzyme used for these measurements was in the native, active form, cleavage of the DNA molecules by NgoMIV was also assessed. Addition of the enzyme to a DNA sample containing the reaction buffer (Multi-Core from Promega) resulted in fast disappearance (a few seconds after the addition of enzyme) of the FRET fluorescence signal and the cross-correlation between Cy3 and Cy5 detection channels. The remaining fluorescence bursts were due to the fast diffusion of the short pieces of DNA labeled with Cy3.
The NgoMIV complexes detected are the result of sequence specific interactions
When the same enzyme concentrations were added to DNA molecules that were identical to the ones described above except for several base substitutions within the restriction sites (see Methods for a detailed description), only the zero peak population in the FRET efficiency distributions was observed at any enzyme concentration (data not shown). This indicates that the appearance of the other geometrically distinct populations of complexes in the measurements (centered at 30% and 10% efficiency) is due to specific interactions between NgoMIV and DNA.
The different DNA/NgoMIV complexes detected have different diffusion times
To investigate the specific binding of NgoMIV to DNA in more detail, fluorescence correlation spectroscopy was used to measure the diffusion time (τd) of the DNA molecules or DNA/protein complexes in solution (Kettling et al., 1998; Rippe, 2000; Zander et al., 1996). For these measurements, again 10−11 M DNA with or without restriction sites was investigated, and the NgoMIV concentration was also varied from 0 to 3 × 10−6 M. The FCS data was fitted to find the diffusion time τd of the molecules diffusing through the beam using the equation:
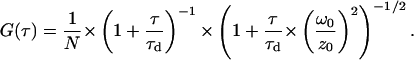 |
N is the average number of molecules in the probe volume, τ is time, τd is diffusion time, ω0/z0 is the ratio of the diameter over the z-dimension (thickness) of the confocal volume (Brand et al., 1997; Hess et al., 2002; Rippe, 2000). The value for ω0/z0 was fixed as previously determined (Daniel et al., 2002).
Results from both autocorrelation (correlation within one channel) and cross-correlation (correlation between the two wavelength channels) measurements (Eigen and Rigler, 1994) were analyzed using the same equation. The fits of the diffusion times from autocorrelation and cross-correlation data were essentially the same. Below, only the results of cross-correlation data will be presented, as they directly represent the properties of the looped DNA/protein complexes. The Cy3 and Cy5 cross-correlation results obtained at three different concentrations of NgoMIV are shown in Fig. 4. At 0 M enzyme concentration, fitting of the cross-correlation from the free DNA molecules resulted in a diffusion time of 0.29 ± 0.02 ms. It should be noted, here, that the small cross-correlation signal observed in this case represents the 15% bleedthrough of Cy3 fluorescence to the Cy5 detection channel and is essentially equivalent to an autocorrelation trace of the Cy3 channel only. Similar diffusion time values previously obtained from autocorrelation measurements of 59-bp dsDNA labeled with FAM or ROX dyes were ∼0.32 and 0.37 ms, respectively (Rippe, 2000).
FIGURE 4.

Fluorescence cross-correlation results and fits at different concentrations of NgoMIV endonuclease. Insert: normalized fits at different concentrations of NgoMIV. The solid line is at an NgoMIV concentration of 3 × 10−6 M, the dashed dot line at a concentration of 10−12 M, and the dashed line at 0 M.
Upon addition of the enzyme at 10−12 M concentration to the DNA containing restriction sites (final DNA concentration 10−11 M), an increase in the magnitude of the cross-correlation signal is observed (see Fig. 4). This occurs due to FRET between the Cy3 and Cy5 molecules, as described above. At the same time, there is an increase in the diffusion time to 0.38 ± 0.02 ms. However, if the DNA molecules do not contain the restriction sites, the addition of the enzyme to the sample does not result in a change in either the auto- or cross-correlation curves and associated diffusion times. An increase in the diffusion times of DNA/protein complexes versus free DNA molecules has been observed previously (Rippe, 2000). From the data shown in Fig. 4, one can conclude that addition of the enzyme to the DNA results in the specific binding of the enzyme to the restriction sites, forming a more slowly diffusing complex. The formation of the enzyme-DNA looped complexes occurs within several seconds after the addition of the enzyme (probably limited by the mixing time in these measurements), and NgoMVI remains bound to the DNA at least as long as the time required to diffuse through the beam, i.e., a few tenths of a millisecond.
