

Abstract
Molecular dynamics simulations are presented for a Thermus aquaticus (Taq) DNA polymerase I complex (consisting of the protein, the primer-template DNA strands, and the incoming nucleotide) subjected to external forces. The results obtained with a force applied to the DNA template strand provide insights into the effect of the tension on the activity of the enzyme. At forces below 30 pN a local model based on the parameters determined from the simulations, including the restricted motion of the DNA bases at the active site, yields a replication rate dependence on force in agreement with experiment. Simulations above 40 pN reveal large conformational changes in the enzyme-bound DNA that may have a role in the force-induced exonucleolysis observed experimentally.
INTRODUCTION
DNA replication, a crucial process for the propagation of the genome, is catalyzed by DNA polymerases (Kornberg and Baker, 1991). These motor enzymes function in repeated cycles during which they move along a partly single-stranded, partly double-stranded DNA chain (Joyce and Steitz, 1994). In each catalytic cycle, a single nucleotide, complementary to the nucleotide on the template strand (if no error is introduced), is added to the primer strand of the double-stranded DNA. Thus, the double-stranded DNA chain with N basepairs changes to one with N + 1 basepairs. Because the fidelity of the base addition is fundamental for genetics and elucidating its mechanism may aid in the design of therapeutic agents against rapidly mutating pathogens, an understanding of the incorporation step performed by the polymerases is important. It is essential for a full description of DNA replication.
Based on sequence similarity, polymerases can be classified in seven different families (Patel and Loeb, 2001). The most extensively studied, both kinetically and structurally, are those in family A (which includes the prokaryotic and archaea Pol I, the eukaryotic polymerases γ and θ, as well as the viral T3, T5, and T7 polymerases) and those in family B (which include the prokariotic Pol II, several eukariotic and archaeal polymerases, as well as the viral adenovirus, HSV, RB69, T4, and T6 polymerases). Despite their diverse biological functions (replication, recombination, repair) the structural and chemical mechanisms of base incorporation by the polymerases in these two families seem to be very similar (Patel and Loeb, 2001; Steitz, 1998; Brautigam and Steitz, 1998). For a number of polymerases in family A (T7 polymerase, Doublié et al., 1998; Taq polymerase I, Li et al., 1998b) and in family B (RB69 polymerase, Franklin et al., 2001) there exist crystal structures for ternary intermediate complexes (i.e., DNA primer/template and incoming nucleotide in complex with the intermediate (active) state of the enzyme). These ternary structures are important for understanding the mechanism of the incoming nucleotide incorporation. The gross features of these DNA polymerases have been likened to a right hand (Ollis et al., 1985), with the fingers interacting with the incoming nucleotide and the template, the palm containing the catalytic site and binding to the incoming nucleotide, and the thumb binding the double-stranded DNA. The incoming nucleotide forms H-bonds with its partner (template base) and is stacked onto the last primer base (see Fig. 1).
FIGURE 1.

Atomic detail of the active site of the open (left) and closed (right) conformations of Taq polymerase (in blue surface representation) complexed with the DNA, depicted in licorice representation (primer in red, template in gray, incoming dCTP in yellow). Only protein regions within 7 Å of the DNA bases shown are represented. The hydrogen atoms on the sugar-phosphate backbone are omitted for clarity.
A common mechanism for these polymerases has been proposed (Patel and Loeb, 2001; Steitz, 1999), based on the available crystal structures. In crystal structures of the apo form (Kim et al., 1995; Nayal et al., 1995) (i.e., with neither DNA nor the nucleotide bound), in those of the binary complexes with dNTP (Ollis et al., 1985; Li et al., 1998a) or with DNA (Beese et al., 1993; Eom et al., 1996; Kiefer et al., 1998; Li et al., 1998b), and in that of a ternary complex (i.e., with DNA and incoming nucleotide; Li et al., 1998b) a relaxed, “open” configuration of the fingers domain has been observed. In other ternary complex structures (Doublié et al., 1998; Li et al., 1998b; Franklin et al., 2001), this domain is in a “closed” configuration. In going from the open structure to the closed structure, the fingers domain closes by ∼40–60° and the template DNA base that is to pair with the incoming nucleotide rotates inward by >90° (see Fig. 2), placing the incoming nucleotide in an optimal alignment for its subsequent chemical incorporation into the DNA primer. Although the details of the transition are not entirely understood, this major conformational change takes place before chemistry can occur, and is believed to be the rate-limiting step in a kinetic mechanism (Patel et al., 1991; Dahlberg and Benkovic, 1991) for the overall reaction. Additionally, this mechanism is consistent with an early suggestion (Bryant et al., 1983) (i.e., one proposed before the availability of crystal structures) that dNTP first binds to a polymerase/primer/template complex independently of the template information, and that only after the rate-limiting step (believed to be the closure of the fingers) does tight binding become template dependent. Among the polymerases for which ternary closed structures exist (i.e., the intermediate (active) ternary structures mentioned above with the fingers in the closed configuration) the large fragment of Taq polymerase I (Li et al., 1998b) (Klentaq1) is particularly appropriate for a computational analysis because, in addition to the closed ternary complex structure with ddCTP bound, a ternary open structure with ddCTP bound has been reported (Li et al., 1998b). Both open and closed structure crystals diffracted to similar resolution (2.3 Å) in the same space group, and were grown using identical crystallization procedures, with the exception that the open form was obtained from a selenomethionine-substituted protein and, after crystallization, was incubated with a washing solution that partially depleted the population of bound nucleotide. This procedure is believed (Li et al., 1998b) to have caused the transformation of an originally closed crystal structure to the open form. Closed ternary structures have been determined also for ddATP-, ddTTP-, and ddGTP-trapped complexes, in addition to that with the ddCTP nucleotide (Li and Waksman, 2001), but no open ternary structure with the last three nucleotides is available.
FIGURE 2.

“Minimalist” representation of the open and closed conformational change of Taq-DNA complex in the immediate vicinity of the active site; this can be compared with Fig. 1. The ds DNA template extending to the 3′ side comprises nucleotides labeled +1, +2, …; template nucleotides on the 5′ side are labeled 0, −1, …; the primer strand is in red. In the open complex, the incoming nucleotide (in yellow) encounters a Tyr (blue licorice) whereas its destined partner base (gray licorice, at 0) is tilted sideways and inaccessible. In the closed complex, the Tyr is displaced by the change in the position of the O helix (blue cylinder) and the partner base is flipped into a bonding configuration in the space previously occupied by the Tyr.
Recently, the structural and kinetic data for family A polymerases have been supplemented by single-molecule experiments, in which it was shown that the rate of the replication reaction catalyzed by the DNA polymerases is altered when a force is applied to the template strand (Wuite et al., 2000; Maier et al., 2000). T7 DNA polymerase was studied with an optical trap (Wuite et al., 2000) and the large (Klenow) fragment of Escherichia coli, its 3′–5′exonuclease deficient mutant (Klenow exo−), and a 3′–5′exonuclease deficient mutant of T7 DNA polymerase (Sequenase) were studied by use of a magnetic trap (Maier et al., 2000). The two sets of experiments showed similar behavior. It was observed that the replication rate decreases at high forces and appears to increase at low forces. The experimental uncertainties are such that, although the rate decrease at high applied force is unequivocal, the rate increase at low force is within the error bars. The qualitative demonstration that the rate of polymerization depends strongly on the applied force f is of interest per se; i.e., it shows that the limiting (rate-determining) step involves work by the polymerase complex (and therefore motion) against an external force. However, a quantitative interpretation of the experimental results requires introduction of a model expressing the replication rate in terms of the force-induced work.
It is possible, in principle, that the application of the force transforms a nonlimiting step (e.g., translocation) into a limiting step. Therefore, the fact that rate depends on force does not always mean that motion is involved in the limiting step. However, the opposite is true: if there is no rate-force dependence, then there is no motion in the rate-limiting step. For RNA polymerases (Wang et al., 1998) and DNA helicases (V. Croquette, ENS, Paris, personal communication), analogous single-molecule experiments were crucial in showing that the rate-limiting step is independent of an applied force.
The models proposed previously to interpret the single-molecule experiments are phenomenological in nature and assume Arrhenius behavior (Wang et al., 1998), as in other studies of the effect of external forces on molecular reactions, such as protein unfolding (Carrion-Vazquez et al., 1999) or ligand unbinding (Moy et al., 1994). The rate constant in the absence of the force, k(0), is related to that in the presence of the force, k(f), by the equation
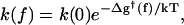 |
(1) |
with Δg†(f) = ΔG†(f) − ΔG†(0) the force-induced barrier change relative to the zero-force activation barrier, ΔG†(0). Equation 1 implicitly assumes that k(0) also has an Arrhenius-type temperature dependence and that the magnitude of the force is such that the system is not in the so-called diffusive limit (Izrailev et al., 1997; Evans and Ritchie, 1997). Using Eq. 1, two types of models have been proposed. Both assume the closed conformation of the enzyme complex can be taken as a surrogate for the transition state. One model (Wuite et al., 2000; Maier et al., 2000), which we refer to as “global,” evaluates the force-induced activation barrier from experimentally determined extension versus force curves for “bare” single-stranded (ss) and double-stranded (ds) DNA polymers, thousands of basepairs long and not interacting with enzyme. The other model, referred to as “local” (Goel et al., 2001, 2002), focuses solely on conformational changes of just two DNA segments (each involving one nucleotide) in the neighborhood of the active site of the enzyme.
From the global model it was concluded that the best fit to the replication rate data was obtained if, in the rate-determining step, more than one (n = 2 or 4, depending on the enzyme) of the single-stranded nucleotides at the end of the duplex were converted from ss to ds geometry, only to have n − 1 of them (i.e., all but the one that becomes part of the ds DNA) return to ss geometry before the next catalytic cycle. If correct, this conclusion would have important mechanistic implications. The local model also obtained fair agreement with the rate data, but assumed that only one ss base (n = 1) of the two bases considered in the ss portion of the template is converted to ds geometry in the closed state of the enzyme complex. This is consistent with the ternary crystal structures (Doublié et al., 1998; Li et al., 1998b; Franklin et al., 2001) that “catch” the polymerase in the act of incorporating the incoming nucleotide; these structures indicate that only one template base becomes part of the ds DNA. It has been argued (Wuite et al., 2000), however, that the n = 2 interpretation remains tenable because not only the first template segment, but also the second one is ordered in the closed state crystal structures and the interphosphate distance in the second is close to that in ds DNA.
Neither model is capable of calculating the effect of the force on the rate-determining step without introducing certain assumptions. To provide first-principle-based information that makes possible a more complete understanding of the experimental results, we use all-atom molecular dynamics simulations and explore the dynamical processes in the open and closed structures as a function of the applied force. Given these results, we evaluate some of the parameters of the rate-force models used to describe experimental data. Specifically, we address the issue of the geometry of the DNA segments at the active site by monitoring distance and angle time series obtained during the molecular dynamics simulations at different values of the applied force, and use these to calculate the force-dependent barrier of the reaction.
In what follows, we begin with a brief review of the global and local models. We then present the results of the molecular dynamics simulations in the presence of an external force and use them to evaluate the parameters that appear in the local model. This permits us to refine the local model and present a restricted-cone local model (RCLM), which takes into account the restrictions on the ss DNA motion due to the presence of the protein. An evaluation of the results is given in the Concluding Discussion section. It is followed by a brief Methods section, which describes the molecular dynamics simulation protocol.
THE GLOBAL AND LOCAL MODELS
Global model
The global model (GM) (Wuite et al., 2000; Maier et al., 2000) for the rate-force dependence assumes that the force-dependent part of the activation free energy, i.e., Δg†(f) in Eq. 1, has the form
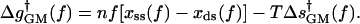 |
(2) |
The first term on the right-hand side is the force-dependent part of the activation enthalpy, which is equal to the reversible work required to convert n bases from the ss geometry to the ds geometry. The values of xss(f), xds(f), measured in different experiments, are the extensions (along the direction of the force) per base, at force f, for ss and ds DNA chains, respectively; the values are taken from data on stretching of ss or ds polymers (Smith et al., 1996; Maier et al., 2000). The second term, involving the force-dependent entropy of activation, is evaluated, in the GM version of Wuite et al. (2000), from the areas under the experimental curves by plotting the forces fss,ds versus the extensions xss,ds produced by those forces,
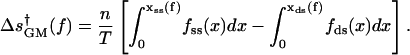 |
(3) |
This term is neglected in the GM version of Maier et al. (2000). Both versions of the GM implicitly assume that the ds geometry is formed in the transition state leading to the closed state.
The model is global in the sense that the functions xss(f) and xds(f), which govern both terms in Eq. 2, pertain to the contributions per base to the end-to-end extension of the entire ss or ds polymers on which the pulling experiments are performed. These polymers typically consist of over 10,000 basepairs, whereas the portion of the DNA interacting with the polymerase consists of only ∼10 basepairs (Doublié et al., 1998; Li et al., 1998b) of the duplex and ∼4 ss bases of the template (Turner et al., 2003). In effect, data on the elasticity of ss and ds DNA, obtained from experiments in the absence of an enzyme, are used to calibrate the amount of work involved in converting a single ss DNA base to ds geometry, thereby slightly shortening the template. This calibration is then assumed to apply as well in the presence of the enzyme. Accordingly, the activation barrier has no direct contribution from enzyme-DNA interactions; the only parameter having to do with the enzyme is n, the number of ss DNA bases converted to ds geometry in the transition state. Fitting this model to the observed variations of replication rates with tension indicated, on the basis of the calibrated elasticity changes, shortenings that correspond to n = 2 for T7 DNAp (Wuite et al., 2000) and Sequenase (Maier et al., 2000) and n = 4 for the Klenow fragment (Maier et al., 2000).
Molecular dynamics (MD) simulations are well suited to test both conceptual and specific assumptions of the global model. The prime factors employed in the model, xss(f) and xds(f), the projections of DNA segments on the direction of the force, are averages over thousands of residues. Consequently, there is no distinction between the orientation, relative to the direction of the applied force, of the DNA segments at the active site and the orientation of segments far away from the active site. This is inconsistent with a key structural feature, incorporated in the MD simulations. One of the two DNA segments at the active site of the closed complex (see Figs. 2 and 3) is clearly kinked, approximately perpendicular to the axis of the duplex. Because ds DNA is extremely stiff compared with ss DNA, the applied force is essentially along the axis of the duplex, and therefore, being approximately perpendicular to the ss overhang, the resulting torque (that tries to move the ss overhang from a perpendicular to a parallel direction relative to the axis) will contribute significantly to the energetics.
FIGURE 3.

