

Abstract
Single-molecule measurements have revealed that what were assumed to be identical molecules can differ significantly in their static and dynamic properties. One of the most striking examples is the hairpin ribozyme, which was shown to exhibit two to three orders of magnitude variation in folding kinetics between molecules. Although averaged behavior of single molecules matched the bulk solution data, it was not possible to exclude rigorously the possibility that the variations around the mean values arose from different ways of interacting with the surface environment. To test this, we minimized the molecules' interaction with the surface by encapsulating DNA or RNA molecules inside 100- to 200-nm diameter unilamellar vesicles, following the procedures described by Haran and coworkers. Vesicles were immobilized on a supported lipid bilayer via biotin-streptavidin linkages. We observed no direct binding of DNA or RNA on the supported bilayer even at concentrations exceeding 100 nM, indicating that these molecules do not bind stably on the membrane. Since the vesicle diameter is smaller than the resolution of optical microscopy, the lateral mobility of the molecules is severely constrained, allowing long observation periods. We used fluorescence correlation spectroscopy, nuclease digestion, and external buffer exchange to show that the molecules were indeed encapsulated within the vesicles. When contained within vesicles, the natural form of the hairpin ribozyme exhibited 50-fold variation in both folding and unfolding rates in 0.5 mM Mg2+, which is identical to what was observed from the molecules tethered directly on the surface. This strongly indicates that the observed heterogeneity in dynamic properties does not arise as an artifact of surface attachment, but is intrinsic to the nature of the molecules.
INTRODUCTION
Single-molecule spectroscopy can be used to explore biological phenomena directly, without the temporal and population averaging that occurs in conventional ensemble studies (Weiss, 1999). The ability to track asynchronous time evolution of individual biological molecules provides unique insight into detailed reaction kinetics and pathways. These measurements require prolonged observation periods in biologically relevant environments. Such measurements are frequently made using macromolecules that are tethered on a glass surface. However, there have been reports of variability of surface environment, and suspicion that observed heterogeneity of dynamic properties in single molecules might be an artifact of the local surface. Talaga et al. observed different end-to-end distances between free and immobilized proteins caused by protein denaturation on the surface (Talaga et al., 2000). Yasuda et al. reported that more than 99% of the F1-ATPase molecules bound on a glass surface did not show activity in single-molecule fluorescence measurements (Yasuda et al., 2003). These observations suggest that biological molecules can be subject to significant alteration upon surface fixation.
It is possible to bypass surface problems altogether via “burst spectroscopy.” Fluorescent molecules freely diffusing in solution can emit bursts of photons as the molecules traverse the tightly focused laser beam (Deniz et al., 1999, 2000; Rothwell et al., 2003; Schuler et al., 2002; Talaga et al., 2000). Although completely free of surface effects, the observation period is limited by the molecules' residence time in the laser focal volume, and it is very difficult to detect conformational changes that occur in millisecond or longer timescales. For this reason, several immobilization methods have been employed. These include specific single-point attachment on glass (Wennmalm et al., 1997) or polymer-coated surfaces (Ha et al., 2002), and confinement inside the porous polyacrylamide (Dickson et al., 1997) and agarose (Lu et al., 1998) gel matrices. Although extremely useful in obtaining novel biological information, these methods are still susceptible to behavioral alteration of biological molecules because of potential interactions with their immediate environment.
Another promising approach is to restrict translational motion within a small container. Chiu et al. monitored chemical reactions in individual vesicles and suggested that the technique is well-suited for mimicking in vivo reaction conditions of biological molecules (Chiu et al., 1999). As a proof of concept, Haran and coworkers trapped fluorescently labeled single protein molecules inside 100-nm vesicles that were then anchored on the surface (Boukobza et al., 2001). Fluorescence polarization measurements suggested that encapsulated molecular motions were similar to the free solution behavior. Using the same scheme, Rhoades et al. were able to measure the equilibrium folding-unfolding fluctuations of single adenylate kinase molecules (Rhoades et al., 2003). Since the vesicles are small compared to the resolution of optical microscopy, lateral motion of the biological molecules is operationally prevented, and provided that the interaction with the membrane is minimal, the molecules would behave essentially as if they are free in solution.
