

Abstract
A laser trap was used to compare the load-dependent binding kinetics between truncated P- and L-selectin to their natural ligand, P-selectin glycoprotein ligand-1 (PSGL-1) over the predicted physiological range of loading rates. Human PSGL-1 was covalently coupled to polystyrene beads. Chimeric selectins were adsorbed to nitrocellulose-coated glass beads on a coverslip. A PSGL-1 bead was held in a laser trap and touched to a vertical surface of the glass bead, allowing a bond to form between selectin and ligand. The surface was moved away from the microsphere, applying load at a constant rate until bond rupture. Rupture force for both selectins increased with loading rate, but significant differences in rupture force between P- and L-selectin were observed only above 460 pN/s. These data are best represented as two energy barriers to unbinding, with the transition from the low to high loading rate regime at 260–290 pN/s. The data also allow the first estimate of a two-dimensional specific on-rate for binding of these two selectins to their natural ligand (1.7 μm2/s). These data suggest that P- and L-selectin lectin domains have very similar kinetics under physiological conditions.
INTRODUCTION
Leukocyte and endothelial adhesion molecules govern the trafficking of cells in ischemia-reperfusion injury (Granger et al., 1995; Rabb et al., 1997), thermal injuries and transplantation (Gute and Korthius, 1995), inflammation and immunity (Springer, 1995), cancer metastasis (Albelda, 1993), atherosclerosis, and wound healing (Grinnell, 1992). Extravasation of immune cells from the blood to a site of infection or inflammation is a multistep process limited to postcapillary venules. Inflammation causes surrounding cells to release inflammatory mediators that activate endothelium cells. Capture of leukocytes results from the formation of intermolecular bonds between leukocyte and vascular endothelium, and occurs under shear stress from the pressure-driven flow of blood. If new bonds are formed before the initial bond breaks, rolling occurs (Ley, 1999; Springer, 1995). Leukocyte rolling may allow stable leukocyte arrest (firm adhesion), mediated by integrins. It is believed that adhesion under shear stress requires adhesion molecules with rapid association rates (on-rates), resulting in rapid formation of bonds.
Leukocyte capture and rolling are modulated by selectins, a family of mammalian lectins found on the surfaces of leukocytes, endothelial cells, and platelets. Selectins have rates of association comparable to those of integrins, but vastly higher rates of bond dissociation (Labadia et al., 1998; Nicholson et al., 1998). This makes selectins especially well suited to mediate cell rolling (Chang et al., 2000). There are three selectins: E, P, and L. They show >60% identity in amino acid sequence. These similarities in structure indicate that the selectins share a common ancestry and function (Forrest and Paulson, 1995). All selectins share a modular structure (Ley, 1999; Somers et al., 2000) including a calcium-dependent N-terminal lectin domain, an epidermal growth factor domain, several short consensus repeat (CR) domains, a single membrane spanning domain, and a cytoplasmic tail (Forrest and Paulson, 1995; Ley, 1999). Adhesion occurs through calcium-dependent interactions between the lectin domain and the glycan ligand on opposing cells. The other domains are thought to modulate function (Kansas et al., 1993); however, the function of these domains is, as of yet, unknown (Somers et al., 2000).
Structural similarities between the selectins suggest that they bind similar or even identical ligands. In fact, all three selectins recognize and bind to sialyl Lewis (sLe), a rigid polysaccharide; in particular, sLex and sLea (Brunk and Hammer, 1997; Forrest and Paulson, 1995; Greenberg et al., 2000; Rodgers et al., 2000). However, only one glycoprotein has been fully characterized as a physiological selectin ligand. P-selectin glycoprotein ligand-1 (PSGL-1) is constitutively expressed on almost all leukocytes and a few other cells (Laszik et al., 1996). It exists as a homodimer with significant posttranslational modification. In addition to binding P-selectin, PSGL-1 can also bind L-selectin (Spertini et al., 1996; Guyer et al., 1996) and may bind E-selectin (Xia et al., 2002; Snapp et al., 1997; Asa et al., 1995).
P- and L-selectins play very different pathophysiological roles. Most notably, L-selectin is important in lymphocyte homing to peripheral and mesenteric lymph nodes (Arbones et al., 1994; Lewinsohn et al., 1987) and mediating leukocyte-leukocyte interactions (Eriksson et al., 2001). L-selectin is constitutively expressed by most leukocytes, whereas P-selectin is stored in secretory granules of endothelial cells and platelets. All the selectins can initiate leukocyte rolling in vivo (Ley et al., 1995; Mayadas et al., 1993).
Despite the structural similarities between the molecules, faster rolling occurs with L- than with P- or E-selectin, which implies different bond kinetics for these two molecules (Puri et al., 1997; Smith et al., 1999). The cytoplasmic domain of L-selectin, which is ∼17-amino-acids long, plays an important role in adhesion regulation. Removing 11 of these amino acids abolishes rolling (Kansas et al, 1993; Dwir et al., 2001), and proteolytic shedding of L-selectin modulates rolling velocity in vivo (Hafezi-Moghadam et al., 2001). Thus, if the apparent off-rate is a combination of bond breakage and proteolytic shedding, then the true off-rate may not be different for P- and L-selectin. An L-selectin mutant incapable of shedding, however, has been shown not to differ from wild-type cells in adhesion kinetics (Smith et al., 1999). Thus, delineating the differences in kinetics between the selectins has proved difficult.
Leukocytes are often used in parallel plate flow chamber assays to compare the kinetics of selectins (Smith et al., 1999). However, the densities, deformability, rheology, roughness, and cell signaling are not easily controlled! It is difficult then to definitively examine single receptor-ligand bonds. Protein-coated beads provide an alternative system (Brunk and Hammer, 1997; Greenberg et al., 2000; King and Hammer, 2001). Unfortunately, the accuracy of the experiment is limited due to leverage effects. After bond formation, the bead will rotate about the bond until it touches the surface, thus leveraging the bond. Therefore, to calculate the effect of the leverage, the length of the receptor-ligand pair must be accurately known (Park et al., 2002).
Several groups have measured the mechanokinetics (Fig. 1) of single E-, L-, and P-selectin/ligand bonds using a variety of experimental techniques, including force probes. Fritz et al. (1998) studied the mechanics of chimeric P-selectin and PSGL-1 using an atomic force microscope. Long et al. (2001), Evans et al. (2001), and Hanley et al. (2003) studied the mechanics of L- or P-selectin bonds using variations on a biomembrane force probe. Finally, Tees et al. (2001) studied the mechanics of chimeric E-selectin and sLex using a glass microcantilever device. Given the variety of molecular constructs, ligands, and technologies, one is left without a defensible comparison of the mechanokinetics of any two selectin classes.
FIGURE 1.

Schematic of Bell's proposal on bond strengths. When unloaded, the free energy landscape of a bond is represented as a Lennard-Jones-like function. The ligand approaches the receptor, reaching an equilibrium position where the free energy is minimized. As load (F) is applied (relative values next to the individual lines), work is done on the bond (F × bond separation) that tilts the energy landscape. The free energy change for escape from the bond, ɛF, is lower compared to the unloaded case ɛ0. Thus, the bond fails in less time.
The current study tests the hypothesis that the mechanokinetics of P- and L-selectin ligand-binding domains can explain differences observed between P- and L-selectin in vivo. The specific aim was to determine how the kinetic properties of bond lifetime and rupture force for P- and L-selectin depend on loading rate within the physiological range. This was done using a laser trap force transducer. We found that chimeric P- and L-selectin bonds with native PSGL-1 have similar load-dependent kinetics. Thus the functional differences between these two selectins lie beyond the lectin domains, and require either the full-length molecules or the native cellular environment for their full functional expression. These data also underscore the importance of directly measuring selectin bond loading rates during leukocyte rolling.
METHODS
Proteins
PSGL-1 was purified from HL60 cell lysates by affinity chromatography using KLP1 mAb coupled to CNBr-activated Sepharose (Park et al., 2002). Chimeric P- and L-selectins were obtained commercially (Glycotech, Gaithersburg, MD). The chimeras include the lectin, epidermal growth factor, and two of the CR domains connected to an immunoglobulin Fc domain. Native L-selectin chimera has two CRs whereas the P-selectin has nine.