(Top panel) Top view (down the axis of the double helix) showing the kinked ss DNA protruding outside the helical boundaries as a consequence of the interactions with the polymerase in both the open (left) and closed (right) structures (same color code as in Fig. 2; protein not shown for clarity). Because tension is applied in the direction coming out of the plane of the article, the resulting torque, acting to align the overhang from an in-plane to an out-of-plane orientation, is expected to have a significant effect on the kinked DNA region. (Bottom panel) Side view of the open and closed structures, (same format as top panel). The direction of the applied force is illustrated by the dotted line. Again, it is apparent that the ss overhang is the “lever arm” of a significant torque contribution forcing the ss segments to orient from a vertical to horizontal position.
The MD simulations, which evaluate the range of fluctuations in the orientations of the segments at the active site, demonstrate that for both the kinked segment and others, the averaged projections on the force direction differ markedly from the “generic” results given by xss(f) and xds(f). This shows that the basic “calibration” procedure adopted in the global model is not appropriate.
The inference from the global model that n > 1 in Eq. 2 for the three enzymes studied is therefore based on assumptions that are not appropriate, particularly because they do not take into account the importance of the specific (kinked) geometry of the enzyme-DNA interactions.
Local model
The fact that the global model ignores the specific geometry and interactions of the DNA segments at the active site, other than that a number n of bases are converted from ss to ds geometry, led Goel et al. (2001, 2002) to suggest a local, “structurally guided” model for the rate-force dependence. In this model, the force-dependent activation energy depends on the behavior of two DNA segments neighboring the active site. Vectors a and b in Fig. 4 are introduced by connecting equivalent atoms in adjacent bases along the template DNA strand associated with the +1, 0 segment and with the 0, −1 segment (in the local model (LM), the vectors connecting neighboring C1′ atoms were used); both the orientation and length of a and b can change in going from the open to the closed state. The additional activation enthalpy barrier in the presence of the force was assumed to equal the average mechanical work done by the enzyme against the external force f, in the process of converting the two segments a, b from their conformation in the open form of the enzyme complex (the reactant state), to their conformation a′, b′ in the closed complex (the transition state); i.e.,
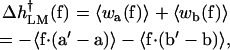 |
(4) |
where  indicates an average over the probability distribution functions for the scalar products of f with the base-associated vectors.
indicates an average over the probability distribution functions for the scalar products of f with the base-associated vectors.
FIGURE 4.

Views of open and closed Taq polymerase complexes, focused on DNA segments at the exit from the protein. Arrows (a, b in open, a′, b′ in closed) show the designation of the segment vectors between adjacent C5′ atoms and the direction of the applied force f (parallel to axis of the ds DNA helix). Other DNA segments than these two schematized here do not change significantly in going from their open to close states. Portions of the protein (blue) within 10 Å of the segments a or a′ are shown; the atoms in this part of the protein are the main contributors to the restriction of orientations available to the DNA segments. The explicit solvent and ions included in the simulation are omitted.
The local model was introduced to test whether a model that assumes only one base changes geometry from ss to ds, could fit the replication rate data. In the local model it was assumed, in accord with the structural data, that, upon going from the open to the closed complex, the a vector, connecting bases 0 and +1, goes from ss to ds geometry whereas the b vector, connecting −1 and 0, retains ss geometry, although the orientation of b does change (see Fig. 5). Thus only one nucleotide changes its conformation from ss-like to ds-like geometry to be incorporated into the DNA duplex, although the motion of two nucleotides (i.e., 0, −1) is actually involved. Consequently, the distinction between n = 1 as used to describe the LM model (Goel et al., 2001) and n = 2 is somewhat arbitrary and the essential part is that the displacements of two nucleotides are included. The lengths of the a and b vectors (Fig. 2) were constrained to agree approximately with the average contour lengths per residue, Lss = 7 Å and Lds = 2.6 Å, consistent with structural data, as well as the stretching curves used in the global model.
FIGURE 5.

Views of first five bases in the open (left) and closed (right) template DNA strand at the active site: bases −1, 0, +1, +2, +3 in blue, red, gray, orange, and yellow, respectively. Clearly, base 0 changes between the open and closed configuration by stacking onto the double-stranded part of the template (i.e., onto base + 1).
The duplex bound to the polymerase is in B-DNA form except for the three pairs toward the ss junction, which are in A-DNA form. Although the B-form is more stable for free DNA, the DNA segments at the active site of family A polymerases and HIV-1 reverse transcriptase are observed to be in the A-form (Patel and Loeb, 2001). The a segment is thus in A-form, which justifies the A-form value of lds = 2.6 Å rather than the value of 3.4 Å for B-form DNA.
In Eq. 4 the work terms contributing to the force dependence of the activation barrier then become
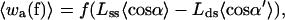 |
(5a) |
and
 |
(5b) |
Here the angles α and β specify the orientation, with respect to the direction of f, of the a and b vectors for the open complex; α′ and β′ denote the same for the closed conformation (see Fig. 6). The external force is assumed constant and locally directed along the duplex axis, as a consequence of the long persistence length of ds DNA. The averages over the LM angular orientations in Eqs. 5a and 5b correspond to the GM projections in Eq. 2; thus, 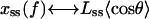 for θ = α, β, β′ and
for θ = α, β, β′ and 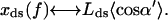 These angular orientations were evaluated using the freely jointed chain (FJC) approximation, which averages over a Boltzmann distribution corresponding to the force-dependent potential energy of the FJC segments (Bueche, 1962). Over most of the pertinent range of f, the FJC results are fairly similar to the experimental xss(f) and xds(f) functions. The FJC angular averages are given by the Langevin formula,
These angular orientations were evaluated using the freely jointed chain (FJC) approximation, which averages over a Boltzmann distribution corresponding to the force-dependent potential energy of the FJC segments (Bueche, 1962). Over most of the pertinent range of f, the FJC results are fairly similar to the experimental xss(f) and xds(f) functions. The FJC angular averages are given by the Langevin formula,
 |
(6) |
for  or β′, where
or β′, where 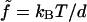 with kB the Boltzmann constant, T temperature, and
with kB the Boltzmann constant, T temperature, and 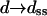 or dds the polymer Kuhn length (the Kuhn length is the characteristic length scale describing the flexibility of the chain, and equals twice the persistence length); dss = 14 Å and dds = 1000 Å were used, conventional values (Rouzina and Bloomfield, 2001) in accord with the experimental stretching curves. With the FJC model, simple analytic formulas can be obtained (Goel et al., 2002) for
or dds the polymer Kuhn length (the Kuhn length is the characteristic length scale describing the flexibility of the chain, and equals twice the persistence length); dss = 14 Å and dds = 1000 Å were used, conventional values (Rouzina and Bloomfield, 2001) in accord with the experimental stretching curves. With the FJC model, simple analytic formulas can be obtained (Goel et al., 2002) for 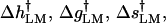 analogous to Eqs. 2 and 3, with each the sum of contributions from the a and b segments.
analogous to Eqs. 2 and 3, with each the sum of contributions from the a and b segments.
FIGURE 6.

