

Abstract
A major result of incoherent elastic neutron-scattering experiments on protein powders is the strong dependence of the intramolecular dynamics on the sample environment. We performed a series of incoherent elastic neutron-scattering experiments on lyophilized human butyrylcholinesterase (HuBChE) powders under different conditions (solvent composition and hydration degree) in the temperature range from 20 to 285 K to elucidate the effect of the environment on the enzyme atomic mean-square displacements. Comparing D2O- with H2O-hydrated samples, we were able to investigate protein as well as hydration water molecular dynamics. HuBChE lyophilized from three distinct buffers showed completely different atomic mean-square displacements at temperatures above ∼200 K: a salt-free sample and a sample containing Tris-HCl showed identical small-amplitude motions. A third sample, containing sodium phosphate, displayed highly reduced mean-square displacements at ambient temperature with respect to the other two samples. Below 200 K, all samples displayed similar mean-square displacements. We draw the conclusion that the reduction of intramolecular protein mean-square displacements on an Ångstrom-nanosecond scale by the solvent depends not only on the presence of salt ions but also on their type.
INTRODUCTION
During the last decade several proteins have been investigated by incoherent elastic neutron-scattering (IENS) in a powder or a solution environment (reviewed by Gabel et al., 2002). Experimental parameters that have been varied in such experiments have included hydration, solvent composition, and temperature. These investigations have yielded important information concerning the dependence of internal protein dynamics on the protein environment on a pico- to nanosecond timescale. Hydration and temperature, in particular, affect protein flexibility strongly, as has been shown, e.g., in the case of bacteriorhodopsin in the purple membrane (reviewed by Zaccai, 2000). Hydrated samples, in general, undergo a so-called dynamical transition in the temperature range 180–230 K, which has been proposed to be related to biological function (Rasmussen et al., 1992; Ferrand et al., 1993). Above the dynamical transition temperature, nonharmonic motions occur which are not observed below it (Zaccai, 2000). It has been postulated that above the dynamical transition protein atoms are able to explore conformational substates which are inaccessible at lower temperatures (Frauenfelder et al., 1988). This transition is not observed for dry samples. Trehalose and glycerol have also been used to modify protein powder environments (Cordone et al., 1999; Réat et al., 2000; Tsai et al., 2000; Paciaroni et al., 2002). These cryoprotectants have been shown to suppress nonharmonic dynamics up to ambient temperature (Cordone et al., 1999), reduce mean-square displacements (MSDs) with respect to dry samples up to ∼300 K, and shift the transition temperature (Tsai et al., 2000) to higher temperatures. Although these experiments have shed light on the influence of the molecular environment on protein internal dynamics, an important biological parameter of the molecular environment has not been investigated fully by this technique to date—namely the influence of different types of salt. However, this issue is of major biological importance, since physiological protein environments essentially consist of saline aqueous solutions and in vitro biochemical studies are always under buffered conditions. Tehei et al. (2001) examined the effect of high salt concentration on the internal dynamics of halophilic proteins and found results which are probably related to the specific molecular adaptation of these proteins to an extreme environment. In the present work we found the surprising result that buffer salt composition influenced protein internal dynamics, even in hydrated powder samples, in which bulk solvent was absent.
The strong dependence of protein dynamics on the environment has aroused controversy as to whether protein dynamics is “slaved” to the dynamics of the environment (Hong et al., 1990; Fenimore et al., 2002; Paciaroni et al., 2002), resulting in the dynamics of the solvent molecules and the protein atoms being correlated over a wide temperature and time range.
Thermal neutrons provide a unique probe for investigating protein molecular dynamics on an Ångstrom length scale and pico- to nanosecond timescale. They have energies of the order of thermal molecular motions (several meV) and wavelengths comparable to intramolecular distances (several Ångstroms). They are able to exchange a measurable amount of both their intrinsic energy and momentum in a single scattering event with nuclei in the sample, and can penetrate deeply into bulk matter due to their neutrality. Since the incoherent scattering cross section of the hydrogen (H) atom exceeds the scattering cross sections (coherent as well as incoherent) of all other nuclei typically abundant in biological samples (mainly C, O, N, P, and S) as well as that of the hydrogen isotope deuterium (D) by at least an order of magnitude, total scattering is dominated by the incoherent scattering of the hydrogen nuclei. Since the latter are uniformly distributed in biological macromolecules, the scattering by a biological sample represents mean global hydrogen dynamics in the space-time window selected by instrument characteristics (energy resolution, wave vector transfer), typically representing protein side-chain and backbone dynamics on a pico- to nanosecond timescale and on an Ångstrom length scale (Smith, 1991).
Beginning in the late 1980s with work of Doster et al. (1989), deuterated water (D2O) has been used as a hydration solvent for neutron-scattering experiments on protein powders. D2O has been preferred over the natural solvent, H2O, so as to reduce contributions from the solvent to the scattering signal. In the present article, we investigate the influence of solvent composition on protein internal dynamics as well as on hydration water dynamics by comparing incoherent neutron-scattering data from protein powders hydrated in H2O and D2O. There are very few incoherent elastic neutron-scattering studies comparing H2O- with D2O-hydrated samples (Settles and Doster, 1996; Tehei et al., 2001) and, to our best knowledge, no systematic study investigating the influence of solvent composition on both protein internal dynamics and hydration water in powder samples has been carried out up to date.
To elucidate the role of the solvent composition on global protein molecular dynamics on an Ångstrom-nanosecond scale, we performed a series of IENS experiments on human serum butyrylcholinesterase (HuBChE; E.C. 3.1.1.8) powders, comparing samples under various experimental conditions (hydrated vs. dry; salt-containing vs. salt-free).
HuBChE is a homotetrameric sialoglycoprotein synthesized in the liver, whose crystal structure was recently solved (Nicolet et al., 2003). The tetramer contains 574 amino acids and nine n-glycan chains per catalytic subunit monomer (Lockridge et al., 1987), carbohydrate making up ∼27% of the dry weight (von Haupt et al., 1966). It is a pharmacologically and toxicologically relevant enzyme that is known to act as a catalytic scavenger for certain natural and synthetic carboxyl-esters, such as cocaine (Sun et al., 2001), heroin (Lockridge et al., 1980), and aspirin (Masson et al., 1998). It also reacts as a stoichiometric scavenger with organophosphates (Ashani et al., 1991; Doctor, 2003), thus having the capacity to provide prophylactic protection against highly toxic chemical warfare nerve agents, such as soman and VX (Raveh et al., 1997). Data presented by Burgess and Lister (1988) and Burgess and Oxendine (1993) suggest that the resistance of HuBChE, and of the structurally very similar enzyme, acetylcholinesterase (AChE, E. C. 3.1.1.7), against acid and thermal inactivation depends strongly on the presence and type of solvent ions. Protection against inactivation follows the Hofmeister series (von Hippel and Schleich, 1969), with kosmotropic ions being more efficient than chaotropic ions. The molecular mechanism underlying the protective capacity of kosmotropic ions is, however, unclear.
MATERIALS AND METHODS
Enzyme purification
Four batches of HuBChE were purified to homogeneity from outdated human plasma according to modification of the procedure of Grunwald et al. (1997). The three-step procedure employed uses filtration, pH adjustment, batch adsorption, and/or column chromatography on procainamide-sepharose gel followed by ion exchange chromatography. The highly purified HuBChE was stored after lyophilization from a 10 mM sodium phosphate buffer (pH 8.0) in the solid form. Enzyme activities ranged typically from 650 to 700 U/mg (Ellman et al., 1961) for the samples before neutron-scattering experiments. SDS polyacrylamide gel electrophoresis (T = 10%; Laemmli, 1970) revealed a dominant band at ∼85 kDa, assigned to the HuBChE monomer and a weaker band at ∼170 kDa assigned to residual HuBChE dimers. Contamination by other proteins was negligible.
Sample preparation for neutron-scattering
The four samples from the same batch of enzyme preparation were dialyzed against ammonium acetate, sodium phosphate, or Tris-HCl buffer solutions, lyophilized and used for neutron-scattering experiments. Two samples were modified (see Samples in Detail) after the neutron-scattering experiments, and subjected to a second cycle of neutron-scattering data collection. The lyophilized powders (typically ∼100–200 mg) were placed in aluminum sample holders (30 × 40 × 0.2 mm3), dried for two days at atmospheric pressure over silica gel, and weighed to determine their dry weight (h = 0 g water per g dry powder, denoted by g/g in the following). Hydration of samples was carried out by water (D2O or H2O) vapor exchange during 2–3 days over either pure water (100% relative humidity) or saturated aqueous KNO3 (93% relative humidity) at 25°C. The water content was determined by weighing shortly before closing the sample holders and was close to 0.45 g/g for all samples. To verify that no loss of material had occurred, samples were weighed both before and after the neutron-scattering experiments. No losses were detected for any sample. The samples were assayed before and after neutron-scattering experiments by the Ellman procedure (Ellman et al., 1961), with butyrylthiocholine iodide (1 mM) as the substrate. Sample activity was reduced by ∼20% or less after neutron-scattering experiments for all samples measured. SDS-polyacrylamide gel electrophoresis (as described under Enzyme Purification) showed the same single polypeptide band before and after neutron-scattering experiments.
