

Abstract
Single-stranded DNA (ssDNA) is an essential intermediate in various DNA metabolic processes and interacts with a large number of proteins. Due to its flexibility, the conformations of ssDNA in solution can only be described using statistical approaches, such as flexibly jointed or worm-like chain models. However, there is limited data available to assess such models quantitatively, especially for describing the flexibility of short ssDNA and RNA. To address this issue, we performed FRET studies of a series of oligodeoxythymidylates, (dT)N, over a wide range of salt concentrations and chain lengths (10 ≤ N ≤ 70 nucleotides), which provide systematic constraints for testing theoretical models. Unlike in mechanical studies where available ssDNA conformations are averaged out during the time it takes to perform measurements, fluorescence lifetimes may act here as an internal clock that influences fluorescence signals depending on how fast the ssDNA conformations fluctuate. A reasonably good agreement could be obtained between our data and the worm-like chain model provided that limited relaxations of the ssDNA conformations occur within the fluorescence lifetime of the donor. The persistence length thus estimated ranges from 1.5 nm in 2 M NaCl to 3 nm in 25 mM NaCl.
INTRODUCTION
Single-stranded DNA (ssDNA) is an essential intermediate in many DNA metabolic processes such as replication, recombination, repair, and transcription and is specifically recognized by many proteins. It is therefore important to understand the general physical properties of ssDNA, especially its conformational flexibility. Such information is also of interest for RNA studies because stretches of unpaired bases are ubiquitous in RNA. Due to its intrinsic flexibility, the structures of isolated ssDNA in solution are not well defined and thus statistical descriptions are necessary to characterize them. However, there is limited quantitative data available that can be used to assess existing models for ssDNA flexibility, especially for short oligodeoxynucleotides that are in common use for studies of ssDNA and their interactions with proteins.
Early attempts to assess and describe the flexibility of polymeric ssDNA and RNA (Achter and Felsenfeld, 1971; Eisenberg and Felsenfeld, 1967; Inners and Felsenfeld, 1970) were hampered by the inability to obtain monodisperse samples. More recent studies of ssDNA flexibility have used a variety of experimental approaches including mechanical stretching of natural ssDNA (Maier et al., 2000; Smith et al., 1996), transient electric birefringence (Mills et al., 1999), fluorescence resonance energy transfer (FRET) (Deniz et al., 2001), fluorescence recovery after photobleaching (Tinland et al., 1997), thermal melting profiles of DNA hairpins (Kuznetsov et al., 2001), and atomic force microscopy (Rivetti et al., 1998). The above-mentioned measurements were made on both long (≫100 nucleotides) polydisperse sequences (Maier et al., 2000; Smith et al., 1996) and short (<100 nucleotides) monodisperse sequences (Deniz et al., 2001; Kuznetsov et al., 2001; Mills et al., 1999; Rivetti et al., 1998). The data were interpreted using flexible polymer models such as freely jointed chain (Flory, 1969) or worm-like chain (Kratky and Porod, 1949) models, yielding estimates of the persistence length. Although such models and their modified forms have proven useful for characterizing long ssDNA (Dessinges et al., 2002; Koch et al., 2002; Montanari and Mezard, 2001; Smith et al., 1996; Storm and Nelson, 2003; Zhang et al., 2001), as well as very long duplex DNA (Bustamante et al., 1994; Hagerman, 1988), their validity for describing the conformational statistics of short ssDNA where the chain length, unit base length, and persistence length are similar, has not been well established. Oligodeoxythymidylates (oligo-dT) do not have basepairing capabilities under the conditions used in our experiment (Saenger, 1984), and thus should provide a better example of a flexible polymer, and are widely used for studying ssDNA-protein interactions. In this work, we have used single-molecule FRET measurements of fluorescently labeled oligo-dT to examine the conformational flexibility of ssDNA over a wide range of NaCl concentrations (from 25 mM to 2 M) and chain lengths (10–70 nucleotides) to obtain a comprehensive set of data against which the applicability of various statistical models can be tested.
FRET between donor and acceptor fluorophores is widely used to examine the conformational properties of biological molecules to which they are attached (Selvin, 2000). FRET applied at the single-molecule level (Ha et al., 1996) provides a powerful means of observing the dynamic structural changes of biomolecules as well as subpopulations in a heterogeneous mixture (Brasselet et al., 2000; Deniz et al., 2000; McKinney et al., 2003; Rothwell et al., 2003; Schuler et al., 2002; Zhuang et al., 2000). The energy transfer efficiency, E, defined experimentally as the fraction of the excitation of the donor that results in the excitation of the acceptor, is related to the characteristic Förster distance, R0, and the distance, R, between the two fluorophores by
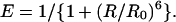 |
(1) |
We measured R0 experimentally and this allowed us to relate FRET measurements directly to the distance. The direct benefits of performing a single-molecule measurement include the ability to discriminate against inactive acceptor molecules (Deniz et al., 1999).