Pictorial representation of a, b in open, a′, b′ in closed states of the local model.
By design, this implementation of the LM facilitates comparison with the GM. In effect, the GM postulates n terms like 〈wa(f)〉 in Eq. 5, corresponding to n segments, each of which shrinks in length 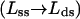 between the open and closed complex. Instead, the LM assumes that only the leading segment a shrinks, but the neighboring segment b nonetheless can contribute, despite no shrinkage in length, if in 〈wb(f)〉 the averaged angular motion with respect to f differs appreciably between the open and closed complex. This point was illustrated by computing k(f)/k(0) for two limiting cases (Goel et al., 2001, 2002). In Case I, the angular fluctuation of the b segment was considered unhindered in both the open and closed complex, so 〈cos β〉 = 〈cos β′〉; in Case II, the fluctuations remained unhindered in the open complex but are strongly restricted by interaction with the enzyme to be near 90° in the closed complex, resulting in 〈cos β′〉 = 0. Case I (based on the free energy of activation) gave results nearly identical to the GM with n = 1, as expected, whereas those for case II resembled the GM with n = 2–4.
between the open and closed complex. Instead, the LM assumes that only the leading segment a shrinks, but the neighboring segment b nonetheless can contribute, despite no shrinkage in length, if in 〈wb(f)〉 the averaged angular motion with respect to f differs appreciably between the open and closed complex. This point was illustrated by computing k(f)/k(0) for two limiting cases (Goel et al., 2001, 2002). In Case I, the angular fluctuation of the b segment was considered unhindered in both the open and closed complex, so 〈cos β〉 = 〈cos β′〉; in Case II, the fluctuations remained unhindered in the open complex but are strongly restricted by interaction with the enzyme to be near 90° in the closed complex, resulting in 〈cos β′〉 = 0. Case I (based on the free energy of activation) gave results nearly identical to the GM with n = 1, as expected, whereas those for case II resembled the GM with n = 2–4.
This LM approach appears to have served its heuristic purpose, demonstrating that the effect of tension on replication rate need not involve “extra” length shrinkage, as inferred from the GM (i.e., n > 1), but might instead be strongly influenced by angular conformational changes induced by the enzyme. The LM in this form cannot otherwise be useful, however. The FJC approximation and other expedient assumptions, although of some use in describing “generic” or global behavior, are not appropriate for local interactions. A realistic version of the LM requires the correct description of the effect of the enzyme-DNA interactions. A purpose of the MD simulations is to address this issue.
Before ending this section, we consider the relation between the LM and GM, and a linear-response model proposed by Bell (1978) and refined by Evans et al. (Evans and Ritchie, 1997). This model has been used for interpreting pulling experiments on ligand-receptor unbinding (Moy et al., 1994), on protein unfolding (Carrion-Vazquez et al., 1999) or DNA unzipping (Strick et al., 2000). In the linear-response model, the amount by which the barrier is changed is Δg† = χf, where χ is the width of the activation barrier (i.e., distance along reaction coordinate from the “reactant” minimum to the top of the barrier) and can also be looked at as a characteristic length over which the force acts. This linear form for the barrier change leads to a force-dependent rate equal to
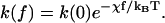 |
(7) |
This is to be compared to the LM result in the limit of large tension
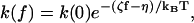 |
(8) |
obtained by adding up Eqs. 4 and 5 in Goel et al. (2001), where ζ and η are positive constants calculated from the model. Equation 8 indicates that, for relatively large forces (higher than ∼7 pN), the LM is in the linear-response regime, but it includes a constant offset η to the barrier change (see Table IV in Goel et al., 2002). In the GM, by contrast, the rate expression, when put in the form of Eq. 7, yields a value for χ that depends on f as estimated from the experimental curves of free DNA.
MOLECULAR DYNAMICS SIMULATIONS
We present a molecular dynamics analysis of the DNA polymerase binding site in this section. By simulating the system at atomic detail in the presence of an external force, we have direct access to the various parameters assumed by the LM, and, as it turns out, are able to introduce a refined version called the RCLM as described in “The restricted-cone local model” section.
This study employed protocols developed for “all-atom” simulations, making full use of the data available from crystal structures for both the open and closed conformations of the Taq polymerase complexed with DNA (Li et al., 1998b) as well as techniques to include solvation and dielectric screening of ionic interactions (as described in Methods). This resulted in a simulation system explicitly treating over 12,000 atoms, of which over 5000 atoms, located within an explicit water sphere of 25-Å radius centered on the binding site, are allowed to move.
The closing of the finger domain is assumed not to be affected by the force; this “first-order” approximation is expected to be reasonable because the force is applied on the DNA and not the protein (Wuite et al., 2000; Maier et al., 2000). In other words, the force-dependent work term depends on f only through the force-dependent properties of the DNA segments (i.e., its orientations) and not through any force-dependent properties of the protein.
Also, in common with the phenomenological treatments, the observed closed structure before the incorporation of the base (with the incoming nucleotide in Watson-Crick pairing with the 0-th template base situated on segment a′, see lower panel in Fig. 2), is taken to correspond to the transition state; the open structure corresponds to the reactant state. Consequently, the force-dependent change in the barrier height is calculated between these two states, i.e., between the open and the closed structures. The absence of a force dependence of the protein transition is consistent with the fact that, in the x-ray structures, the ss DNA overhang is free to sweep inside the space of a cone centered at the active site, without steric hindrance from the surrounding protein atoms (see Fig. 1); this unhindered motion has been observed in the molecular dynamics simulations.
The key change of the DNA determining the overall reaction is the 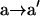 transformation; i.e., the change in the position of the 0-th DNA base from an ss to a ds configuration, after which the chemical bond-forming reaction is a relatively fast exothermic process. The open and closed crystal structures for Klentaq1 show that this transition involves a rotation of the a segment by ∼20° toward the helical axis to allow the ss base at position 0 to flip in and hydrogen-bond with the incoming nucleotide. Because the force acts directly on the DNA alone, the assignment of the closed DNA geometry as the transition state geometry is expected to be a good approximation for the calculation of the force-dependent barrier. Even if the transition state for the conformational change were at an intermediate position between the open and the closed states in the absence of an external force, the force-dependent change of the activation energy from the reactant to this transition state could be well approximated by the energy change from the reactant to the closed state. This assumption comes from arguments invoking the Brønsted relationship (Brønsted, 1928), which belongs to the broader class of linear free energy relationships, in which the logarithm of a rate constant is a linear function of the logarithm of an equilibrium constant; a model explaining this observation has been put forth by Evans and Polanyi (1938).
transformation; i.e., the change in the position of the 0-th DNA base from an ss to a ds configuration, after which the chemical bond-forming reaction is a relatively fast exothermic process. The open and closed crystal structures for Klentaq1 show that this transition involves a rotation of the a segment by ∼20° toward the helical axis to allow the ss base at position 0 to flip in and hydrogen-bond with the incoming nucleotide. Because the force acts directly on the DNA alone, the assignment of the closed DNA geometry as the transition state geometry is expected to be a good approximation for the calculation of the force-dependent barrier. Even if the transition state for the conformational change were at an intermediate position between the open and the closed states in the absence of an external force, the force-dependent change of the activation energy from the reactant to this transition state could be well approximated by the energy change from the reactant to the closed state. This assumption comes from arguments invoking the Brønsted relationship (Brønsted, 1928), which belongs to the broader class of linear free energy relationships, in which the logarithm of a rate constant is a linear function of the logarithm of an equilibrium constant; a model explaining this observation has been put forth by Evans and Polanyi (1938).
Analysis of origin of force dependence
In Eq. 4, the key variables are the vectors a, b, a′, b′ (see Fig. 6). They are replaced in Eq. 5 of the LM by the force-dependent average cosines and the lengths of the vectors. In Goel et al. (2001), the average cosines were calculated using a FJC model in an external field, whereas the lengths of the segments were ascribed constant values equal to the interbase distances of ss and ds DNA measured from the crystal structures. Because molecular dynamics simulations allow us to give a time-dependent description of the key variables, we can calculate  in Eq. 4 directly. To do this, we run constant-temperature molecular dynamics in the open state and in the closed state in the presence of a range of applied forces. This type of simulation, in which the timescale is orders of magnitude shorter than the experimental timescale, has been shown to be useful for mapping out energy profiles for protein folding and unfolding (Paci and Karplus, 2000), ligand-receptor binding (Izrailev et al., 1997; Merkel et al., 1999) or nucleic acid structural transitions (Konrad and Bolonick, 1996; MacKerell and Lee, 1999). To our knowledge, the work presented here is the first simulation of an applied force acting on a protein-DNA complex.
in Eq. 4 directly. To do this, we run constant-temperature molecular dynamics in the open state and in the closed state in the presence of a range of applied forces. This type of simulation, in which the timescale is orders of magnitude shorter than the experimental timescale, has been shown to be useful for mapping out energy profiles for protein folding and unfolding (Paci and Karplus, 2000), ligand-receptor binding (Izrailev et al., 1997; Merkel et al., 1999) or nucleic acid structural transitions (Konrad and Bolonick, 1996; MacKerell and Lee, 1999). To our knowledge, the work presented here is the first simulation of an applied force acting on a protein-DNA complex.
The local force exerted on the DNA in the enzyme complex is assumed to be adequately approximated by the force applied to the entire DNA strand and to be directed parallel to the axis of the double helix portion. Whereas the local instantaneous force acting on a string of arbitrary shape is expected to be directed along the local tangent, we seek to model an average global tension as it is measured in the experiments (and as it is modeled in the GM and LM); its direction is parallel to the double helix axis. Also, the force is assumed to remain constant during the open to closed transition. Actually, the instantaneous force felt by the leading segments at the active site will fluctuate and thereby differ from the global tension applied to the entire DNA strand. The timescale for such fluctuations can be estimated (Goel et al., 2001) from the Zimm model (Grosberg and Khokhlov, 1994) in terms of the Kuhn lengths and the solvent viscosity. This indicates that the timescale for fluctuations of individual Kuhn segments of ds DNA is vastly longer than our computation intervals of 3 ns. Accordingly, the helix axis remains practically stationary in our MD simulations. The stiffness of ds DNA likewise ensures that for it the local and global f are nearly the same (except for very weak forces). The fluctuation timescale for Kuhn segments of ss DNA are, in contrast, substantially shorter than 3 ns. These fluctuations thus are averaged over in the MD simulations. In the simulation, a point was placed along the direction of the double helix axis 40 Å away from the O5′ atom of the −1 nucleotide (see Fig. 7) and a constraint force with the desired magnitude was applied to this atom, which is the outmost nonhydrogen atom of the modeled template; i.e., the O5′ atom of the b or b′ segments. (Details concerning the way the force was simulated are given in the Methods section.)
FIGURE 7.

Force direction (shown here in the open state). The incoming nucleotide is in green, the template in gray, and the primer in red. The dotted line points to the position of the fictitious particle used as reference for the application of the force. The direction of the force is parallel to the direction of the DNA helix. For clarity, only the protein within 10 Å of segment a is shown (in blue) and the explicit solvent and ions included in the simulation are omitted.
Fig. 8 specifies the coordinate system and angles employed in the MD simulations to describe the orientation of the DNA segments. For both the open and closed complexes, the z axis is along the axis of the duplex helix; the polar angle θ = α, β, α′, β′ then specifies the angle between the z-direction and the segment vectors. The azimuthal orientation of the segments about the duplex axis is specified by φa, φb, φ′a, φ′b, measured counterclockwise from the x axis, which is chosen to lie in a fixed plane that contains the z axis and the most probable direction of the a vector in the open complex. We use the distances between consecutive C5′ atoms; this choice is employed in all subsequent analysis reported in this article. The C5′ distances are preferable to the C1′ distances originally used in the local model (Goel et al., 2001). This is because the C1′ atoms are not aligned with the sugar-phosphate backbone to which the pulling force is applied in the simulations and can rotate about that backbone. However, the simulations are rather insensitive to the particular choice of defining points; choosing the distances between the P atoms or the O5′ atoms or between the centroid of the sugar-phosphate backbone all give similar results.
FIGURE 8.

Coordinate system used to describe angular orientation of DNA segments a and b in MD simulations. The z azis is along the axis of the ds DNA helix. The x axis lies in a fixed plane containing the z axis and the most probable direction of the a vector in the open complex. Polar angles α and β are measured from the z-direction; azimuthal angles φa and φb are measured counterclockwise from the x axis to the projection of the segment vector in the x-y plane. The Cartesian axes x, y, z remain the same for the closed complex, wherein the segment vectors become a′ and b′.
We performed two independent runs, Run I and Run II, for forces up to 20 pN, in steps of 2 pN. (The constant-force experiments cover the range 0–20 pN for Klenow and Sequenase (Maier et al., 2000), and 0–15 pN for T7 DNAp (Wuite et al., 2000); some constant-elongation data points are available for up to 35 pN for T7 DNAp (Wuite et al., 2000).) At each force value, 3-ns simulations were run and the last configuration at a given force was used as the starting configuration for the next (higher) force. In these calculations, the length of a(t), b(t) (in the open state), and a′(t), b′(t) (in the closed state) were computed between the C5′ atoms of the template nucleotides +1−0, and from 0 to −1, respectively (see Fig. 2). Fig. 9 shows the time series of the angles made by the two DNA segments with the direction of the force, over the range between 0 and 20 pN, and Fig. 10 plots the corresponding lengths of the two segments. We note that, even in the open state, a is much more restricted in its motion (by its interactions with the surrounding enzyme) than b. More importantly, we observe that the angular orientations of a, a′, b′, are less influenced by the external force, whereas the orientation of b is strongly affected by forces larger than 8–10 pN.
FIGURE 9.

