

Abstract
Fluorescence correlation spectroscopy (FCS) is a versatile tool for investigating the mobilities of fluorescent molecules in cells. In this article, we show that it is possible to distinguish between freely diffusing and membrane-bound forms of biomolecules involved in signal transduction in living cells. Fluorescence correlation spectroscopy was used to measure the mobility of phytochrome, which plays a role in phototropism and polarotropism in protonemal tip cells of the moss Ceratodon purpureus. The phytochrome was loaded with phycoerythrobilin, which is fluorescent only in the phytochrome-bound state. Confocal laser scanning microscopy was used for imaging and selecting the xy measuring position in the apical zone of the tip cell. Fluorescence correlation was measured at ancient z-positions in the cell. Analysis of the diffusion coefficients by nonlinear least-square fits showed a subcellular fraction of phytochrome at the cell periphery with a sixfold higher diffusion coefficient than in the core fraction. This phytochrome is apparently bound to the membrane and probably controls the phototropic and polarotropic response.
INTRODUCTION
Many light-dependent developmental effects in plants are controlled by the phytochrome photoreceptors (Kendrick and Kronenberg, 1994). The intracellular distribution of phytochrome has been monitored by conventional, electron, and fluorescence microscopy (Pratt, 1994; Kircher et al., 1999; Hisada et al., 2000). With these techniques, it has been possible to detect phytochrome in the cytosole and in the nucleus, but information about the mobility of phytochrome molecules is lacking.
Fluorescence correlation spectroscopy (FCS) is a sensitive and noninvasive technique with which it is possible not only to measure fluorescence intensity but also to learn about the mobility of particles within a femtoliter volume on the single molecule level in living cells through analysis of the intensity fluctuations.
The diffusibility of biomolecules in specific subcellular compartments can be determined by combining laser scanning microscopy (LSM) with local FCS measurements. The methodological aim of this work was to show that it is possible to distinguish between a freely diffusing biomolecule in the cytosole and a membrane-associated form of the same by determining the molecular mobility in the center and the cell edge using FCS. FCS has previously been used to characterize the membrane association of signaling molecules in mammalian cells (Brock et al., 1999). In this work, we used phytochrome as an example for a signal transduction protein to examine its role in phototropism and polarotropism in plant cells.
Phytochromes are homodimeric photochromic chromoproteins. Each subunit carries one bilin chromophore; in plants, this is usually phytochromobilin (Rüdiger and Thümmler, 1994). Phytochrome action is triggered by photoconversion from the red-light absorbing form, Pr, into the far-red-light absorbing form, Pfr. In seed plants grown in the dark, phytochrome is generally found in the cytosole, but upon photoconversion it is partially translocated into the nucleus (Nagy et al., 2000) where it is involved in the regulation of gene expression.
Despite evidence that phytochrome can act in the nucleus, nature has provided many examples where phytochrome exerts its action at the cell periphery: in green algae, mosses, and ferns, phytochrome controls cellular vectorial responses such as chloroplast movement and phototropism of protonemal tip cells (Kraml, 1994). Experiments with polarized light have suggested that the active phytochrome molecules are attached to or located closely at the plasmalemma and that the transition dipole moment of the Pr form is oriented parallel to the longitudinal axis of the cell (Kraml, 1994).
Our aim was to gain further insight into the intracellular distribution of phytochrome in protonemal tip cells of the moss Ceratodon purpureus. These cells display a phytochrome-controlled phototropic response. As in other cryptogam species, the effect of polarized red light implies that phytochrome acts at the cell periphery (Hartmann et al., 1983; Esch et al., 1999). On the other hand, the major part of extracted phytochrome is found in the soluble fraction (Lamparter et al., 1995) and antibodies detected phytochrome in the cytosole (T. Lamparter, unpublished). The fraction of phytochrome, which controls phototropism and polarotropism, may be small. Using conventional techniques, the phytochrome at the cell periphery appears to be hidden behind the bulk of cytosolic phytochrome. Since membrane-associated molecules have a lower mobility than those in the cytosole, it should be possible to separate membrane-bound from cytosolic phytochrome by FCS, which provides an extremely small femtoliter element.
To obtain fluorescent phytochrome, we used direct labeling by chromophore replacement with phycoerythrobilin (PEB). Because there is only one double bond difference between the PEB adduct and the natural phytochromobilin adduct, the PEB method does probably not influence the surface binding properties of the phytochrome protein we were interested in. Other groups labeled phytochrome by expressing it as a fusion protein with green fluorescent protein (GFP) (Kircher et al., 1999; Yamaguchi et al., 1999), but a protein fusion with GFP entails the danger of affecting the protein activity, e.g., by modifying surface binding.
Apophytochromes assemble with PEB in vitro (Li et al., 1995) and in vivo (Murphy and Lagarias, 1997). The fluorescence of free PEB is negligible, but the phytochrome adduct is highly fluorescent with a quantum yield of ∼0.7–0.8, an absorbance maximum at ∼575 nm, and an emission maximum at ∼585 nm (Murphy and Lagarias, 1997). In contrast, the fluorescence quantum yield of the natural adduct with the phytochromobilin chromophore is ∼10−3–10−4 (Sineshchekov, 1995) and cannot be used for detecting phytochrome at the cellular level.