MATERIALS AND METHODS
Oligodeoxynucleotides
Oligodeoxynucleotide sequences were 5′-Cy5 GCCTCGCTGCCGTCGCCA-3′-Biotin and 5′-TGGCGACGGCAGCGAGGC(T)N Cy3-3′ where N ranged from 10 to 70 and were synthesized and purified as described (Cheng et al., 2001). Cy3 and Cy5 were incorporated in phosphoramidite forms and biotin was added as BiotinTEG CPG (all three from Glen Research, Sterling, VA) during automated synthesis. The DNA was gel purified and annealed at 1:1.5 ratio of Cy3:Cy5 strands in 10 ∼ 30 μM concentrations in 20 mM Tris, pH8.0, and 500 mM NaCl.
Fluorescence spectra
Fluorescence emission spectra were measured in Tris 12 ∼ 20 mM (original 20 mM Tris buffer was diluted upon adding appropriate amount of 5 M NaCl solution, reaching 12 mM Tris for data obtained at 2 M NaCl). We independently confirmed that this range of Tris concentrations does not have detectable effects on the FRET data if [NaCl] > 100 mM), pH 8.0 at 24(±1)°C and the [NaCl] specified in the text using 532 nm excitation (Jobin Yvon, Edison, NJ). The donor quantum yield was measured relative to rhodamine 101 in ethanol. Absorption spectra were obtained using a Perkin-Elmer spectrophotometer (Shelton, CT) and fluorescence anisotropies were measured using an ISS fluorometer (Champaign, IL).
Single-molecule fluorescence
Single-molecule fluorescence data were taken at 24(±1)°C with a prism-type total internal reflection microscope (Funatsu et al., 1995) based on an inverted microscope (Olympus IX70) with 60× water objective (Olympus, Melville, NY) and an intensified CCD camera (Intensified Pentamax, Roper Scientific, Trenton, NJ). DNA was added at 10 pM to the sample cell coated by bovine serum albumin-biotin and streptavidin. Measurements were taken in a buffer of Tris 12 ∼ 20 mM pH 8.0, containing an oxygen scavenging system (0.1 mg/ml glucose oxidase, 0.02 mg/ml catalase, 1% β-mercaptoethanol, and 7% (w/w) β-D-glucose), and [NaCl] between 25 mM and 2 M. The donor and acceptor emission were collected simultaneously using a 100-ms bin time by separating the two wavelengths with dichroic beam splitters (635DCLP, Chroma, Rockingham, VT) and redirecting the beams onto separate sides of the CCD face. 10 frames of data were averaged to yield single molecule donor and acceptor intensities, ID and IA. The experimental configuration results in three corrections that have been made to the raw data: a small background fluorescence signal, small contributions of the signal occur from direct excitation of the acceptor, and some donor fluorescence is detected in the acceptor channel (together typically accounting for ∼15% of the donor signal). These contributions were subtracted from the raw data to obtain a corrected value of E = IA/(IA + ID), where IA and ID are the corrected intensities of the sensitized emission of the acceptor and the donor emission, respectively (Ha et al., 1999). The correction process can yield negative values for IA or ID if FRET is very high or very low, respectively, which in turn can result in E values slightly >1 or <0.
Simulations
Metropolis Monte Carlo simulations (Metropolis et al., 1953) as described in the Results section were performed using MATLAB (Mathworks, Natick, MA) to compare the experimental data to the worm-like chain model. The probability distribution function of the end-to-end distance of a polymer as reported by Thirumalai and Ha (1998) was used to determine whether or not each randomly generated step in the simulation should be accepted.
RESULTS
Fig. 1 shows a schematic of the DNA constructs used in our experiments. Each DNA contains an 18 basepair duplex DNA possessing a 3′-(dT)N tail of varying length (N = 10–70 nucleotides). The donor fluorophore, Cy3, was covalently attached at the 3′ end of the (dT)N. The acceptor fluorophore, Cy5, was covalently attached at the 5′ end of the complementary strand of the duplex DNA, near the base of the (dT)N tail. The whole DNA molecule including the fluorophores is denoted here as dT10, … , dT70 depending on the tail length N. This geometry allows us to probe the end-to-end distance of the (dT)N tail, while maintaining nearly identical local environments of the fluorophores for all tail lengths.
FIGURE 1.

Schematic of the dTN tailed, fluorescently labeled DNA. The lengths of the 3′-single-stranded DNA ranged from N = 10–70 dT with the donor (Cy3) at the 3′ end of the tail and the acceptor (Cy5) attached to the 5′ end of the complementary strand. The molecule was labeled with biotin at the 3′ end of the complementary DNA strand that forms the duplex region to immobilize it on a streptavidin-coated quartz surface. The whole constructs including the fluorophores are denoted here as dT10, dT15, … for N = 10, 15, ….