Essentially the same diffusion time value is observed over a range of enzyme concentrations from 10−12 to 10−8 M. However, there is an increase in the cross-correlation amplitude with increasing enzyme concentration to 10−9 M, indicating increasing concentration of the bound DNA/protein complexes. At even higher enzyme concentrations, from 10−7 to 3 × 10−6 M, much slower diffusion times are observed, ranging from 0.6 to 1.5 ms (see Fig. 3). This increase in the diffusion time at very high concentrations of the enzyme suggests that under these conditions two NgoMIV molecules can be bound to the same DNA molecule and possibly between DNA molecules, generating large aggregates that diffuse more slowly (see below).
Burst size distribution analysis
Burst size distributions at 10−12 and 10−7 M enzyme concentrations were calculated to evaluate the average number of fluorophores (DNA molecules) per protein/DNA complex (see Fig. 5). To calculate these distributions, bins were selected from the Cy3 detection channel that had more that 10 counts per 1 ms. Adjacent bins that met this criterion where considered to be part of the same burst. The burst size was calculated as the total number of counts in a burst. The burst size distributions shown in Fig. 5 were derived from data collected over a 160-s interval. The results in Fig. 5 clearly show two main differences at low and high enzyme concentrations. First, at the 10−12 M NgoMIV concentration, the total number of qualifying bursts is significantly smaller than at 10−7 M. Also, the number of high intensity bursts is markedly smaller at the lower enzyme concentration. The burst size distribution at 10−12 M enzyme concentration is very similar to that of free DNA labeled with Cy3 (not shown) indicating that the bursts detected predominantly correspond to complexes containing a single Cy3 fluorophore rather than complexes containing multiple Cy3 molecules. This is expected because the DNA concentration is 10−11 M, 10-fold greater than the NgoMIV concentration. However, at 10−7 M enzyme concentration the number of high intensity bursts is much greater (Fig. 5), which suggests that these bursts arise from the presence of several dye molecules per complex in the focal volume. Multifluorophore particles in the focal volume give a higher probability to detect more fluorescence, which would also explain the increase of the total number of bursts. Thus, the burst size distribution at 10−7 M NgoMIV concentration also supports the hypothesis of DNA/protein aggregate formation.
Comparison of FRET distances to structural models
The results described above indicate that the addition of NgoMIV restriction endonuclease to DNA fragments containing appropriate restriction sites results in the formation of DNA/protein complexes in which FRET between the dyes at the two ends of the DNA can be detected at the single molecule level. This conclusion is supported by the observed increase in the diffusion times of DNA molecules (Fig. 4), as well as the FRET efficiency distributions (Fig. 3). The FRET results allow one to estimate the distance between the dye molecules. Gaussian fits of the FRET efficiency distributions result in a center value of 28 ± 2% (see above). If one assumes an R0 of 53 Å for the Cy3/Cy5 FRET pair (Ishii et al., 1999) and an average value for the orientation factor κ2 of 2/3, a distance of 62 ± 1 Å is obtained. The distance between DNA helices determined from the structure of DNA bound to the NgoMIV enzyme is ∼55 Å. The angle between DNA helices at this point is ∼60° (Deibert et al., 2000) (see Figs. 1 and 6). The labeled DNA molecules were created to extend an additional 4 basepairs beyond the restriction site on either end (see Materials and Methods). The DNA molecules used in the crystal structure had a palindromic sequence, which made it impossible to predict the actual direction of the looped DNA helices (see Fig. 1). Assuming the binding occurs in such a way that the two DNA helices extend in the same direction but at an angle of 60° (this would be the configuration with the most relaxed bend in the DNA), the distance between the ends of the DNA fragment used can be calculated to be ∼65 Å. Though, in principle, the length of the linker between DNA bases and dye molecules could affect the distance between dye molecules attached to the ends of the DNA, the fact that the dye is attached on each end to the 5′ phosphate group by a labile C3 linker should result in an average distance between the dyes that is approximately the same as the distance between the ends of the DNA. Using the same assumptions about the structure of the linker and the fluorophore position relative to the ends of the DNA, the 15-bp control DNA molecule that had Cy3 attached to one end and Cy5 to the other gave the expected distance upon analysis of its single molecule FRET emission (see above).