From Ceratodon, mutants with a defect in chromophore biosynthesis have been isolated. These mutants are characterized by their aphototropic growth and low chlorophyll content. The mutants, termed ptr class 1 mutants, can be rescued by biliverdin, a precursor of phytochromobilin. This finding implied that the mutants are defective in heme-oxygenase, an enzyme that converts heme into biliverdin. This suggestion has been confirmed for the class 1 mutant ptr116 by microinjection studies (Brücker et al., 2000). The Ceratodon class 1 mutants offer a valuable tool for labeling phytochrome by PEB feeding, because the chromophore-free phytochrome in the cell will assemble with PEB from the medium. The PEB adducts do not undergo photoconversion into the Pfr form. In fluorescence measurements, photoactive phytochrome would soon be converted into Pfr; therefore, PEB phytochrome are advantageous for measuring the intracellular distribution in the ground state.
Fluorescence autocorrelation spectroscopy
Conventional fluorimetry measures the average fluorescence intensity of a bulk sample and can be used to obtain data about concentration, quenching, and fluorescence resonance energy transfer (FRET) efficiency. However, the pattern of intensity fluctuations in very small ensembles of fluorescent molecules, which cannot be obtained from averaged signals, also yields a wealth of additional information. A method for quantitative analysis of such fluctuations in equilibrium was introduced in 1972 (Madge et al., 1972; Elson and Madge, 1974) and termed fluorescence correlation spectroscopy. In the simplest case, the intensity fluctuations are induced by fluorophores entering and leaving the illuminated region by Brownian motion and are analyzed by calculating the autocorrelation function of the fluorescence signal with high temporal resolution. This autocorrelation analysis reveals characteristic timescales of the fluorescence fluctuations by self (auto)-comparison of the fluctuating intensity signal for statistical repetitions.
The full potential of FCS has been exploited since 1991. The introduction of confocal detection optics, which focuses a laser beam through a microscope (Fig. 1), makes measurements in an open femtoliter volume element possible. This has led to an increased signal/background ratio (Qian and Elson, 1991; Rigler et al., 1993). In combination with highly sensitive avalanche photodiodes, stable laser sources, and precise optics, it has been possible to analyze fluorophores on a single molecule level (Rigler et al., 1992); e.g., 1 fl nanomolar solution contains on average of 0.6 molecules that produce intensity fluctuations by entering and leaving the focal volume. The measured time-dependent fluctuations allow the diffusional parameters to be calculated by autocorrelation analysis. In addition to revealing the molecular residence or diffusion time τD (Fig. 2 c, info 1), this analysis determines the average number of simultaneously observed particles in the focus (Fig. 2 c, info 2) (indicating the molecular brightness) and the short-lived average times of dark states of the fluorophore, such as triplet state or blinking (Fig. 2 c, info 3). FCS has been used for the analysis of fluorescently labeled biomolecules in solution (Qian et al., 1992; Schwille et al., 1997), on membranes (Bacia et al., 2002; Schwille et al., 1999b), and in living cells (Widengren and Rigler, 1998; Brock et al., 1998; Schwille et al., 1999a).
FIGURE 1.

Setup. Schematic representation of the ConfoCor 2 setup for autocorrelation analysis. The 543-nm laser line of a helium-neon laser was reflected by a dichroic mirror (main beam splitter 543) and focused through an objective onto the sample. The fluorescence emission collected by the same objective was passed through a bandpass filter, focused, and recorded with an avalanche photodiode while reducing out-of-plane fluorescence by a pinhole. The fluorescence signal was software correlated and displayed online.
FIGURE 2.

Principle of fluctuation analysis (a); representative data of a fluorophore containing a fluorescent trace of Alexa 488 (b); and a model autocorrelation curve (c). Entering and leaving of a fluorophore induces intensity fluctuations that are processed by autocorrelation analysis resulting in a curve such as info 1 for a diffusion time of τD = 0.2 ms. The average number of particles in the focus is found in G(t)−1 (info 2). The autocorrelation curve resulting from the diffusion is shown shaded. Internal dynamics such as triplet state or blinking give signals that result in a second curve (info 3). This adds up to the diffusion curve.
MATERIALS AND METHODS
Bilin chromophores
The production of the bilin chromophores PEB and phycocyanobilin (PCB) is described in detail in Lamparter et al. (2001). The bilins were extracted from Porphyridium cruentum and Spirulina geitlerie, respectively, by boiling methanolysis, purified by high-performance liquid chromatography (HPLC), and stored as methanol stock solutions (∼5 mM) at −80°C.