Microspheres
PSGL-1 was covalently coupled to 2-μm carboxylated polystyrene beads (Polysciences, Warrington, PA) using carbodiimide crosslinking (Brunk and Hammer, 1997). In these reactions, a carboxyl-modified bead interacts with a water-soluble carbodiimide to form an O-acylisourea intermediate. This intermediate reacts with an available amine on the ligand. This reaction results in a ligand covalently coupled to a bead. A detailed description of this protocol can be found in Polysciences Technical Data Sheet 238F (Polysciences, 1999).
Blocking with Tween-20 (Park et al., 2002) reduced nonspecific interactions. The cross-linked beads were incubated in the 1% Tween in PBS for 30 min. Unfortunately, the use of such blockers does not entirely eliminate nonspecific interactions (King and Hammer, 2001; Pierres et al., 1995; Stout, 2001). Therefore we account for nonspecific interactions through multiple controls, including calcium chelators and monoclonal blockade (see Controls).
Flow cells
Experiments were conducted in 50-μl flow cells prepared in a similar manner to Guilford et al. (1997). Glass coverslip arrays were prepared by applying nine drops of water equidistant from one another to the back of a square petri dish. Glass coverslips (22 mm × 22 mm) were adhered to the petri dish by placing them over each water drop. The glass coverslip arrays are then sprayed with a glass bead (Polysciences) suspension using a thin-layer chromatography sprayer. The bead suspension consisted of 40 μg of beads (3–10 μm in diameter) into 1 ml of water, which is then diluted with 25 ml of ethanol. The coverslips were subsequently pressed through a thin film of nitrocellulose to coat them. The film was made by adding 8–12 drops of 1% nitrocellulose solution in amyl acetate (Ladd Research, Williston, VT) to an ice bucket filled with water. The nitrocellulose layer was allowed to dry for 5 min. The petri dish was then submerged at a slight angle, with the coverslip side down, through the dry nitrocellulose layer. The coated coverslips were dried overnight at room temperature. The coated coverslips were attached with ultraviolet curing optical glue (Norland Products, Cranbury, NJ) to 0.1-mm-thick Mylar shim-stock (Rydens, Washington, PA) and then to an 18-mm × 18-mm coverslip to form a 50-μl channel. The assembled flow cells were cured by exposing them to ultraviolet light for 2 min (ELC-500, Electro-Lite, Danbury, CT).
Laser trap apparatus
A simplified schematic of the laser trap is shown in Fig. 2. Technical details of the laser trap electronics and computer-based control are being published separately. Briefly, the light source for the trap is a fiber laser (SDL, San Jose, CA, Model FL10: 11W CW, TEM0,0, M2 ≈ 1.0, λ = 1112 nm). The randomly polarized beam (0.41-mm diameter, 1/e2) passes through two lenses, L1 and L2, which expand the beam by 15×. A two-dimensional acoustooptic deflector (AOD) (NEOS, Melbourne, FL) diffracts the beam and allows for rapid and precise steering of the laser trap in the specimen plane. The first-order diffracted beam then passes into a periscope (M1 and M2), and through a lens, L3. At this point, a beam sampler (BS) reflects a sample of the beam (5%) to a photodetector (PD) to sample laser power. The beam then passes into a final lens mounted to the side of the microscope, L4. L3 and L4 comprise a second beam expander, which expands the beam by 1.1× to fill the back aperture of the microscope objective (OBJ).
FIGURE 2.

Schematic of the laser trap and the experimental protocol (inset). For these experiments, the bead shown on the left is a glass bead adhered to a surface, and coated with nitrocellulose and selectin. The bead shown on the right is a polystyrene bead with covalently coupled PSGL-1. The PSGL-1 bead is held in a laser trap, and brought into contact with the receptor. The black line represents a selectin-ligand bond. The receptor-ligand bond is loaded by moving the stage (S) under computer control. Legend: fiber laser (FL), Glan laser prism (GLP), lenses (Lx), half-wave plate (1/2), acoustooptic deflector (AOD), periscope (P), beam sampler (BS), photodetector (PD), iris diaphragm (I), dichroic mirrors (DMx), quadrant photodiode (QP), charge-coupled device camera (CCD), digital signal processing card (DSP), and direct digital synthesis subsystem (DDS).
The trap is based on an Olympus (Melville, NY) IX70 inverted microscope with side and laser port adapters. A dichroic mirror, DC1 (Omega Optical, Brattleboro, VT, XF118: reflecting 730–1150 nm), directs the beam to the objective (100× Olympus UplanFL, NA 1.3). Bright field imaging is accomplished using a 100-W halogen light source and a charge-coupled device camera (Pulnix, Sunnyvale, CA).
The position of the bead is detected by back focal plane interferometry. Each bead acts as a lens to partially collimate the trapping laser beam. The microscope condenser collects this light. When the bead moves relative to the trap, the distribution of light changes in the back focal plane of the condenser. This change in light distribution may be monitored as an indication of the position of the bead within the trap (Allersma et al., 1998). The advantage of this technique is that it is simple and independent of the laser trap position in the field of view. The disadvantage is that it is sensitive to fluctuations in laser power.
A 40× air objective with numerical aperture 0.65 (Leitz, Troisdorf, Germany) in a commercial mount (Olympus) was used as the condenser in these experiments. The light is coupled through a dichroic mirror, DC2 (identical to DC1), to a lens, L5, which projects an image of the back focal plane onto a 5-mm germanium quadrant photodiode (QP) (J16QUAD, Judson Technologies, Montgomeryville, PA). These optics are held in a custom mount that fits in place of the DIC slider in the condenser assembly.
Transformation of the photocurrent from the individual quadrant to x and y coordinates for the bead has been reported by others. Our only significant variations were that 1), the germanium QP allows us to use significantly less gain, thus increasing the bandwidth of the system; and 2), the photodetector signal was used to normalize against fluctuations in laser power, rather than the sum of all the QP quadrants. The initial transformation is done through an analog computer, as described by others (Finer et al., 1994), except that a high-speed analog divider was used (AD734, Analog Devices, Norwood, MA).
The laser trap apparatus is connected to an external computer (Dell, Round Rock, TX). This allows the position of the trap to be monitored and precisely manipulated through a digital AOD driver/amplifier (NEOS). Data from the analog computer, above, was collected by a 14-bit, 100 kHz A/D converter and digital signal processing card (Spectrum Signal Processing, Burnaby, B.C., DL3-A1 and Darkar, respectively). The QP data was viewed on a digital oscilloscope (Tektronix, Beaverton, OR, TDS3012). A three-dimensional piezoelectric microscope stage (Nanonics, Jerusalem, Israel) is also controlled by the computer. A custom graphical user interface controls the laser trap and the stage.
Protocol
The selectins were incubated in the flow cells for 15 min at room temperature in a buffer consisting of 150 mM NaCl, 25 mM Imidazole, 1 mM CaCl2, and 1 mM NaN3 at pH 7.4. This is done at a concentration of 0.27 μg/ml for Human P-selectin IgG (Glycotech) and 0.32 μg/ml for Human L-selectin IgG (Glycotech). The protein concentrations were determined by a protein assay (Cytoskeleton, Denver, CO).
To reduce nonspecific interactions, the flow cells were then soaked in a 1% Tween solution in the above buffer for 1 h to block the unbound sites on the nitrocellulose. The bead suspension was then injected into the flow chamber. PSGL-1-coated beads were trapped and held in the laser trap. The PSGL-1-coated bead was then brought into contact with a selectin-coated glass bead on the surface of the flow cell (Fig. 2). The piezoelectric stage was then moved back and forth at constant velocity, with a 2-ms pause when the two beads were in contact. Contact time between the two beads was limited to 2 ms to reduce the probability of forming multiple bonds (Benoit et al., 2000).