One of the most fascinating and potentially important discoveries afforded by single-molecule spectroscopy is the static and dynamic heterogeneity in molecular behavior in some systems. The first such example for enzymes was reported by Lu and Xie for cholesterol oxidase (Lu et al., 1998). Perhaps the most striking example was observed in studies of the minimal form of the hairpin ribozyme, for which four distinct populations were detected with different unfolding rates spanning three orders of magnitude (Zhuang et al., 2002). Our previous studies on the natural form of the hairpin ribozyme that includes the four-way junction also revealed significantly heterogeneous folding kinetics, with both the folding and unfolding rates varying 50-fold between molecules (Tan et al., 2003). The average values from these single-molecule measurements agreed with the bulk solution data, but the possibility that the heterogeneous kinetics were caused by the interactions with the immobilization medium could not be ruled out. To investigate how intrinsic the observed heterogeneities are, we employed the vesicle encapsulation method.
In these studies, we have encapsulated DNA and RNA molecules inside phospholipid vesicles, confirming their encapsulation by fluorescence correlation spectroscopy (FCS), nuclease digestion, and buffer exchange. The vesicles were immobilized on supported bilayers (Fig. 1) and single-molecule analysis was performed for DNA molecules and hairpin ribozymes. We have observed the same degree of heterogeneity in folding kinetics from the vesicle-encapsulated hairpin ribozyme as that observed from surface immobilized hairpin ribozymes. This clearly shows that the heterogeneous dynamics were not the consequence of surface interactions, but are intrinsic to the structure of the ribozyme. This study further validates the standard approach of using bovine serum albumin (BSA)-biotin attachment for nucleic acid studies, and indicates that the vesicle encapsulation method is suitable for studying the dynamic changes of single DNA and RNA molecules free of surface perturbations.
FIGURE 1.

Schematic to illustrate an encapsulated, fluorescently labeled hairpin ribozyme molecule, drawn to scale. A 30-nm diameter unilamellar vesicle is tethered on a quartz-supported bilayer via a streptavidin molecule. The encapsulated hairpin ribozyme is shown in its folded conformation as observed in the crystal (Rupert and Ferre-D'Amare, 2001), with Cy3 and Cy5 attached to the 5′ termini of the A and B arms. For the actual experiments, 100-nm or 200-nm diameter vesicles were used.
MATERIALS AND METHODS
L-α-phosphatidlycholine (egg, chicken) (eggPC) and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl) (16:0 biotinyl cap PE) were purchased from Avanti Lipids (Alabaster, AL). DiIC18(3) was purchased from Molecular Probes (Eugene, OR). DNaseI and RNaseA were purchased from Sigma Aldrich (St. Louis, MO). For the oxygen-scavenging system, catalase, glucose oxidase, and 2-mercaptoethanol were purchased from Roche Diagnostic (Indianapolis, IN), Sigma Aldrich (St. Louis, MO), and Acros Organics (Morris Plains, NJ), respectively.
Single-stranded DNA and hairpin ribozyme
5′-Cy5-GCCTCGCTGCCGTCGCCA-3′ biotin and 5′-TGGCGACGGCAGCGAGGC-Tn-Cy3-3′ DNA oligos were a gift of Tim Lohman from Washington University. The two strands were annealed and an 18-base pair partial duplex was formed with Cy5 at the junction and Cy3 attached at the end of the 3′-ssDNA (dT)n tail (Fig. 2 a) Hairpin ribozyme was prepared as described elsewhere (Tan et al., 2003).
FIGURE 2.

(a) Representation of the DNA partial duplex used in this experiment. This comprises a duplex region of 18 base pairs with a single-stranded 3′ extension of 19 thymine nucleotides. (The thymine tail lengths were arbitrarily chosen for the experiments.) (b) Size exclusion chromatography of the DNA following lipid encapsulation. The concentration of DNA in each fraction was estimated by comparing the fluorescence intensities of the fractions with that of a known DNA concentration under the same conditions. The vesicles and free DNA have different elution volumes owing to their different molecular sizes. Two peaks are apparent in the elution profile from the Sepharose 4B column. Vesicles encapsulating the DNA elute earlier (first peak) and the free DNA elutes later (second peak). (c) Fluorescence autocorrelation spectroscopy of free tetramethylrhodamine and the early and late fractions from the size exclusion chromatography in standard lipid buffer. Diffusion coefficients obtained from the fits are shown for each.