Ensemble fluorescence spectra in bulk solution
Fig. 2 A shows three fluorescence emission spectra of DNA molecules of the type shown in Fig. 1 with N = 23 (dT23) at [NaCl] = 50 mM, 400 mM, and 2 M, displaying clearly anticorrelated, [NaCl]-dependent changes in donor and acceptor signals without significant changes in the individual spectral shapes. We used the ratio between the peak intensities of acceptor and donor spectra (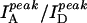 ) only as a relative measure of FRET in bulk solution because of the presence of inactive acceptor species (see Fig. 3 A). Fig. 2 B shows that the values of
) only as a relative measure of FRET in bulk solution because of the presence of inactive acceptor species (see Fig. 3 A). Fig. 2 B shows that the values of 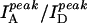 increase steeply with [NaCl], saturating around 1 M NaCl. Also shown in Fig. 2 B are the total intensities,
increase steeply with [NaCl], saturating around 1 M NaCl. Also shown in Fig. 2 B are the total intensities, 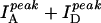 , which remain relatively constant (within 13%) for all [NaCl], reconfirming the anticorrelated nature of the fluorescence changes. These FRET changes are consistent with a reduction in the average end-to-end distance of the dT23 at higher [NaCl] as expected from additional Na+ binding to the dT23 and/or screening of electrostatic repulsion between the negative charges on the DNA backbone. However, if R0 were to vary with changes in the [NaCl], a change in FRET would be observed even without a conformational change in the molecule. R0 is defined as (Clegg, 1992),
, which remain relatively constant (within 13%) for all [NaCl], reconfirming the anticorrelated nature of the fluorescence changes. These FRET changes are consistent with a reduction in the average end-to-end distance of the dT23 at higher [NaCl] as expected from additional Na+ binding to the dT23 and/or screening of electrostatic repulsion between the negative charges on the DNA backbone. However, if R0 were to vary with changes in the [NaCl], a change in FRET would be observed even without a conformational change in the molecule. R0 is defined as (Clegg, 1992),
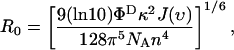 |
where ΦD is the quantum yield of the donor, J(ν) is the spectral overlap between the donor's emission and the acceptor's absorption, n is the index of refraction of the medium, and κ is determined by the relative orientation of the two dipole moments.
FIGURE 2.

FRET changes a function of [NaCl] reflect changes in average distance. (A) Bulk solution fluorescence emission spectra of 20 nM dT23 for several [NaCl]. Anticorrelated behavior is seen between the donor and acceptor intensities as the [NaCl] is changed. (B) The total intensity, 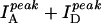 , does not vary much with [NaCl]; however their ratio,
, does not vary much with [NaCl]; however their ratio, 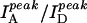 , increases with [NaCl] more drastically indicating changes in FRET. (C) Quantum yield (ΦD) of Cy3-dT23 and absorbance of Cy5-dT23 (170 nM) at various [NaCl]. (D) Anisotropy of Cy3-dT23 and Cy5-dT23 at various [NaCl].
, increases with [NaCl] more drastically indicating changes in FRET. (C) Quantum yield (ΦD) of Cy3-dT23 and absorbance of Cy5-dT23 (170 nM) at various [NaCl]. (D) Anisotropy of Cy3-dT23 and Cy5-dT23 at various [NaCl].
FIGURE 3.

(A) Single-molecule FRET efficiency histograms for dT17, dT27, and dT40 in 200 mM NaCl. The peaks at FRET ∼0 are due to DNA molecules possessing inactive Cy5, whereas the other peaks are due to FRET. Also shown are the fits to Gaussian curves. The widths of the histograms are within the measurement noise. (B) Average FRET efficiencies determined for a variety of tail lengths, N (10–70), and [NaCl] (from 25 mM to 2 M). The lines are simulations using the worm-like chain model with indicated persistence lengths, ranging from 1.5 nm to 3 nm.
The photophysical parameters that influence R0 have been measured in bulk solution (Fig. 2). The quantum yield of Cy3-dT23 (dT23 with Cy3, but without Cy5) varies by 18%, and the absorbance of Cy5-dT23 (dT23 with Cy5, but without Cy3) also varies by 18%. The rotational mobility of each dye at all [NaCl] was examined by measuring the fluorescence polarization anisotropy, which was 0.23 (±14%) for Cy3-dT23, and 0.30 (±8%) for Cy5-dT23 (Fig. 2 D). Although Cy3 and Cy5 dyes are charged and their interaction with the DNA may be affected by the changes in the salt concentration, relatively constant anisotropy values indicate that such an effect is not significant here. Although the index of refraction does not change greatly with [NaCl] (Weast, 1972), the effects of n were still taken into account when calculating ΦD (Lakowicz, 1999). We assumed that κ2 = 2/3 here to estimate the overall contribution of [NaCl] to R0. Because the value of R0 calculated using the measured values of ΦD, n, and J at each [NaCl] ranged from 59 to 61 Å (only 3.4% change), we conclude that the uncertainties in these parameters are not significant enough to change R0, and the observed change in FRET is mostly due to a change in distance, R. The assumption of κ2 = 2/3 is strictly valid only if the fluorophores freely rotate much more rapidly than the fluorescence lifetimes, hence, it is only an approximation in our studies. Although these control experiments were done for dT23 only, the conclusion is likely to hold for other ssDNA molecules because the local environment of each dye is expected to be similar regardless of the tail length.