Samples in detail
HuBChE-H2O
A first batch of purified HuBChE, purified as described above, was dialyzed against 25 mM ammonium acetate (150 ml) pH 7.0, with six changes of the dialysis buffer in three days and lyophilized. Since ammonium acetate is volatile, this should result in a salt-free HuBChE preparation. Indeed, lyophilization of a sample of 1 M ammonium acetate alone left no solid residue. The lyophilized sample was hydrated (H2O) to 0.44 g/g and taken for neutron-scattering measurements.
HuBChE-dry
The HuBChE-H2O sample as described above was dried over silica gel at atmospheric pressure for 48 h and resubmitted to neutron-scattering.
HuBChE-Phos-D2O
A second batch of purified HuBChE as described above was lyophilized from 20.1 ml of 10 mM sodium phosphate, pH 8.0. The lyophilized powder was hydrated in an atmosphere of pure D2O to 0.46 g/g.
HuBChE-Phos-H2O
A third batch of purified HuBChE was lyophilized twice (total volume of 62.7 ml) from 5 mM sodium phosphate, pH 8.0. The powder was hydrated over pure H2O, followed by saturated aqueous KNO3, to 0.43 g/g.
HuBChE-D2O
A fourth batch of purified HuBChE was lyophilized from 25 mM ammonium acetate, pH 8.0. The powder was hydrated under an atmosphere of D2O to 0.47 g/g.
HuBChE-Tris-D2O
The major part of the HuBChE-D2O sample was redissolved in 63 ml of 10 mM Tris-HCl, pH 8.0 buffer solution. After lyophilization, the sample was hydrated by D2O vapor exchange to 0.43 g/g and resubmitted to neutron-scattering.
Ion concentrations in samples
The samples lyophilized from an ammonium acetate buffer (HuBChE-H2O, HuBChE-dry, and HuBChE-D2O) are salt-free, whereas the samples HuBChE-Phos-D2O, HuBChE-Phos-H2O, and HuBChE-Tris-D2O all contain salt. For the samples lyophilized from the phosphate and Tris buffers, the amounts of ions were calculated on the basis of buffer volume, buffer pH, and salt concentration. The amount of protein was then determined by subtracting the amount of salt from the dry weight of the lyophilized sample. The amounts of protein, salt, and water in each sample are listed in Table 1. To allow a comparison of the neutron-scattering results, all salt-containing samples were prepared from solutions at pH 8.0. At this pH, and assuming a pK2 of 7.2, the Henderson-Hasselbalch equation yields a ratio of 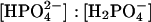 of ∼4:1. For HuBChE-Phos-D2O, the number of sodium ions per protein molecule was estimated by determination of sodium by flame spectrometry (Corning 480 flame photometer, CIBA Corning, Sergy-Pontoise, France), multicalurione (140 Na/5K, Chiron Diagnostics, Halstead, UK) used as a gauge and by assaying the protein using bicinchoninic acid (Smith et al., 1985). A ratio of ∼2600 Na+ ions per tetramer was thus obtained. This value is considerably higher than that calculated based on the salt/protein mass ratio (Table 1); i.e., ∼1650 Na+ ions per tetramer. For the HuBChE-Phos-H2O, Table 1 gives a ratio of 700 Na+ ion per tetramer. At 25°C and pH 8.0, assuming a pKa of 8.15, the Tris-HCl buffer consists of a mixture of cationic Tris·H+ and neutral Tris in a ratio [Tris]:[Tris·H+] of ∼0.7. The HuBChE-Tris-D2O buffer volume (63 ml) and concentration (10 mM) before lyophilization were chosen so that the number of Cl− ions (equal to the number of Tris·H+ ions) per tetramer was identical to the total number of anions per tetramer of the HuBChE-Phos-D2O sample, resulting in a ratio of ∼1350 Tris-H+ ions per tetramer.
of ∼4:1. For HuBChE-Phos-D2O, the number of sodium ions per protein molecule was estimated by determination of sodium by flame spectrometry (Corning 480 flame photometer, CIBA Corning, Sergy-Pontoise, France), multicalurione (140 Na/5K, Chiron Diagnostics, Halstead, UK) used as a gauge and by assaying the protein using bicinchoninic acid (Smith et al., 1985). A ratio of ∼2600 Na+ ions per tetramer was thus obtained. This value is considerably higher than that calculated based on the salt/protein mass ratio (Table 1); i.e., ∼1650 Na+ ions per tetramer. For the HuBChE-Phos-H2O, Table 1 gives a ratio of 700 Na+ ion per tetramer. At 25°C and pH 8.0, assuming a pKa of 8.15, the Tris-HCl buffer consists of a mixture of cationic Tris·H+ and neutral Tris in a ratio [Tris]:[Tris·H+] of ∼0.7. The HuBChE-Tris-D2O buffer volume (63 ml) and concentration (10 mM) before lyophilization were chosen so that the number of Cl− ions (equal to the number of Tris·H+ ions) per tetramer was identical to the total number of anions per tetramer of the HuBChE-Phos-D2O sample, resulting in a ratio of ∼1350 Tris-H+ ions per tetramer.
TABLE 1.
Masses (in mg) of the components of the various samples
| Protein | Salt | Water | |
|---|---|---|---|
| HuBChE-Phos-D2O | 80 | 28 | 50 |
| HuBChE-Phos-H2O | 183 | 27 | 94 |
| HuBChE-dry | 117 | — | — |
| HuBChE-H2O | 117 | — | 52 |
| HuBChE-D2O | 145 | — | 68 |
| HuBChE-Tris-D2O | 98 | 75 | 75 |
Incoherent elastic neutron scattering
Considering only incoherent scattering from the sample hydrogen atoms, the theoretical scattering law can be written in the form
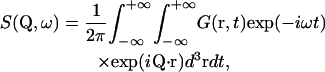 |
(1) |
with the intermediate scattering function
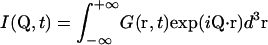 |
(2a) |
and the hydrogen atom autocorrelation function
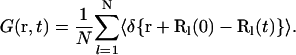 |
(2b) |
Q is the wave vector transfer in a scattering event, i.e., the neutron momentum exchange in units of ℏ. Splitting I(Q,t) into a time-independent and a time-dependent part (see e.g., Bée, 1988) gives
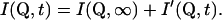 |
(3a) |
The scattering law can be separated into the elastic incoherent scattering factor (EISF) and a quasielastic contribution due to the time-dependence of the atomic motions,
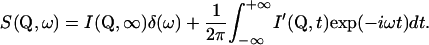 |
(3b) |
The finite instrumental resolution is taken into account by a convolution of the theoretical scattering law with the instrumental resolution function,
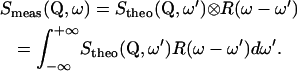 |
(4) |
The elastic intensity (EI) measured in an elastic scan is given by
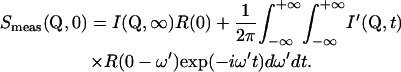 |
(5) |
It is composed of an instrument-independent part and a term depending on the instrumental resolution function stemming from quasielastic motions. The instrument-independent part (the EISF) can be attributed to the time-independent part of the atomic equilibrium distribution. The integral making up the time-dependent part is related to the instrumental time resolution: The better the energy resolution, the larger the associated time resolution. This means that part of the measured EI is also due to the time-dependent motions for an instrument with a finite energy resolution. The measured EI is, therefore, not equivalent to the EISF. For confined motions complying with the condition
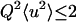 |
(6) |
(where 〈u2〉 are the MSDs), the EI can be approximated by
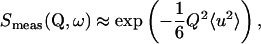 |
(7) |
the so-called Gaussian approximation, which is in analogy to the Guinier approximation (Guinier and Fournet, 1955) in small-angle scattering (SAS).
Instrumental aspects and data analysis
Neutron-scattering experiments were carried out on the backscattering spectrometer IN16 (energy resolution ΔE = 0.9 μeV, corresponding to a time window of ∼730 ps; accessible Q-range: 0.19 to 1.93 Å−1) in the temperature range from 20 to 285 K at the Institut Laue-Langevin, Grenoble, France.
Transmission values varied between 0.9 and 0.95 according to the sample. The scattered signals were corrected for container scattering, absorption, and instrumental resolution (determined by separate runs of the empty container and of a vanadium sample) by a standard FORTRAN 77 program Iq0 based on the correction formula of Paalman-Pings coefficients (Bée, 1988). Data were not corrected for multiple scattering.
To assure the absence of (salt or water) crystalline structures in the samples, we used the IN16 option to record diffraction spectra in parallel to the elastic spectra. We did not observe Bragg peaks corresponding to crystalline water or salt in any of the samples at any of the measured temperature points.