Vesicle preparation and encapsulation
We used the extrusion technique to prepare small unilamellar vesicles (SUV) (Johnson et al., 2002). Regular SUVs were used to form supported bilayers (Boxer, 2000; Brian and McConnell, 1984) on which DNA/RNA-containing SUVs were immobilized via biotin-streptavidin linkers.
16:0 biotinyl cap PE was mixed with eggPC in 1:100 molar ratio in chloroform, and was then dried in a stream of nitrogen. Lipid films were left in a desiccator for 2 h to remove residual chloroform. Regular vesicles were prepared by hydrating eggPC (with 1% biotinylated DPPE) lipid film with 10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA (standard lipid buffer), which induces swelling of the film to form a final 5 mg/mL lipid concentration of multilamellar vesicles (MLV). DNA/RNA-containing vesicles were prepared by hydrating with DNA/RNA in the desired buffer conditions. RNA samples were prepared in 10 mM Tris-HCl, pH 7.4, 50 mM NaCl, 0.4% glucose, and 0.5 mM Mg2+. Freeze-thaw cycles were repeated 5 times to break down the MLVs and to help unilamellar vesicle formation and encapsulation. The MLV preparation was then extruded through 100- to 200-nm pores to induce SUV formation (Avanti Mini Extruder). SUV preparations were stored at 4°C and usually used within 2 days.
Size exclusion chromatography
After preparation of vesicles encapsulating nucleic acids, the vesicles were separated from the free molecules by means of size exclusion chromatography. A glass column (Glass Econo-Column from Bio-rad, Hercules, CA) was packed with 6 mL of Sepharose 4B agarose beads (Sigma Aldrich). The beads were allowed to settle for 5 h resulting in ∼4 mL of gel matrix. The matrix was then equilibrated by passage of at least 15 mL of the same buffer in which the encapsulation sample had been prepared. The vesicle sample (typically 100 μL) was applied and eluted under gravity. The eluate was collected in 4-drop (∼190 μL) fractions. After use the column was washed in water and stored in 10 mM Tris-HCl, pH 8.0, at 4°C until reuse.
Total internal reflection microscopy
A quartz cover slide and a glass coverslip were taped to each other and the 100-μm channel in between the two surfaces was used as a flow cell for microscopy. Five mg/ml SUV (eggPC with 1% biotinylated DPPE) was incubated in the flow cell for up to 2 h to allow supported bilayer formation (Boxer, 2000; Brian and McConnell, 1984). Stray vesicles were washed away with the standard lipid buffer. The sample chamber was then treated with 0.2 mg/ml streptavidin for 10 min. After washing away excess streptavidin, a vesicle preparation encapsulating the nucleic acid was added at a low concentration to achieve a surface coverage for single-molecule imaging. Imaging was performed under the same buffer conditions used for vesicle preparation (unless otherwise specified) with oxygen-scavenging system (0.1 mg/ml glucose oxidase, 0.02 mg/mL catalase, 1% (v/v) 2-mercaptoethanol) to prevent rapid photobleaching of fluorophores. The surface was imaged by wide-field objective type (Yildiz et al., 2003) and prism type (Ha et al., 2002; Tan et al., 2003; Zhuang et al., 2000) total internal reflection (TIR) microscopy for control measurements and data acquisition, respectively.
Nuclease digestion
Samples were incubated with 0.2 mg/mL DNaseI or RNaseA for 5 min in the appropriate buffers as specified by the manufacturers. The flow chamber was washed with the imaging buffer and was monitored via TIR microscopy (see above) afterwards. Nucleic acids attached on the surface were removed upon nuclease digestion if the molecules were not protected by vesicles and were directly attached on the surface.