As the stage moves, the bead in the trap is displaced from trap center only if a bond has formed between the two beads. A sample interaction is seen in Fig. 3. Baseline noise is a measurement of the Brownian motion of the bead in the trap with no external force applied to the bead. A trace showing no interaction is included for comparison. As the beads approach one another, there is occasionally optical interference from the glass bead on the coverslip. This optical interference and any slight compression of the beads against one another is seen as a dip in the trace before the interaction. The portion of the plot seen in Fig. 3 that has a linear increase in slope represents the bond response. As the bond is stretched by the stage moving away, the bead in the trap is displaced. When the bond breaks, the trapped bead snaps back into the center of the trap, seen as an almost immediate return to baseline. The most frequently recorded rupture force in this technique depends upon the loading rate (Evans, 2001), which in turn was determined by the rate of separation of the two beads, and the stiffness of the laser trap.
FIGURE 3.

Sample selectin-ligand interaction. The portion of the plot that has a sharp increase in slope represents the bead displacement in the trap as the bond is stretched. Once it breaks, the trace returns to baseline. Brownian motion of the bead in the trap causes the baseline noise. A control bead/plate trace is shown below.
Bond durations were measured from the time when the trapped bead displacement crosses baseline (immediately after any negative displacement, if present) to the time when the highest peak of the interaction occurs. Negative displacements may mask the beginning of bond loading, resulting in a slight but consistent underestimate of bond duration for that bead pair. When no negative displacement was observed, the beginning of the positive displacement event was estimated based on the slope of the linear segment.
Calibration and statistics
QP calibration (in nm/V) was determined by step response (Svoboda and Block, 1994). Briefly, the trap was rapidly displaced a predetermined distance using the AOD. While the trapped bead slowly creeped to the new trap center, the initial displacement of the trap relative to the bead caused a spike in the QP output, indicative of the QP response to a known microsphere deflection. Since this calibration step was not always possible, calibration was routinely confirmed based on the initial slope of each interaction, which is a direct measure of the predetermined rate of withdrawal of the target bead from the trapped bead. Beads were trapped, on average, ∼2.5-μm from the coverslip surface.
The trap stiffness (α) was determined using the Stokes' force method (Svoboda and Block, 1994), and ranged from 0.004 to 0.05 pN/nm, depending on the desired loading rate. Bond lifetimes were measured directly, and rupture force calculated from the trap stiffness.
Statistical errors were propagated. Comparisons were performed using a two-tailed z-test with p < 0.05. Each data point is derived from 21 to 60 binding events.
Controls
A number of control experiments were performed to ensure that the reported kinetics represented single, selectin-mediated interactions. Controls were performed without protein, in the form of protein-free beads and flow cells.
Additionally, the PSGL-1 beads are treated with a monoclonal anti-human PSGL-1 function-blocking antibody (anti-PSGL-1) (Ancell, Bayport, MN). In these experiments, PSGL-1 beads were incubated in 0.02 mg/ml anti-PSGL-1 for 30 min. Because the presence of anti-PSGL-1 blocks PSGL-1 function, the number of interactions should be reduced dramatically. However, the antibody does not bind to all the available PSGL-1. Therefore, remaining interactions would be expected to be normal in terms of magnitudes and durations.
Similar function blocking controls were performed against the selectins. Selectins were bound to the flow cells as described previously. The flow cell was then blocked, and an antibody solution added to concentrations of 0.016 mg/ml and 0.034 mg/ml for monoclonal anti-human P- and L-selectin, respectively (Ancell). The antibody solution was incubated for 30 min at room temperature.
Selectin function is calcium-dependent. Thus, as a final control, EGTA was added to a concentration of 1 mM and CaCl2 omitted (Benoit et al., 2000).
Site density determination
In addition to the control experiments, site density determinations were performed to ensure that the reported data reflects single molecular events. The method used for site density determination was time-resolved fluorescence detection of Eu-labeled streptavidin (Suonpaa et al., 1992). The flow cells were treated with selectin and blocked by the protocol described previously. Control flow cells were made by incubation with blocking solution alone. The flow cells were then rinsed and the antibody solution (0.034 mg/ml) incubated for 30 min at room temperature. Flow cells were then rinsed with 300 μl buffer, and 1.0 mg/ml biotinylated goat anti-mouse IgG (Oncogene Research Products, Darmstadt, Germany) added for 30 min. After a final wash, the flow cells were split and placed in small plastic petri dishes so that the protein-coated nitrocellulose side faced up. A quantity of 1 ml of 0.1 μg/ml Eu-labeled streptavidin solution was added to each plate and incubated at room temperature for 30 min. The dishes were washed with six sequential rinses of 0.5% Tween 20 in PBS, and 1.8 ml DELFIA enhancement solution (PerkinElmer, Boston, MA) added at room temperature for 5 min. All of the liquid was then collected from each dish and placed into 96 well plates, 0.3 ml per well. The same method was employed to determine the site density of PSGL-1 on microspheres, save that centrifugal removal of microspheres from suspension was used between solutions, and anti-PSGL-1 used in place of anti-selectin antibody. After secondary antibody incubation, the concentration of beads was measured using a Coulter Multisizer (Beckman Coulter, Fullerton, CA). Based on this concentration, a volume of beads with 1-cm2 bead surface area was used to perform the remainder of the protocol.
Time-resolved fluorescence was measured with a SPECTRAmax Gemini XS dual-scanning microplate spectrofluorimeter (Molecular Devices, Sunnyvale, CA) at 360-nm excitation, 610-nm emission, during a 250–1050-μs time interval after a 1-μs excitation flash. The mass of bound Eu-streptavidin was then determined from calibration curves using known concentrations of Eu-streptavidin. For the determination of site densities, one-to-one binding was assumed between the selectin:primary antibody, the primary antibody:secondary antibody, and the secondary antibody:Eu-streptavidin.
To estimate the number of molecules available to interact during bead contact, we estimated the surface area over which two spheres in contact might be able to form a bimolecular bond of length l based on the geometry of sphere-sphere intersections (Kern and Bland, 1948). Referring to Fig. 4 a, there is a single point at which a bond may form if the two spherical surfaces are exactly l apart. If we assume that the molecules on the spherical surfaces are sparse or compressible such that the spheres may be brought into direct contact, then there exist two hemispherical “caps” that are within distance l of one another (Fig. 4 b). The area of a hemispherical cap is given by
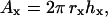 |
(1) |
where hx is the height of the cap on sphere x, and rx is the radius of the sphere. As illustrated, if we constrain the bond to be parallel to the x axis and the spheres are of different radii, the cap heights will differ (Fig. 4 c). Simple algebraic manipulation yields the cap height for one sphere given the radii and bond length, l. Thus,
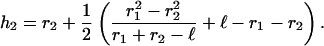 |
(2) |
The second cap height is given by h1 = l − h2. We estimate l as the sum of the lengths of PSGl-1 (50 nm; Li et al., 1996) and P-selectin (48 nm; Erlandsen et al., 2001). The PSGL-1-coated microspheres were consistently of 1-μm radius. The glass spheres bearing selectins were of highly variable diameter, but averaged ∼2 μm in radius. These values yield interaction areas of 0.40 and 0.41 μm2 for the PSGL-1 and selectin beads, respectively. The reaction volume may be estimated as the free space between the two microspheres that is encompassed by a cylinder of length l (Fig. 4 c),
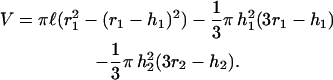 |
(3) |
FIGURE 4.

Model of microsphere surface areas available to form bimolecular bonds. (a) A single point for potential bond formation occurs when the spherical surfaces are one bimolecular bond length (l) apart. (b) When the spheres are brought into contact, l encompasses a hemispherical cap on each of the spheres, delineating the areas over which bonds may form. (c) h1 and h2 indicate the height of the hemispherical cap on each sphere. Cap heights are determined by the radii of the spheres (r1 and r2), and l. A volume (V) exists between the spheres within which the binding reactions will occur.
Models
The Bell Model was first published in 1978 and is commonly used in evaluating protein-protein interactions under load (Bell, 1978). This model is based on the presumption that the free energy available will vary with the separation of the receptor and ligand. When a bond is experimentally loaded, work is done on the bond. The total energy, which is the sum of the bond energy and work due to the applied force, results in lowered energy barriers as seen in Fig. 1. This raises the rate constant for dissociation of the bond, but because bond rupture is probabilistic, bonds appear to strengthen with increased load. Nonetheless, bonds with smaller barriers always break faster (Evans, 2001).