FIGURE 6.

Possible geometries of NgoMIV binding to a double-stranded DNA molecule. (a) Double-stranded DNA molecule with two NgoMIV restriction sites and no enzyme added. (b) DNA looping when a small concentration of NgoMIV is added. (c) Two DNA molecules bound by one enzyme, and (d) aggregates formed by linking multiple DNA molecules when the NgoMIV concentration is very high.
Thus, the distance between the donor and acceptor calculated from the FRET measurements matches that predicted by the structure. This implies that the 30% population in the FRET efficiency distribution (Fig. 3 b) is due to energy transfer events between the dye molecules bound to the ends of a DNA strand complexed with the NgoMIV enzyme in a configuration consistent with the crystal structure. These events correspond to DNA looping by NgoMIV, as this 30% population is observed even at very low concentrations of the enzyme and DNA, when binding of two DNA molecules to one enzyme molecule is highly unlikely.
At high enzyme concentrations, a new population with FRET efficiency of ∼10% is observed (Fig. 3, c and d). At the same enzyme concentrations, cross-correlation FCS results indicate that the diffusion time of the DNA/protein complexes increases severalfold (Fig. 4). These two effects are also accompanied by a significant increase of the amplitude of fluorescence bursts (see Fig. 5). This is consistent with the concept presented above that at high concentrations large aggregates of NgoMIV and DNA are formed with multiple fluorophores, and therefore increased amounts of fluorescence are detected (see Fig. 6 d). The presence of these large complexes (also called trans complexes) was previously proposed from the gel-shift studies of SfiI endonuclease binding to linear DNA molecules (Watson et al., 2000). As observed with SfiI, it is likely that two NgoMIV molecules bind to the same DNA molecule at high concentrations of the enzyme (see Fig. 6, c and d). Then, in principle, the large complexes can form as the enzymes link together different DNA molecules. The formation of the large complexes is supported by the increase of diffusion times from cross-correlation measurements as well as the increase of fluorescence signal, which implies the existence of several dye molecules in a single particle passing through the probing volume (multiple DNA molecules per particle).
Taking all these facts into account, there could be several reasons for the 10%–15% FRET efficiency population. One possibility is that the aggregation of protein/DNA complexes at high enzyme concentration is such that there are a mixture of geometries, with some Cy3 and Cy5 molecules in close proximity and others separated, resulting in an intermediate average FRET efficiency or even a range of efficiencies (at the highest enzyme concentrations, there seems to be a rather broad distribution of efficiencies). Alternatively, the 10% FRET efficiency population could arise from a specific DNA/protein binding geometry in these large complexes. Two different DNA strands could bind to the same enzyme molecule, but in opposing directions, similar to what has been shown for SfiI endonuclease (Halford et al., 2000; Watson et al., 2000) and is represented in Fig. 6, c and d. In this case, from the NgoMIV structure, the angle between the DNA ends would be 120° instead of 60°, and the distance between the DNA ends would be ∼75 Å, which should correspond to ∼10% FRET efficiency. However, it is not possible to distinguish between these possibilities from the present data.
CONCLUSIONS
Our results show that individual looped DNA-NgoMIV complexes can be detected using single molecule FRET. Very importantly, geometrically different populations of complexes in a mixture can be distinguished, lending insight into the binding states and geometries utilized at different enzyme/DNA concentrations. A key feature of these results is the ability to follow the distribution of multiple geometries in the sample and this is possible because the data were collected at the single molecule level. This system is now poised for future measurements of DNA/protein dynamics associated with the binding and looping processes, and similar approaches could be applied to other enzymes and binding proteins that result in looping of DNA (for example, wrapping of DNA around the histone core particles of nucleosomes). To generate dynamic data on the NgoMIV system, it will be necessary to immobilize the DNA because the complex between the enzyme and the DNA is apparently considerably longer-lived than the diffusion time of the complex through the laser beam. This work is in progress.
Acknowledgments
This research was supported by USDA grant 98-35306-6396. The authors thank Carole Flores for critical reading of the manuscript.