Fluorescence correlation spectroscopy
A 520-nm line of a Kr/Ar ion laser (Melles Griot, Carlsbad, CA) was focused within the sample chamber by a 60× objective lens (Olympus, Melville, NY; NA = 1.2, water). The emission was collected through the same objective. Emission and excitation light were separated by a 530-nm cutoff longpass dichroic mirror (Chroma, Rockingham, VT), and a 550-nm cutoff longpass filter (Chroma) was used to eliminate the excitation light. The emission light was then focused on a 75-μm diameter pinhole to reject the out-of-focus photons. Finally, the fluorescence signal was divided in two by a 50:50 beamsplitter cube (Melles Griot) (or a dichroic mirror) and then focused on two avalanche photodiodes (Perkin Elmer Optoelectronics, Fremont, CA). A bandpass filter centered at 580 nm and with 60-nm width (Chroma) and a 660-nm cutoff longpass filter (Chroma) were placed in front of the avalanche photodiodes used to detect Cy3 and Cy5 emission, respectively. An ISS data acquisition card (Champaign, IL) was used, and the Globals data fitting program (Laboratory of Fluorescence Dynamics, University of Illinois at Urbana-Champaign) was used to perform the autocorrelation analysis. The diffusion coefficients were determined by fitting the data with the autocorrelation function 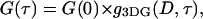 where:
where:
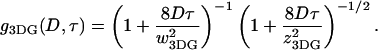 |
(1) |
The subscript 3DG (3D-Gaussian) refers to the geometry of the excitation volume. D is the diffusion coefficient, 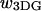 is the width, and
is the width, and 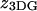 is the height of the excitation spot.
is the height of the excitation spot. 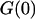 is the value of the autocorrelation at
is the value of the autocorrelation at 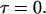
RESULTS AND DISCUSSION
Encapsulation of DNA and RNA molecules inside phospholipid vesicles
For these experiments we employed an 18-base pair duplex, with a 3′ (dT)19 tail, labeled with donor and acceptor fluorophores (Cy3 and Cy5). The DNA was encapsulated within 100-nm diameter vesicles in standard lipid buffer, as described in Materials and Methods. Briefly, dried lipid film was hydrated with buffer containing DNA and vesicles of definite size were made via extrusion through a polycarbonate membrane containing 100-nm diameter cylindrical holes. For lipid hydration, 3 μM DNA was used. The vesicles were separated from free DNA by means of size exclusion chromatography. DNA concentration in each fraction was determined by measuring fluorescence intensities of the fractions and comparing the measured values with those of DNA samples of known concentrations (Fig. 2 b). The elution profile displays two distinct peaks. As larger species are more excluded we assigned the faster-eluting (early) material to the DNA trapped in the vesicles and the second peak (late) to free DNA.
Analysis of encapsulated DNA species by fluorescence correlation spectroscopy
This assignment, for the two peaks of the elution profile, was tested using FCS, to explore the diffusional properties of the two species. In an FCS experiment, temporal changes in the fluorescence intensity are monitored within an optically illuminated volume defined by a focused laser beam (Maiti et al., 1997). The changes in the fluorescence signal are caused by excitation of fluorescent molecules diffusing in and out of the laser beam or other effects that change the fluorophores' brightness. Information about chemical kinetics, diffusion coefficients, and equilibrium concentrations can be extracted from the autocorrelation of the temporal fluctuations of the detected signal. We found that the material eluting within the two peaks exhibited different diffusional behavior. Fig. 2 c shows the autocorrelation data and fits to the autocorrelation function (Materials and Methods) for tetramethylrhodamine, and early and late fractions from the size exclusion column. The diffusion coefficient (D) of free rhodamine in water is well known (280 μm2/s) and its autocorrelation was used to determine the lateral 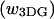 and axial
and axial 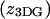 dimensions of the focal volume. Autocorrelation gave a significantly slower decay for the early fraction, showing that this was the more slowly diffusing species. Indeed, the fit to data revealed diffusion coefficients of 5.2 μm2/s and 68 μm2/s for the early and late fractions, respectively. The diffusion coefficient for a 100-nm diameter spherical object can be estimated by using the Stokes-Einstein formula:
dimensions of the focal volume. Autocorrelation gave a significantly slower decay for the early fraction, showing that this was the more slowly diffusing species. Indeed, the fit to data revealed diffusion coefficients of 5.2 μm2/s and 68 μm2/s for the early and late fractions, respectively. The diffusion coefficient for a 100-nm diameter spherical object can be estimated by using the Stokes-Einstein formula:
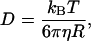 |
(2) |
where kB is the Boltzmann constant, T is the temperature in Kelvin, η is the viscosity, and R is the radius of the spherical object. We obtained D = 4.9 μm2/s using the viscosity of water, η = 0.89 × 10−3 kg/m/s, at room temperature. We also measured the diffusion coefficient of the same size vesicles prepared with 0.05% of membrane-specific dyes (DiIC18(3)) via their fluorescence, and obtained 4.3 μm2/s. These observations support the assignment of the first peak as that comprising DNA contained within vesicles.