Single-molecule fluorescence
To estimate absolute values of the FRET efficiency, E, and to discriminate against any subpopulation of DNA molecules without active acceptors, we performed single-molecule FRET measurements. Fig. 3 A shows three representative histogram plots of data for dT17, dT27, and dT40 in 200 mM NaCl. The values of E that we report were calculated by fitting the second peak to a Gaussian distribution and finding the center. The first peak centered at zero FRET is due to a population of DNA molecules containing donors, but inactive acceptors (Deniz et al., 1999). The second peak shifts to higher E values for shorter tails. Interestingly, the presence of the donor-only subpopulation serves as a control from which we can calculate the leakage of the donor signal to the acceptor detector channel. It also provides an alternative method of measuring E via donor quenching (by using E = 1 − ID / IDo, where ID and IDo are average intensities of donor for each population with and without the active acceptor, respectively).
Fig. 3 B shows the average E vs. N obtained at several [NaCl]. The average E was determined for the two E values obtained using the two different methods described above. The two methods are in good agreement with each other with the largest difference being ∼10%. The average E values along with their relative errors are given in Table 1. Because the properties of the dyes do not change significantly with [NaCl], these FRET changes mainly reflect the average change in distance between the two ends of the (dT)N.
TABLE 1.
FRET measurements calculated by IA/(IA + ID) and 1 − IDA/ID and averaged
| N | 25 mM | 50 mM | 100 mM | 200 mM | 400 mM | 800 mM | 1 M | 2 M |
|---|---|---|---|---|---|---|---|---|
| 10 | 0.84 ± 0.05 | 0.84 ± 0.07 | 0.86 ± 0.04 | 0.87 ± 0.03 | 0.87 ± 0.04 | 0.88 ± 0.04 | – | 0.90 ± 0.02 |
| 15 | 0.66 ± 0.06 | 0.68 ± 0.07 | 0.68 ± 0.08 | 0.74 ± 0.05 | 0.76 ± 0.05 | 0.80 ± 0.02 | – | 0.84 ± 0.04 |
| 17 | 0.59 ± 0.002 | 0.60 ± 0.06 | 0.62 ± 0.05 | 0.67 ± 0.04 | 0.68 ± 0.07 | 0.74 ± 0.04 | 0.74 ± 0.04 | 0.81 ± 0.02 |
| 23 | 0.38 ± 0.06 | 0.45 ± 0.05 | 0.50 ± 0.08 | 0.54 ± 0.07 | 0.61 ± 0.04 | 0.66 ± 0.04 | 0.68 ± 0.04 | 0.72 ± 0.02 |
| 27 | 0.29 ± 0.05 | 0.35 ± 0.07 | 0.38 ± 0.06 | 0.42 ± 0.04 | 0.49 ± 0.06 | 0.55 ± 0.05 | 0.58 ± 0.05 | 0.65 ± 0.03 |
| 40 | 0.10 ± 0.03 | 0.12 ± 0.04 | 0.15 ± 0.03 | 0.21 ± 0.07 | 0.256 ± 0.02 | 0.39 ± 0.04 | 0.41 ± 0.03 | – |
| 70 | – | 0.07 | 0.082 | 0.10 | 0.15 | 0.18 | 0.19 | 0.21 |
The error associated with each measurement is calculated by the spread in these two measurements.
Estimating the contour length of ssDNA
Before making comparison to theoretical models, we need to estimate the unit length of each base b0 so that the entire length of a fully stretched ssDNA of N bases, or the contour length L, is given by L = b0N. To estimate b0, we used the average of distances between adjacent phosphorus atoms determined from five crystal structures of ssDNA-protein complexes and obtained b0 = 6.3 (±0.8) Å (Fig.4 A). The protein data bank identifications are 1A1V, 1UAA, 1EYG, 1J4W, and 1JMC corresponding to the structures of HCV NS3 helicase bound to oligo-dU (Kim et al., 1998), Escherichia coli Rep helicase bound to oligo-dT (Korolev et al., 1997), E. coli SSDNA binding protein bound to oligo-dT (Raghunathan et al., 2000), Fbp protein bound to mixed sequence ssDNA (Braddock et al., 2002), and human RPA bound to poly-dC (Bochkarev et al., 1997). Although it is possible that interactions with the proteins can significantly distort the dimension of each nucleotide, 6.3 Å is consistent with the interphosphate distances of ssDNA that can range between 5.9 Å and 7 Å depending on the sugar puckering (Olson, 1975; Saenger, 1984). The 6.3 Å we used is also close to 7 Å used to fit force versus extension curve by Smith et al. (1996) and is consistent with 5–7 Å estimated by Mills et al. (1999).