Moss strains and cultures
The wild-type strain wt4 and the mutant ptr116 of the moss C. purpureus were used for this study (Lamparter et al., 1996). Protonemal filaments were grown on petri dishes containing 1b medium with 1.1% agar (Lamparter et al., 1996). Protonemal tissue was inoculated on small cellophane squares that were placed on top of the agar medium. Thereafter, the filaments were grown for 5 or 6 days at 20°C in the dark, keeping the petri dishes in a vertical position. Under these conditions, the filaments grow upwards. The tip cells were located at a distance ∼5 mm above the inoculation zone. For bilin feeding, stock solutions were mixed with the agar medium before cooling and hardening of the agar. The final concentrations were 4 μM for PEB and 10 μM for PCB. Moss filaments were transferred to that medium 1 day before the fluorescence assays and kept under the same growth conditions as before. The moss plates were transferred to the microscope room in black boxes before single filament samples were taken for microscopy. Sample handling was performed under dim white light, keeping the time of light exposure as short as possible. For measurements on intracellular green fluorescent protein, the pBASGFP expression plasmid was injected into ptr116 tip cells that had been grown as above (Brücker et al., 2000). Fluorescence measurements were performed 2 days after the injection. In vitro FCS measurements with 30 nM GFP or with 30 nM PEB adduct of phytochrome Cph1 from the cyanobacterium Synechocystis PCC 6803 (Lamparter et al., 2001) were performed in buffer (50 mM Tris/HCl, 5 mM EDTA, 100 mM NaCl, pH 7.6).
Experimental setup for LSM and FCS
Confocal LSM and FCS were performed on a commercial ConfoCor 2 LSM 510 combination system (Zeiss, Jena, Germany) in an autocorrelation configuration (Fig. 1). The 543-nm laser line of a helium-neon laser was reflected by a dichroic mirror (main beam splitter 543) and focused through a Zeiss C-Apochromat 40×, NA 1.2 water immersion objective onto the sample. The excitation power at the location of the sample was 13 μW. The fluorescence emission collected by the same objective was passed through a 560–615-nm bandpass filter and recorded with an avalanche photodiode. Out-of-plane fluorescence was reduced by a pinhole with a diameter of 80 μm. The fluorescence signal was software correlated and displayed online. The detection volume had a 1/e2 lateral radius of ≅0.20 μm as determined by calibration measurements with Alexa 546 (Molecular Probes, Eugene, OR); the axial 1/e2 radius was ≅1.1 μm. The desired position for intracellular measurements was selected in the LSM image, using the automated stage positioning of the ConfoCor 2 system. During data acquisition, the entire sample was covered by a black lid. Therefore, the cells were only irradiated by measuring light. Data were evaluated by Levenberg-Marquardt nonlinear least-square fitting to the appropriate model equations using Origin Software 7.0 (OriginLab, Northampton, MA).
Control measurements with GFP were performed with a similar setup. The 488-nm laser line of an argon-ion laser was reflected by a main beam splitter 488 nm; the emitted light was passed through a 505–550-nm bandpass filter. With this setting, the detection volume had a 1/e2 lateral radius of ≅0.18 μm and an axial 1/e2 radius of ≅0.98 μm as determined by calibration measurements with Alexa 488 (Molecular Probes).
Mathematically, FCS analyzes the fluctuations of the fluorescence emission signal for statistical regularities by autocorrelation analysis using the general correlation function (Elson and Madge, 1974):
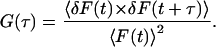 |
(1) |
In a confocal setup (Qian and Elson, 1991), a focal spot is obtained with the laser intensity decreasing to 1/e2 defined as ω0; z0 is the 1/e2 parameter in axial direction. The structure parameter SP of this detection volume is defined as z0/ω0 (Schwille, 2001).
For the observation of translational 3D diffusion, the autocorrelation function G(τ) is defined as follows (Aragon and Pecora, 1976; Meseth et al., 1999; Schwille et al., 1997):
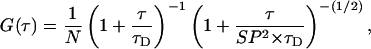 |
(2) |
with the characteristic diffusion time τD:
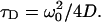 |
(3) |
D is the diffusion coefficient of the fluorophore and N the average number of fluorescent particles in the detection volume.
The diffusion time τD of a fluorophore can be determined by Levenberg-Marquardt nonlinear least-square fitting of the observed autocorrelation curve. The diffusion time of a second component obtained by binding of the fluorophore in a complex with a higher molecular weight can be determined by introducing a second independent diffusion form into the fit function as (Rigler et al., 1993)
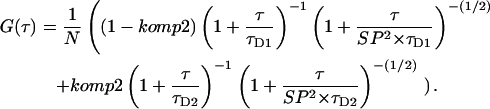 |
(4) |
RESULTS AND DISCUSSION