Analysis of encapsulated DNA species by nuclease digestion
In a further test of the assignment we explored the accessibility of the DNA in the two fractions to enzymatic degradation. We carried out DNaseI digestion of the early and late fractions in the FCS apparatus. Upon addition of DNaseI to the sample cell that contained the early fraction, there was no significant change in the diffusion coefficient. However, after the DNaseI treatment of the late fraction, the diffusion coefficient increased from its original value of 67.7 μm2/s to 167 μm2/s. These observations indicate that the late fraction contains free DNA that is accessible to the nuclease, whereas the early fraction contains DNA encapsulated in vesicles that is thus protected from digestion.
In further experiments we examined the nuclease accessibility of two fractions from the gel exclusion chromatography when immobilized on the surface. After dilution, the late fraction was applied to a glass slide that was surface-coated with biotinylated BSA and streptavidin. No binding was observed in the absence of streptavidin. Fluorescently labeled DNA molecules appeared as bright spots on the surface when imaged with objective-type TIR microscopy. Nuclease incubation resulted in the disappearance of all spots from the surface, indicating efficient digestion of the DNA. In contrast, in corresponding experiments using samples prepared from the early fraction, fluorescent spots were resistant to DNaseI treatment (data not shown). Taken together, our results indicate that the DNA is completely encapsulated within the vesicles and that vesicle-encapsulated molecules can be separated from free molecules.
To test potential interaction between DNA and membrane, 100 nM of a partial duplex DNA with a (dT)23 tail (Materials and Methods) was injected in the flow chamber after formation of a supported bilayer on a quartz slide. No nonspecific binding of DNA on the eggPC bilayer was observed, suggesting that the DNA did not bind stably on membrane and the interaction between the membrane and the DNA was not significant. Similar observations were made for the hairpin ribozyme.
Changing intravesicular salt concentration by variation of the osmotic pressure
Since the lipid vesicles do not allow solvent passage to occur it is not possible to carry out exchange of solutes. However, salt concentration can be altered indirectly by changing the volume of the vesicle by alteration of osmotic pressure. Fig. 3, a and b, shows single-molecule fluorescence resonance energy transfer (FRET) histograms of surface-tethered vesicles encapsulating the (dT)23-tailed partial duplex prepared in the standard lipid buffer. The apparent FRET efficiency Eapp is IA/(IA + ID), where IA and ID are fluorescence intensities for the donor and the acceptor, respectively, averaged over 1 s. Eapp peaks at 0.56 (Fig. 3 a) for isotonic (150 mM NaCl) and at 0.81 for hypertonic (2 M NaCl) imaging buffer conditions. This change of the FRET efficiency probably arises from a change in the mean distance between Cy3 and Cy5 dyes separated from each other by a single-stranded chain of 23 thymine nucleotides. It is known that FRET measured between two ends of a ssDNA increases with elevation of NaCl concentration (Deniz et al., 2001; Murphy et al., 2004) and that this effect is due to a shortening of end-to-end distance rather than changes in the photophysical properties of the dyes, including fluorescence anisotropy and quantum yield (Murphy et al., 2004). This effect most probably results from the screening of electrostatic repulsion between DNA backbone phosphates, which would lower the stiffness of the ssDNA (the oligothymine tail in this particular case) at high salt concentrations. Since the membrane is impermeable to ions, the effective salt concentration inside the vesicle must have increased due to the osmotic shrinking of the vesicle when the extravesicular condition was hypertonic and vice versa. The vesicles were treated with DNaseI after the data was taken in the hypertonic buffer and the molecules remained thereafter, indicating that the vesicles were still intact.
FIGURE 3.

Single-molecule analysis of encapsulated DNA species. Vesicles encapsulating partial duplex with a 3′ (dT)23 tail were immobilized on the surface and imaged using the TIR microscope. (a and b) Histograms of the number of observed molecules displaying a given apparent FRET efficiency (Eapp) under particular conditions. (a) Isotonic extravesicular buffer conditions (standard lipid buffer, 150 mM NaCl). The (peak) FRET efficiency is Eapp ∼0.56 under these conditions. The peak at Eapp ∼0 represents the molecules that have only the donor fluorophore (Cy3) being active. Negative Eapp values are due to noise around the background level when the acceptor intensity is low. (b) Hypertonic solution (2 M NaCl). Under these conditions, the vesicles shrink due to the osmotic pressure, and the intravesicular salt concentration consequently increases, leading to an increase in Eapp. (c) The Poisson distribution of the number of molecules inside a vesicle. (d and e) Representative time records for two tethered vesicles. The number of encapsulated DNA molecules can be determined by counting the digital photobleaching events. In trace d the Cy5 intensity reduces to its background level in two discrete steps, which implies that there are two molecules entrapped within this particular vesicle, whereas in trace e photobleaching occurs in a single step, indicating occupancy by a single DNA molecule.