FIGURE 4.

Parameters for worm-like chain model. (A) A histogram of the distances between successive ssDNA phosphorus atoms obtained from various ssDNA-protein crystal structures with the average distance being b0 = 6.3 (±0.8) Å. We assumed that the contour length of a ssDNA with N nucleotides is given by b0N. (B) Root mean square deviation between E and Esim averaged over all NaCl concentrations versus the random walk step size δ (•). The line is a second order polynomial fit that shows a minimum near δ = 5.5 Å.
Comparison to the worm-like chain model
Thirumalai and Ha (1998) derived an analytical expression for the probability distribution p(r) of the end-to-end distance of a polymer that obeys the worm-like chain model.
 |
where the normalization constant A is
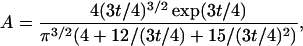 |
and t is the contour length L in multiples of the persistence length Lp, t = L/Lp, and r is the end-to-end distance R normalized to the contour length, r = R/L.
Because no time-dependent FRET changes were observed with time resolutions down to 8 ms (data not shown), we can assume that the dynamic motion of the ssDNA is fully equilibrated within 1 s averaging time used for the single-molecule FRET histograms. If the ssDNA is rigid and immobile within the fluorescence lifetime of the donor, the time-averaged FRET efficiency can be obtained by
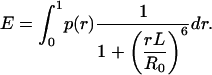 |
The other extreme case entails conformational fluctuations rapid enough to be averaged within the fluorescence lifetime of the donor. The instantaneous energy transfer rate, kr(R0/R)6 where kr is the fluorescence decay rate of the donor in the absence of energy transfer, becomes much larger than kr if the two dyes are very close to each other (R ≪ R0). Therefore, even if the probability of close approach is small, sufficiently fast conformational fluctuations will allow the energy transfer to occur before the donor decay and would yield FRET of 100% regardless of N or salt concentrations. This is clearly not the case in our studies. Nevertheless, there may be limited, but significant sampling of the phase space via intramolecular motions within the donor lifetime and this effect can influence FRET.
To compare our data to the worm-like chain model, we performed Metropolis Monte-Carlo simulations (Metropolis et al., 1953) of the end-to-end distance (R = rL) time trajectories. R is allowed to undergo a biased random walk between 0 and L taking steps of size δ (varied between 0 and 1.2 nm) every one-hundredth of the donor lifetime. Because the lifetime of Cy3 is ∼1 ns, this corresponds to a random walk in the radial coordinate R with diffusion coefficient ranging from 0 to 7 × 10−4 cm2/s. We use the probability function p(r) as the weighting function that determines whether or not each randomly generated step is accepted. For each step of the random walk, the energy transfer and donor fluorescence decay were allowed to occur with rates of kr(R0/R)6 and kr respectively, and the random walk continued until energy transfer or donor fluorescence decay occurred. Three-thousand such random walks were performed and energy transfer efficiency Esim was determined as the fraction of the random walks that ended with energy transfer.
For each NaCl concentration, the persistence length was assigned so that the root mean square deviation between the experimental E and Esim, 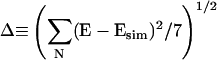 , was minimized. Here the summation is over all seven values of ssDNA length N. This process was performed for various values of the random walk step size δ, and the values of Δ averaged over all NaCl concentrations is shown in Fig. 4 B as a function of δ. The deviation is minimized at δ = 0.55 nm and grows if δ is increased or decreased. Therefore, we fixed δ at 0.55 nm and obtained the persistent length for each NaCl concentration by minimizing Δ. The persistence length thus obtained decreased from 3 nm at low salt (25 mM NaCl) to 1.5 nm at high salt (2 M NaCl) (Fig. 5). Corresponding Esim vs. N curves are shown in Fig. 3 B overlaid with the experimental E values.
, was minimized. Here the summation is over all seven values of ssDNA length N. This process was performed for various values of the random walk step size δ, and the values of Δ averaged over all NaCl concentrations is shown in Fig. 4 B as a function of δ. The deviation is minimized at δ = 0.55 nm and grows if δ is increased or decreased. Therefore, we fixed δ at 0.55 nm and obtained the persistent length for each NaCl concentration by minimizing Δ. The persistence length thus obtained decreased from 3 nm at low salt (25 mM NaCl) to 1.5 nm at high salt (2 M NaCl) (Fig. 5). Corresponding Esim vs. N curves are shown in Fig. 3 B overlaid with the experimental E values.
FIGURE 5.

Persistence length decreases with increasing salt concentration. The graph shows the persistence lengths versus [NaCl] determined by fitting the experimental data to the worm-like chain model. It was assumed that the distance between the two fluorophores can change via random walk with an elementary step size of 5.5 Å every one-hundredth of the donor lifetime.