References
- Bickle, T. A., and D. H. Kruger. 1993. Biology of DNA restriction. Microbiol. Rev. 57:434–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand, L., C. Eggeling, C. Zander, K. H. Drexhage, and C. A. M. Seidel. 1997. Single-molecule indentifications of coumarin-120 by time resolved fluorescence detection: comparison of one and two photon excitation in solution. J. Phys. Chem. 101:4313–4321. [Google Scholar]
- Dahan, M., A. A. Deniz, T. Ha, D. S. Chemla, P. G. Schultz, and S. Weiss. 1999. Ratiometric measurements and identification of single diffusing molecules. Chem. Phys. 247:85–106. [Google Scholar]
- Daniel, D. C., M. Thompson, and N. W. Woodbury. 2002. DNA-binding interactions and conformational fluctuations of Tc3 transposase DNA binding domain examined with single molecule fluorescence spectroscopy. Biophys. J. 82:1654–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deibert, M., S. Grazulis, G. Sasnauskas, V. Siksnys, and R. Huber. 2000. Structure of the tetrameric restriction endonuclease NgoMIV in complex with cleaved DNA. Nat. Struct. Biol. 7:792–799. [DOI] [PubMed] [Google Scholar]
- Deniz, A. A., M. Dahan, J. R. Grunwell, T. Ha, A. E. Faulhaber, D. S. Chemla, S. Weiss, and P. G. Schultz. 1999. Single-pair fluorescence resonance energy transfer on freely diffusing molecules: observation of Förster distance dependence and subpopulations. Proc. Natl. Acad. Sci. USA. 96:3670–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich, A., V. Buschmann, C. Muller, and M. Sauer. 2002. Fluorescence resonance energy transfer (FRET) and competing processes in donor-acceptor substituted DNA strand: a comparative study of ensemble and single-molecule data. J. Biotechnology. 82:211–231. [DOI] [PubMed] [Google Scholar]
- Eigen, M., and R. Rigler. 1994. Sorting single molecules: application to diagnostics and evolutionary biotechnology. Proc. Natl. Acad. Sci. USA. 91:5740–5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Embleton, M. L., V. Siksnys, and S. E. Halford. 2001. DNA cleavage reactions by type II restriction enzymes that require two copies of their recognition sites. J. Mol. Biol. 311:503–514. [DOI] [PubMed] [Google Scholar]
- Friedhoff, P., R. Lurz, G. Luder, and A. Pingoud. 2001. Sau3AI, a monometric type II restriction endonuclease that dimerizes on the DNA and thereby induces DNA loops. J. Biol. Chem. 276:23581–23588. [DOI] [PubMed] [Google Scholar]
- Ha, T., A. Y. Ting, J. Liang, W. B. Caldwell, A. A. Deniz, D. S. Chemla, P. G. Schultz, and S. Weiss. 1999a. Single-molecule fluorescence spectroscopy of enzyme conformational dynamics and cleavage mechanism. Proc. Natl. Acad. Sci. USA. 96:893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha, T., X. Zhuang, H. D. Kim, J. W. Orr, J. R. Williamson, and S. Chu. 1999b. Ligand-induced conformational changes observed in single RNA molecules. Proc. Natl. Acad. Sci. USA. 96:9077–9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford, S. E., D. M. Gowers, and R. B. Sessions. 2000. Two are better than one. Nat. Struct. Biol. 7:705–707. [DOI] [PubMed] [Google Scholar]
- Hess, S. T., S. Huang, A. A. Heikal, and W. W. Webb. 2002. Biological and chemical applications of fluorescence correlation spectroscopy: a review. Biochemistry. 41:697–705. [DOI] [PubMed] [Google Scholar]
- Ishii, Y., T. Yoshida, T. Funatsu, T. Wazawa, and T. Yanagida. 1999. Fluorescence resonance energy transfer between single fluorophores attached to a coiled-coil protein in aqueous solution. Chem. Phys. 247:163–173. [Google Scholar]