Determination of the number of molecules per vesicle
The vesicle encapsulation is a Poisson process with a probability distribution
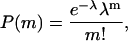 |
(3) |
where m is the number of molecules in a vesicle, P(m) is the probability of having m molecules inside one vesicle, and λ is the average probability of encapsulation. We prepared an encapsulation sample with 200 nM (dT)23-tailed DNA in 200-nm diameter vesicles. After vesicle immobilization, Cy5 molecules were excited directly with a 632-nm He-Ne laser (Coherent, Santa Clara, CA) for a long enough period to photobleach almost all the fluorophores. We could determine the number of molecules per vesicle by counting the number of steps in digital photobleaching events (Fig. 3, d and e) and the histogram of the number of photobleaching steps is shown in Fig. 3 c. The distribution could be fitted to a Poisson distribution with λ = 0.46 (Fig. 3 c); note that this distribution does not include m = 0 because it was not possible to count the vesicles that did not encapsulate any molecules. The distribution was normalized accordingly. For this particular sample preparation, we assumed that the average number of encapsulated molecules per vesicle would simply be proportional to the ratio of the interior volume of one vesicle to the average volume occupied by one molecule at a certain concentration. Thus, we used a DNA concentration and a vesicle diameter (200 nM and 200 nm, respectively) to obtain an average of 0.48 DNA per vesicle (λ = 0.48, arbitrarily chosen) according to the equation
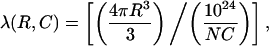 |
(4) |
where C denotes the concentration in nanomolar within a vesicle of radius R (μm) and N is Avogadro's number. However, it should be noted that our assumption is probably oversimplified and may not be very accurate as the encapsulation process is affected by many parameters (Walde and Ichikawa, 2001; Colletier et al., 2002), such as number of freeze-thaw cycles, interaction between the molecule to be encapsulated and the phospholipid bilayer, and the method used for vesicle preparation.
Heterogeneous folding kinetics is intrinsic to the hairpin ribozyme
The hairpin ribozyme consists of a four-way RNA junction with an internal loop on each of two adjacent arms (Fig. 5, Murchie et al., 1998). A minimal form of the ribozyme lacking the four-way junction is also active. In both the complete ribozyme, and the simple junction derived from it, the arms of the junction are coaxially stacked in pairs, A on D and B on C (Murchie et al., 1998; Rupert and Ferre-D'Amare, 2001). Both species undergo transitions between an extended and an antiparallel conformation the rate of which depends on the concentration of divalent metal ions (Tan et al., 2003, Zhuang et al., 2002). The active form of the ribozyme is generated by the docking of the internal loops, creating the local environment in which catalysis can occur (Murchie et al., 1998; Walter et al., 1998; Rupert and Ferre-D'Amare, 2001.) This form is stabilized by the binding of Mg2+ ions.
FIGURE 5.

The dynamic properties of hairpin ribozymes as encapsulated species. The ribozymes have been studied in their natural four-way junction form, encapsulated within 200-nm vesicles in 0.5 mM Mg2+ at room temperature. (a–d) Four representative time traces of Eapp clearly show significant variation in the kinetics of folding-unfolding transitions between molecules. (a) This molecule spends most of the time in the unfolded state, but occasionally adopts the folded state for a brief period. (b) In marked contrast, this molecule remains in the folded conformation for most of the time. (c) A molecule undergoing continuous unfolding-folding fluctuations. (d) This molecule also exhibits continuous unfolding-folding transitions, but with shorter dwell times for both the folded and the unfolded states. (e) Scatter plot for the algebraic average of unfolded and folded dwell times for many molecules, calculated from their time traces. The distribution is broad and both the unfolding-folding times exhibit up to 50-fold variance. Encapsulation samples prepared using ribozymes without (circles) and with (squares) biotin are shown on the same plot, and they are similarly distributed. The points corresponding to traces a–d are indicated in the plot (filled circles). (f) Scatter plot for the surface-immobilized molecules adapted from Tan et al. (2003).