Run I (top two panels) and Run II (bottom two panels) time series of the DNA segment polar angles during the pulling simulations for the open (α,β) and closed (α′,β′) complexes. In each window (indicated by the vertical grid lines) the value of the force is indicated above the upper border.
FIGURE 10.

Time series for length of DNA segments (a in black, b in red) during the pulling simulations; format as in Fig. 9. Results of run I are shown; run II yields similar plot, with nearly identical mean and standard deviation, and is not shown.
Fig. 11 plots histograms displaying MD results for the azimuthal angles, defined as in Fig. 8. As with the polar angles, the range of azimuthal orientations is seen to be much more constrained for the a vector than the b vector, in both the open and closed conformations. For forces up to 20 pN (and even at higher tensions) the azimuthal orientations of the a and a′ vectors remain within a few degrees of their orientations at f = 0. In contrast, for the open conformation, the most probable azimuthal angle for b, which occurs near 55° for f = 0, shifts markedly to near 90° for f = 20 pN (and remains there at higher forces). In the closed conformation, the histogram of the azimuthal angle for b′ is trimodal for f = 0, with a prominent peak near 30° and other sizable ones near 80° and 100°. For f = 20 pN (and higher forces), the most probable azimuthal angle for b′ is the most prominent peak, which remains in the vicinity of 30°. These results indicate that in the force range 0–20 pN, the changes in azimuthal orientations of the DNA segments are roughly comparable to those for the polar angles.
FIGURE 11.

Histograms for the DNA segment azimuthal angles for small and large applied pulling forces: f = 0 (blue), 20 pN (red), 40 pN (green), and 60 pN (violet); dashed lines for segment a (or a′ in the bottom panel), continuous for b (or b′ in the bottom panel). The limits of the available azimuthal angles for all simulated forces up to 20 pN indicate that the ratio 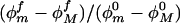 is approximately unity and thus there is little contribution to the force-dependent thermodynamic functions from the azimuthal angles.
is approximately unity and thus there is little contribution to the force-dependent thermodynamic functions from the azimuthal angles.
Under the assumptions of ergodicity, we can calculate for each force f that we apply in the simulation, the ensemble averaged work 〈w〉, as required for Eq. 4, from the difference of time averages,
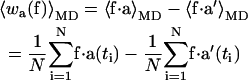 |
(9a) |
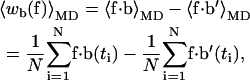 |
(9b) |
where the first sum on the right-hand side of each equation is calculated from the trajectory in the open state, and the second sum is from the trajectory in the closed state.
The results of the simulations were used to calculate 〈wa(f)〉MD and 〈wb(f)〉MD in Eqs. 9a and 9b. From these values, 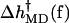 was calculated with
was calculated with
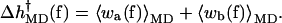 |
(10) |
Fig. 12 shows the various contributions and the predicted values of 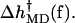 Although there are significant differences between the two runs, particularly for the more flexible open state, the overall trends are clear. For low forces (2–8 pN) the
Although there are significant differences between the two runs, particularly for the more flexible open state, the overall trends are clear. For low forces (2–8 pN) the 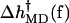 values are negative, corresponding to an increase of rate, but for higher forces
values are negative, corresponding to an increase of rate, but for higher forces 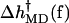 is positive and the rate decreases.
is positive and the rate decreases.
FIGURE 12.

Dependence on pulling force of enthalpy of activation for converting the open-to-closed complex, obtained from MD simulations determining work done by the enzyme on DNA segments. Data represented with lines are averages from Runs I and II; diamonds and squares represent separately Δh from Run I and Run II, respectively, with the error bars indicating standard deviations. For segments a, a′, and b′, at forces up to 20 pN, the dependence of the work on force is nearly linear, indicating the orientation of these segments is relatively insensitive to the force; however, segment b undergoes a marked reorientation near 10 pN that shifts the enthalpy of activation from negative to positive values.
This sign change is due to orientation, relative to the force direction 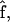 of the most mobile segment, i.e., b in the open state. In the crystal structure as well as at low forces, the angle between
of the most mobile segment, i.e., b in the open state. In the crystal structure as well as at low forces, the angle between  and b is obtuse, so the average projection
and b is obtuse, so the average projection 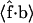 is negative, whereas larger forces are able to orient b to make an acute angle with f so that
is negative, whereas larger forces are able to orient b to make an acute angle with f so that 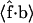 becomes positive (see Fig. 9). By contrast, for the more rigid closed state,
becomes positive (see Fig. 9). By contrast, for the more rigid closed state, 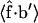 has negative signs throughout the entire force range; the orientation of b′ is significantly affected only for forces larger than 40 pN, as described in the “Higher force regime” section.
has negative signs throughout the entire force range; the orientation of b′ is significantly affected only for forces larger than 40 pN, as described in the “Higher force regime” section.
Fig. 13 shows the values of k(f)/k(0) calculated from 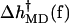 in Eq. 1 assuming that TΔs†(f) is negligible, as it appears to be in the LM. The figure also shows the experimentally measured values for the Klenow fragment and the results of the RCLM model described in the next section. It can be seen that there is good agreement with the experimental decrease in the rate for forces between 8 and 20 pN. The apparent increase in the measured rate for two related enzymes is also qualitatively reproduced, although the value and position of the peak in the k–f curve are somewhat different from those of the experiments. As noted in the Introduction, the observed increase in rate at low force is within the experimental error bars; the calculations suggest that the behavior is real.
in Eq. 1 assuming that TΔs†(f) is negligible, as it appears to be in the LM. The figure also shows the experimentally measured values for the Klenow fragment and the results of the RCLM model described in the next section. It can be seen that there is good agreement with the experimental decrease in the rate for forces between 8 and 20 pN. The apparent increase in the measured rate for two related enzymes is also qualitatively reproduced, although the value and position of the peak in the k–f curve are somewhat different from those of the experiments. As noted in the Introduction, the observed increase in rate at low force is within the experimental error bars; the calculations suggest that the behavior is real.
FIGURE 13.

Dependence of the enzyme-catalyzed replication rate constant at T = 300 K on force applied to the DNA template strand. Experimental data for T7 DNA polymerase from Wuite et al. (2000), with squares (k(0) = 130 s−1) and for Klenow from Maier et al. (2000), with diamonds (k(0) = 13.5 s−1); theoretical results from MD simulations (circles) and restricted cone local model (red curve).
Fig. 9 leads to the following picture for how the force affects the rate through the orientation of the segments. As explained above, the increase in the rate is due to the change of the sign of 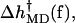 which is negative for low forces and positive for forces larger than ∼8 pN. The interactions that keep b at angles β above 90° in the open state are overcome by forces higher than 8–10 pN. By contrast, in the closed state, the corresponding angle β′ remains at the values close to the zero-force case, for this range of forces. The two distinct behaviors of the angles β and β′ above 8 pN, combined with the relatively constant contributions of angles α and α′, yield a force dependence of
which is negative for low forces and positive for forces larger than ∼8 pN. The interactions that keep b at angles β above 90° in the open state are overcome by forces higher than 8–10 pN. By contrast, in the closed state, the corresponding angle β′ remains at the values close to the zero-force case, for this range of forces. The two distinct behaviors of the angles β and β′ above 8 pN, combined with the relatively constant contributions of angles α and α′, yield a force dependence of 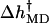 for this force regime that explains the MD-calculated rate-force curve. Moreover, for comparison, our MD estimate of 8–10 pN for the force required to “break” b free from its position in the open structure, is within the range of experimental forces needed to break DNA hairpins (9 ± 3 pN for A-T and 20 ± 3 pN for G-C hairpins; Rief et al., 1999).
for this force regime that explains the MD-calculated rate-force curve. Moreover, for comparison, our MD estimate of 8–10 pN for the force required to “break” b free from its position in the open structure, is within the range of experimental forces needed to break DNA hairpins (9 ± 3 pN for A-T and 20 ± 3 pN for G-C hairpins; Rief et al., 1999).
Comparison with GM and LM
In Fig. 14 we contrast the variation with force of the average cosines determined from the MD simulations with experimental force-extension data, x(f)/L, used in the GM and with the similar FJC curves from Eq. 6 used in the LM treatment. Such functions, in effect, restrict θ to acute angles and typically give values of 〈cos θ〉 greatly different in magnitude and/or sign from the MD results; in Case I of the LM, the segments are free to rotate in a solid angle of 4π, so their most favorable orientation is along f (angles of 0° are most favorable), which yields large average cosines for a, b, a′, and b′. By contrast, in the force regime between 0 and 20 pN, the MD values are always very small (〈cos θ〉 ≤ .15). In Case II, the average cosines for a, b, a′ are large (angles close to 0°), whereas the average cosine for b′ is postulated by the LM to be 0 (due to what is called in the LM the kink, which keeps the respective angle at 90°). There is a similar contrast with respect to segment lengths. In the GM and the LM, shrinkage accompanying the conversion of an ss segment to a ds segment has a major role; the values used in the LM are Lss = 7 and Lds = 2.6 Å (see above). However, whereas the mean interbase distances do show such shrinkage for free DNA strands, the differences obtained from MD are much smaller and even opposite in sense for segments near the active site when DNA is in an enzyme complex. In the MD simulations the DNA segment lengths do not differ much between the open and closed complexes; moreover, the nominally ss segments, b and b′, are actually shorter than the a and a′ segments. The agreement of the MD results and the experimental rate-force curve is due to the force-dependent orientation of the segments in the open relative to the closed state. In particular, the main contributor for the 0–30 pN range is the b segment, which due to its orientation, makes a negative contribution for the force-dependent barrier for forces lower than 10 pN and a positive contribution, for forces larger than 10 pN, which orient this segment along the direction of the applied force. These comparisons show that the apparent agreement between the GM or LM and experiment must be attributed to compensating errors in the distance and angular parameters used in the models.
FIGURE 14.

Variation with force of average cosines relating orientation of two DNA segments to the force direction for the open and closed enzyme complexes. Each panel shows points obtained from MD simulations (Run I (○) and Run II (X), Figs. 6–8). Also included are curves pertaining to the global model, derived from experimental stretching data for ss DNA (a,b,b′) or ds DNA (a′) (•••); curves for the freely joined chain approximation used in Case I of the local model (green line); and curves from the restricted-cone local model, with parameters from Table 1 (red line).
Higher force regime
Although the experiments show that the rate of incorporation goes to zero at 25 pN, we extended the simulations to higher forces. A second domain of large force-induced change was found in the MD simulations extended for forces above the stalling force for polymerization. Results obtained for the polar angles, using conditions of Run I with 3-ns simulation intervals, are shown in Figs. 15 and 16 for forces up to 80 pN. In the closed complex, the segment b′ undergoes a major excursion: β′ first being obtuse, reaching values around 160° by 30 pN, abruptly plunges by 80° to become acute by ∼40 pN. Moreover, in both the open and closed complex, at 40 pN the other segment also shifts significantly: α and α′ both become more acute, shifting closer to the direction of f by ∼10°.
FIGURE 15.

Time series of the angles β and β′ describing the orientation of the last ss DNA segment, b and b′, respectively, during the pulling simulations at large forces (30–60 pN), for the open state (top panel) and closed state (bottom panel). For the open state the segment b has oriented toward the direction of the external force at smaller forces (between 8 and 10 pN; see Fig. 9). In the bottom panel of this figure, for the closed state, the evolution of the β′-angle shows that b′ experiences this “crossover” phenomenon at forces larger than 40 pN.
FIGURE 16.