- Kettling, U., A. Koltermann, P. Schwille, and M. Eigen. 1998. Real-time enzyme kinetics monitored by dual-color fluorescence cross-correlation spectroscopy. Proc. Natl. Acad. Sci. USA. 95:1416–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H. D., G. U. Nienhaus, T. Ha, J. W. Orr, J. R. Williamson, and S. Chu. 2002. Mg2+-dependent conformational change of RNA studies by fluorescence correlation and FRET on immobilized single molecules. Proc. Natl. Acad. Sci. USA. 99:4284–4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milsom, S. E., S. E. Halford, M. L. Embleton, and M. D. Szczelkun. 2001. Analysis of DNA looping interactions by type II restriction enzyme that require two copies of their recognition sites. J. Mol. Biol. 311:515–527. [DOI] [PubMed] [Google Scholar]
- Nobbs, T. J., and S. E. Halford. 1995. DNA cleavage at two recognition sites by the Sfi I restriction endonuclease: salt dependence of cis and trans interactions between distant DNA sites. J. Mol. Biol. 252:399–411. [DOI] [PubMed] [Google Scholar]
- Norman, D. G., R. J. Grainger, D. Uhrin, and D. M. J. Lilley. 2000. Location of cyanine-3 on double stranded DNA: importance for fluorescence resonance energy transfer. Biochemistry. 39:6317–6324. [DOI] [PubMed] [Google Scholar]
- Oram, M., J. F. Marko, and S. E. Halford. 1997. Communications between distant sites on supercoiled DNA from non-exponential kinetics for DNA synapsis by resolvase. J. Mol. Biol. 270:396–412. [DOI] [PubMed] [Google Scholar]
- Pace, C. N., and F. X. Schmid. 1997. How to determine the molar absorbance coefficient of a protein. In Protein Structure, a Practical Approach. T. E. Creighton, editor. IRL Press, Oxford, UK. 253–259.
- Rippe, K. 2000. Simultaneous binding of two DNA duplexes to NtrC-enhancer complex studied by two-color fluorescence cross-correlation spectroscopy. Biochemistry. 39:2131–2139. [DOI] [PubMed] [Google Scholar]
- Schleif, R. 1992. DNA looping. Annu. Rev. Biochem. 61:199–223. [DOI] [PubMed] [Google Scholar]
- Siksnys, V., R. Skirgaila, G. Sasnauskas, C. Urbanke, D. Cherny, S. Grazulis, and R. Huber. 1999. The Cfr 10I restriction enzyme is functional as a tetramer. J. Mol. Biol. 291:1105–1118. [DOI] [PubMed] [Google Scholar]
- Voet, D., and J. G. Voet. 1995. Biochemistry. Wiley & Sons, New York.
- Watson, M. A., D. M. Gowers, and S. E. Halford. 2000. Alternative geometries of DNA looping: an analysis using the SfiL endonuclease. J. Mol. Bio. 289:758–797. [DOI] [PubMed] [Google Scholar]
- Weiss, S. 1999. Fluorescence spectroscopy of single biomolecules. Science. 283:1676–1683. [DOI] [PubMed] [Google Scholar]
- Weiss, S. 2000. Measuring conformational dynamics of biomolecules by single molecule fluorescence spectroscopy. Nat. Struct. Biol. 7:724–729. [DOI] [PubMed] [Google Scholar]
- Wentzell, L. M., T. J. Nobbs, and S. E. Halford. 1995. The SfiI restriction endonuclease makes a four-strand DNA break at two copies of its recognition sequence. J. Mol. Biol. 248:581–595. [DOI] [PubMed] [Google Scholar]
- Widengren, J., E. Schweinberger, S. Berger, and C. A. M. Seidel. 2001. Two new concepts to measure fluorescence resonance energy transfer via fluorescence correlation spectroscopy: theory and experimental realizations. J. Phys. Chem. A. 105:6851–6866. [Google Scholar]
- Ying, L., M. I. Wallace, S. Bakasubramanian, and D. Klenerman. 2000. Rotiometric analysis of single-molecule fluorescence resonance energy transfer using logical combinations of threshold criteria: a study of 12-mer DNA. J. Phys. Chem. B. 104:5171–5178. [Google Scholar]
- Zander, C., M. Sauer, K. H. Drexhage, D. S. Ko, A. Schulz, J. Wolfrum, L. Brand, C. Eggeling, and C. A. M. Seidel. 1996. Detection and characterization of single molecules in aqueous solution. Appl. Phys. B. 63:517–523. [Google Scholar]
- Zhuang, X., L. E. Bartley, H. P. Babcock, R. Russell, T. Ha, D. Herschlag, and S. Chu. 2000. A single-molecule study of RNA catalysis and folding. Science. 288:2048–2051. [DOI] [PubMed] [Google Scholar]