In this work, fluorescence correlation spectroscopy was used to distinguish between cytosolic and the membrane-associated form of a biomolecule in living cells using phytochrome as an example. In the class 1 mutant ptr116 of C. purpureus, biosynthesis of the phytochrome chromophore is blocked at the point of biliverdin formation (Brücker et al., 2000). After feeding with phycoerythrobilin at appropriate concentrations, phytochrome molecules will incorporate PEB as chromophore. Ceratodon has three phytochrome genes, CerpuPhy1 to CerpuPhy3 (Thümmler et al., 1992; Hughes et al., 1996; Mittmann, 2003). The mRNA level of CerpuPhy1 in protonemal tissue is very weak (Pasentsis et al., 1998), and the protein level is below the detection limit of western blots (Lamparter et al., 1995). The abundance of CerpuPhy2, which has been detected on the mRNA and protein level (Lamparter et al., 1995; Pasentsis et al., 1998), correlates with spectrally measurable phytochrome (Lamparter et al., 1995; Esch and Lamparter, 1998), but detailed information about CerpuPhy3 is as yet lacking. The PEB stain will thus reflect the intracellular distribution of CerpuPhy2 alone or together with CerpuPhy3. In wild-type filaments, only a minor fraction of phytochrome molecules should incorporate PEB from the growth medium, because the cells produce normal levels of phytochromobilin, the natural phytochrome chromophore (Zeidler et al., 1998), which competes with PEB. Initially, PEB-loaded ptr116 and wild-type protonemal tip cells were analyzed by confocal laser scanning microscopy, with spectral parameters set for the PEB adduct of phytochrome. A strong fluorescence was seen in ptr116 tip cells (Fig. 3 a); without PEB, no significant fluorescence was detected under the conditions selected (data not shown). Assembly studies have shown that recombinant oat phytochrome incorporates PCB with a kinetic constant of 0.25 s−1 and PEB with 0.054 s−1 (Li et al., 1995). Since the PCB adduct is nonfluorescent, a simultaneous PCB/PEB feeding at comparable concentrations should lower the PEB signal. Indeed, when 4 μM PEB was fed together with 10 μM PCB, the fluorescence signal was reduced to one-third of the level without PCB competition (Fig. 3 b and Table 1). Similarly, when wild-type cells that produce the natural phytochromobilin chromophore were incubated with 4 μM PEB, the PEB signal was about one-fourth as compared to ptr116 (Fig. 3 c and Table 1). The specificity of the fluorescence signal shows that it is related to the PEB adduct of phytochrome. Comparable results have been obtained with Arabidopsis wild-type and chromophore-deficient-mutant seedlings, for which the phytochrome/PEB labeling has been established (Murphy and Lagarias, 1997). From the LSM images, it appears that in the protonemal tip cell phytochrome molecules are located in the cytosole. No fluorescence signal was observed in plastids and vacuoles. Usually, there was also no signal detected in the nucleus.
FIGURE 3.

Fluorescence images of protonemal tip cells of C. purpureus after PEB/PCB feeding, apical region. (a and b) The phytochrome-chromophore deficient mutant ptr116, (a) 4 μM PEB, (b) 4 μM PEB and 10 μM PCB, and (c) wild-type, 4 μM PEB.
TABLE 1.
Fluorescence intensity of PEB- and PCB-loaded Ceratodon tip cells
| Counts per second | |
|---|---|
| ptr116 PEB | 1800 ± 300 |
| ptr116 PEB/PCB | 600 ± 100 |
| Wild-type PEB | 440 ± 70 |
Images as shown in Fig. 3 were used for quantification of the signal. From each cell, pixel values of a line across the cell were averaged. Mean values from six cells ± SE.
The relative mobility of the fluorescing particles was measured with fluorescence correlation spectroscopy on PEB-labeled wild-type and ptr116 cells. Preliminary experiments were performed to optimize the duration of measurement, intensity of exciting light, and spectral parameters. Under the conditions selected (see Materials and Methods section), bleaching of the PEB fluorophore was negligible. The xy position of the confocal detection volume was chosen in the LSM image. All measurements were performed in the apical dome of the tip cell (see Fig. 3), which is the growing zone of the cell and the site of light perception for the phototropic stimulus (Esch et al., 1999). Again, the signal intensity was smaller in wild-type cells, but otherwise qualitatively similar results were obtained for both strains. To compare the mobility of phytochrome at the cell periphery and at the core of the cell, FCS measurements were performed at different z-positions that were adjusted by a stepper motor (Fig. 4). Before each FCS-measurement, an automated z-scan of the fluorescence intensity within the confocal volume was performed in 0.5-μm steps. This procedure gives typical intensity profiles, which mark the upper and lower edges of the cell. These profiles also allow corrections for possible z-movements of the cell between subsequent FCS measurements. An intensity profile of a ptr116 cell is shown in Fig. 5 a. This example was measured close to the tip, and it shows a gradual transition from the membrane region to the core of the cell. Normalized FCS curves taken at five different z-positions of the same cell are shown in Fig. 5 b using a logarithmic timescale. The FCS traces taken at the cell periphery are shifted to much higher diffusion times τD, which reveals a restricted mobility of phytochrome. The characteristic mobility difference between periphery and center was found for every cell analyzed (n = 12). Fitting the FCS curves from the periphery to the autocorrelation function required two components, which suggests that in this region of the cell two populations with different mobility are detected within the confocal volume. We attempted to find two fixed diffusion times that could be used to fit all autocorrelation curves. To that end, FCS curves were fitted to Eq. 4 (with two components) by Levenberg-Marquardt nonlinear least-square fits. From measurements of the core of a cell, a mean τD value of 0.75 ± 0.05 ms was obtained for the faster component. This corresponds to an average diffusion coefficient of D = 1.3×10−7 cm2/s based on the calibrated detection volume. The diffusion time of the slower component was obtained from measurements at the cell periphery; in this case, the mean value was 4.8 ± 1 ms, which corresponds to an average diffusion coefficient of D = 2.1×10−8 cm2/s. This sixfold decrease of the diffusion coefficient indicates the binding of phytochrome to a huge cellular structure.
FIGURE 4.