Histogram of the α, α′-angles of segments a, a′ in the open (top) and closed complex (bottom) for low (up to 40 pN) and high external forces (40 pN and up). Both run I and II were used for f < 20 pN. Forces larger than 40 pN shift the angle distribution by 10°; of particular interest is the orientation α′ of a′, because this segment is the one opposite the incoming nucleotide. It is possible that forces in this range change the specificity of interactions with the correct nucleotide and could trigger exonucleolysis.
The striking polar angular shifts seen at 40 pN, especially for α′, suggest a possible connection to exonucleolysis. In the pulling experiments on T7 DNAp, the polymerase activity was observed to stall at 34 ± 8 pN and a force-induced 100-fold increase in exonucleolysis activity began above 40 ± 3 pN (Wuite et al., 2000). Although the Taq polymerase has no detected exonuclease activity, this activity is well characterized for analogous family A polymerases. Exonucleolysis occurs at a different site that, in the Klenow fragment (which has the same overall structure as Klentaq1, 50% homology and 36% amino acid identity) is ∼30 Å away from the polymerization site (Beese et al., 1993). Thus, it is of interest to consider whether changes in the geometrical alignments at the polymerase site for Klentaq1 at high force may be akin to those that foster exonucleolysis. Improper orientation of the a′ and b′ segments in the closed conformation could trigger the 3′–5′ proofreading activity of the enzyme. This is in accord with current views of the preservation of nucleotide insertion fidelity, which emphasize geometric selection by induced fit at the active site (Steitz, 1999; Kunkel and Bebenek, 2000). Particularly important for the chemical reaction of incorporating the incoming nucleotide is the orientation of the a′ segment in the closed structure. A value of α′ around 72° (distributions to the right in the lower panel of Fig. 16) corresponds to the correct positioning for the polymerization step, whereas values around 60° (distributions to the left in Fig. 16) are suboptimal and could trigger the exonucleolysis mode. Two-state behavior consistent with this bimodal hypothesis has been observed for T7 DNAp; near the stalling force, a competition between polymerization and exonucleolysis was observed (Fig. 5 in Wuite et al., 2000).
In summary, the MD simulations reveal how the two leading DNA segments (of which only one changes its geometry from an ss to a ds one) at the polymerase active site respond to competition between enzyme-DNA interactions and the external force. The segment lengths, corresponding to interbase distances between adjacent nucleotides, change by ∼7% between the open and closed conformations of the enzyme, but remain virtually unchanged by external forces up to 80 pN. The segment angular orientations with respect to the duplex axis direction (along which the external force is exerted) undergo large changes, in response to both the open to closed transition and to the external force. In contrast, the azimuthal orientations about the duplex axis change greatly in the open to closed transition, but are almost unaffected by the external force. As displayed in Fig. 17, the major effect of the applied force occurs for the outermost DNA segment. In the open complex, a force of 10 pN suffices to tilt b by 45° toward f, whereas in the closed complex, increasing force at first has no effect on b′. However, at ∼40 pN there is an abrupt change with the force reorienting b′ by 80° toward f. Above 40 pN, in both the open and closed complexes, the inner segment a is also shifted further toward f by 10°, as seen in Fig. 16. These shifts, which change the geometry of the binding site, might inhibit base incorporation and foster exonuclease activity.
FIGURE 17.

Representation of all polar angles for the open (in black and green) and the closed (in red and blue) states as a function of the force applied in the molecular dynamics simulations; error bars (standard deviations) are shown for several data points. Notice the abrupt decrease to acute values of β at 10 pN and of β′ after 30 pN.
THE RESTRICTED-CONE LOCAL MODEL
Although in the LM all orientations of the DNA segments considered in the model are assumed to be accessible, the molecular dynamics results show that steric constraints exclude a significant amount of the space in both the open and the closed structures. To take account of this fact in a simple way, we introduce the restricted-cone local model. As in the LM, the RCLM focuses on the DNA vectors (a, b in the open and a′ and b′ in the closed states), which are treated as “dipoles” orienting in an external force field f and a heat bath of temperature T (as in conventional FJC models). The essential point of the RCLM, as implied by its name, is that, due to the presence of the protein, not all angular orientations Ω(θ, φ) are allowed for the DNA segments, as illustrated in Fig. 18. By writing down the partition function for the protein-restricted segments in the presence of the external force, we derive the corresponding enthalpy and free-energy barriers. The LM is recovered as a particular case of the RCLM by removing the angular restrictions.
FIGURE 18.

Schematic representation of the RCLM components modeling the spatial restriction shown in Fig. 4. The shaded region depicts the volume excluded by the presence of the enzyme and that restricts the orientation available for the a and b vectors. Regions A and B contain the rest of the DNA degrees of freedom that the RCLM is neglecting: for example, region A contains the guanine base that, when the reaction proceeds toward the closed state, flips in to pair with the incoming cytosine nucleotide.
To introduce the RCLM, we assume that the potential energy of a segment d (d = a, b, a′, b′) in the presence of the protein has the form:
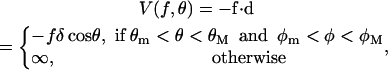 |
(11) |
where δ = |d| is the length of the vector segment. The indices m and M denote the minimum and maximum angles relative to the direction of f allowed by the enzyme. The angular partition function in the presence of the external force f is
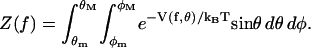 |
(12) |
This reduces in the zero-force case to the free diffusion-in-a-cone partition function. In the diffusion-in-a-cone picture, a characteristic segment is allowed to diffuse freely in a conical region of solid angle Ω(θ, φ). Thus the zero-force partition function, Z(0), is
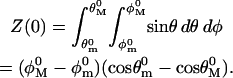 |
(13) |
The superscripts added to the limiting angles indicate that these pertain to zero-force, and may differ, in principle, from the limits appropriate when the force is applied. From Eqs. 12 and 13, the partition function ratio z(f) between that in the presence of the force and the zero-force case is
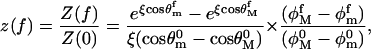 |
(14) |
with ξ = fδ/kBT and the superscript f indicating the possible dependence of the limiting angles on the force. We assume, in what follows, that the maximum and minimum values of the angles are those observed in the MD simulations. The actual values are given in Table 1.
TABLE 1.
Parameters describing the orientation (θ, φ) and length (δ) of DNA segments
| Segment | a | b | a′ | b′ |
|---|---|---|---|---|
| θ0[°] | 82.5 | 140.0 | 71.4 | 124.3 |
| θm[°] | 63.0 | (105.0, 57.7) | 53.6 | (96.8, 56.4) |
| θM[°] | 102.1 | (178.1, 105.0) | 89.9 | (179.3, 104.8) |
| φ0[°] | 0 | 101.4 | −1.9 | 79.7 |
| δ[Å] | 6.4 | 5.5 | 6.2 | 5.2 |
| f̃[pN·Å] | 6.5 | 7.5 | 6.7 | 8.0 |
Subscript zero denotes most probable value in absence of external force; subscripts m and M refer to minimum and maximum values used in the RCLM. The polar angles made by b and b′ are found to have two sets of limiting values, depending on the force range. For b the first value in each pair in the parentheses for θm and θM is for forces up to 10 pN, and the second value is for forces above 10 pN. For b′, the first and second values in the pairs are for forces below and, respectively, above 40 pN. Also shown are the force scale factors 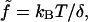 with kBT = 41.42 pN·Å = 0.5962 kcal/mol at 300 K.
with kBT = 41.42 pN·Å = 0.5962 kcal/mol at 300 K.
Given Eq. 14, we can derive all thermodynamic functions of the DNA segments as a function of the external force. The force-dependent contributions to the enthalpy for a segment d are given by
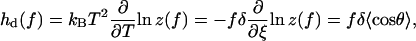 |
(15) |
with
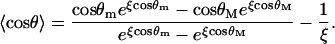 |
(16) |
In the RCLM, the enthalpy does not involve the azimuthal orientation because in Eq. 15 only the factors dependent on temperature (or ξ) contribute. Note also that in the limit 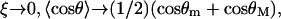 whereas in the limit
whereas in the limit 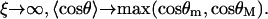 In the corresponding force-dependent contribution to the free energy,
In the corresponding force-dependent contribution to the free energy,
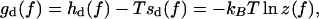 |
(17) |
all factors in Eq. 14 enter because they contribute to the entropy. However, the azimuthal orientation contributes to ln z(f) only to the extent that the ratio 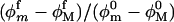 differs from unity. At least for forces below 20 pN, where the RCLM is likely to be most useful, we find from the MD results (Fig. 11) that this ratio is nearly unity. In computing differences, Δgd(f), between the open and closed complexes, both the residual azimuthal contributions and those from the force-independent term,
differs from unity. At least for forces below 20 pN, where the RCLM is likely to be most useful, we find from the MD results (Fig. 11) that this ratio is nearly unity. In computing differences, Δgd(f), between the open and closed complexes, both the residual azimuthal contributions and those from the force-independent term, 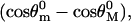 tend to cancel. If these terms are neglected, Δgd(f) is given simply by
tend to cancel. If these terms are neglected, Δgd(f) is given simply by
 |
(18) |
and the corresponding entropy term by
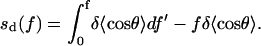 |
(19) |
These formulas were used in evaluating the RCLM results included in Figs. 13 and 19. Table 1 lists values assigned to the segment lengths and limiting angles and Fig. 14 displays the corresponding 〈cos θ〉 functions obtained from Eq. 16, together with results from the MD simulations and the FJC approximation used in the original LM.
FIGURE 19.