DISCUSSION
Our choice of studying oligo-dT was made to eliminate any potential basepairing and minimize base-stacking interactions within the ssDNA structure. In contrast, heterogeneous DNA sequences were used for mechanical measurements of force versus extension curves from ssDNA and it was necessary to incorporate hairpin formations into theoretical models to account for the experimental data (Dessinges et al., 2002; Montanari and Mezard, 2001; Zhang et al., 2001). These mechanical measurements were performed on the timescales of milliseconds and seconds during which the available conformational space of the DNA is likely to be fully sampled. In contrast, fluorescence measurements have an additional internal clock, the radiative lifetime of the donor, and fluorescence signals can be influenced depending on how quickly the relative distance and orientation between the fluorophores fluctuate compared to the donor lifetime. Therefore we incorporated limited relaxations of the ssDNA conformation into the worm-like chain model to compare with the experimental data.
The best agreement could be obtained with the random walk step size δ of 0.55 nm with the stepping time of ∼0.01 ns. This corresponds to a diffusion coefficient of ∼3 × 10−4 cm2/s (= [0.55 nm]2/2/[0.01 ns]), and simulations assuming no diffusional motion during the donor lifetime or much faster diffusional motion yielded worse agreements. This diffusion coefficient could be an overestimate, however; additional relaxation processes may exist. For instance, it is likely that some relative rotations occur between the donor and acceptor during the fluorescence lifetime because the measured fluorescence anisotropy values are <0.3. If relative angles that yield large κ2 values are reached frequently, this could have an analogous effect as having very small distances, providing an additional source for fluorescence signal averaging.
Overall, the worm-like chain model can adequately describe our fluorescence data if it is assumed that some relaxation mechanisms are operating during the donor lifetime, be it diffusional motion of the ssDNA itself or fluorophores' rotational motion. The persistence length thus estimated changed by a factor of two from 3 nm to 1.5 nm when NaCl concentration was varied from 2 M to 25 mM. It is probable that the screening of the electrostatic repulsion between the negatively charged phosphate groups is responsible for the increased flexibility at higher salt concentrations.
There have been three previous studies that estimated the persistence length of oligo-dT. Rivettie et al. (1998) performed atomic force microscope imaging of DNA molecules with oligo-dT gaps up to (dT)20. They estimated the persistence length of 1.3 nm for gaps up to five nucleotides and the persistence length between 2 and 4 nm for larger gaps. Mills et al. (1999) estimated the persistence length in the range of 2 ∼ 3 nm from transient electric birefringence studies of two DNA duplexes connected by an oligo-dT gap (12 or 24 mer) in 3 ∼ 8 mM Mg2+ at ∼4°C. Kuznetsov et al. (2001) used equilibrium DNA hairpin melting profiles to obtain the persistence length of 1.4 nm in 100 mM NaCl. Therefore, our results (1.5 ∼ 3 nm) reasonably agree with previous estimates that utilized different techniques.
In a previous single-molecule FRET study by Deniz et al. (2001) it was suggested that oligo-dT in [NaCl] ≥ 100 mM is more compact than an ideal polymer. This was based on the observation that R2/N was a decreasing function of N. In an ideal polymer, 〈R2〉 scales linearly with N hence 〈R2〉/N should be a constant. Indeed, if we calculate R using Eq. 1 and experimentally determined E values, our data also show that R2/N is a decreasing function of N (data not shown). However, this procedure is unlikely to be valid because what is being averaged in FRET measurements is not R2 and the apparent deviation from the ideal polymer behavior in their analysis may have originated from the way the data were compared to the theoretical model.
CONCLUSION
This work probed the conformational flexibility of oligo-dT, in the range between 10 and 70 nucleotides from 0.025 to 2 M NaCl using FRET, a natural spectroscopic ruler. The data can be described using the worm-like chain model under the assumption that the ssDNA can partially sample the available phase space within the donor lifetime. The persistence length thus determined is a moderate function of salt concentration and decreases from 3 nm at 25 mM NaCl to 1.5 nm at 2 M. This range of persistence length is in general agreement with other studies of oligo-dT that utilized different approaches.
Acknowledgments
We thank Bob Clegg for useful suggestions, Sean McKinney for help in C++ coding, and Patrick Miller for experimental help. Also, we thank the Laboratory of Fluorescence Dynamics and Sangmin Jeon for their help in bulk solution measurements.
This work was funded by the National Institutes of Health (T.M.L.), the NIH, National Science Foundation, Research Corporation, and Searle Scholars Award (T.H.), and NIH Institutional NRSA in Molecular Biophysics (5T32GM08276, M.C.M).