Schematical drawing of the experimental setup for FCS measurements. The protonemal tip cells of Ceratodon (not drawn to scale) have a diameter of 10–13 μm and are ∼200 μm long. The double-cone symbol indicates the location of the confocal volume for the autocorrelation measurements. Arrows show how the sample is moved during a z-scan. The xy position is chosen from the LSM image.
FIGURE 5.

(a) Fluorescence-intensity profile of a z-scan through the apical region of a PEB-loaded ptr116 cell; (b) normalized FCS curves at four different z-positions of the cell (positions indicated in a). (c) Examples for fit functions obtained for diffusion times of 0.75 and 4.8 ms.
With these two time constants, all phytochrome measurements could be fitted well to Eq. 4. Examples for fit functions are shown in Fig. 5 c. From these fits, the relative contribution of each component was calculated for all positions along the cross section of a cell. In Fig. 6 a, fluorescence intensities and the calculated relative contributions of the slower component obtained from three different cells are plotted against the measuring position (z-position).
FIGURE 6.

(a) Fluorescence intensity and fraction of phytochrome with low mobility of three different cells plotted against the relative z-position. The fluorescence intensity is measured in the same focus used for FCS analysis; the profile gives the cell dimension in z-direction. The low-mobility fraction was determined by autocorrelation analysis and nonlinear least-square fit. Fixed diffusion times were used in a fit function with two components (see text). The relative contribution of the high- and low-mobility component were obtained from these fits. For cell 1 and cell 2, the measuring position was ∼6 μm behind the tip, where the cell diameter in z-direction is ∼10 μm. The measuring position of cell 3 was closer to the tip region; therefore, the diameter was only ∼5 μm. (b) Fraction of low-mobility phytochrome, determined as in a, plotted against the relative intensity (see text). Data from seven different cells.
As the analyzed cell profiles varied in size and maximal fluorescence intensity, we chose a second approach for comparing and presenting data from different cells. For this purpose, we plotted the fraction of the slower component, calculated as above, against the relative intensity (Fig. 6 b). As a 100% reference for the intensity, we chose the mean value from 10 data points of the cell center. Results from data points where the relative intensity was 100% or above are plotted at the 100% position in the center of Fig. 6 b. All other data points are plotted either to the left or to the right of this position, depending on whether the measuring point was in the upper or in the lower half of the cell, respectively. Since a decrease in the fluorescence intensity indicates that the measuring position is close to the edge of the cell, the left and right region of the plot will represent the situation of the upper and lower edge of the cells.
From Fig. 6, a and b, it is evident that the fraction of lower mobility can make up to 80% measured phytochrome in the cell periphery. We propose that the peripheral fraction with lower mobility represents phytochrome bound to the plasma membrane of the tip cell. This is consistent with the calculated diffusion coefficient of D = 1.3×10−7 cm2/s for freely diffusing phytochrome and of D = 2.1×10−8 cm2/s for phytochrome at the cell periphery. The small low-mobility fraction in the core of the cell might originate from a nonspecific background autofluorescence of intracellular fluorescent proteins with a high molecular weight (see Brock et al., 1998). On the other hand the mean τD value for the faster component was fixed, so small deviations may manifest in a second component.
To check whether the increase of τD could result from particular optical conditions at the cell periphery, we performed control measurements through microinjection of a GFP expression vector into ptr116 tip cells. All GFP FCS traces, including those from the cell periphery, fitted well with autocorrelation functions with a single component (Eq. 2). The τD values calculated from these fits varied between 0.12 and 0.35 ms with a mean value of 0.22 ± 0.03 ms, corresponding to a calculated diffusion coefficient of D = 3.89 × 10−7 cm2/s for freely diffusing GFP. There was no specific increase of τD at the cell periphery (Fig. 7). The diffusion coefficient of GFP is three times higher than that of the mobile fraction of phytochrome. GFP is a 27-kDa monomer (Prasher et al., 1992), whereas plant phytochromes are homodimers with ∼120 kDa for each subunit (Kendrick and Kronenberg, 1994) and have therefore a ∼10-fold higher molecular weight. For equally shaped molecules of equal density, the diffusion time is proportional to the radius of the molecule, which in turn depends on the third square root of the molecular weight. Thus, a ratio of 3 between both diffusion coefficients would be equivalent to a ratio of ∼27 between the molecular weights. This discrepancy might be explained by the different shape of the molecules. Whereas GFP is a more or less globular protein (Ormo et al., 1996), phytochromes deviate from the spherical form and appear ellipsoid or Y shaped (Jones and Quail, 1989; Quail, 1997; Zeidler et al., 1998). To test for the influence of molecule shape on FCS measurements, we compared recombinant GFP with the PEB adduct of cyanobacterial Cph1 from Synechocystis PCC 6803 in vitro. The τD values were 0.086 ± 0.001 ms for GFP and 0.300 ± 0.02 ms for PEB-Cph1, corresponding to diffusion coefficients of D = 9.35 × 10−7 cm2/s and D = 3.27 × 10−7 cm2/s, respectively, and a D ratio of 2.8. This value is slightly smaller than the value above which arose from the comparison of Ceratodon PEB phytochrome and GFP in vivo. This correlates with the molecular size of the Cph1 dimer of 170 kDa, which is slightly smaller than that of plant phytochromes. Thus, the in vitro measurements support the assumption that the relatively small diffusion coefficient of phytochrome results from the shape of the molecule.