Guinier plots and the determination of atomic mean-square displacements
The EI of each sample at a given temperature T was normalized to its EI at the lowest temperature (∼20 K) for each detector, and the values so-obtained were plotted in a semilogarithmic plot versus Q2 (we refer to these plots in the following as Guinier plots due to their analogy to the Guinier plots in SAS), i.e., as a function of the detector angle. MSD were extracted from two linear regimes in these plots. The plots can be interpreted in analogy to Guinier plots of polydisperse dilute solutions in SAS. In these experiments, linear regimes can be assigned to populations of distinct radii of gyration (Guinier and Fournet, 1955). In analogy, if linear regimes are present in IENS Guinier plots, they can be associated to dynamically different atomic populations displaying distinct MSD values. For all the samples that we measured, a more or less pronounced kink appeared at ∼200 K. Accordingly, two linear fit zones were selected, above and below the kink (Figs. 1–3): A low Q-range, 0.19 Å−2 < Q2 < 1.13 Å−2 and a high Q-range, 1.13 Å−2 < Q2 < 2.46 Å−2, which we attributed, respectively, to a large-amplitude population and a small-amplitude population. The criterion for the validity of the Gaussian approximation (Eq. 6) was verified a posteriori.
FIGURE 1.

Guinier plots of the logarithms of the normalized (to 20 K) EIs of HuBChE-H2O, HuBChE-D2O, HuBChE-Phos-H2O, and HuBChE-Phos-D2O as a function of Q2 at 200 K.
FIGURE 3.

Guinier plots of the logarithms of the normalized (to 20 K) EI of HuBChE-D2O, HuBChE-Tris-D2O, HuBChE-Phos-D2O, and HuBChE-dry as a function of Q2 at 280 K.
A linear regime in a Guinier plot can be expressed mathematically by
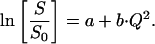 |
(8) |
S0 is the EI at the lowest temperature (normalization temperature) and S at an arbitrary measured temperature. The value b is the slope leading to the MSDs and a is the intersection with the ordinate. For a single dynamical population lacking quasielastic contributions (i.e., confined motions that display no time-dependence on the scale of the instrumental time-resolution), a is 0. However, as soon as a population displaying, e.g., large-scale diffusive motions (not complying to the Gaussian approximation) is present in the sample, the normalized EI is shifted to lower values in the Guinier plots and the intersection with the ordinate of linear regressions in a linear regime renders a < 0 (Gabel, unpublished). The same holds for the situation where two or more linear regimes (due to dynamically different populations) are present in the Guinier plots. In both cases, the situation can be expressed mathematically by the following equation:
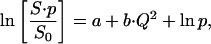 |
(9) |
where p′ = 1−p can be considered as a measure for contributions not complying to the Gaussian approximation in the Q-range where the linear regression is carried out (assuming a = 0). We interpret p′ in terms of a very-large-amplitude population which may comprise large-amplitude, confined motions complying to the Gaussian approximation (Eq. 7) in a lower Q-range and/or motions of quasielastic/diffusive character. We stress that Eq. 9 is a phenomenological equation that is justified here (within the interpretation given in the preceding sentence) by its ability to fit the experimental data. The loss p of the elastic intensity at higher temperatures with respect to the normalization temperature expressed by Eq. 9 has, in general, two origins: an increase of the amplitude of the motions described by a modification of the EISF (which is independent of the instrumental energy resolution) as well as the onset of quasielastic, timescale-dependent motions that are dependent on the instrumental resolution (compare to Becker and Smith, 2003). The numerical value of p′ is obtained by the intersection of the extrapolation of the linear regression of the low Q-range (0.19 Å−2 < Q2 < 1.13 Å−2) with the Guinier plot y axis (Fig. 2; numerical values for the investigated samples are presented in Table 2). This information about the non-Gaussian character of motions would be lost by focusing exclusively on the MSD, without taking the underlying Guinier plots into account.
FIGURE 2.

Guinier plots of the logarithms of the normalized (to 20 K) EI of HuBChE-H2O, HuBChE-D2O, HuBChE-Phos-H2O, and HuBChE-Phos-D2O as a function of Q2 at 280 K. The linear fits in the low (0.19 Å−2 < Q2 < 1.13 Å−2) and high Q-ranges (1.13 Å−2 < Q2 < 2.46 Å−2) (compare to Materials and Methods) are traced by continuous lines for the HuBChE-H2O sample. The extrapolation of the low fit range toward the ordinate having served to extract the very-large-amplitude populations according to Eq. 9 (compare to Materials and Methods) is also traced for HuBChE-H2O.
TABLE 2.
“Very-large-amplitude” populations p′ according to Eq. 9 for the various samples; extracted by the intersection of the extrapolation of the linear regression of the low Q-range with the y axis (Fig. 2)
| Very-large-amplitude populations p′ | |
|---|---|
| HuBChE-Phos-D2O | 17% |
| HuBChE-Phos-H2O | 28% |
| HuBChE-D2O | 35% |
| HuBChE-H2O | 41% |
| HuBChE-Tris-D2O | 37% |
| HuBChE-dry | 7% |
Contributions to the scattering signal of the various sample components (Table 3)
TABLE 3.
Contributions of sample components as a percentage of the total sample scattering signal
| Protein | Sugars | Water | Salt | |
|---|---|---|---|---|
| HuBChE-H2O | 45% (93%) | 13% (94%) | 42% (97%) | — |
| HuBChE-D2O | 75% (89%) | 16% (83%) | 9% (0%) | — |
| HuBChE-Phos-H2O | 42% (93%) | 13% (94%) | 44% (97%) | 1% (78%) |
| HuBChE-Phos-D2O | 69% (89%) | 16% (83%) | 13% (0%) | 2% (16%) |
| HuBChE-dry | 77% (93%) | 23% (94%) | — | — |
| HuBChE-Tris-D2O | 40% (89%) | 10% (83%) | 9% (0%) | 41% (86%) |
In parentheses: percentage of the contributions due to incoherent scattering from H-atoms of the components.
To determine the contributions of the various components of the samples to the total signal, we calculated the scattering due to the salt molecules, the hydration water, the polysaccharide chains, and the protein for each sample based on their composition, atomic-scattering cross sections taken from the literature (Bée, 1988), and the molecular weights of the components.
The unglycosylated HuBChE monomeric subunit (C2962H4466N772O849S19) has a molecular weight of 65,084 Da (Swiss Prot entry P06276) and a total scattering cross section of ∼394,000 barns (1 barn = 10−24 cm2). It has 1134 potentially exchangeable H-atoms (amide backbone and side-chain hydrogens; calculated from Swiss Prot entry P06276 and Wüthrich, 1986), corresponding to ∼25% of all protein hydrogen atoms. For the samples hydrated by D2O vapor exchange over 24 h or more, we assume that a major part of the exchangeable H-atoms were replaced by D. As shown by Schinkel et al. (1985) on lyophilized lysozyme powders, ∼80% of all potentially exchangeable hydrogen atoms are exchanged after 24 h under conditions of equilibrium vapor exchange. In the samples investigated, it can, therefore, be assumed that a major part of the nonbackbone, surface H-atoms, as well as part of the backbone amide atoms, are exchanged. We estimate, therefore, that 20% of the totality of protein hydrogen atoms (or ∼900) are replaced by D resulting in a total scattering cross section of 327,000 barns for the D-exchanged, unglycosylated HuBChE monomer.
The contribution of the oligosaccharides was estimated on the basis of glycosylated equine butyrylcholinesterase (EqBChE) (Saxena et al., 1997). The chemical formula of a single oligosaccharide chain is given by C110H148O77N6, with 66 exchangeable H-atoms. The totality of nine glycosylation sites per monomer has, therefore, a total of 594 exchangeable H-atoms, and a chemical formula C990H1332O693N54 (25,056 Da). The total cross section of the nine nonexchanged oligosaccharide chains per monomer is 118,000 barns and 74,000 barns in the case of D-exchange. The preceding calculations are based on the assumption that all potentially exchangeable H-atoms of the sugars are indeed exchanged, due to their high exposure to the solvent.
The sodium phosphate molecules can be represented by the chemical formulae Na2HPO4 and NaH2PO4, with molecular weights of 142 Da and 120 Da, respectively (143 Da and 122 Da in the case of H-D-exchange). Assuming a “pH memory” for lyophilized compounds (Costantino et al., 1997), a value of pH 8.0 for the solution leads, via the Hendersson-Hasselbalch equation, to an ionic ratio of 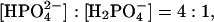 i.e., the divalent anion concentration is fourfold higher than that of the monovalent anion. The total cross sections of the nonexchanged Na2HPO4 and NaH2PO4 are 109 and 187 barns, respectively. For the exchanged molecules, the values are 35 and 39 barns, respectively.
i.e., the divalent anion concentration is fourfold higher than that of the monovalent anion. The total cross sections of the nonexchanged Na2HPO4 and NaH2PO4 are 109 and 187 barns, respectively. For the exchanged molecules, the values are 35 and 39 barns, respectively.
The total cross section per H2O molecule (18 Da) is 168 barns and per D2O molecule (20 Da) is 19 barns.
Tris-HCl (C4H11NO3) has five exchangeable H-atoms. The total scattering cross section of Tris after an H-D-exchange, is therefore, 575 barns.
In Table 3, the scattering contributions of each component of each sample are given as a percentage of the total scattered intensity. Additionally, the percentage of incoherent scattering of each component to its total scattering is given in parentheses.