References
- Achter, E. K., and G. Felsenfeld. 1971. The conformation of single-strand polynucleotides in solution: sedimentation studies of apurinic acid. Biopolymers. 10:1625–1634. [DOI] [PubMed] [Google Scholar]
- Bochkarev, A., R. A. Pfuetzner, A. M. Edwards, and L. Frappier. 1997. Structure of the single-stranded-DNA-binding domain of replication protein A bound to DNA. Nature. 385:176–181. [DOI] [PubMed] [Google Scholar]
- Braddock, D. T., J. M. Louis, J. L. Baber, D. Levens, and G. M. Clore. 2002. Structure and dynamics of KH domains from FBP bound to single-stranded DNA. Nature. 415:1051–1056. [DOI] [PubMed] [Google Scholar]
- Brasselet, S., E. J. G. Peterman, A. Miyawaki, and W. E. Moerner. 2000. Single-molecule fluorescence resonant energy transfer in calcium concentration dependent cameleon. J. Phys. Chem. B. 104:3676–3682. [Google Scholar]
- Bustamante, C., J. F. Marko, E. D. Siggia, and S. Smith. 1994. Entropic elasticity of lambda-phage DNA. Science. 265:1599–1600. [DOI] [PubMed] [Google Scholar]
- Cheng, W., J. Hsieh, K. M. Brendza, and T. M. Lohman. 2001. E. Coli Rep oligomers are required to initiate DNA unwinding in vitro. J. Mol. Biol. 310:327–350. [DOI] [PubMed] [Google Scholar]
- Clegg, R. M. 1992. Fluorescence resonance energy transfer and nucleic acids. Methods Enzymol. 211:353–388. [DOI] [PubMed] [Google Scholar]
- Deniz, A. A., M. Dahan, J. R. Grunwell, T. J. Ha, A. E. Faulhaber, D. S. Chemla, S. Weiss, and P. G. Schultz. 1999. Single-pair fluorescence resonance energy transfer on freely diffusing molecules: observation of Förster distance dependence and subpopulations. Proc. Natl. Acad. Sci. USA. 96:3670–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniz, A. A., T. A. Laurence, G. S. Beligere, M. Dahan, A. B. Martin, D. S. Chemla, P. E. Dawson, P. G. Schultz, and S. Weiss. 2000. Single-molecule protein folding: diffusion fluorescence resonance energy transfer studies of the denaturation of chymotrypsin inhibitor 2. Proc. Natl. Acad. Sci. USA. 97:5179–5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniz, A. A., T. A. Laurence, M. Dahan, D. S. Chemla, P. G. Schultz, and S. Weiss. 2001. Ratiometric single-molecule studies of freely diffusing biomolecules. Annu. Rev. Phys. Chem. 52:233–253 [Review]. [DOI] [PubMed] [Google Scholar]
- Dessinges, M. N., B. Maier, Y. Zhang, M. Peliti, D. Bensimon, and V. Croquette. 2002. Stretching single stranded DNA, a model polyelectrolyte. Phys. Rev. Lett. 89:248102. [DOI] [PubMed] [Google Scholar]
- Eisenberg, H., and G. Felsenfeld. 1967. Studies of the temperature-dependent conformation and phase separation of polyriboadenylic acid solutions at neutral pH. J. Mol. Biol. 30:17–37. [DOI] [PubMed] [Google Scholar]
- Flory, P. J. 1969. Statistical Mechanics of Chain Molecules. Interscience Publishers, New York, NY.
- Funatsu, T., Y. Harada, M. Tokunaga, K. Saito, and T. Yanagida. 1995. Imaging of single fluorescent molecules and individual ATP turnovers by single myosin molecules in aqueous solution. Nature. 374:555–559. [DOI] [PubMed] [Google Scholar]
- Ha, T., T. Enderle, D. F. Ogletree, D. S. Chemla, P. R. Selvin, and S. Weiss. 1996. Probing the interaction between two single molecules: fluorescence resonance energy transfer between a single donor and a single acceptor. Proc. Natl. Acad. Sci. USA. 93:6264–6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha, T. J., A. Y. Ting, J. Liang, W. B. Caldwell, A. A. Deniz, D. S. Chemla, P. G. Schultz, and S. Weiss. 1999. Single-molecule fluorescence spectroscopy of enzyme conformational dynamics and cleavage mechanism. Proc. Natl. Acad. Sci. USA. 96:893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman, P. J. 1988. Flexibility of DNA. Annu. Rev. Biophys. Biophys. Chem. 17:265–286. [DOI] [PubMed] [Google Scholar]
- Inners, L. D., and G. Felsenfeld. 1970. Conformation of polyribouridylic acid in solution. J. Mol. Biol. 50:373–389. [DOI] [PubMed] [Google Scholar]
- Kim, J. L., K. A. Morgenstern, J. P. Griffith, M. D. Dwyer, J. A. Thomson, M. A. Murcko, C. Lin, and P. R. Caron. 1998. Hepatitis C virus Ns3 RNA helicase domain with a bound oligonucleotide: the crystal structure provides insights into the mode of unwinding. Structure. 6:89–100. [DOI] [PubMed] [Google Scholar]
- Koch, S. J., A. Shundrovsky, B. C. Jantzen, and M. D. Wang. 2002. Probing protein-DNA interactions by unzipping a single DNA double helix. Biophys. J. 83:1098–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korolev, S., J. Hsieh, G. H. Gauss, T. M. Lohman, and G. Waksman. 1997. Major domain swiveling revealed by the crystal structures of complexes of E-Coli Rep helicase bound to single-stranded DNA and ATP. Cell. 90:635–647. [DOI] [PubMed] [Google Scholar]
- Kratky, O., and G. Porod. 1949. Recl. Trav. Chim. 68:1106–1122. [Google Scholar]
- Kuznetsov, S. V., Y. Q. Shen, A. S. Benight, and A. Ansari. 2001. A semiflexible polymer model applied to loop formation in DNA hairpins. Biophys. J. 81:2864–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz, J. R. 1999. Principles of Fluorescence Spectroscopy. Kluwer Academic/Plenum, New York, NY.