FIGURE 7.

Fluorescence measurements with GFP in tip cells of the ptr116 mutant. The measurements were performed in the apical dome of GFP expressing tip cells ∼5 μm from the tip. (a) Normalized autocorrelation traces from selected positions of the tip cell. See b for the position within the cell. (b) Fluorescence intensity profiles and autocorrelation τD values along the z axis of two different cells.
The high diffusion times found for peripheral phytochrome result indeed from restricted mobility, which in turn indicates association of phytochrome to the plasmamembrane. Other studies measured ligand binding to receptors on the surface of cells by autocorrelation analysis (Rigler et al., 1999; Pramanik and Rigler, 2001). In these experiments, diffusion coefficients for membrane-bound particles ranged from D = 2 × 10−7 cm2/s to D = 2 × 10−9 cm2/s (calculated from the determined  and
and 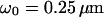 (Aragon and Pecora, 1976; Krichevsky and Bonnet, 2002), which is in accordance with our results.
(Aragon and Pecora, 1976; Krichevsky and Bonnet, 2002), which is in accordance with our results.
For many cryptogam species, including the moss C. purpureus analyzed here, a particular orientation of active phytochrome molecules has been postulated from action dichroism studies (Hartmann et al., 1983; Kraml, 1994; Esch et al., 1999). It has long been proposed that active phytochrome molecules in cryptogams are either attached to the plasma membrane or to structures close by. Membrane-associated phytochrome has not been directly detected in the cell yet, because the background of cytosolic phytochrome makes it difficult to specifically detect a membrane-bound subfraction.
In this article, we show that this problem can be solved by FCS, which provides an extremely small femtoliter volume element suitable for highly sensitive measurements. With FCS, the mobility of biomolecules in living cells can be determined in a noninvasive manner. In this way, a peripheral fraction of phytochrome with low mobility was detected at the cell periphery despite the existence of the bulk cytosolic phytochrome displaying normal mobility.
The fact that peripheral phytochrome appears still mobile on the plasma membrane does not conflict with its proposed dichroic orientation. The mode of phytochrome-membrane interaction is as yet only poorly understood, but membrane-diffusible anchor proteins could hold the phytochrome dimer in a particular orientation with the transition dipole of each chromophore at a particular angle to the cell surface. Moreover, it is possible that another subfraction of membrane-associated phytochrome is overseen with our technique: FCS is insensitive for immobile molecules. Although LSM images (Fig. 3) and intensity scans (Fig. 6 a) show that the concentration of phytochrome in the plasmamembrane region is not above the average, we consider it possible that an immobile membrane-bound fraction is hidden from FCS detection.
With FCS, it will be possible to test for membrane-associated phytochrome in other cryptogams and in angiosperms. Since membrane-associated phytochrome has generally been found in cell extracts (Rubinstein et al., 1969; Lamparter et al., 1992; Esch and Lamparter, 1998), it has to be checked with the help of FCS whether membrane association of phytochrome might be a general phenomenon, which is not only restricted to cryptogam species. Autocorrelation studies on GFP-labeled phytochromes may allow a distinction between different phytochrome species and mutants thereof, if the GFP fusion does not affect the surface binding properties of the phytochrome. Finally, revealing the membrane association of a biomolecule through FCS may be a powerful tool with which to examine the regulation of membrane binding of other signal transduction proteins in living cells.
Acknowledgments
We thank Cornelia Görick for the purification of PEB, Gerhard Brücker for microinjection of the GFP plasmid, Viola Eckl for technical help, members of the Experimental Biophysics group, especially Kirsten Bacia and Elke Haustein, for helpful discussions, and Timo Thoms for correcting the English.
This work was supported by the Deutsche Forschungsgemeinschaft, La 799/6-1, and by the German Ministry for Education and Research (Biofuture program). The ConfoCor 2 LSM 510 combination system was kindly provided by Carl Zeiss.