RESULTS
Guinier plots
Figs. 1 and 2 show the natural logarithm of the normalized (to lowest temperature) EI of the samples HuBChE-H2O, HuBChE-D2O, HuBChE-Phos-H2O, and HuBChE-Phos-D2O, as a function of Q2, at 200 and 280 K, respectively. At 200 K (Fig. 1), data points for all four samples are virtually superimposable, decreasing slightly in intensity over the low and high Q-ranges under investigation. The same is true at temperatures below 200 K (not shown): The ln(S/S0) curves (logarithm of the normalized EI) for all four samples are qualitatively similar (superimposed and slightly decreasing with increasing Q-value) (not shown). In contrast, at 280 K (Fig. 2), the curves for the four samples differ in slope and are y-shifted with respect to each other. The contributions of the very-large-amplitude populations (see Materials and Methods) for all four samples at 280 K, determined by the intersection of the linear regressions (Eq. 9) of the low Q-range with the y axis, are presented in Table 2. In Fig. 2, the extrapolation of the linear regression is traced for HuBChE-H2O.
For both the HuBChE-Phos-H2O and HuBChE-Phos-D2O samples at 280 K, the slopes are almost identical (Fig. 2) in both the low and the high Q-ranges used for the linear fits. However, they are shifted mutually, a fact not taken into account by MSDs: From the intersection of the linear regression of the low Q-range with the y axis and utilizing Eq. 9, one obtains a 17% very-large-amplitude population for HuBChE-Phos-D2O. For HuBChE-Phos-H2O the corresponding value is 28% (Table 2).
An almost Q-independent y-shift is observed between the plots of the logarithms of the normalized EI for HuBChE-H2O and HuBChE-D2O, which is similar to that between the two HuBChE-Phos samples (Fig. 2). However, the slope is larger than for the HuBChE-Phos samples, and a pronounced kink is present at ∼1.1 Å−2, indicating that the low and high Q-ranges represent two distinct dynamical populations. The very-large-amplitude populations obtained from the lowQ-range represent 35% and 41% of the total populations for HuBChE-D2O and HuBChE-H2O, respectively.
A surprising result is the large difference in position on the y axis between the curves for HuBChE-D2O and HuBChE-Phos-D2O, which must be due exclusively to nonexchangeable protein H-atoms. However, both for samples in D2O and in H2O, the presence of sodium phosphate results in a change in slope, especially in the low Q-range.
In Fig. 3, the logarithm of the normalized EI is traced as a function of Q2 for HuBChE-D2O, HuBChE-Phos-D2O, HuBChE-Tris-D2O, and HuBChE-dry. At 280 K, the dry sample displays the smallest y-shift, with a very-large-amplitude population of only 7%. However, its slope is identical to that of the HuBChE-Phos-D2O sample. The logarithm of the normalized EI of the HuBChE-Tris-D2O sample displays the largest negative shift on the y axis, indicating a notable loss of the EI at 280 K (very-large-amplitude population of 37% for the low Q-range). In the high Q-range, the slope for the HuBChE-Tris-D2O sample is similar to that for the HuBChE-D2O sample, whereas in the low Q-range it is larger.
Mean-square displacements
HuBChE-H2O, HuBChE-D2O, and HuBChE-dry: low Q-range (Fig. 4 A)
FIGURE 4.

(A) MSDs extracted by linear regression from the Guinier plots in the low Q-range (0.19 Å−2 < Q2 < 1.13 Å−2) for HuBChE-H2O, HuBChE-D2O, and HuBChE-dry. (B) MSDs extracted by linear regression from the Guinier plots in the high Q-range (1.13 Å−2 < Q2 < 2.46 Å−2) for HuBChE-H2O and HuBChE-D2O.
In the low Q-range, the dry sample displays a linearly increasing MSD through to 300 K (with a slight kink at 120 K), whereas its hydrated counterparts undergo a dynamical transition at ∼220 K, similar to that observed for other proteins (Doster et al., 1989; Ferrand et al., 1993). The deviation from a constant linear increase in MSDs sets in for both the H2O- and the D2O-hydrated samples at ∼220 K. Above the dynamical transition, the MSDs for HuBChE-D2O increases somewhat more slowly than for HuBChE-H2O. This difference is most pronounced above 270 K.
HuBChE-H2O and HuBChE-D2O: high Q-range (Fig. 4 B)
The main difference between both samples in the highQ-range is that the transition for HuBChE-D2O is shifted to higher temperatures and more pronounced than for its HuBChE-H2O counterpart. The MSDs remain at a very low level for HuBChE-D2O. At the highest measured temperatures, the MSDs for both samples merge again.
HuBChE-Phos-H2O, HuBChE-Phos-D2O, and HuBChE-dry: low Q-range (Fig. 5)
FIGURE 5.

MSDs extracted by linear regression from the Guinier plots in the low Q-range (0.19 Å−2 < Q2 < 1.13 Å−2) for HuBChE-Phos-H2O, HuBChE-Phos-D2O, and HuBChE-dry.
Whereas low Q-range MSDs for HuBChE-Phos-D2O and HuBChE-dry are almost identical up to the highest measured temperature, those for HuBChE-Phos-H2O begin to deviate from linearity above 240 K. However, this increase in slope ends at ∼265 K. Above 280 K, MSDs for HuBChE-Phos-H2O are similar to those for HuBChE-Phos-D2O.
HuBChE-H2O and HuBChE-Phos-H2O: low and high Q-range (Fig. 6)
FIGURE 6.

MSDs extracted by linear regression from the Guinier plots in the low (0.19 Å−2 < Q2 < 1.13 Å−2) and in the high Q-range (1.13 Å−2 < Q2 < 2.46 Å−2) for HuBChE-H2O and HuBChE-Phos-H2O.
The separation into two fit-ranges reveals two populations above 200 K for HuBChE-H2O, reflecting the onset of a dynamical transition at that temperature. Choosing a single fit-range comprising both zones would have masked this effect, resulting in averaged MSDs for the two populations. Below 200 K, no distinction can be made between small- and large-amplitude populations. In the low Q-range, MSDs for HuBChE-H2O deviate quite rapidly from linear behavior beyond 200 K, the MSDs for HuBChE-Phos-H2O are smaller above 200 K, and undergo a dynamical transition only at 240 K. Above 265 K, their values plateau.
HuBChE-D2O, HuBChE-Phos-D2O, and HuBChE-Tris-D2O: high Q-range (Fig. 7)
FIGURE 7.

MSDs extracted by linear regression from the Guinier plots in the high Q-range (1.13 Å−2 < Q2 < 2.46 Å−2) for HuBChE-D2O, HuBChE-Tris-D2O, and HuBChE-Phos-D2O.
When focusing only on dynamics of the nonexchangeable protein hydrogen atoms by using heavy water as a hydration medium, the same general behavior is observed as in the H2O-hydrated samples, i.e., protein atomic motions of HuBChE-Phos-D2O are reduced at higher temperatures (> 250 K) relative to HuBChE-D2O. Below this temperature, the presence of sodium phosphate does not seem to influence the atomic motions in the accessible space-time window. In contrast to MSDs for HuBChE-Phos-D2O, those for HuBChE-Tris-D2O are not reduced above 250 K with respect to MSDs for HuBChE-D2O.
HuBChE-D2O, HuBChE-Phos-D2O, and HuBChE-Tris-D2O: low Q-range (Fig. 8)
FIGURE 8.

MSDs extracted by linear regression from the Guinier plots in the low Q-range (0.19 Å−2 < Q2 < 1.13 Å−2) for HuBChE-D2O, HuBChE-Tris-D2O, and HuBChE-Phos-D2O.
The temperature-dependence of MSDs for HuBChE-D2O and HuBChE-Phos-D2O in the low Q-range is qualitatively similar to that in the high Q-range, the HuBChE-D2O MSDs being significantly larger above 250 K than their HuBChE-Phos-D2O counterparts. However, MSDs for HuBChE-Tris-D2O increase very rapidly above 240 K, reaching values twice as large as those for HuBChE-D2O at 285 K.
DISCUSSION
Effects of the solvent on HuBChE intramolecular dynamics
The MSDs, as well as the normalized EIs (Figs. 3, 7, and 8) of the three D2O-hydrated samples, reflect a solvent-specific influence on HuBChE intramolecular dynamics above ∼250 K. Below this temperature, the presence of salt ions in the solvent does not influence HuBChE molecular dynamics on an Ångstrom-nanosecond scale. Since neither HuBChE-D2O nor HuBChE-Phos-D2O contain hydrogen atoms in the solvent, they can be directly compared, whereas care must be taken in interpreting data collected for the HuBChE-Tris-D2O sample, since the nonexchangeable H-atoms of the Tris contribute ∼40% to the total scattering cross section of the sample. We discuss the HuBChE-Tris-D2O sample at the end of this section.