- Maier, B., D. Bensimon, and V. Croquette. 2000. Replication by a single DNA polymerase of a stretched single-stranded DNA. Proc. Natl. Acad. Sci. USA. 97:12002–12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney, S. A., A. C. Declais, D. M. J. Lilley, and T. Ha. 2003. Structural dynamics of individual Holliday junctions. Nat. Struct. Biol. 10:93–97. [DOI] [PubMed] [Google Scholar]
- Metropolis, N., A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller, and E. Tell. 1953. Equation of state calculations by fast computing machines. J. Chem. Phys. 21:1087–1092. [Google Scholar]
- Mills, J. B., E. Vacano, and P. J. Hagerman. 1999. Flexibility of single-stranded DNA: use of gapped duplex helices to determine the persistence lengths of poly(dT) and poly(dA). J. Mol. Biol. 285:245–257. [DOI] [PubMed] [Google Scholar]
- Montanari, A., and M. Mezard. 2001. Hairpin formation and elongation of biomolecules. Phys. Rev. Lett. 86:2178–2181. [DOI] [PubMed] [Google Scholar]
- Olson, W. K. 1975. Configurational statistics of polynucleotide chains. A single virtual bond treatment. Macromolecules. 8:272–275. [DOI] [PubMed] [Google Scholar]
- Raghunathan, S., A. G. Kozlov, T. M. Lohman, and G. Waksman. 2000. Structure of the DNA binding domain of E. coli SSB bound to ssDNA. Nat. Struct. Biol. 7:648–652. [DOI] [PubMed] [Google Scholar]
- Rivetti, C., C. Walker, and C. Bustamante. 1998. Polymer chain statistics and conformational analysis of DNA molecules with bends or sections of different flexibility. J. Mol. Biol. 280:41–59. [DOI] [PubMed] [Google Scholar]
- Rothwell, P. J., S. Berger, O. Kensch, S. Felekyan, M. Antonik, B. M. Wohrl, T. Restle, R. S. Goody, and C. A. Seidel. 2003. Multiparameter single-molecule fluorescence spectroscopy reveals heterogeneity of HIV-1 reverse transcriptase:primer/template complexes. Proc. Natl. Acad. Sci. USA. 100:1655–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saenger, W. 1984. Principles of Nucleic Acid Structure. Springer-Verlag, New York, NY.
- Schuler, B., E. A. Lipman, and W. A. Eaton. 2002. Probing the free-energy surface for protein folding with single-molecule fluorescence spectroscopy. Nature. 419:743–747. [DOI] [PubMed] [Google Scholar]
- Selvin, P. R. 2000. The renaissance of fluorescence resonance energy transfer. Nat. Struct. Biol. 7:730–734 [Review]. [DOI] [PubMed] [Google Scholar]
- Smith, S. B., Y. J. Cui, and C. Bustamante. 1996. Overstretching B-DNA: the elastic response of individual double-stranded and single-stranded DNA molecules. Science. 271:795–799. [DOI] [PubMed] [Google Scholar]
- Storm, C., and P. C. Nelson. 2003. Theory of high-force DNA stretching and overstretching. Phys Rev E Stat Nonlin Soft Matter Phys. 67:051906. [DOI] [PubMed] [Google Scholar]
- Thirumalai, D., and B. Y. Ha. 1998. Statistical mechanics of semiflexible chains. In Theoretical and Mathematical Models in Polymer Research. A. Grosberg, editor. Academic Press, San Diego, CA. 1–35.
- Tinland, B., A. Pluen, J. Sturm, and G. Weill. 1997. Persistence length of single-stranded DNA. Macromolecules. 30:5763–5765. [Google Scholar]
- Weast, R. C. 1972. CRC Handbook of Chemistry and Physics. R. C. Weast, editor. Chemical Rubber Co., Cleveland, Ohio.
- Zhang, Y., H. Zhou, and Z. C. Ou-Yang. 2001. Stretching single-stranded DNA: interplay of electrostatic, basepairing, and basepair stacking interactions. Biophys. J. 81:1133–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang, X. W., L. E. Bartley, H. P. Babcock, R. Russell, T. J. Ha, D. Herschlag, and S. Chu. 2000. A single-molecule study of RNA catalysis and folding. Science. 288:2048–2051. [DOI] [PubMed] [Google Scholar]