Force dependence of enthalpy of activation from the molecular dynamics simulation (○). Also shown are the activation energy components for the restricted cone local model (see text in “The restricted-cone local model” section) at T = 300 K.
On removing the angular restrictions  the RCLM formulas reduce to the same form as the FJC approximation; in particular Eq. 16 reduces to Eq. 6. A key conceptual and practical difference remains, in that the RCLM uses actual segment lengths obtained from MD whereas the FJC model deals with Kuhn lengths that characterize the stiffness of the chain. The Kuhn lengths are statistical in nature, meant to describe an average property of a long polymer in a solvent (experimental values, Cocco et al., 2001, are about dss = 15 Å and dds = 1000 Å for free DNA). Such statistical quantities are appropriate for a global model but not for a local model. We have atomic resolution in the simulation and use specific atomic segments, which is the rationale behind taking d = a, b, a′, or b′, rather than the statistical Kuhn segment values. Moreover, the two segments at the enzymatic site are neither purely ss (as in the free ss DNA), nor purely ds (because b′ is the last segment of the ds helix and thus subject to fraying or other end effects (Olmsted et al., 1991)), and therefore ascribing to them Kuhn segment lengths of free ss or ds DNA is artificial.
the RCLM formulas reduce to the same form as the FJC approximation; in particular Eq. 16 reduces to Eq. 6. A key conceptual and practical difference remains, in that the RCLM uses actual segment lengths obtained from MD whereas the FJC model deals with Kuhn lengths that characterize the stiffness of the chain. The Kuhn lengths are statistical in nature, meant to describe an average property of a long polymer in a solvent (experimental values, Cocco et al., 2001, are about dss = 15 Å and dds = 1000 Å for free DNA). Such statistical quantities are appropriate for a global model but not for a local model. We have atomic resolution in the simulation and use specific atomic segments, which is the rationale behind taking d = a, b, a′, or b′, rather than the statistical Kuhn segment values. Moreover, the two segments at the enzymatic site are neither purely ss (as in the free ss DNA), nor purely ds (because b′ is the last segment of the ds helix and thus subject to fraying or other end effects (Olmsted et al., 1991)), and therefore ascribing to them Kuhn segment lengths of free ss or ds DNA is artificial.
We note that the thermodynamic functions (g,h,s) per base are, for forces larger than ∼7 pN, comparable in magnitude and force dependence in the RCLM and the FJC pictures. In addition, the FJC thermodynamic functions can be recovered as a particular case of the RCLM. For instance, the underlying ss-stretching free energy per base for free DNA (i.e., in the absence of an enzyme) was, in the FJC picture, found to be well described by
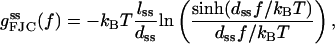 |
(20) |
with lss = 5.6 Å and the Kuhn length dss = 15 Å (Cocco et al., 2001; Rief et al., 1999). We can recover this formula as a particular case of our Eq. 14 by putting θM = 0 and θM = π, such that  Also note that the same formula (Eq. 20) is recovered in the LM picture by adding Eqs. 4 and 3. Moreover, the FJC ds-stretching enthalpy per base for a free DNA duplex in the A-form, with the usual parameters, i.e., a Kuhn length dds = 1000 Å and with lds = 2.6 Å,
Also note that the same formula (Eq. 20) is recovered in the LM picture by adding Eqs. 4 and 3. Moreover, the FJC ds-stretching enthalpy per base for a free DNA duplex in the A-form, with the usual parameters, i.e., a Kuhn length dds = 1000 Å and with lds = 2.6 Å,
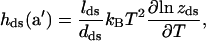 |
(21) |
gives values very close to the f · a′ term we calculate by MD.
Summing up, we have, in the RCLM, that the enthalpy and free-energy barrier changes due to the force for the process of closing the enzyme's fingers are, for the a and b segments only:
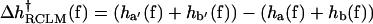 |
(22) |
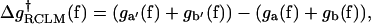 |
(23) |
with h and g calculated from Eqs. 15–17. Regarding the entropic contribution in the RCLM, the TΔs term is much smaller than Δh, as can be seen in Fig. 19, which justifies the use of the  term in the rate-force curve in Fig. 13. The corresponding entropic contribution in both GM and LM is small. We have calculated the force-dependent entropy contribution to the barrier height also from the molecular dynamics simulations using the quasiharmonic approximation (Karplus and Kushick, 1981). Although the noise (error) at each force point is larger than the difference between the open and closed quasiharmonic entropy, the average entropies obtained by linear regression yield a small force-dependent entropic barrier, with a magnitude similar to the RCLM estimate (see Fig. 20).
term in the rate-force curve in Fig. 13. The corresponding entropic contribution in both GM and LM is small. We have calculated the force-dependent entropy contribution to the barrier height also from the molecular dynamics simulations using the quasiharmonic approximation (Karplus and Kushick, 1981). Although the noise (error) at each force point is larger than the difference between the open and closed quasiharmonic entropy, the average entropies obtained by linear regression yield a small force-dependent entropic barrier, with a magnitude similar to the RCLM estimate (see Fig. 20).
FIGURE 20.