Influence of phosphate ions
Diffusive motions are suppressed in HuBChE-Phos-D2O up to 285 K relative to HuBChE-D2O and HuBChE-Tris-D2O. The absence of a nonlinear component for the Q2-dependence of its normalized EI throughout the whole experimental Q-range (Fig. 3) supports the weak diffusive character of the intramolecular motions in HuBChE, and results in almost identical MSDs for the enzyme in the low and high Q-range up to 285 K (compare the relevant traces in Figs. 7 and 8). The absence of a kink in the Guinier plot (Fig. 3) implies that a dynamical heterogeneity of the intramolecular HuBChE motions in the Ångstrom-nanosecond range is suppressed by the presence of sodium phosphate compared to HuBChE-D2O and HuBChE-Tris-D2O, which both display dynamical heterogeneity in this range. How can this effect of the sodium and phosphate ions be interpreted?
The low ratio of only four water molecules per solvent ion (Na+, 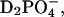
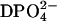 ) in HuBChE-Phos-D2O (derived from the mass ratio, Table 1, and the molecular weights of the salt and water) supports the notion that a large fraction of the water molecules are subject to more or less frequent interactions with the ions. The ions, particularly the phosphate anions, might acquire many of the available water hydrogen bonds normally directed toward protein surface residues (Collins and Washabaugh, 1985). Phosphate ions have been classified as “kosmotropic”—i.e., order-building with water molecules in their vicinity (id.). This effect of orienting water hydrogen bonds away from the (prevalently negatively charged) protein surface is a known effect, “salting out”, by means of which protein precipitation is achieved by increasing the concentration of an appropriate kosmotropic salt. The hydrogen bonds would then be available only partially for the protein itself. As pointed out by Doster and Settles (1999), hydrogen bonding between solvent and protein is considered to be important in facilitating the dynamical transition. The suppression of the dynamical transition in the case of HuBChE-Phos-D2O might thus be ascribed to a lack of accessible hydrogen bonds. Molecular dynamics simulations carried out by Tarek and Tobias (2002) further support the notion that dynamical modifications of the hydration water influence the dynamics of the hydrated protein itself. They introduced an artificial harmonic potential, with a force constant of 0.6 kJ/(mol × Å2), to restrain translational mobility of water O atoms. As a consequence, the dynamics of a fully D2O-hydrated ribonuclease A crystal (0.58 g/g) approach those of a poorly hydrated (0.05 g/g) ribonuclease powder sample at 300 K. The authors ascribed this effect to a reduced translational diffusion coefficient of the hydration water molecules and to a reduced capacity to form water-protein hydrogen bonds. Tournier et al. (2003) carried out molecular dynamics simulations, varying protein and solvent temperature independently, and also reached the conclusion that a reduction of solvent translational mobility suppresses the protein dynamical transition. Fenimore et al. (2002) favored contributions of dielectric interactions over those of hydrogen bonds in interpreting solvent-protein interactions. They divided protein motions into two classes: slaved and nonslaved. Comparing dielectric relaxation rates for solvent with several relaxation rates for myoglobin, they concluded that the energy of dielectric hydration water fluctuations provides the energy to induce protein conformational changes. According to these authors, the underlying physical basis would be the large difference in dielectric constants between water and protein, which exceeds differences in other physical properties such as heat capacity. In the light of the data discussed by these authors and the conclusions which they draw, the reduction in protein MSDs for HuBChE-Phos-D2O relative to HuBChE-D2O might be ascribed to a reduction in the dielectric constant of the solvent resulting in a concomitant decrease in the energy of dielectric solvent fluctuations. Indeed, the dielectric constant of bulk water (ɛ0 = 80) is two-to-threefold reduced at molar electrolyte concentrations (Robinson and Stokes, 1959). Energetically, this would result in reduced interactions with charged or polar moieties of the protein surface residues due to dielectric fluctuations, resulting in a reduction in or freezing of protein residue fluctuations.
) in HuBChE-Phos-D2O (derived from the mass ratio, Table 1, and the molecular weights of the salt and water) supports the notion that a large fraction of the water molecules are subject to more or less frequent interactions with the ions. The ions, particularly the phosphate anions, might acquire many of the available water hydrogen bonds normally directed toward protein surface residues (Collins and Washabaugh, 1985). Phosphate ions have been classified as “kosmotropic”—i.e., order-building with water molecules in their vicinity (id.). This effect of orienting water hydrogen bonds away from the (prevalently negatively charged) protein surface is a known effect, “salting out”, by means of which protein precipitation is achieved by increasing the concentration of an appropriate kosmotropic salt. The hydrogen bonds would then be available only partially for the protein itself. As pointed out by Doster and Settles (1999), hydrogen bonding between solvent and protein is considered to be important in facilitating the dynamical transition. The suppression of the dynamical transition in the case of HuBChE-Phos-D2O might thus be ascribed to a lack of accessible hydrogen bonds. Molecular dynamics simulations carried out by Tarek and Tobias (2002) further support the notion that dynamical modifications of the hydration water influence the dynamics of the hydrated protein itself. They introduced an artificial harmonic potential, with a force constant of 0.6 kJ/(mol × Å2), to restrain translational mobility of water O atoms. As a consequence, the dynamics of a fully D2O-hydrated ribonuclease A crystal (0.58 g/g) approach those of a poorly hydrated (0.05 g/g) ribonuclease powder sample at 300 K. The authors ascribed this effect to a reduced translational diffusion coefficient of the hydration water molecules and to a reduced capacity to form water-protein hydrogen bonds. Tournier et al. (2003) carried out molecular dynamics simulations, varying protein and solvent temperature independently, and also reached the conclusion that a reduction of solvent translational mobility suppresses the protein dynamical transition. Fenimore et al. (2002) favored contributions of dielectric interactions over those of hydrogen bonds in interpreting solvent-protein interactions. They divided protein motions into two classes: slaved and nonslaved. Comparing dielectric relaxation rates for solvent with several relaxation rates for myoglobin, they concluded that the energy of dielectric hydration water fluctuations provides the energy to induce protein conformational changes. According to these authors, the underlying physical basis would be the large difference in dielectric constants between water and protein, which exceeds differences in other physical properties such as heat capacity. In the light of the data discussed by these authors and the conclusions which they draw, the reduction in protein MSDs for HuBChE-Phos-D2O relative to HuBChE-D2O might be ascribed to a reduction in the dielectric constant of the solvent resulting in a concomitant decrease in the energy of dielectric solvent fluctuations. Indeed, the dielectric constant of bulk water (ɛ0 = 80) is two-to-threefold reduced at molar electrolyte concentrations (Robinson and Stokes, 1959). Energetically, this would result in reduced interactions with charged or polar moieties of the protein surface residues due to dielectric fluctuations, resulting in a reduction in or freezing of protein residue fluctuations.
Dependence of protein dynamics on solvent type
Concentrating on small-amplitude MSDs (Fig. 7), the presence of Tris-HCl in the solvent, in contrast to that of sodium phosphate, does not modify HuBChE dynamics with respect to the salt-free HuBChE. The nature of the solute thus plays an important role. The smaller protein MSDs in the HuBChE-Phos-D2O sample, relative to those in the HuBChE-Tris-D2O and HuBChE-D2O samples, is in accordance with the proposed protective capacity of a phosphate buffer with respect to a Tris-HCl buffer against HuBChE inactivation in particular (Masson et al., 1994; Froment et al., 1998) and in enzyme stabilization in general (von Hippel and Schleich, 1969). The increased rigidity of the enzyme prohibits large conformational changes that are considered necessary in the denaturation process. This interpretation is in agreement with the observed longevity of HuBChE lyophilized from a phosphate buffer, which remains stable and active for several years when stored at 4°C (Grunwald et al., 1997). However, the large differences observed in the low Q-range between HuBChE-D2O and HuBChE-Tris-D2O (Fig. 8) are difficult to explain without taking into account diffusive motions of the Tris solute molecules which, possessing diffusion coefficients of the order of 10−11 m2 s−1, should contribute notably to the total scattered intensity and MSDs due to their nonexchangeable H-atoms (Gabel, unpublished).