Bell used a theory from the strength of solids to describe bond lifetime (Zhurkov, 1965),
 |
(4) |
where γ relates to the degree of dislocation in metal crystals and polymers, f is the load applied to the bond, Eo is the bond energy, and τ and τo are the loaded and unloaded bond lifetimes, respectively. Bell related this model to dissociation rates as
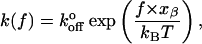 |
(5) |
where 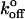 is the unloaded dissociation rate, and xβ is equivalent to γ in the Zhurkov model. Relating the value γ to a physical structure in biological polymers and bonds is problematic. Nonetheless, the Bell model has become standard in the interpretation of biological bond lifetime experiments. By applying various loads to biological bonds, one may estimate the unloaded bond lifetime.
is the unloaded dissociation rate, and xβ is equivalent to γ in the Zhurkov model. Relating the value γ to a physical structure in biological polymers and bonds is problematic. Nonetheless, the Bell model has become standard in the interpretation of biological bond lifetime experiments. By applying various loads to biological bonds, one may estimate the unloaded bond lifetime.
Unfortunately, in experiments the externally applied force is not a single, instantaneous load. The applied load increases with time. Therefore, load-dependent dissociation is better expressed in terms of a loading rate (Yuan et al., 2000; Tees et al., 2001) as
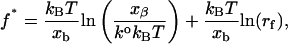 |
(6) |
where rf is the rate of applied load and f* is the characteristic rupture force. It is clear that the model predicts a linear relationship between rupture force and the logarithm of the loading rate. It is important to note, however, that the characteristic rupture force is effectively the peak of the bond rupture force distribution, or the mode of the distribution, not the mean of the rupture forces.
Based on the Bell model, the common prediction is that with increasing loading rates, bond lifetime decreases whereas bond strength increases. Evans and Ritchie (1997) propose that due to the complex nature of the macromolecule, there are multiple energy minima. Bond energy then depends on the nature of the bonds and physical barriers of the receptor. Merkel et al. (1999) found evidence of multiple energy wells. Data from a biomembrane force probe on biotin-streptavidin bonds showed the predicted linear relationship between loading rate and rupture force. However, this data suggests not a single linear regime, but a series of linear regimes representing individual energy barriers.
An alternative approach to fitting Bell parameters to the data is to model the mean rupture force due to a single energy barrier from reliability theory (Tees et al., 2001),
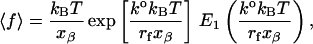 |
(7) |
where E1 is the exponential integral.
Both the multiple-barrier model of Evans and Ritchie (1997) and the single barrier model of Tees et al. (2001) were employed in our analysis. Linear fits to the f* versus ln(rf) data were accomplished using SigmaPlot (SPSS Software, Chicago, IL), which uses the Levenberg-Marquardt algorithm. Equation 4 was fitted to the data using a simplex algorithm (Press et al, 1997) with an X2 estimator using software developed in Delphi (Borland, Scotts Valley, CA).
RESULTS
Controls
P- and L-selectin kinetics were examined over a range of loading rates from 25 to 600 pN/s. To ensure that the data presented here represent single bonds, the number of interactions was compared to the number of touches. Typically, one interaction out of nine is sufficiently low to indicate unitary binding events (Evans et al., 2001). On average, 1 in 20 ±5 and 23.3 ±4 touches yielded a bond interaction for P- and L-selectin, respectively. The use of function blocking anti-PSGL-1 reduced the number of interactions from 1 in 20 to 1 out of 100 for both P- and L-selectin. The few interactions were comparable in magnitude and duration to experimental values. The use of anti-L-selectin reduced the number of interactions to 1 in 96 whereas the use of anti-P-selectin reduced the interactions to 1 in 76.
Control beads, control flow cells, and EGTA controls reduced both the magnitude of the interactions and the probability of forming a bond on any given bead-bead contact from ∼0.05 to 0.027. It is often difficult to differentiate between these small, nonspecific interactions and optical interference from the two beads approaching one another. Nonetheless, characteristic rupture forces for control beads against P- and L-selectin surfaces are shown in Fig. 6. Control rupture forces were significantly less than experimental rupture forces at all loading rates, and did not increase with increased loading rate.
FIGURE 6.

Characteristic rupture force as a function of loading rate for chimeric L- (•) and P- (○) selectins interacting with PSGL-1. Rupture forces for chimeric L- (▾) and P- (▿) selectins interacting with a protein-free surface are also shown. Error bars indicate standard error of the mean. When not visible, error bars are within the symbol. N for each point range between 21 and 60 measured interactions.
Site density
The average mass of Eu-streptavidin on the flow cells was 0.53 ng. Accounting for the surface area in the flow cell, this mass was translates into a site density for the selectins of 26.5 μm−2. Similarly, the mass of Eu-streptavidin on the PSGL-1 beads was 0.0044 ng. This translates to a site density of 1.08 μm−2 for PSGL-1 on the 2-μm polystyrene beads. Using these site densities and the interaction areas calculated in the Methods, we estimate the average number of molecules available to form bonds on bead contact as 0.4 PSGL-1 and 11 selectins. Thus, PSGL-1 densities are limiting in these experiments. The probability of being able to form a bond is <1 for any given contact, in agreement with the experimental frequency of bond formation.
Effects of loading rate
Off-rate increases with increasing loading rate, as seen in Fig. 5. From 25 to 270 pN/s, there is no statistical difference between P- and L-selectin. At 460 and 600 pN/s, koff is significantly higher for L-selectin compared to P-selectin.
FIGURE 5.

The off rate as a function of loading rate for chimeric L- (•) and P- (○) selectins interacting with PSGL-1. Error bars indicate the standard error of the mean. When not visible, error bars are within the symbol. N for each point ranges between 21 and 60 measured interactions.
Characteristic and mean rupture forces also increase with loading rate (Fig. 6). There is no significant difference between P- and L-selectin from 25 to 460 pN/s. At 600 pN/s, P-selectin has a significantly higher mean rupture force of 47.4 pN compared to 37.6 pN for L-selectin.
Fitted parameters
Interpreted within the context of an Evans and Richie-like model, there appear to be two energy barriers to unbinding. The characteristic (modal) rupture forces, f*, appear to fall into two linear regimes on a f* versus ln(rf) plot. xβ was determined from the slope of linear regression to each of these segments as
 |
(8) |
where m is the slope of the linear fit. Substituting these xβ values into the equation for the intercept yields 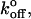 as
as
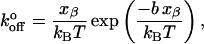 |
(9) |
where b is the slope of the linear fit. Calculated values of xβ and 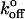 for the low and high regimes are given in Table 1. According to this analysis, xβ and
for the low and high regimes are given in Table 1. According to this analysis, xβ and 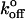 are very similar for both selectins, and over both ranges of loading rates. The transition-point in loading rate separating the inner and outer barriers is between 260 and 290 pN/s.
are very similar for both selectins, and over both ranges of loading rates. The transition-point in loading rate separating the inner and outer barriers is between 260 and 290 pN/s.
TABLE 1.