Quasiharmonic entropy, at various forces, in the open and the closed state; all atoms of the two segments have been included in the calculation. Each data point is an average of both Run I and II. The straight lines have been obtained by linear regression through the two sets of point. A qualitative interpretation is evident: the open state has higher entropy than the closed one. The difference between the two abscisae (open and closed) at any force (i.e., ΔS†(f)) and the distance between the two intercepts (i.e., ΔS†(0)) yields the entropic barrier Δs†(f) that, although difficult to estimate exactly due to the numerical error of the calculation, seems to have a small magnitude, in accord with the result of Eq. 19 plotted in Fig. 19. We also note that the MD-calculated ΔS(0) compares favorably with the melting entropy of DNA, i.e., with the entropy difference per base between ds and ss DNA, of ∼0.012 cal/mol·K (Rouzina and Bloomfield, 2001).
In Fig. 19 we plot the force-dependent enthalpy (work) for forces extending to 60 pN, using the MD data (together with the enthalpy, free energy, and entropy of activation computed using the RCLM). Chiefly because of the drastic tilt of β′ into the acute range, Δg† becomes negative above 40 pN. Accordingly, in that range Eq. 4 predicts that the replication rate would speed up; for sufficiently high force it would substantially exceed k(0), the rate in the absence of an external force. As noted, experiments show that above 40 pN exonucleolysis overwhelms replication. Despite agreeing well with experiment for forces below 30 pN, the MD analysis using just two DNA segments proves quite inadequate in the higher force regime. To treat replication in that regime it is clearly essential to include other degrees of freedom. In particular, it will be interesting to explore with MD whether, as a consequence of the geometrical changes induced by high forces, the rate-limiting transition state for replication is no longer the closed enzyme conformation but rather becomes completion of the pairing reaction between the incoming nucleotide and the base attached to segment a′ (which, in the open state, is flipped out by ∼90° relative to the closed state; see Fig. 2).
Consistent with the MD results up to 30 pN, the determining force effect in the RCLM is on the open-closed conformational change (the rate-determining step), and the magnitude of the activation barrier is mainly determined by the force-dependent orientations of the two DNA segments.
CONCLUDING DISCUSSION
Molecular dynamics simulations of a ternary complex of Taq DNA polymerase, ss and ds DNA strands and the incoming nucleotide have been performed in the presence of an external force applied to the template strand. It is generally agreed that the conformational change of the enzyme from the open to the closed form is the rate-determining step of the overall incorporation reaction and that the closed state is a good model for the transition state (see text). Consequently, a comparison of molecular dynamics simulation results for the effect of an external force on the open and the closed states of the polymerase-DNA complex can be used to provide an estimate of the effect of the force on the activation free energy. From the structural data, it appears that only three nucleotides play an essential role; these are the last ds residue and the two first ss residues of the template strand. Vectors (a, b), for the open state, and (a′, b′) for the closed one, were introduced to denote the two DNA segments that these three nucleotides delineate. We refer to these two segments as the “inner” (a or a′) and the “outer” (b or b′) segments, respectively, in light of their position relative to the polymerase site.
Although only the nucleotide 0 changes from a ds to an ss geometry (n = 1), the nucleotide −1 also undergoes a significant change in position, even if it does not go to a ds geometry. Moreover, both segments play an energetic role in determining the effect of the force so that the notation “n = 1” or “n = 2” does not provide a definitive description of the actual situation.
For forces up to 30 pN, the range of interest for the single-molecule experiments, the primary effect of the external forces was to reorient the outmost ss segment with respect to the force direction, tilting it markedly toward f in the open state, while the inner segment tilts only slightly from its initial orientation. Thus, in fact, the ss to ds transition involving the inner segment appears to play only a minor role. The determining force effect on the replication rate arises from the work done by the force during the open-close conformational change of the enzyme complex; this is mainly governed by the angular orientations of the two DNA segments. Although the details of incoming nucleotide incorporation are complex and depend on many interactions at the enzymatic site, the overall character of the molecular dynamics results and the experimental data for the force dependence of the replication rate can be described well by a simple model. We refer to this model as the restricted-cone local model. It involves the two DNA segments nearest the active site (i.e., the “inner” and the “outer” ones) and restricts their angular ranges to represent enzyme-imposed steric constraints.
Previous interpretations of pulling experiments on biomolecules have employed phenomenological models. The force dependence of the activation barrier, resulting from work done against the force, is usually ascribed to distance changes rather than angular orientations. Most widely used is the linear response model (Bell, 1978; Evans and Ritchie, 1997), which has been applied to ligand-receptor unbinding (Moy et al., 1994), protein unfolding (Carrion-Vazquez et al., 1999), and DNA unzipping (Strick et al., 2000). This construes the force-dependent contribution to the free energy of activation as given by Δg† = χf, where χ is a characteristic distance along the reaction coordinate. We have evaluated two types of models (the global, GM, and local, LM) which are based on phenomenological analyses. The GM, applied in single-molecule studies of polymerase activity under tension (Wuite et al., 2000; Maier et al., 2000), employs experimental stretching data for free ss and ds DNA to relate the work done by the force to changes in interbase distances that accompany conversion of ss to ds DNA segments. Interpretations based on such models appear untenable from the perspective of our molecular dynamics results, as well as the RCLM model. Distance changes of the kind usually invoked are seen to have, at most, a minor role. The major effects of the external force involve angular orientations and steric constraints in the vicinity of the active site. The LM of Goel et al. (2001) takes into account the angular orientations. However, as shown by the molecular dynamics simulations, the angular contributions are incorrect and the overall agreement obtained by the LM involves the cancellation of two sets of errors, i.e., the error in the average orientations is cancelled by the choice of distance parameters Lss and Lds. The RCLM amends the LM by assigning angular distribution consistent with crystal structures and with the actual dynamics of the relevant DNA segments in the protein matrix. It uses the actual DNA segments (rather than statistical Kuhn lengths) and makes clear the importance of the force-controlled orientation of the segments for the estimation of the force-dependent barrier height.
From our study, the main contributors to this barrier change seem to be the two ss DNA segments as they move in the active site. Therefore, mutation of those residues that border the “cone” of motion of these residues should affect the result of single-molecule pulling experiments.
Another experimentally testable prediction for this low-force regime has to do with DNA polymerase β, which belongs to a structurally distinct class of polymerases, family X, and for which controversy surrounding the identity of its rate-limiting step appears to exist because of two seemingly incongruent results. Although crystal structures of DNA polymerases from several polymerase families (including family A of the DNA polymerase we studied) show that the palm subdomains (that bind dNTP and harbor the residues responsible for catalysis) are structurally homologous, a stark exception is pol β of family X, which has a rather unique palm domain topology. Additionally, its overall fold is structurally distinct from family A or B in that pol β is smaller and has a left-hand shape; its “fingers” domain stabilizes the duplex DNA while the “thumb” binds the incoming nucleotide (unlike family A, which binds the nucleotide in the “palm” while the “thumb” latches onto the duplex). One study by Arndt et al. (2001), using stopped-flow kinetic experiments, suggests that there is no detectable rate-limiting conformational change step for pol β and suggests that chemistry might be rate limiting, whereas another study, by Berg et al. (2001) shows (also using pre-steady-state kinetics) the existence of a rate-limiting conformational change triggered by the closed conformation. This second study also establishes that chemistry is fast for pol β, therefore non-rate-limiting, i.e., in contradiction with the Arndt et al. (2001) work. Because there are no reported studies on the rate-force dependence of polymerases from family X, the above-mentioned incongruency points to this system as a useful subject for future single-molecule experiments or simulation studies. If for family X the open-close transition would not be rate limiting, such pulling studies on pol β should show no rate-force dependence; if the contrary is true, then an exponential dependence (as in the family A case) should be observed.
It is true that force could in principle affect the chemical step by distortion of the active site. However, the exponential decay of the rate with the force is usually indicative of the fact that motion is involved. This can be understood by the fact that the barrier change due to the force goes as −f · x, where x is a typical length. If force distorts the chemical step, it is more likely that an all-or-nothing effect at the active site occurs (which is seen, as discussed above, for RNA polymerases and DNA helicases).
In the higher force regime (f > 30 pN), for the closed state, the outermost DNA segment b′, which had been aimed well away from the direction of the force, undergoes a “dynamical” transition at 40 pN. This force is large enough to cause a sharp reorientation of b′ in the direction of the force f, while at the same force the inner segment, vector a′, already closer to f, also abruptly tilts even closer (cf. Figs. 15 and 16). These angular shifts are of particular interest in view of the experimental observation of the onset at 40 pN of force-induced exonucleolysis in T7 DNAp (Wuite et al., 2000). The simulation results suggest that the changes in the DNA geometry due to the force inhibit the nascent pairing between the incoming nucleotide and the base attached to the inner DNA segment, thereby inhibiting or preventing replication. Although the polymerase modeled computationally here, Klentaq1, has no detected exonuclease activity, this activity is well characterized for other analogous family A polymerases, whose exonuclease site is located in a separate structural domain at a distance of ∼30 Å from the polymerization site (Beese et al., 1993). Because we do not attempt to model the actual process of migration to the exonuclease site, the lack of exonuclease activity does not affect the following energetic argument, which involves the relative free energies of the complex with the incoming nucleotide bound at the polymerization site, and of the complex with the primer strand bound at the exonuclease site. A force above 40 pN could raise the free energy of a correctly bound nucleotide sufficiently to equal or exceed that of an incorrect nucleotide at the polymerase site, thus leading to exonucleolysis if the exo site would be active. The present RCLM is not appropriate for exploring further such aspects of the higher force regime. We are undertaking molecular dynamics simulations focused on the effects of applied force on the nascent basepairing in the active site.
Our MD estimates of the forces at which major changes occur compare well with experimental estimates. In the lower force regime, the force of 8–10 pN required to “break” the outer DNA segment loose from its initial position in the open complex is similar to that needed to break DNA hairpins (Rief et al., 1999). Likewise, in the higher force regime, 40 pN is comparable (Gurrieri et al., 1999) to the forces that cause ds DNA to fray. Fraying at the 3′ end of the primer DNA is regarded as a likely precursor to exonucleolysis. The 3′ terminus of the primer is thought to shuttle between the polymerase site and the exonuclease site with or without dissociation of the DNA from the protein (Joyce, 1989). By exposing the tensed primer/template strands to E. Coli exonuclease I (which attacked only ss DNA at the 3′ end) and comparing the force-dependent rate of base removal for this enzyme with the T7 DNAp exonuclease rate, Wuite et al. (2000) inferred that fraying is not rate limiting for the T7 DNAp exonucleolysis. Instead, they suggested that the force-induced exonucleolysis initiated at 40 pN is a specific property of the enzyme-DNA complex, perhaps due to ds DNA deformation at the 3′ end. The MD simulations support this hypothesis by showing that the orientation of the a′ segment changes significantly due to forces in this high range. Also, we note that the change seen in our simulations at high forces in the orientation of the inner, nominally ds segment in the closed conformation relative to the helix axis, resembles S-shape conformational changes of ds DNA, which occur above 60 pN (Cluzel et al., 1996; Smith et al., 1996).
Outward base flipping from the double strand, as found in the DNA polymerases (see, for instance, left panel in Fig. 5 depicting the flipped-out position of base 0 in the open state), often occurs when enzymes need access to DNA or RNA (Roberts and Cheng, 1998), an example being DNA repair enzymes. Many other biomolecular rate processes are recognized or suspected to involve angular reorientations, and these offer prospects for computational and experimental studies in the presence of external forces. Based on a molecular dynamics simulation, it has been suggested recently that a slow internal rotation of an arginine residue may play a role in the rate-limiting open-close transition of DNA polymerase β (Yang et al., 2002), belonging to the X family of polymerases, which is structurally distinct from family A or B (as explained above).
A corresponding Arg residue exists at position 660 in the O helix of the Taq polymerase. Mutating this residue was found (Li et al., 1999) to selectively reduce ddGTP incorporation, mainly due to an additional hydrogen bond that Asp-600 makes with ddGTP relative to another three incoming nucleotides (Li and Waksman, 2001). It is not known whether, as for pol β, Arg-660 is important in the rate-limiting step of Taq polymerase. This could be examined by means of simulations corresponding to those described here, with a focus on the interaction of Arg-660 with the nascent basepair and its dependence on applied forces.
Simulations of applied forces can elucidate kinetic mechanisms, by mapping out free-energy profiles with and without the force, if a particular reaction coordinate is suspected to be involved in the rate-determining step. Although DNA polymerases in vivo do not usually perform their function on DNA molecules under tensions in the high-force range presented here, significant tensions might be encountered by the polymerases due to supercoiling at the replication fork or after chromosome disentangling. Moreover, for some DNA-binding proteins, large forces or torques act onto the DNA in vivo; examples are RecA-DNA filaments (Hegner et al., 1999) and the nucleosomes in chromatin fibers (Hansen, 2002). For such systems, simulations of the type presented here have the potential of offering new insights into their biological activity.
METHODS
Simulation system
As starting model structures for the all-atom simulations we have used the 2.3-Å resolution crystal structures of the large fragment of Thermus aquaticus DNA polymerase I (Klentaq1)-DNA/incoming nucleotide complex in the open and closed states (PDB codes 2ktq and 3ktq, respectively). The complex system included the protein, the primer/template DNA (5′–AGGGCGCCGTGGTC–3′ for the template and 3′–CGCGGCACCAG–5′ for the primer) and we modeled the incoming nucleotide as well as the 3′-end base of the primer as deoxi-riboses (they are dideoxi-riboses in the crystal structure to prevent the polymerase reaction) to emulate the biologically active conditions. All simulations were performed with the CHARMM program, version 29, with the combined protein-DNA parameter set 27.