Dynamics of the protein hydration water: H2O- versus D2O-hydrated samples
Hydration water dynamics at ambient temperature
Because the hydrogen atoms of water in the H2O-containing samples account for ∼40% of the total scattering cross section (Table 3), their contribution must be taken into account when comparing these samples to their D2O-hydrated counterparts. Since all curves are normalized to the lowest temperature, i.e., to the number of scatterers in the samples, the Q-independent shift of the normalized EI for the H2O-hydrated samples with respect to their D2O-hydrated counterparts at 280 K (Fig. 2) can be interpreted as a very-large-amplitude population leaving the instrumental observation window in the Q-range investigated (see Materials and Methods). These populations (Table 2) can be partly assigned to the H2O molecules in the hydration shell in agreement with the values in Table 3. Indeed, data extracted from quasielastic neutron-scattering experiments on H2O-hydrated D-phycocanine at 0.4 g/g (Bellissent-Funel et al., 1996) yielded a diffusion coefficient of ∼1 × 10−9 m2 s−1 for the hydration water at 298 K, approximately twofold lower than the value for bulk water (2.30 × 10−9 m2 s−1) at that temperature. Bon et al. (2002) have reported a long-range diffusion coefficient for the first hydration layer water molecules in lysozyme five-to-sixfold lower than for bulk water. The values reported for the diffusion coefficient of the hydration water in both of these studies are such that the hydration water at temperatures close to ambient should make only a negligible contribution at the chosen energy resolution and in the Q-range of our experiments (Gabel, unpublished). Describing hydration water motions at ambient temperature in an oversimplified way by a free translational diffusion law, the associated half-width at half-maximum (HWHM) Γ of the scattering law associated with free translational diffusive movements is
 |
(10) |
and in the investigated Q-range (0.19 Å−2 < Q2 < 2.46 Å−2) varies in the range 5 μeV < Γ < 170 μeV. Even in the unfavorable case (5 μeV), the HWHM is several times larger than the instrumental energy resolution (0.9 μeV) employed for the lowest Q-values used in the experiment (Q2 = 0.19 Å−2): The diffusive motions of the hydration water at ambient temperature can be considered as too fast to be resolved by the instrument, and contribute merely as a small, quasiflat background, thus explaining the Q-independent shift of the normalized EI observed between H2O- and D2O-hydrated samples (Fig. 2). This assessment is supported by a 10-ns molecular dynamics simulation performed on the hydration water molecules of AChE by Henchman and McCammon (2002) at ambient temperature. They found that the majority of the water molecules had residence times <500 ps and jump distances of 2–4 Å. Due to these short residence times and large jump distances, most hydration water molecules would not, therefore, contribute notably to MSDs determined with a ∼1 μeV instrumental energy resolution, and would constitute a negligible background. However, Settles and Doster (1996) extracted hydration water diffusion coefficients at ambient temperature ranging from 2 × 10−11 to 1 × 10−10 m2 s−1 using myoglobin hydrated in either D2O or H2O. If these values are applied to our experimental situation, according to Eq. 10, the smallest HWHM would be ∼0.3 μeV at the lowest Q-value (Q2 = 0.19 Å−2) and would thus contribute to both EI and MSD values. In the low Q-range, diffusive motions of hydration water would not, therefore, be separated from intramolecular protein motions, whereas, in the large Q-range, hydration water motions would again make a negligible contribution to the EI and not affect the measured MSDs. Protein atomic motions, in contrast, can be described by diffusion coefficients of the order of 6 × 10−12 m2 s−1 (Kneller and Smith, 1994) at ambient temperatures; the HWHM associated with these protein atomic motions would thus be of the order of the instrumental energy resolution (0.08 μeV < Γ < 1 μeV) in the Q-range investigated, resulting in a substantial contribution to the measured MSDs.
Hydration water dynamics below ∼230 K
Unfortunately, neutron-scattering experiments of hydration water below 0°C are scarce. Information concerning this temperature range is, therefore, derived mainly from molecular dynamics simulations. Bizzarri et al. (2000) carried out simulations of plastocyanin hydration water (0.39 g/g) at 100–150 K in a 200-ps time window corresponding to typical instrumental resolutions of several μeV. In this temperature range, the authors found effective diffusion coefficients of 5–20 × 10−11 m2 s−1. The HWHMs corresponding to these diffusion coefficients in the Q-range which we investigated (0.19 Å−2 < Q2 < 2.46 Å−2) vary in the range 0.8 μeV < ΔE < 40 μeV. Therefore, in the temperature range of 100–150 K, the quasielastic broadening is comparable to the instrumental energy resolution employed (0.9 μeV), especially in the small Q-range. This means that elastic scattering from hydration water motions should contribute to MSDs at low temperatures (20–150 K). However, the fact that we could not detect differences in MSD values for any of the H2O- and D2O-hydrated samples <200 K is a strong indication that protein hydration water dynamics in this temperature range is comparable to protein atomic dynamics itself on an Ångstrom-nanosecond scale.
The comparison of the temperature dependence of protein hydration water and its link to the temperature dependence of protein internal dynamics is best summarized by Fig. 4 B: At low temperatures (T < 200 K), protein internal dynamics and hydration water dynamics are indistinguishable. In an intermediate temperature range (200 K < T < 260 K), the H2O-hydrated sample displays larger MSDs than the D2O-hydrated sample, whereas at the highest measured temperatures (T > 260 K), MSDs of both samples seem to be very similar again (Fig. 4 B). In combination with data from the Guinier plots (Figs. 1 and 2), we conclude that, in the intermediate temperature range, hydration water becomes mobile, leaves the instrumental energy-space resolution window, and no longer contributes to MSDs at high temperatures. This point is discussed further and related to work recently published by Becker and Smith (2003) and Hayward et al. (2003) in Remarks on Diffusive Populations, below. A natural conclusion of our interpretation would be that, in the high Q-range, protein internal dynamics and hydration water dynamics display different dynamical transition temperatures, with the hydration water transition temperature (∼170 K) lying below the protein transition temperature (∼250 K).
Although D2O is known to stabilize HuBChE against denaturation with respect to H2O by ∼10 K (Masson and Laurentie, 1988), the large observed shift of the transition temperature by ∼80 K between H2O- and D2O-hydrated samples makes us believe that an isotopic effect of the hydration water on protein internal dynamics plays a minor role in our experiments.
Influence of salt ions on hydration water dynamics
The Q-independent shift of the normalized EI values between HuBChE-D2O and HuBChE-H2O and between HuBChE-Phos-D2O and HuBChE-Phos-H2O (Fig. 2) indicates that possible modifications of the dynamics of the hydration water molecules due to the presence of phosphate ions are not noticeable on an Ångstrom-nanosecond scale. Otherwise, there would have been a change in slope between HuBChE-Phos-D2O and HuBChE-Phos-H2O. How can this absence of a difference be reconciled with the known effect of the addition of ions to water on the physicochemical properties of adjacent water molecules (Robinson and Stokes, 1959; Collins and Washabaugh, 1985; Gabel, 2000)? The lyophilization process concentrates the initially low salt concentrations (10 mM) to ratios of salt ions (for example, cations plus anions) per water molecule of ∼4 (derived from the weight ratios, Table 1, and the molecular weights) corresponding to molar concentrations. At such high salt concentrations, the dynamic properties of water molecules are known to be substantially modified. For example, for a 3 M solution of H2SO4, a strong kosmotrope, the self-diffusion coefficient is reduced by 30% relative to free water (McCall and Douglass, 1965). Apart from such reductions in the self-diffusion coefficient, residence times of the water molecules are increased by up to fivefold (Leung and Safford, 1970). One may ask how these modifications in the behavior of bulk solvent apply to protein hydration water which, itself, is already subject to dynamical restrictions (see preceding section). If the two effects were additive, they would produce a reduction in the self-diffusion coefficients and associated HWHM of ∼30%. Such a reduction would not have a noticeable effect in the instrumental space-time resolution employed. The argument made in the preceding paragraph remains, therefore, valid: Hydration water molecules contribute to MSDs at low temperatures but, at ambient temperatures, are not within the range of instrumental resolution for either HuBChE-Phos-H2O or HuBChE-H2O. However, a small difference does seem to exist between both samples (Tables 2 and 3): Whereas a very-large-amplitude population (41%) corresponding to the scattering contribution of hydration water of HuBChE-H2O (42%) leaves the instrumental energy window, a very-large-amplitude population (28%) corresponds only to ∼65% of the amount of hydration water (44%) in the case of HuBChE-Phos-H2O. This may be an indication that part of the hydration water in the latter sample displays reduced MSDs, but may also be attributed to a reduction in internal quasielastic/diffusive motions of the protein.
Small- and large-amplitude intramolecular dynamical populations
HuBChE-H2O and HuBChE-D2O
The splitting of the MSDs into large- and small-amplitude populations in the HuBChE-H2O sample >200 K (Fig. 6) is due to the appearance of a kink in the Guinier plot at that temperature. Both populations must be associated with protein atoms, rather than with hydration water molecules, since similar splitting is observed for the HuBChE-D2O sample (compare corresponding traces in Fig. 4, A and B). Without specific labeling, the protein hydrogens associated with these populations cannot be assigned definitively. Possible assignments are a backbone population and a side-chain population, or a rigid protein core population, and a flexible surface side-chain population.
In both Q-ranges, the H2O-hydrated sample displays larger MSDs than the D2O-hydrated sample, with a single exception in the high Q-range for T > 270 K (Fig. 4, A and B). This difference is probably due to the onset of diffusive motions of the water molecules at 200 K. These motions increase global MSDs of the H2O samples as long as the width of their associated scattering law is of the order of the instrumental resolution (Gabel, unpublished). Once it has become much larger, it represents a small background and no longer contributes to the MSD values. This is probably the case for the high Q-values, where the curves of the H2O-hydrated and D2O-hydrated samples merge again at 260 K (Fig. 4 B).
HuBChE-Phos-H2O and -D2O
The presence of the salt ions shifts the onset of the splitting into two dynamically different populations to higher temperatures and suppresses its extent, in contrast to what is observed for the salt-free samples (Fig. 6). In combination with the absence of a splitting into two different populations in the case of HuBChE-Phos-D2O, it can be concluded that the presence of salt residues in the hydration shell restricts the motions of the protein atoms as well as of the hydration water molecules above 200 K.