Fitted parameters based on the Bell-derived models of Evans and Richie (1997) and Tees et al. (2001), organized by selectin and range of loading rates
| Evans and Richie
|
Tees et al.
|
||||
|---|---|---|---|---|---|
| Selectin | rf (pN/s) | xβ (nm) | ko (s−1) | Xβ (nm) | ko (s−1) |
| P | 0–260 | 0.63 | 1.39 | 0.21 | 3.76 |
| 260–600 | 0.14 | 4.30 | |||
| L | 0–260 | 0.75 | 1.35 | 0.27 | 2.52 |
| 260–600 | 0.15 | 5.03 | |||
The mean rupture forces (〈fbreak〉) were also analyzed assuming a single energy barrier according to Tees et al. (2001). Here, Eq. 4 was fit to 〈fbreak〉 versus rf. The results of this analysis are also shown in Table 1 and Fig. 7. The parameters xβ and 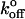 were each of similar magnitude to the inner barrier deduced from the previous analysis. However, the quality of the fit (Q ≡ 1−P) based on the X2 estimator is rather poor for the P-selectin data (Q = 0.45) compared to the L-selectin data (Q = 0.89), as is obvious from the graph. This may be due to the inclusion of the lowest loading rate data, which can lead to poor estimation of the Bell parameters (Tees et al., 2001). However, when these data are omitted from the fits, an unreasonable
were each of similar magnitude to the inner barrier deduced from the previous analysis. However, the quality of the fit (Q ≡ 1−P) based on the X2 estimator is rather poor for the P-selectin data (Q = 0.45) compared to the L-selectin data (Q = 0.89), as is obvious from the graph. This may be due to the inclusion of the lowest loading rate data, which can lead to poor estimation of the Bell parameters (Tees et al., 2001). However, when these data are omitted from the fits, an unreasonable  value (9.1 s−1) is determined for P-selectin. In contrast, omitting from the fits the highest loading rate P-selectin, we find values for xβ and
value (9.1 s−1) is determined for P-selectin. In contrast, omitting from the fits the highest loading rate P-selectin, we find values for xβ and 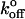 (0.15 nm, and 4.3 s−1, respectively) very similar to the inner barrier determined by the previous method. This suggests that it is the highest loading rate data for P-selectin that are not well represented by the Tees model.
(0.15 nm, and 4.3 s−1, respectively) very similar to the inner barrier determined by the previous method. This suggests that it is the highest loading rate data for P-selectin that are not well represented by the Tees model.
FIGURE 7.

Mean rupture force as a function of loading rate for chimeric L- (•) and P- (○) selectins interacting with PSGL-1, together with fits of Eq. 7 to data for L- (solid) and P- (dashed) selectin. Error bars indicate standard error of the mean. When not visible, error bars are within the symbol.
DISCUSSION
P- and L-selectin kinetics are remarkably similar over the range of loading rates we studied. There is no significant difference between the two selectin chimeras with regard to bond lifetime and rupture force for loading rates 0–460 pN/s. P- and L-selectin exhibit somewhat different characteristics only at the very highest loading rates. This is the first reported comparison of two selectins using the relatively new technique of dynamic force spectroscopy.
Strong similarities in the mechanokinetics of P- and L-selectin is perhaps not a surprising result, given the predicted structural similarity of two molecules. Bajorath and Aruffo (1995) used the known structure of E-selectin as a basis for molecular dynamics prediction of the L-selectin structure. If one aligns the known P-selectin structure (Somers et al., 2000) and a predicted L-selectin structure (Bajorath and Aruffo, 1995), the root mean-square difference between the backbones is only 0.48 Å, and perhaps one less opportunity for hydrogen-bonding by L-selectin to the ligand. Thus, one might expect the mechanokinetics of P- and L-selectin to be similar, considering the lectin-binding domain in isolation.
These experiments were designed to cover the predicted physiological range of loading rates (see Physiological Loading Rate). Thus we must conclude that the slight differences in mechanokinetics of the lectin domains of P- and L-selectin do not explain differences in tether bond lifetime observed in rolling cells. Neutrophils rolling in vitro on sparse substrates undergo transient pauses that are thought to represent the formation and breaking of individual adhesive bonds. Pause durations of neutrophils rolling on L-selectin demonstrate a much stronger dependence on shear stress than when rolling on P-selectin (Smith et al, 1999). This was interpreted as a threefold-greater bond separation distance (xβ) than in P-selectin, with no significant change in 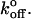 This difference does not manifest itself in our data. There are several possible explanations. First, the loading rate in rolling cells may be significantly higher than those applied here. Unfortunately, the in vivo loading rate on these bonds is not known. Second, the cellular context or membrane environment may be vital to the physiological behavior of the molecule, through formation of membrane tethers or cytoskeletal associations. Third, the full-length molecules may be necessary for full physiological behavior. The chimeras used here lack the cytoplasmic and membrane spanning domains, and the P-selectin chimera lacks some of the consensus repeats found in the native molecule. Finally, P-selectin may be able to form multivalent interactions with PSGL-1 that are not possible with the limiting protein concentrations used here.
This difference does not manifest itself in our data. There are several possible explanations. First, the loading rate in rolling cells may be significantly higher than those applied here. Unfortunately, the in vivo loading rate on these bonds is not known. Second, the cellular context or membrane environment may be vital to the physiological behavior of the molecule, through formation of membrane tethers or cytoskeletal associations. Third, the full-length molecules may be necessary for full physiological behavior. The chimeras used here lack the cytoplasmic and membrane spanning domains, and the P-selectin chimera lacks some of the consensus repeats found in the native molecule. Finally, P-selectin may be able to form multivalent interactions with PSGL-1 that are not possible with the limiting protein concentrations used here.
Nonetheless, our data agrees well with the work of Evans et al. (2001) on L-selectin. Although we were restricted to a lower loading rate regime, our higher loading rate rupture forces overlap seamlessly with their lowest loading rate data, and we were able to more closely approach the unloaded condition.
A number of groups have analyzed the mechanokinetics of P-selectin. In Table 2 we compare the Bell model parameters from these studies. Our value for xβ is similar to that generated by other force-probe studies (Hanley et al., 2003; Fritz et al., 1998), but differs somewhat from those determined by cell rolling and surface-binding techniques (Mehta et al., 1998; Smith et al., 1999; Ramachandran et al., 2001). In contrast, our value for  is very similar to those generated by cell rolling and surface-binding techniques, whereas force-probe studies return
is very similar to those generated by cell rolling and surface-binding techniques, whereas force-probe studies return 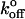 values an order-of-magnitude lower. We believe these differences stem from the range and accuracy of applied loading rates in these various studies.
values an order-of-magnitude lower. We believe these differences stem from the range and accuracy of applied loading rates in these various studies.
TABLE 2.
Comparison of Bell model kinetic parameters measured for P-selectin from various studies, using various experimental techniques
| Method | xβ (nm) |
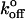 (s−1) (s−1) |
Reference |
|---|---|---|---|
| Laser trap | 0.14 | 4.3 | Current study |
| Surface plasmon resonance | – | 1.4 | Mehta et al., 1998 |
| Rolling cells | 0.04 | 2.4 | Smith et al., 1999 |
| Rolling cells | 0.07 | 1.1 | Ramachandran et al., 2001 |
| Molecular force probe | 0.12 | 0.19 | Hanley et al., 2003 |
| Atomic force microscope | 0.25 | 0.022 | Fritz et al., 1998 |
Force-probe techniques that focus on higher loading rates may accurately estimate xβ, since forced dissociation models are more sensitive to xβ than to 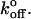 The laser trap may allow us to more accurately estimate
The laser trap may allow us to more accurately estimate 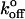 because it allows accurate application of low loading rates. Low loading rates obviously approach more closely the unloaded condition (
because it allows accurate application of low loading rates. Low loading rates obviously approach more closely the unloaded condition (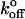 being the unloaded dissociation rate). In intact cells, loading rates must be estimated based on assumed lever models. Because all forced dissociation models are more sensitive to xβ than to
being the unloaded dissociation rate). In intact cells, loading rates must be estimated based on assumed lever models. Because all forced dissociation models are more sensitive to xβ than to 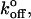 it is not surprising that this parameter might systematically vary when loading rate is not precisely known.
it is not surprising that this parameter might systematically vary when loading rate is not precisely known.
Limitations
In the study of adhesive mechanisms, it is often difficult to prove that the data reflects the characteristics of a single, specific molecular bond rather than multiple bonds or nonspecific interactions. The specificity of the interactions was confirmed using controls with blank beads, blank flow cells, and EGTA, each of which dramatically reduced both the probability of observing an adhesive event and the magnitude of those events. Further evidence of specific interactions is provided by the antibody controls. Both of the anti-PSGL-1 and anti-selectin controls dramatically reduced the number of interactions, but did not alter the characteristics of the events that did occur. This is expected, since function-blocking antibodies simply provide a second, competing association reaction. Thus, the observed rate of association will be reduced, but the reverse rate (koff), and therefore the binding event magnitude and duration, will be unaffected.