Although both a′ and b′ segments are present in the crystal structure of the closed state, the b segment is missing from the crystal structure of the open state, indicative of the fact that it is more mobile. To find a suitable starting position for the b segment, we have built the atoms of the b segment using the coordinates of the corresponding atoms present in the closed structure. Two Mg2+ ions are included in each structure; one of the Mg2+, absent in the open structure was built in the position it had in the closed structure and was then allowed to equilibrate subsequently. Hydrogens were built with HBUILD. A 10-residue loop, missing from the crystal structure of the open complex (but present in the closed structure), was built using the equilibrium values of the bonds and angles found in the parameter set. We centered a 25-Å explicit water sphere from a large box of equlibrated water on the C1′ atom of the 0-th DNA base on the template; water molecules present in the crystal structure were included, as well. The spherical constraint potential of the stochastic boundary simulation method ((Brooks and Karplus, 1983) was used on the water sphere; there were 951 water molecules in the open structure and 911 in the closed one. To ensure charge neutrality of the spherical region, we placed 13 Na+ counterions by replacing 13 water molecules with the highest electrostatic energy on their oxygen atom and the additional condition that no two ions are closer than 5 Å to each other. All the coordinates of the atoms originally present in the crystal structure were fixed and extensive minimization and equilibration of the built atoms was performed. Subsequently, constraints were removed and 2.4 ns of equilibration were performed for the closed complex and 10 ns of equilibration were performed for the open structure. The longer time for the open structure was required to permit the convergence of the β-angle. During the subsequent production runs all coordinates of protein and DNA atoms outside the water sphere were kept fixed at their crystal structure values. The approximation that the atoms outside the sphere are fixed, as opposed to being allowed to move, is expected to be satisfactory because the coordinates of those atoms do not change significantly between the open and closed structures and the major conformational change takes place in the sphere.
The external force was simulated by placing a reference point at l = 40 Å from the position of the O5′ atom of the outmost ss segment in each starting structure along the helical axis direction (see Fig. 7). A force resulting from a harmonic constraint of the desired magnitude f, was applied between this point and the O5′ atom by choosing the force constant k such that f = kl. We note that the distance of 40 Å is about six times larger than the persistence length of ss DNA (whose value is ∼7 Å). Thus, at this distance any “memory” based on preferential directions of the ss segments is expected to be lost; as a consequence, our calculations do not depend on the value of l as long as it is larger than 40 Å.
The lengths of the vectors a, b, a′, b′ were taken as the distances between consecutive C5′ atoms, as explained in the “Analysis of origin of force dependence” section. As noted there, the results of the simulations do not depend on the choice of the type of consecutive atoms. Moreover, the RCLM we have proposed does not rely on any definition of ds or ss geometry, so the discussion of which internucleotide distance is more appropriate for ds and ss geometry becomes irrelevant.
At each force 3 ns of constant-temperature dynamics at 300 K were performed with the Nosé-Hoover method (Nosé, 1984; Hoover, 1985); a time step of 2 fs was used and bonds to hydrogen atoms were kept fixed using the SHAKE algorithm (Ryckaert et al., 1977). Data for averaging were accumulated during the last 2.4 ns of each trajectory. For comparison of timescales, we note that the Zimm model (which includes averaged hydrodynamic effects) predicts a relaxation time of 0.7 ns for ss-Kuhn segments, and that this relaxation time decreases with tension (Hatfield and Quake, 1998).
Treatment of water-screened electrostatic interactions
Because there is no explicit water outside the 25-Å sphere in our simulation, we make use of a charge scaling method that has been successfully applied in classical free-energy calculations (Simonson et al., 1997), as well as in a hybrid quantum-mechanical/molecular-mechanical calculation of activation energies (Dinner et al., 2001). All ionic residues outside the spherical reaction region are scaled to mimic the dielectric screening effect of the bulk solvent. Both the outer ionic protein residues and the outer basepairs are scaled. This is done by solving numerically for the electrostatic potential field Φ(r) from the Poisson-Boltzman equation
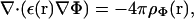 |
(24) |
where
 |
(25) |
with ρ the fixed source charge density, v a dimensionless excluded volume potential (v is 0 in regions accessible to mobile ions, and ∞ in inaccessible regions), and n ± the bulk number densities of ions. The numerical solution uses the CHARMM implementation by B. Roux (unpublished) of a three-step multigrid relaxation technique for an ionic concentration of 0.1 M (which is a typical experimental value).
Using the solution of Eq. 24, the scaling factors αi for all charged residues outside the sphere are calculated according to
 |
(26) |
where the average potentials in vacuum (ε = 1) and water (ε = 80) are obtained by setting the charges of all ionic sides chains of the outer region to zero, except for the charge of the i-th one, which is left unmodified, writing the potentials onto a grid covering the inner region and then averaging over the points of that grid. The evaluation of ε(r) in the Poisson-Boltzmann equation is done by determining the solvent excluded regions using a spherical probe with radius 1.4 Å and standard atomic radii.
Acknowledgments
We thank Aaron Dinner for help with the charge scaling procedure. We also thank the National Energy Research Scientific Computing Center of the Department of Energy for allocation of computer resources. The molecular graphics were created with VMD (Humphrey et al., 1996) and PovRay.
Partial support for the research was provided by the National Institutes of Health (M.K.) and the National Science Foundation (grant PHY-0210437 to D.H.). A.G. is grateful for support from the Klivingston Fellowship for Biodynamics provided by the Fetzer Institute.
Ioan Andricioaei's present address is Dept. of Chemistry, University of Michigan, Ann Arbor, MI.
References
- Arndt, J. W., W. Gong, X. Zhong, A. K. Showalter, J. Liu, C. A. Dunlap, Z. Lin, C. Paxson, M.-D. Tsai, and M. K. Chan. (2001). Insight into the catalytic mechanism of DNA polymerase: structures of intermediate complexes. Biochemistry. 40:5368–5375. [DOI] [PubMed] [Google Scholar]
- Beese, L., V. Derbshire, and T. Steitz. 1993. Structure of DNA polymerase I Klenow fragment bound to duplex DNA. Science. 260:352–355. [DOI] [PubMed] [Google Scholar]
- Bell, G. I. 1978. Models for the specific adhesion of cells to cells. Science. 200:618–627. [DOI] [PubMed] [Google Scholar]
- Berg, B. V., W. Wilson, and S. Wilson. 2001. DNA structure and aspartate 276 influence nucleotide binding to human DNA polymerase β. J. Biol. Chem. 276:3408–3416. [DOI] [PubMed] [Google Scholar]
- Brautigam, C., and T. Steitz. 1998. Structural and functional insights provided by crystal structures of DNA polymerases and their substrate complexes. Curr. Opin. Struct. Biol. 8:54–63. [DOI] [PubMed] [Google Scholar]
- Brønsted, N. 1928. Acid and basic catalysis. Chem. Rev. 5:231–338. [Google Scholar]
- Brooks III, C. L., and M. Karplus. 1983. Deformable stochastic boundaries in molecular dynamics. J. Chem. Phys. 79:6312–6325. [Google Scholar]
- Bryant, F., K. Johnson, and S. Benkovic. 1983. Elementary steps in the DNA polymerase I reaction pathway. Biochemistry. 22:3537–3546. [DOI] [PubMed] [Google Scholar]
- Bueche, F. (1962). Physical Properties of Polymers. Interscience, New York.
- Carrion-Vazquez, M., A. Oberhauser, S. Fowler, P. Marszalek, S. Broedel, J. Clarke, and J. Fernandez. 1999. Mechanical and chemical unfolding of a single protein: a comparison. Proc. Natl. Acad. Sci. USA. 96:3694–3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cluzel, P., A. Lebrun, C. Heller, R. Lavery, J.-L. Viovy, D. Chatenay, and F. Caron. 1996. DNA: an extensible molecule. Science. 271:792–794. [DOI] [PubMed] [Google Scholar]
- Cocco, S., R. Monasson, and J. Marko. 2001. Force and kinetic barriers to unzipping of the DNA double helix. Proc. Natl. Acad. Sci. USA. 98:8608–8613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlberg, M., and S. Benkovic. 1991. Kinetic mechanism of DNA-polymerase-I (Klenow fragment): identification of a 2nd conformational change and evaluation of the internal equilibrium-constant. Biochemistry. 30:4835–4843. [DOI] [PubMed] [Google Scholar]
- Dinner, A., G. Blackburn, and M. Karplus. 2001. Uracil-DNA glycosylase acts by substrate autocatalysis. Nature. 413:752–755. [DOI] [PubMed] [Google Scholar]
- Doublié, S., S. Tabor, A. Long, C. Richardson, and T. Ellenberger. 1998. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 A resolution. Nature. 391:251–258. [DOI] [PubMed] [Google Scholar]
- Eom, S., J. Wang, and T. Steitz. 1996. Structure of Taq polymerase with DNA at the polymerase active site. Nature. 382:278–281. [DOI] [PubMed] [Google Scholar]
- Evans, M., and M. Polanyi. 1938. Inertia and driving force of chemical reactions. Trans. Faraday Soc. 34:11–29. [Google Scholar]
- Evans, E., and K. Ritchie. 1997. Dynamic strength of molecular adhesion bonds. Biophys. J. 72:1541–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin, M., J. Wang, and T. Steitz. 2001. Structure of the replicating complex of a pol α family DNA polymerase. Cell. 105:657–667. [DOI] [PubMed] [Google Scholar]
- Goel, A., T. Ellenberger, M. Frank-Kamenetskii, and D. Herschbach. 2002. Unifying themes in DNA replication: reconciling single-molecule kinetic studies with structural data on DNA polymerases. J. Biomol. Struct. Dyn. 19:571–584. [DOI] [PubMed] [Google Scholar]
- Goel, A., M. Frank-Kamenetskii, T. Ellenberger, and D. Herschbach. 2001. Tuning DNA “strings”: modulating the rate of DNA replication with mechanical tension. Proc. Natl. Acad. Sci. USA. 98:8485–8489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosberg, A., and A. Khokhlov. (1994). Statistical Physics of Macromolecules. American Institute of Physics Press, College Park, MD.
- Gurrieri, S., S. Smith, and C. Bustamante. 1999. Trapping of megabase-sized DNA molecules during agarose gel electrophoresis. Proc. Natl. Acad. Sci. USA. 96:453–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, J. 2002. Conformational dynamics of the chromatin fiber in solution: determinants, mechanisms, and functions. Annu. Rev. Biophys. Biomol. Struct. 31:361–392. [DOI] [PubMed] [Google Scholar]
- Hatfield, J., and S. Quake. 1998. Dynamic properties of an extended polymer in solution. Phys. Rev. Lett. 82:3548–3551. [Google Scholar]
- Hegner, M., S. Smith, and C. Bustamante. 1999. Polymerization and mechanical properties of RecA-DNA filaments. Proc. Natl. Acad. Sci. USA. 96:10109–10114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover, W. G. 1985. Canonical dynamics—equilibrium phase-space distributions. Phys. Rev. A. 31:1695–1697. [DOI] [PubMed] [Google Scholar]
- Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- Izrailev, S., S. Stepaniants, M. Balsera, Y. Oono, and K. Schulten. 1997. Molecular dynamics study of unbinding of the avidin-biotin complex. Biophys. J. 72:1568–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce, C. 1989. How DNA travels between the separate polymerase and 3′-5′-exonuclease sites of DNA polymerase I (Klenow fragment). J. Biol. Chem. 264:10858–10866. [PubMed] [Google Scholar]
- Joyce, C., and T. Steitz. 1994. Function and structure relationships in DNA polymerases. Annu. Rev. Biochem. 63:777–822. [DOI] [PubMed] [Google Scholar]
- Karplus, M., and J. Kushick. 1981. Method for estimating the configurational entropy of macromolecules. Macromolecules. 14:325–332. [Google Scholar]
- Kiefer, J., C. Mao, J. Braman, and L. Beese. 1998. Visualizing DNA replication in a catalytically active Bacillus DNA polymerase crystal. Nature. 391:304–307. [DOI] [PubMed] [Google Scholar]
- Kim, Y., S. Eom, J. Wang, D. Lee, S. Suh, and T. Steitz. (1995). Crystal structures of Thermus aquaticus DNA polymerase. Nature, 376. [DOI] [PubMed]
- Konrad, M., and J. Bolonick. 1996. Molecular dynamics simulation of DNA stretching is consistent with the tension observed for extension and strand separation and predicts a novel ladder structure. J. Am. Chem. Soc. 118:10989–10994. [Google Scholar]
- Kornberg, A., and T. Baker. (1991). DNA Replication. W. H. Freeman, New York.
- Kunkel, T., and K. Bebenek. 2000. DNA replication fidelity. Annu. Rev. Biochem. 69:497–529. [DOI] [PubMed] [Google Scholar]
- Li, Y., Y. Kong, S. Korolev, and G. Waksman. 1998a. Crystal structures of the Klenow fragment of Thermus aquaticus DNA polymerase I complexed with deoxyribonucleoside triphosphates. Protein Sci. 7:1116–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y., S. Korolev, and G. Waksman. 1998b. Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: structural basis for nucleotide incorporation. EMBO J. 17:7514–7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y., V. Mitaxov, and G. Waksman. 1999. Structure-based design of Taq DNA polymerase with improved properties of dideoxinucleotide incorporation. Proc. Natl. Acad. Sci. USA. 96:9491–9496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y., and G. Waksman. 2001. Crystal structures of a ddATP-, ddTTP-, ddCTP-, and ddGTP-trapped ternary complex of Klentaq1: insights into nucleotide incorporation and selectivity. Protein Sci. 10:1225–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell, A., Jr., and G. Lee. 1999. Structure, force, and energy of a double-stranded DNA oligonucleotide under tensile loads. Eur. Biophys. J. 28:415–426. [DOI] [PubMed] [Google Scholar]
- Maier, B., D. Bensimon, and V. Croquette. 2000. Replication by a single DNA polymerase of a stretched single-stranded DNA. Proc. Natl. Acad. Sci. USA. 97:12002–12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkel, R., P. Nassoy, A. Leung, K. Ritchie, and E. Evans. 1999. Energy landscapes of receptor-ligand bonds explored with dynamic force spectroscopy. Nature. 397:50–53. [DOI] [PubMed] [Google Scholar]
- Moy, V., E.-L. Florin, and H. Gaub. (1994). Intermolecular forces and energies between ligands and receptors. Science. 266:257–259. [DOI] [PubMed] [Google Scholar]
- Nayal, S. K. M., W. Barnes, E. D. Cera, and G. Waksman. 1995. Crystal structure of the large fragment of Thermus aquaticus DNA polymerase I at 2.5 Å resolution: structural basis for thermostability. Proc. Natl. Acad. Sci. USA. 92:9264–9268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosé, S. 1984. A unified formulation of the constant temperature molecular-dynamics methods. J. Chem. Phys. 81:511–519. [Google Scholar]
- Ollis, D., P. Brick, R. Hamlin, N. Xuong, and T. Steitz. 1985. Structure of the large fragment of Escherichia coli DNA polymerase I complexed with dTMP. Nature. 313:762–766. [DOI] [PubMed] [Google Scholar]
- Olmsted, M., C. Anderson, and M. Record, Jr. 1991. Importance of oligoelectrolyte end effects for the thermodynamics of conformational transitions of nucleic acids oligomers: a grand canonical Monte Carlo analysis. Biopolymers. 31:1593–1604. [DOI] [PubMed] [Google Scholar]
- Paci, E., and M. Karplus. 2000. Unfolding proteins by external forces: the importance of topology and energetics. Proc. Natl. Acad. Sci. USA. 97:6521–6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, P., and L. A. Loeb. 2001. Getting a grip on how DNA polymerases function. Nat. Struct. Biol. 8:656–659. [DOI] [PubMed] [Google Scholar]
- Patel, S., I. Wong, and K. Johnson. 1991. Pre-steady-state kinetic analysis of processive DNA replication including complete characterization of an exonuclease-deficient mutant. Biochemistry. 30:511–525. [DOI] [PubMed] [Google Scholar]
- Rief, M., H. Clausen-Schaumann, and H. Gaub. 1999. Sequence-dependent mechanics of single DNA molecules. Nat. Struct. Biol. 6:346–349. [DOI] [PubMed] [Google Scholar]
- Roberts, R., and X. Cheng. 1998. Base flipping. Annu. Rev. Biochem. 67:181–198. [DOI] [PubMed] [Google Scholar]
- Rouzina, I., and V. Bloomfield. 2001. Force-induced melting of the DNA double helix. 1. Thermodynamic analysis. Biophys. J. 80:882–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert, J. P., G. Ciccotti, and H. J. C. Berendsen. 1977. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23:327–341. [Google Scholar]
- Simonson, T., G. Archontis, and M. Karplus. 1997. Continuum treatment of long-range interactions in free energy calculations. Application to protein-ligand binding. J. Phys. Chem. B. 101:8349–8362. [Google Scholar]
- Smith, S., Y. Cui, and C. Bustamante. 1996. Overstretching B-DNA: the elastic response of individual double-stranded and single-stranded DNA molecules. Science. 271:795–799. [DOI] [PubMed] [Google Scholar]
- Steitz, T. 1998. A mechanism for all polymerases. Nature. 391:231–232. [DOI] [PubMed] [Google Scholar]
- Steitz, T. 1999. DNA polymerases: structural diversity and common mechanisms. J. Biol. Chem. 274:17395–17398. [DOI] [PubMed] [Google Scholar]
- Strick, T., J.-F. Allemand, D. Bensimon, and V. Croquette. 2000. Stress-induced structural transitions in DNA and proteins. Annu. Rev. Biophys. Biomol. Struct. 29:523–543. [DOI] [PubMed] [Google Scholar]
- Turner, R., Jr., N. Grindley, and C. Joyce. 2003. Interaction of DNA polymerase I (Klenow fragment) with the single-stranded template beyond the site of synthesis. Biochemistry. 42:2373–2385. [DOI] [PubMed] [Google Scholar]
- Wang, M., M. Schnitzer, H. Yin, R. Landick, J. Gelles, and S. Block. 1998. Force and velocity measured for single molecules of RNA polymerase. Science. 282:902–907. [DOI] [PubMed] [Google Scholar]
- Wuite, G., S. Smith, M. Young, D. Keller, and C. Bustamante. 2000. Single-molecule studies of the effect of template tension on T7 DNA polymerase activity. Nature. 404:103–106. [DOI] [PubMed] [Google Scholar]
- Yang, L., W. Beard, S. Wilson, S. Broyde, and T. Schlick. 2002. Polymerase β simulations suggest that Arg258 rotation is a slow step rather than large subdomain motions per se. J. Mol. Biol. 317:651–671. [DOI] [PubMed] [Google Scholar]