Remarks on diffusive populations, the role of the instrumental energy resolution on the observable motions, multiple scattering, and exchangeable H-atoms
The interpretation of a shift to smaller intensities in the Guinier plots in terms of a purely diffusive population is not unequivocal. One possible alternative is an intramolecular population displaying a superposition of confined motions with diffusive motions. As has been stressed in Materials and Methods in context with Eq. 9, a shift in the Guinier plots to smaller elastic intensities may have two origins: A change in amplitude of the motions (expressed by a change of the EISF) or effects due to quasielastic motions and the influence of the instrumental resolution width on their elastic contributions. Our findings are in qualitative agreement with work recently published by Becker and Smith (2003) and Hayward et al. (2003), who analyze the instrumental energy resolution effects on observable motions in elastic scans. The first article states that deviations from the Gaussian approximation may originate from dynamical heterogeneity or from non-Gaussian behavior of single atoms, two effects that are not easily separable by elastic scans. Furthermore, Becker and Smith (2003) showed that a finite instrumental energy resolution may induce an apparent dynamical transition for motions whose relaxation times change with temperature. These findings as well as the interpretation of data from proteins in solution by Hayward et al. (2003) are in good qualitative agreement with our comparison and interpretation (in terms of Guinier plots and MSDs) of H2O- and D2O-hydrated samples (Figs. 1, 2, and 4 B). At very low temperatures (T < 200 K), protein hydration water has very long relaxational times and displays similar dynamics as protein atoms. In an intermediate temperature range (200 K < T < 260 K), the relaxation times of the protein hydration water strongly increase (dynamical transition) and the signal leaves the instrumental energy-space window. At the highest measured temperatures (T > 260 K), hydration water no longer contributes to MSDs extracted by linear fits in the chosen Q-range.
However, another feature that needs to be taken into account in interpreting this shift is multiple scattering. As pointed out by Settles and Doster (1997), even at transmission values as high as 0.9, ∼20% of neutrons are scattered twice or more. They distinguished a Q-independent inelastic background for second scattering events of the elastic-inelastic type. This background may account for part of the Q-independent loss of EI observed for all samples even at low temperatures (Fig. 1) and, in the case of the dry samples, at high temperatures (Fig. 3), at which large-scale diffusive motions are not supposed to occur.
Throughout this study, we have compared H2O- and D2O-hydrated samples as if the exchangeable H atoms displayed, on the average, the same dynamical behavior as the nonexchangeable H-atoms on an Ångstrom-nanosecond scale. Differences observed between H2O- and D2O-hydrated samples (Fig. 4 B) could, in principle, be due partly to exchangeable protons. However, as can be calculated from Table 3 and Materials and Methods, although protein hydration water contributes in the H2O-hydrated samples to ∼40% of the total scattering cross section of a sample, the exchangeable protons contribute to only ∼10%. The differences in dynamics observed between H2O- and D2O-hydrated samples are therefore likely to be dominated by the hydration water. To estimate their influence more precisely, experiments on deuterated proteins should be carried out, which, unfortunately, cannot yet be performed with HuBChE.
CONCLUSIONS
A series of incoherent elastic neutron-scattering experiments probing molecular dynamics on an Ångstrom-nanosecond scale were carried out on lyophilized HuBChE samples. Atomic MSDs were determined as a function of temperature, degree of hydration, type of hydration water (D2O, H2O), and solvent composition. Otherwise identical HuBChE samples lyophilized from a nonvolatile sodium phosphate buffer or from a volatile ammonium acetate buffer displayed large differences in atomic dynamics at temperatures >200 K. Protein atomic and, to a smaller degree, hydration water MSDs, on an Ångstrom-nanosecond scale, were suppressed in the sample obtained from sodium phosphate buffer. This was attributed to the presence of the strongly kosmotropic phosphate ions. We interpret the suppression of large-amplitude protein atomic movements as being due to the preferred orientation of the water hydrogen bonds toward the phosphate anions depriving the surface protein residues of hydrogen bonds. In a control experiment with HuBChE lyophilized from a nonvolatile Tris-HCl buffer, no effect on MSDs was observed in a range where diffusive motions are small, underlining the importance of the type of salt ion. These results may be correlated with the suggested protective effect of salts on the stability of BChE in aqueous solution (Burgess and Lister, 1988) and lyophilized state (Doctor, 2003), which appears to follow the Hofmeister series, as has been observed for other proteins (Jensen et al., 1995). Our results complement a number of studies carried out on proteins in different solvent environments (Cordone et al., 1999; Réat et al., 2000; Tsai et al., 2000). They show that the presence of kosmotropic salt ions in the solvent has a comparable effect on protein internal dynamics (i.e., a reduction of MSDs and a suppression of diffusive motions) as have different cryoprotectants such as trehalose or glycerol. In addition, our results show that protein hydration water becomes mobile at a significantly lower temperature than the protein dynamical transition.
Acknowledgments
The authors thank Jacques Colletier for useful discussions on cholinesterase biochemistry as well as the IN16 instrumental team (Bernhard Frick, Thilo Seydel, and Matthias Elender) and the IN13-CRG team (Marc Bée, Francesca Natali, and Sebastien Vial) at the Institute Laue-Langevin, Grenoble, France.
References
- Ashani, Y., S. Shapira, D. Levy, A. D. Wolfe, B. P. Doctor, and L. Raveh. 1991. Butyrylcholinesterase and acetylcholinesterase prophylaxis against soman poisoning in mice. Biochem. Pharmacol. 41:37–41. [DOI] [PubMed] [Google Scholar]
- Becker, T., and J. C. Smith. 2003. Energy resolution and dynamical heterogeneity effects on elastic incoherent neutron-scattering from molecular systems. Phys. Rev. E. 67:0219041–0219047. [DOI] [PubMed] [Google Scholar]
- Bée, M. 1988. Quasielastic Neutron-Scattering. D. Millen, editor. Adam Hilger, Bristol, UK and Philadelphia, PA.
- Bellissent-Funel, M., J.-M. Zanotti, and S. Chen. 1996. Slow dynamics of water molecules on the surface of a globular protein. Faraday Discuss. 103:281–294. [Google Scholar]
- Bizzarri, A., A. Paciaroni, and S. Cannistraro. 2000. Glasslike dynamical behavior of the plastocyanin hydration water. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Topics. 62:3991–3999. [DOI] [PubMed] [Google Scholar]
- Bon, C., A. J. Dianoux, M. Ferrand, and M. S. Lehmann. 2002. A model for water motion in crystals of lysozyme based on an incoherent quasielastic neutron-scattering study. Biophys. J. 83:1578–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess, S. K., and A. K. Lister. 1988. An investigation of the protective effect of various salts on the acid inactivation of human serum butyrylcholinesterase (E.C. 3.1.1.8). J. Protein Chem. 7:641–654. [DOI] [PubMed] [Google Scholar]
- Burgess, S. K., and S. L. Oxendine. 1993. Thermal inactivation of butyrylcholinesterase and acetylcholinesterase. J. Protein Chem. 12:651–658. [DOI] [PubMed] [Google Scholar]
- Collins, K., and M. Washabaugh. 1985. The Hofmeister effect and the behavior of water at interfaces. Q. Rev. Biophys. 18:323–422. [DOI] [PubMed] [Google Scholar]
- Cordone, L., M. Ferrand, E. Vitrano, and G. Zaccai. 1999. Harmonic behavior of trehalose-coated carbon-monoxy-myoglobin at high temperature. Biophys. J. 76:1043–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino, H. R., K. Griebenow, R. Langer, and A. Klibanov. 1997. On the pH memory of lyophilized compounds containing protein functional groups. Biotechnol. Bioeng. 53:345–348. [DOI] [PubMed] [Google Scholar]
- Doctor, B. P. 2003. Butyrylcholinesterase: its Function and Inhibitors. E. Giacobini, editor. Martin Dunitz, London, UK. 163–177.
- Doster, W., and M. Settles. 1999. The dynamical transition in proteins: the role of hydrogen bonds. In Hydration Processes in Biology. M. Bellissent-Funel, editor. IOS Press, Amsterdam, The Netherlands. 177–191.
- Doster, W., S. Cusack, and W. Petry. 1989. Dynamical transition of myoglobin revealed by inelastic neutron-scattering. Nature. 337:754–756. [DOI] [PubMed] [Google Scholar]
- Ellman, G., K. Courtney, V. Andres, and R. Featherstone. 1961. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 7:88–95. [DOI] [PubMed] [Google Scholar]
- Fenimore, P., H. Frauenfelder, B. McMahon, and F. Parak. 2002. Slaving: solvent fluctuations dominate protein dynamics and functions. Proc. Natl. Acad. Sci. USA. 99:16047–16051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrand, M., A. Dianoux, W. Petry, and G. Zaccai. 1993. Thermal motions and function of bacteriorhodopsin in purple membranes: effects of temperature and hydration studied by neutron-scattering. Proc. Natl. Acad. Sci. USA. 90:9668–9672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauenfelder, H., F. Parak, and R. D. Young. 1988. Conformational substates in proteins. Annu. Rev. Biophys. Biophys. Chem. 17:451–479. [DOI] [PubMed] [Google Scholar]
- Froment, M.-T., O. Lockridge, and P. Masson. 1998. Resistance of butyrylcholinesterase to inactivation by ultrasound: effects of ultrasound on catalytic activity subunit association. Biochim. Biophys. Acta. 1387:53–64. [DOI] [PubMed] [Google Scholar]
- Gabel, F. 2000. Etude des Membranes Biologiques par Diffusion de Neutrons. DEA Report. Universite Joseph Fourier, Grenoble.