References
- Aragon, S. R., and R. Pecora. 1976. Fluorescence correlation spectroscopy as a probe of molecular-dynamics. J. Chem. Phys. 64:1791–1803. [Google Scholar]
- Bacia, K., I. V. Majoul, and P. Schwille. 2002. Probing the endocytotic pathway in live cells using dual-color fluorescence correlation spectroscopy. Biophys. J. 83:1184–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brock, R., M. Hink, and T. M. Jovin. 1998. Fluorescence correlation microscopy of cells in the presence of autofluorescence. Biophys. J. 75:2547–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brock, R., G. Vámosi, G. Vereb, and T. M. Jovin. 1999. Rapid characterization of green fluorescent protein fusion proteins on the molecular and cellular level by fluorescence correlation microscopy. Proc. Natl. Acad. Sci. USA. 96:10123–10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brücker, G., M. Zeidler, T. Kohchi, E. Hartmann, and T. Lamparter. 2000. Microinjection of heme oxygenase genes rescues phytochrome-chromophore-deficient mutants of the moss Ceratodon purpureus. Planta. 210:529–535. [DOI] [PubMed] [Google Scholar]
- Elson, E. L., and D. Madge. 1974. Fluorescence correlation spectroscopy I. Conceptual basics and theory. Biopolymers. 13:1–27. [DOI] [PubMed] [Google Scholar]
- Esch, H., E. Hartmann, D. Cove, M. Wada, and T. Lamparter. 1999. Phytochrome-controlled phototropism of protonemata of the moss Ceratodon purpureus: physiology of wild type and class 2 ptr mutants. Planta. 209:290–298. [DOI] [PubMed] [Google Scholar]
- Esch, H., and T. Lamparter. 1998. Light regulation of phytochrome content in wild-type and aphototropic mutants of the moss Ceratodon purpureus. Photochem. Photobiol. 67:450–455. [DOI] [PubMed] [Google Scholar]
- Hartmann, E., B. Klingenberg, and L. Bauer. 1983. Phytochrome mediated phototropism in protonemata of the moss Ceratodon purpureus BRID. Photochem. Photobiol. 38:599–603. [Google Scholar]
- Hisada, A., H. Hanzawa, J. L. Weller, A. Nagatani, J. B. Reid, and M. Furuya. 2000. Light-induced nuclear translocation of endogenous pea phytochrome A visualized by immunocytochemical procedures. Plant Cell. 2000:1063–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, J., T. Lamparter, and F. Mittmann. 1996. Cerpu; PHY0; 2, a “normal” phytochrome in Ceratodon. Plant Physiol. 112:446. [Google Scholar]
- Jones, A. M., and P. H. Quail. 1986. Quaternary structure of 124-kilodalton phytochrome from Avena sativa L. Biochemistry. 25:2987–2995. [Google Scholar]
- Kendrick, R. E., and G. H. M. Kronenberg, editors. 1994. Photomorphogenesis in Plants, 2nd ed. Kluwer Academic Publishers, Dordrecht, The Netherlands.
- Kircher, S., L. Kozma-Bognar, L. Kim, E. Adam, K. Harter, E. Schäfer, and F. Nagy. 1999. Light quality-dependent nuclear import of the plant photoreceptors phytochrome A and B. Plant Cell. 11:1445–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraml, M. 1994. Light direction and polarisation. In Photomorphogenesis in Plants, 2nd ed. R. E. Kendrick and G. H. M. Kronenberg, editors. Kluwer Academic Publishers, Dordrecht, The Netherlands. 417–446.
- Krichevsky, O., and G. Bonnet. 2002. Fluorescence correlation spectroscopy: the technique and its applications. Rep. Prog. Phys. 65:251–297. [Google Scholar]
- Lamparter, T., H. Esch, D. Cove, J. Hughes, and E. Hartmann. 1996. Aphototrophic mutants of the moss Ceratodon purpureus with spectrally normal and with spectrally dysfunctional phytochrome. Plant Cell Environ. 19:560–568. [Google Scholar]
- Lamparter, T., B. Esteban, and J. Hughes. 2001. Phytochrome Cph1 from the cyanobacterium Synechocystis PCC6803: purification, assembly, and quaternary structure. Eur. J. Biochem. 268:4720–4730. [DOI] [PubMed] [Google Scholar]
- Lamparter, T., P. Lutterbuese, H. A. W. Schneider-Poetsch, and R. Hertel. 1992. A study of membrane-associated phytochrome: hydrophobicity test and native size determination. Photochem. Photobiol. 56:697–707. [Google Scholar]
- Lamparter, T., S. Podlowski, F. Mittmann, H. Schneider-Poetsch, E. Hartmann, and J. Hughes. 1995. Phytochrome from protonemal tissue of the moss Ceratodon purpureus. J. Plant Physiol. 147:426–434. [Google Scholar]
- Li, L., J. T. Murphy, and J. C. Lagarias. 1995. Continuous fluorescence assay of phytochrome assembly in vitro. Biochemistry. 34:7923–7930. [DOI] [PubMed] [Google Scholar]
- Madge, D., E. L. Elson, and W. W. Webb. 1972. Thermodynamic fluctuations in a reacting system-measurement by fluorescence correlation spectroscopy. Phys. Rev. Lett. 29:705–708. [Google Scholar]
- Meseth, U., T. Wohland, R. Rigler, and H. Vogel. 1999. Resolution of fluorescence correlation measurements. Biophys. J. 76:1619–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittmann, F. 2003. Molekularbiologische untersuchungen zum phytochromsystem der moose Physcomitrella patens und Ceratodon purpureus. PhD thesis.
- Murphy, J. T., and J. C. Lagarias. 1997. The phytofluors: a new class of fluorescent protein probes. Curr. Biol. 7:870–876. [DOI] [PubMed] [Google Scholar]
- Nagy, F., S. Kircher, and E. Schäfer. 2000. Nucleo-cytoplasmic partitioning of the plant photoreceptors phytochromes. Semin. Cell Dev. Biol. 11:505–510. [DOI] [PubMed] [Google Scholar]
- Ormo, M., A. B. Cubitt, K. Kallio, L. A. Gross, R. Y. Tsien, and S. J. Remington. 1996. Crystal structure of the Aequorea victoria green fluorescent protein. Science. 273:1392–1395. [DOI] [PubMed] [Google Scholar]
- Pasentsis, K., N. Paulo, P. Algarra, P. Dittrich, and F. Thümmler. 1998. Characterization and expression of the phytochrome gene family in the moss Ceratodon purpureus. Plant J. 13:51–61. [DOI] [PubMed] [Google Scholar]
- Pramanik, A., and R. Rigler. 2001. FCS-analysis of ligand-receptor interactions in living cells. In Fluorescence Correlation Spectroscopy: Theory and Applications. E. L. Elson and R. Rigler, editors. Springer, Berlin, Germany. 101–131.