The likelihood of reporting only individual bonds was increased through dilution (Pierres et al., 1995) and limiting the contact time between the microspheres. We may estimate the rate of complex formation based on solution kinetics,
 |
(10) |
where R, L, and C represent the receptor, ligand, and complex, respectively. Site densities were directly measured, and we estimated the reaction volume between the spheres (Methods). From these values we estimate concentrations of 0.008 and 0.2 μM for PSGL-1 and the selectins, respectively. Assuming a kon of 4.4 × 106 M−1 s−1 (Mehta et al., 1998), we predict that the number of complexes formed during our brief contact time (2 ms) is only 0.0007. The experimental value of 0.04 is higher than this estimate, perhaps because Eq. 10 assumes solution kinetics rather than a semiordered surface.
We may instead apply two-dimensional kinetic theory to derive the two-dimensional  from our data. According to Long et al. (2001), the mean number of bonds formed between two surfaces over a time t is given by
from our data. According to Long et al. (2001), the mean number of bonds formed between two surfaces over a time t is given by
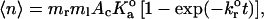 |
(11) |
where mr and ml are the surface densities of the receptor and ligand, respectively; Ac is the contact area; and 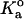 is the equilibrium binding affinity (
is the equilibrium binding affinity (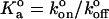 ). Given our estimated contact areas (0.39 μm2), measured site densities, contact time (2 ms), and our fitted value of
). Given our estimated contact areas (0.39 μm2), measured site densities, contact time (2 ms), and our fitted value of 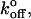 we calculate the two-dimensional
we calculate the two-dimensional 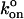 to be 1.7 μm2/s. To our knowledge, this is the first measurement of a two-dimensional specific kon, so direct comparisons with other bimolecular systems is not possible. However, it is of similar magnitude as the two-dimensional kon measured for E-selectin in intact cells, unadjusted for site density (Long et al., 2001).
to be 1.7 μm2/s. To our knowledge, this is the first measurement of a two-dimensional specific kon, so direct comparisons with other bimolecular systems is not possible. However, it is of similar magnitude as the two-dimensional kon measured for E-selectin in intact cells, unadjusted for site density (Long et al., 2001). 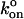 = 1.7 μm2/s must be treated strictly as an estimate, however, since without a study of 〈n〉 versus contact time we are unable to place statistical bounds on this value. Further, we were unable to control for slight compressions of the ligand- and receptor-bearing beads that occur during contact. Using identically sized beads, each held in an independent laser trap, will enable us to control this parameter in the future. Nonetheless, the piezoelectric stage returns to a consistent assumed contact position for 2 ms after each excursion. We may thus reasonably assume that if interactions are observed for some contacts of a given pair of beads, the opportunity to form bonds exists during all contacts of that pair of beads.
= 1.7 μm2/s must be treated strictly as an estimate, however, since without a study of 〈n〉 versus contact time we are unable to place statistical bounds on this value. Further, we were unable to control for slight compressions of the ligand- and receptor-bearing beads that occur during contact. Using identically sized beads, each held in an independent laser trap, will enable us to control this parameter in the future. Nonetheless, the piezoelectric stage returns to a consistent assumed contact position for 2 ms after each excursion. We may thus reasonably assume that if interactions are observed for some contacts of a given pair of beads, the opportunity to form bonds exists during all contacts of that pair of beads.
There is the possibility that the kinetics reported might not reflect the receptor-ligand de-bonding event. The following bonds might break: 1), ligand to polystyrene bead; 2), selectin to ligand; and 3), nitrocellulose to selectin. The PSGL-1 was covalently coupled to the polystyrene beads, and so unlikely to break. It is also unlikely that rupture of the selectin-nitrocellulose bond occurs. Had the selectin-nitrocellulose bond ruptured, selectins would be removed from the surface, and the available population of receptors in a region would be depleted over time. In contrast, the fraction of contacts yielding an interaction remained constant over time for any given pair of beads.
Physiological loading rate
The range of loading rates used in this study is predicted to be in the physiological range. Since the physiological loading rate of selectin-ligand bonds has never been experimentally measured, we made estimates based on known membrane mechanics. Membrane tethers form as cells in flow form transient bonds with the substrate. Tethers can be modeled as a viscoelastic spring that counteracts the drag force acting on the cell through stretching, and they will also moderate the loading rate at the cell-substrate bond. The average shear rate in postcapillary venules is estimated as 400 s−1 (Jung et al., 1998; Ley and Gaehtgens, 1991). If we assume the tether growth rate to be equal to the velocity of cell rolling, this shear rate is then converted to a tether growth rate by linear regression on tether growth rate versus shear rate data (Schmidtke and Diamond, 2000). This yields a tether growth rate as high as 100 μm/s, though typically ≈25 μm/s (Damiano et al., 1996). The spring constant of the tether was estimated from published data to range from 5 pN/μm to >1000 pN/μm (Raucher and Sheetz, 1999; Shao et al., 1998; Park et al., 2002) but is apparently highly nonlinear (Raucher and Sheetz, 1999). Thus, we may calculate a potential range of loading rates at the tip of the membrane tether ranging from 125 to 2500 pN/s. At a molecular level, one would assume that the load is divided among the available bonds, and the loading rates per molecule are considerably lower.
One may also estimate tether stiffness from models of normal membrane deformation in the absence of cytoskeletal components. Tether stiffness may be considered the parallel sum of that due to membrane bending (αb) and membrane stretching (αst). These may be estimated according to
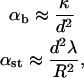 |
(12) |
where κ and λ are the bending and stretching moduli of the membrane, respectively; d is the characteristic width of the membrane deformation; and R is the radius of the cell (Boulbitch, 1998). Assuming a cell radius of 5 μm, d as 0.1 μm, and values for κ and λ of 2 × 10−19 J and 0.001 m, we estimate an initial stiffness for drawing a tether of 16 pN/μm. As the tether forms fully (tether length Ψ > d), membrane bending becomes constant, and the stiffness will fall to 8 pN/μm. Thus, at a tether growth rate of 25 μm/s, we might expect to see an initial loading rate of 400 pN/s, decaying within 1 ms to 200 pN/s.
Raucher and Sheetz (1999) have measured a plateau in the tether force-elongation relationship, perhaps due to the existence of a membrane reservoir. This implies that a brief high loading rate may give way to a very low loading rate or approximately constant load (loading rate ≈ 0) until the reservoir is depleted. Further complicating the matter is the observation that the viscous properties of leukocyte membrane tethers are dependent on the load and rate of elongation (Marcus and Hochmuth, 2002). These uncertainties emphasize the importance of measuring the elastic properties of membrane tethers in leukocytes over a range of controlled physiological elongation rates. As yet, an experimental measurement of physiological loading rate on adhesive bonds in leukocytes is not truly available, and the published data do not allow an accurate estimate.
If the physiological loading rate is significantly higher than the range reported here, our data may accurately project the physiological differences in selectin behavior. Both descriptive models predict that at higher loading rates koff for P-selectin will become progressively less than L-selectin, with correspondingly increased rupture force. In addition, the failure of the Tees model to accurately fit the higher loading rate suggests the beginning of another higher energy barrier (Tees et al., 2001). Direct measures in living leukocytes of bond loading rate are clearly needed to understand the physiological context of leukocyte adhesion molecules.
Acknowledgments
The authors acknowledge Dr. Alexander L. Klibanov of the University of Virginia for his kind assistance in the determination of site densities, and the continuing support of the Cardiovascular Research Center at the University of Virginia.
This work was supported by grants from the National Heart, Lung, and Blood Institute (HL64381) and the National Institute for Arthritis and Musculoskeletal Diseases (AR45604).