- Gabel, F., D. Bicout, U. Lehnert, M. Tehei, M. Weik, and G. Zaccai. 2002. Protein dynamics studied by neutron-scattering. Q. Rev. Biophys. 35:327–367. [DOI] [PubMed] [Google Scholar]
- Grunwald, J., D. Marcus, Y. Papier, L. Raveh, Z. Pittel, and Y. Ashani. 1997. Large-scale purification and long-term stability of human butyrylcholinesterase: a potential bioscavenger drug. J. Biochem. Biophys. Methods. 34:123–135. [DOI] [PubMed] [Google Scholar]
- Guinier, A., and G. Fournet. 1955. Small-Angle Scattering of X-Rays. C. G. Walker, translator. John Wiley and Sons, London; Chapman and Hall, New York.
- Hayward, J. A., J. L. Finney, R. M. Daniel, and J. C. Smith. 2003. Molecular dynamics decomposition of temperature-dependent elastic neutron-scattering by a protein solution. Biophys. J. 85:679–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henchman, R., and J. McCammon. 2002. Structural and dynamic properties of water around acetylcholinesterase. Protein Sci. 11:2080–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, M. K., D. Braunstein, B. R. Cowen, H. Frauenfelder, I. E. Iben, J. R. Mourant, P. Ormos, R. Scholl, A. Schulte, P. J. Steinbach et al. 1990. Conformational substates and motions in myoglobin. External influences on structure and dynamics. Biophys. J. 58:429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen, W. A., J. M. Armstrong, J. De Giorgio, and M. T. Hearn. 1995. Stability studies on maize leaf phosphoenolpyruvate carboxylase: the effects of salt. Biochemistry. 34:472–480. [DOI] [PubMed] [Google Scholar]
- Kneller, G., and J. C. Smith. 1994. Liquid-like side-chain dynamics in myoglobin. J. Mol. Biol. 242:181–185. [DOI] [PubMed] [Google Scholar]
- Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227:680–685. [DOI] [PubMed] [Google Scholar]
- Leung, P., and G. Safford. 1970. A neutron inelastic scattering investigation of the concentration and anion dependence of low frequency motions of H2O. Molecules in solution. J. Phys. Chem. 74:3696–3709. [Google Scholar]
- Lockridge, O., C. Bartels, T. Vaughan, C. Wong, S. Norton, and L. Johnson. 1987. Complete amino acid sequence of human serum cholinesterase. J. Biol. Chem. 262:549–557. [PubMed] [Google Scholar]
- Lockridge, O., N. Mottersraw-Jackson, H. W. Eckerson, and B. N. La Du. 1980. Hydrolysis of diacetylmorphine (heroin) by human serum cholinesterase. J. Pharmacol. Exper. Thera. 215:1–8. [PubMed] [Google Scholar]
- Masson, P., and M. Laurentie. 1988. Stability of butyrylcholinesterase: thermal inactivation in water and deuterium oxide. Biochim. Biophys. Acta. 957:111–121. [DOI] [PubMed] [Google Scholar]
- Masson, P., M.-T. Forment, P.-L. Fortier, J.-E. Visicchio, C. F. Bartels, and O. Lockridge. 1998. Butyrylcholinesterase-catalysed hydrolysis of aspirin, a negatively charged ester, and aspirin-related neutral esters. Biochim. Biophys. Acta. 1387:41–52. [DOI] [PubMed] [Google Scholar]
- Masson, P., P. Gouet, and C. Clery. 1994. Pressure and propylene carbonate denaturation of native and “aged” phosphorylated cholinesterase. J. Mol. Biol. 238:466–478. [DOI] [PubMed] [Google Scholar]
- McCall, D., and D. Douglass. 1965. The effect of ions on the self-diffusion of water. I. Concentration dependence. J. Phys. Chem. 69:2001–2011. [Google Scholar]
- Nicolet, Y., O. Lockridge, P. Masson, J. C. Fontecilla-Camps, and F. Nachon. 2003. Crystal structure of human butyryl cholinesterase and of its complexes with substrate and products. J. Biol. Chem. 278:41141–41147. [DOI] [PubMed] [Google Scholar]
- Paciaroni, A., S. Cinelli, and G. Onori. 2002. Effect of the environment on the protein dynamical transition: a neutron-scattering study. Biophys. J. 83:1157–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen, B. F., A. M. Stock, D. Ringe, and G. A. Petsko. 1992. Crystalline ribonuclease A loses function below the dynamical transition at 220 K. Nature. 357:423–424. [DOI] [PubMed] [Google Scholar]
- Raveh, L., E. Grauer, J. Grunwald, E. Cohen, and Y. Ashani. 1997. The stoichiometry of protection against soman and VX toxicity in monkeys pretreated with human butyrylcholinesterase. Toxicol. Appl. Pharmacol. 145:43–53. [DOI] [PubMed] [Google Scholar]
- Réat, V., R. Dunn, M. Ferrand, J. Finney, R. Daniel, and J. Smith. 2000. Solvent dependence of dynamic transitions in protein solutions. Proc. Natl. Acad. Sci. USA. 97:9961–9966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, R., and R. Stokes. 1959. Electrolyte Solutions. Butterworths, London, UK.
- Saxena, A., L. Raveh, Y. Ashani, and B. P. Doctor. 1997. Structure of glycan moieties responsible for the extended circulatory lifetime of fetal bovine serum acetylcholinesterase and equine serum butyrylcholinesterase. Biochemistry. 36:7481–7489. [DOI] [PubMed] [Google Scholar]
- Schinkel, J., N. Downer, and J. Rupley. 1985. Hydrogen exchange of lysozyme powders. Hydration dependence of internal motions. Biochemistry. 24:352–366. [DOI] [PubMed] [Google Scholar]
- Settles, M., and W. Doster. 1996. Anomalous diffusion of adsorbed water: a neutron-scattering study of hydrated myoglobin. Faraday Discuss. 103:269–279. [Google Scholar]
- Settles, M., and W. Doster. 1997. Iterative Calculation of the Vibrational Density of States from Incoherent Neutron-Scattering Data with the Account of Double Scattering. S. Cusack, H. Büttner, M. Ferrand, P. Langan, and P. Timmins, editors. Adenine Press, Schenectady, NY.
- Smith, J. 1991. Protein dynamics: comparison of simulations with inelastic neutron-scattering experiments. Q. Rev. Biophys. 24:227–291. [DOI] [PubMed] [Google Scholar]
- Smith, P. K., R. I. Krohn, G. T. Hermanson, A. K. Mallia, F. H. Gartner, M. D. Provenzano, E. K. Fujimoto, N. M. Goeke, B. J. Olson, and D. C. Klenk. 1985. Measurement of protein using bicinchoninic acid. Anal. Biochem. 150:76–85. [DOI] [PubMed] [Google Scholar]
- Sun, H., J. El Yazal, O. Lockridge, L. M. Schopfer, S. Brimijoin, and Y.-P. Pang. 2001. Predicted Michaelis-Menten complexes of cocaine-butyrylcholinesterase. J. Biol. Chem. 276:9330–9336. [DOI] [PubMed] [Google Scholar]
- Tarek, M., and D. J. Tobias. 2002. Role of protein-water hydrogen bond dynamics in the protein dynamical transition. Phys. Rev. Lett. 88:1381011–1381014. [DOI] [PubMed] [Google Scholar]
- Tehei, M., D. Madern, C. Pfister, and G. Zaccai. 2001. Fast dynamics of halophilic malate dehydrogenase and BSA measured by neutron-scattering under various solvent conditions influencing protein stability. Proc. Natl. Acad. Sci. USA. 98:14356–14361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournier, A. L, J. Xu, and J. C. Smith. 2003. Translational hydration water dynamics drives the protein glass transition. Biophys. J. 85:1871–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, A., D. Neumann, and L. Bell. 2000. Molecular dynamics of solid-state lysozyme as affected by glycerol and water: a neutron-scattering study. Biophys. J. 79:2728–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Haupt, H., K. Heide, O. Zwisler, and H. G. Schwick. 1966. Isolierung und physikalisch-chemische Charakterisierung der Cholinesterase aus Humanserum. Blut. 14:65–75. [DOI] [PubMed] [Google Scholar]
- von Hippel, P. H., and T. Schleich. 1969. The effects of neutral salts on the structure and conformational stability of macromolecules in solution. In Biological Macromolecules. G. D. Fasman and S. N. Timasheff, editors. Marcel Dekker, New York. 417–584.
- Wüthrich, K. 1986. NMR of Proteins and Nucleic Acids. John Wiley and Sons, New York.
- Zaccai, G. 2000. Moist and soft, dry and stiff: a review of neutron experiments on hydration-dynamics-activity relations in the purple membrane of Halobacterium salinarum. Biophys. Chem. 86:249–257. [DOI] [PubMed] [Google Scholar]