- Prasher, D. C., V. K. Eckenrode, W. W. Ward, F. G. Prendergast, and M. J. Cormier. 1992. Primary structure of the Aequorea victoria green-fluorescent protein. Gene. 111:229–233. [DOI] [PubMed] [Google Scholar]
- Pratt, L. H. 1994. Distribution and localization of phytochrome within the plant. In Photomorphogenesis in Plants, 2nd ed. R. E. Kendrick and G. H. M. Kronenberg, editors. Kluwer Academic Publishers, Dordrecht, The Netherlands. 163–186.
- Qian, H., and E. L. Elson. 1991. Analysis of confocal laser-microscope optics for 3-D fluorescence correlation spectroscopy. Appl. Opt. 30:1185–1195. [DOI] [PubMed] [Google Scholar]
- Qian, H., E. L. Elson, and C. Frieden. 1992. Studies on the structure of actin gels using time correlation spectroscopy of fluorescent beads. Biophys. J. 63:1000–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail, P. H. 1997. An emerging molecular map of the phytochromes. Plant Cell Environ. 20:657–665. [Google Scholar]
- Rigler, R., J. Mets, J. Widengren, and P. Kask. 1993. Fluorescence correlation spectroscopy with high count rate and low background: analysis of translational diffusion. Eur. Biophys. J. 22:169–175. [Google Scholar]
- Rigler, R., A. Pramanik, P. Jonasson, G. Kratz, O. T. Jansson, P. A. Nygren, S. Stahl, K. Ekberg, B. L. Johansson, S. Uhlen, M. Uhlen, H. Jornvall, and H. Wahren. 1999. Specific binding of proinsulin C-peptide to human cell membranes. Proc. Natl. Acad. Sci. USA. 96:13318–13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigler, R., J. Widengren, and Ü. Mets. 1992. Interactions and kinetics of single molecules as observed by fluorescence correlation spectroscopy. In Fluorecence Spectroscopy. New Methods and Applications. O. Wolfbeis, editor. Springer-Verlag, Berlin, Germany. 13–24.
- Rubinstein, B., K. S. Drury, and R. B. Park. 1969. Evidence for bound phytochrome in oat seedlings. Plant Physiol. 44:105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüdiger, W., and F. Thümmler. 1994. The phytochrome chromophore. In Photomorphogenesis in Plants, 2nd edition. R. E. Kendrick and G. H. M. Kronenberg, editors. Kluwer Academic Publishers, Dordrecht, The Netherlands. 51–69.
- Schwille, P. 2001. Fluorescence correlation spectroscopy and its potential for intracellular applications. Cell Biochem. Biophys. 34:383–408. [DOI] [PubMed] [Google Scholar]
- Schwille, P., J. Bieschke, and F. Oehlenschläger. 1997. Kinetic investigation by fluorescence correlation spectroscopy: the analytical and diagnostic potential of diffusion studies. Biophys. Chem. 66:211–228. [DOI] [PubMed] [Google Scholar]
- Schwille, P., U. Haupts, S. Maiti, and W. W. Webb. 1999a. Molecular dynamics in living cells observed by fluorescence correlation spectroscopy with one- and two-photon excitation. Biophys. J. 77:2251–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwille, P., J. Korlach, and W. W. Webb. 1999b. Fluorescence correlation spectroscopy with single-molecule sensitivity on cell and model membranes. Cytometry. 36:176–182. [DOI] [PubMed] [Google Scholar]
- Sineshchekov, V. A. 1995. Photobiophysics and photobiochemistry of the heterogeneous phytochrome system. Biochim. Biophys. Acta. 1228:125–164. [Google Scholar]
- Thümmler, F., M. Dufner, P. Kreisl, and P. Dittrich. 1992. Molecular cloning of a novel phytochrome gene of the moss Ceratodon purpureus which encodes a putative light-regulated protein kinase. Plant Mol. Biol. 20:1003–1017. [DOI] [PubMed] [Google Scholar]
- Widengren, J., and R. Rigler. 1998. Fluorescence correlation spectroscopy as a tool to investigate chemical reactions in solution and on cell surfaces. Cell. Mol. Biol. 44:857–879. [PubMed] [Google Scholar]
- Yamaguchi, R., M. Nakamura, N. Mochizuki, S. A. Kay, and A. Nagatani. 1999. Light-dependent translocation of a phytochrome B-GFP fusion protein to the nucleus in transgenic Arabidopsis. J. Cell Biol. 145:437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidler, M., T. Lamparter, J. Hughes, E. Hartmann, A. Remberg, S. Braslavsky, K. Schaffner, and W. Gärtner. 1998. Recombinant phytochrome of the moss Ceratodon purpureus: heterologous expression and kinetic analysis of Pr→Pfr conversion. Photochem. Photobiol. 68:857–863. [DOI] [PubMed] [Google Scholar]