References
- Albelda, S. M. 1993. Biology of disease role of integrins and other cell adhesion molecules in tumor progression and metastasis. Lab. Invest. 68:4–17. [PubMed] [Google Scholar]
- Allersma, M. W., F. Gittes, M. J. deCastro, R. J. Stewart, and C. F. Schmidt. 1998. Two-dimensional tracking of NCD motility by back focal plane interferometry. Biophys. J. 74:1074–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbones, M. L., D. C. Ord, K. Ley, H. Ratech, C. Maynard-Curry, G. Otten, D. J. Capon, and T. F. Tedder. 1994. Lymphocyte homing and leukocyte rolling and migration are impaired in L-selectin-deficient mice. Immunity. 1:247–260. [DOI] [PubMed] [Google Scholar]
- Asa, D., L. Raycroft, L. Ma, P. A. Aeed, P. S. Kaytes, A. P. Elhammer, and J. G. Geng. 1995. The P-selectin glycoprotein ligand functions as a common human leukocyte ligand for P- and E-selectins. J. Biol. Chem. 270:11662–11670. [DOI] [PubMed] [Google Scholar]
- Bajorath, J., and A. Aruffo. 1995. A template for generation and comparison of three-dimensional selectin models. Biochem. Biophys. Res. Commun. 216:1018–1023. [DOI] [PubMed] [Google Scholar]
- Bell, G. I. 1978. Models for the specific adhesion of cells to cells: a theoretical framework for adhesion mediated by reversible bonds between cell surface molecules. Science. 200:618–627. [DOI] [PubMed] [Google Scholar]
- Benoit, M., D. Gabriel, G. Gerisch, and H. E. Gaub. 2000. Discrete interactions in cell adhesion measured by single-molecule force spectroscopy. Nat. Cell Biol. 2:313–317. [DOI] [PubMed] [Google Scholar]
- Boulbitch, A. A. 1998. Deflection of a cell membrane under application of a local force. Phys. Rev. E. 57:2123–2128. [Google Scholar]
- Brunk, D. K., and D. A. Hammer. 1997. Quantifying rolling adhesion with a cell-free assay: E-selectin and its carbohydrate ligands. Biophys. J. 72:2820–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, K., D. F. Tees, and D. A. Hammer. 2000. The state diagram for cell adhesion under flow: leukocyte rolling and firm adhesion. Proc. Natl. Acad. Sci. USA. 97:11262–11267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiano, E. R., J. Westheider, A. Tozeren, and K. Ley. 1996. Variation in the velocity, deformation, and adhesion energy density of leukocytes rolling within venules. Circ. Res. 79:1122–1130. [DOI] [PubMed] [Google Scholar]
- Dwir, O., D. S. Kansas, and R. Alon. 2001. Cytoplasmic anchorage of L-selectin controls leukocyte capture and rolling by increasing the mechanical stability of the selectin tether. J. Cell Biol. 155:145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson, E. E., X. Xie, J. Werr, P. Thoren, and L. Lindbom. 2001. Importance of primary capture and L-selectin-dependent secondary capture in leukocyte accumulation in inflammation and atherosclerosis in vivo. J. Exper. Med. 194:205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlandsen, S. L., A. G. Bittermann, J. White, A. Leith, and M. Marko. 2001. High-resolution CryoFESEM of individual cell adhesion molecules (CAMs) in the glycocalyx of human platelets: detection of P-selectin (CD62P), GPI-IX complex (CD42a/CD42b α,bβ), and integrin GPIIbIIIa (CD41/CD61) by immunogold labeling and stereo imaging. J. Histochem. Cytochem. 49:808–819. [DOI] [PubMed] [Google Scholar]
- Evans, E., and K. Ritchie. 1997. Dynamic strength of molecular bonds. Biophys. J. 72:1541–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, E. 2001. Probing the strengths of molecular bonds. Annu. Rev. Biophys. Biomed. Struct. 30:105–128. [DOI] [PubMed] [Google Scholar]
- Evans, E., A. Leung, D. Hammer, and S. Simon. 2001. Chemically distinct transition states govern rapid dissociation of single L-selectin bonds under force. Proc. Natl. Acad. Sci. USA. 98:3784–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finer, J. T., R. M. Simmons, and J. A. Spudich. 1994. Single myosin molecule mechanics: piconewton forces and nanometre steps. Nature. 368:113–119. [DOI] [PubMed] [Google Scholar]
- Forrest, M., and J. C. Paulson. 1995. Selectin family of adhesion molecules. In Physiology and Pathophysiology of Leukocyte Adhesion. D. N. Granger and G. W. Schmid-Schonbein, editors. Oxford University Press, New York. 323–338.
- Fritz, J., F. Katopodis, F. Kolbingerm, and D. Anselmetti. 1998. Force-mediated kinetics of single O-selectin/ligand complexes observed by atomic force microscopy. Proc. Natl. Acad. Sci. USA. 95:12283–12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger, D. N., I. Kurose, and P. R. Kvietys. 1995. Modulation of leukocyte adherence and emigration during ischemia and reperfusion. In Physiology and Pathophysiology of Leukocyte Adhesion. D. N. Granger and G. W. Schmid-Schonbein, editors. Oxford University Press, New York. 323–338.
- Greenberg, A. W., D. K. Brunk, and D. A. Hammer. 2000. Cell-free rolling mediated by L-selectin and Sialyl Lewisx reveals the shear threshold effect. Biophys. J. 79:2391–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinnell, F. 1992. Wound repair, keratinocyte activation and integrin modulation. J. Cell Sci. 101:1–5. [DOI] [PubMed] [Google Scholar]
- Guilford, W. H., D. E. Dupuis, G. Kennedy, J. Wu, J. G. Patlak, and D. M. Warshaw. 1997. Smooth muscle and skeletal muscle myosins produce similar unitary forces and displacements in the laser trap. Biophys. J. 72:1006–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gute, D., and R. J. Korthius. 1995. Role of leukocyte adherence in reperfusion-induced microvascular dysfunction and tissue injury. In Physiology and Pathophysiology of Leukocyte Adhesion. D. N. Granger and G. W. Schmid-Schonbein, editors. Oxford University Press, New York. 359–380.
- Guyer, D. A., K. L. Moore, E. B. Lynam, C. M. G. Schammel, S. Rogelj, R. P. McEver, and L. A. Sklar. 1996. P-selectin glycoprotein ligand-1 (PSGL-1) is a ligand for L-selectin in neutrophil aggregation. Blood. 88:2415–2421. [PubMed] [Google Scholar]
- Hafezi-Moghadam, A., K. L. Thomas, A. J. Prorock, Y. Huo, and K. Ley. 2001. L-Selectin shedding regulates leukocyte recruitment. J. Exp. Med. 193:863–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley, W., O. McCarty, S. Jadhav, Y. Tseng, D. Wirtz, and K. Konstantopoulos. 2003. Single molecule characterization of P-selectin/ligand binding. J. Biol. Chem. 278:10556–10561. [DOI] [PubMed] [Google Scholar]
- Jung, U., K. E. Norman, K. Scharffetter-Kochanek, A. L. Beaudet, and K. Ley. 1998. Transit time of leukocytes rolling through venules controls cytokine-induced inflammatory cell recruitment in vivo. J. Clin. Invest. 102:1526–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kansas, G. S., K. Ley, M. Munro, and T. F. Tedder. 1993. Regulation of leukocyte rolling and adhesion to high endothelial venules through the cytoplasmic domain of L-selectin. J. Exp. Med. 177:833–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern, W. F., and J. R. Bland. 1948. Solid Mensuration with Proofs. Wiley, New York.
- King, M., and D. Hammer. 2001. Multiparticle adhesive dynamics: hydrodynamic recruitment of rolling leukocytes. Proc. Natl. Acad. Sci. USA. 98:14919–14924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labadia, M. E., D. D. Jeanfavre, G. O. Caviness, and M. M. Morelock. 1998. Molecular regulation of the interaction between leukocyte function-associated antigen-1 and soluble ICAM-1 by divalent metal cations. J. Immunol. 161:836–842. [PubMed] [Google Scholar]
- Laszik, Z., P. J. Jansen, R. D. Cummings, T. F. Tedder, R. P. McEver, and K. L. Moore. 1996. P-selectin glycoprotein ligand-1 is broadly expressed in cells of myeloid, lymphoid, and dendritic lineage and in some nonhematopoietic cells. Blood. 88:3010–3021. [PubMed] [Google Scholar]
- Lewinsohn, D. M., R. F. Bargatze, and E. C. Butcher. 1987. Leukocyte-endothelial cell recognition: evidence of a common molecular mechanism shared by neutrophils, lymphocytes, and other leukocytes. J. Immunol. 138:4313–4321. [PubMed] [Google Scholar]
- Ley, K. 1999. Adhesion of leukocytes from flow: the E-selectins and their ligands. Vasc. Adhes. Mol. Inflam.
- Ley, K., D. C. Bullard, M. L. Arbones, R. Bosse, D. Vestweber, T. F. Tedder, and A. L. Beaudet. 1995. Sequential contribution of L- and P-selectin to leukocyte rolling in vivo. J. Exp. Med. 181:669–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley, K., and P. Gaehtgens. 1991. Endothelial, not hemodynamic, differences are responsible for preferential leukocyte rolling in rat mesenteric venules. Circ. Res. 69:1034–1041. [DOI] [PubMed] [Google Scholar]
- Li, F., H. P. Erickson, J. A. James, K. L. Moore, R. D. Cummings, and R. P. McEver. 1996. Visualization of P-selectin glycoprotein ligand-1 as a highly extended molecule and mapping of protein epitopes for monoclonal antibodies. J. Biol. Chem. 271:6342–6348. [DOI] [PubMed] [Google Scholar]
- Long, M., H. Zhao, K. Huang, and C. Zhu. 2001. Kinetic measurements of cell surface E-selectin/carbohydrate ligand interactions. Ann. Biomed. Eng. 29:935–946. [DOI] [PubMed] [Google Scholar]
- Marcus, W. D., and R. M. Hochmuth. 2002. Experimental studies of membrane tethers formed from human neutrophils. Ann. Biomed. Eng. 30:1273–1280. [DOI] [PubMed] [Google Scholar]
- Mayadas, T. N., R. C. Johnson, H. Rayburn, R. O. Hynes, and D. D. Wagner. 1993. Leukocyte rolling and extravasation are severely compromised in P-selectin-deficient mice. Cell. 74:541–554. [DOI] [PubMed] [Google Scholar]
- Mehta, P., R. D. Cummings, and R. P. Evers. 1998. Affinity and kinetic analysis of P-selectin binding to P-selectin glycoprotein ligand-1. J. Biol. Chem. 273:32506–32513. [DOI] [PubMed] [Google Scholar]
- Merkel, R., P. Nassoy, A. Leung, K. Ritchie, and E. Evans. 1999. Energy landscapes of receptor-ligands bonds explored with dynamic force spectroscopy. Nature. 397:50–53. [DOI] [PubMed] [Google Scholar]
- Nicholson, M. W., A. N. Barclay, M. S. Singer, S. D. Rosen, and P. A. van der Merwe. 1998. Affinity and kinetic analysis of L-selectin (CD62L) binding to glycosylation-dependent cell-adhesion molecule-1. J. Biol. Chem. 273:763–770. [DOI] [PubMed] [Google Scholar]
- Park, E. Y. H., M. J. Smith, E. S. Stropp, K. R. Snapp, J. A. DiVietro, W. F. Walker, D. W. Schmidtke, S. L. Diamond, and M. B. Lawrence. 2002. Comparison of PSGL-1 microbead and neutrophil rolling: microvillus elongation stabilizes P-selectin bond clusters. Biophys. J. 82:1835–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierres, A., A. Benoliel, and P. Bongrand. 1995. Measuring the lifetime of bonds made between surface-linked molecules. J. Biol. Chem. 270:26586–26592. [DOI] [PubMed] [Google Scholar]
- Press. W. H., S. A. Teukolsky, W. T. Vetterling, and B. P. Flannery. 1997. Numerical Recipes in C. Cambridge University Press, New York.
- Puri, K. D., E. B. Finger, and T. A. Springer. 1997. The faster kinetics of L-selectin than of E-selectin and P-selectin rolling at comparable binding strength. J. Immunol. 158:405–413. [PubMed] [Google Scholar]
- Rabb, H., Y. O'Meara, P. Maderna, P. Coleman, and H. Brady. 1997. Leukocytes, cell adhesion molecules and ischemic acute renal failure. Kidney Int. 51:1463–1468. [DOI] [PubMed] [Google Scholar]
- Ramachandran, V., T. Yago, T. K. Epperson, M. M. A. Kobzdej, M. U. Nollert, R. D. Cummings, C. Zhu, and R. P. McEver. 2001. Dimerization of a selectin and its ligand stabilizes cell rolling and enhances tether strength in shear flow. Proc. Nat. Acad. Sci. USA. 98:10166–10171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raucher, D., and M. P. Sheetz. 1999. Characteristics of a membrane reservoir buffering membrane tension. Biophys. J. 77:1992–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers, S. D., R. T. Camphausen, and D. A. Hammer. 2000. Siayl Lewisx-mediated, PSGL-1-independent rolling adhesion on P-selectin. Biophys. J. 79:694–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtke, D. W., and S. L. Diamond. 2000. Direct observation of membrane tethers formed during neutrophil attachment to platelets or P-selectin under physiological flow. J. Cell Biol. 149:719–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao, J. Y., H. P. Ting-Beall, and R. M. Hochmuth. 1998. Static and dynamic lengths of neutrophils microvilli. Proc. Natl. Acad. Sci. USA. 95:6797–6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, M., E. Berg, and M. Lawrence. 1999. A direct comparison of selectin-mediated transient, adhesive events using high temporal resolution. Biophys. J. 77:3371–3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapp, K. R., A. J. Wagers, R. Craig, L. M. Stoolman, and G. S. Kansas. 1997. P-selectin glycoprotein ligand-1 is essential for adhesion to P-selectin but not E-selectin in stably transfected hematopoietic cell lines. Blood. 89:896–901. [PubMed] [Google Scholar]
- Somers, W. S., J. Tang, and G. Shaw. 2000. Insights into the molecular basis of leukocyte tethering and rolling revealed by structure of P- and E-selectin bound to Slex and PSGL-1. Cell. 103:467–479. [DOI] [PubMed] [Google Scholar]
- Spertini, O., A. S. Cordey, N. Monai, L. Giuffre, and M. Schapira. 1996. P-Selectin glycoprotein ligand 1 is a ligand for L-selectin on neutrophils, monocytes, and CD34+ hematopoietic progenitor cells. J. Cell Biol. 135:523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer, T. A. 1995. Traffic signals on endothelium for lymphocyte recirculation and leukocyte emigration. Annu. Rev. Physiol. 57:827–872. [DOI] [PubMed] [Google Scholar]
- Stout, A. L. 2001. Detection and characterization of individual intermolecular bonds using optical tweezers. Biophys. J. 80:2976–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suonpaa, M., E. Markella, T. Stahlberg, and I. Hemmila. 1992. Eu-labeled streptavidin as a universal label. Indirect time-resolved immunofluorometry of FSH and LH. J. Immunol. Methods. 149:247–253. [DOI] [PubMed] [Google Scholar]
- Svoboda, K., and S. M. Block. 1994. Biological applications of optical forces. Annu. Rev. Biophys. Biomol. Struct. 23:247–285. [DOI] [PubMed] [Google Scholar]
- Tees, D. F. J., R. E. Waugh, and D. A. Hammer. 2001. A microcantilever device to assess the effect of force on the lifetime of selectin-carbohydrate bonds. Biophys. J. 80:668–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, L., M. Sperandio, T. Yago, J. M. McDaniel, R. D. Cummings, S. Pearson-White, K. Ley, and R. P. McEver. 2002. P-selectin glycoprotein ligand-1-deficient mice have impaired leukocyte tethering to E-selectin under flow. J. Clin. Invest. 109:939–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, C., A. Chen, P. Kolb, and V. Moy. 2000. Energy landscape of streptavidin-biotin complexes measured by atomic force microscopy. Biochemistry. 39:10219–10232. [DOI] [PubMed] [Google Scholar]
- Zhurkov, S. N. 1965. Kinetic concept of the strength of solids. Int. J. Fracture Mech. 1:311–322. [Google Scholar]