The minimal form of the ribozyme (lacking the junction) exhibited heterogeneity in the dwell times of the folded state unveiled by single-molecule spectroscopy (Bokinsky et al., 2003; Zhuang et al., 2002). We found that heterogeneity was also characteristic of the natural form of the ribozyme (including the junction) (Tan et al., 2003), with 50-fold heterogeneity in the dwell times of both folded and unfolded states (see Fig. 5 f, adapted from Tan et al. (2003)). It was proposed that the loop-loop interaction was responsible for the heterogeneity (Tan et al., 2003; Zhuang et al., 2002) and this was supported by the observation that the RNA four-way junction alone showed far less variation in its conformational fluctuation rates (Tan et al., 2003). In both cases, the ribozymes were immobilized directly on the surface via biotin-streptavidin linker. Although some evidence was presented that the surface immobilization did not alter the average behaviors of molecules, the possibility that the observed heterogeneity might have arisen from different ways of interacting with the surface could not be excluded.
We therefore encapsulated hairpin ribozymes inside 200-nm vesicles to test for surface effects. Ribozymes with or without biotin terminally attached to helix C were used to prepare two separate samples. The sample containing biotinylated ribozymes was purified via size exclusion chromatography and the vesicle (early) fraction was used for the measurements. The ribozyme sample without biotin was used without purification because we did not observe any nonspecific binding of the ribozymes on the supported bilayer. Both samples were treated with RNaseA upon surface immobilization and the fluorescence spots were retained, indicating that the vesicle encapsulation efficiently protected the ribozymes from digestion (Fig. 4). None of the fluorescent spots was mobile, probably because of multiple attachments of streptavidin-biotin linkers (Boukobza et al., 2001). However, we could demonstrate that the bilayer itself is fluid, because adding free ribozymes with biotin resulted in mobile fluorescent spots that could later be removed by RNaseA digestion.
FIGURE 4.

Encapsulated hairpin ribozyme molecules are protected against ribonuclease digestion. Each panel shows an image of a surface on which fluorescent hairpin ribozyme molecules are attached, either directly or encapsulated. (a) Ribozyme molecules directly immobilized on the surface before treatment with ribonuclease (RNaseA). (b) Ribozyme molecules directly immobilized on the surface after treatment with ribonuclease. The absence of fluorescence indicates that the ribozymes have been digested by the enzyme, allowing free diffusion of the fluorophores away from the surface. (c) Encapsulated ribozymes following treatment with ribonuclease. Unlike b, the encapsulated molecules are clearly not accessible to the enzyme, and thus the surface-bound fluorescence is preserved.
The folding of the hairpin ribozyme depends strongly on Mg2+ concentration. The ribozyme spends most of its time in the folded state above 1 mM Mg2+ but the unfolded state becomes substantially populated at submillimolar Mg2+ (Tan et al., 2003). The folded and unfolded states can be distinguished by different FRET efficiencies between donor and acceptor fluorophores attached to the termini of the loop-carrying arms (Fig. 5). In agreement with our earlier studies, we observed Eapp ∼0.25 for the unfolded and Eapp ∼0.9 for the folded state (Tan et al., 2003). In the presence of Mg2+ concentration of 0.5 mM, ribozymes fluctuate between the two forms as can be seen in the single-molecule time records (Fig. 5, a–d). Although molecules are nominally identical and reside in the same buffer conditions, their behaviors are highly heterogeneous with widely differing rates both for unfolding and folding. For instance, Fig. 5 a shows a molecule that remained unfolded except for three brief excursions into the folded state. In contrast, another molecule shown in Fig. 5 b exhibited just the opposite behavior. Molecules shown in Fig. 5, c and d, underwent fluctuations between low and high FRET states, spending approximately equal amount time in each state, but with markedly different rates. These heterogeneities are remarkable in that they are very persistent. A molecule appears to have its own clock and fluctuates for minutes or longer at the same rate despite undergoing many transitions in that time.
Average dwell times of folded and unfolded states from each molecule are recorded on a scatter plot in Fig. 5 e. The dwell times exhibited 50-fold variation for both states, and the plot is essentially identical to that obtained from the ribozymes directly immobilized on the surface (Fig. 5 f). Because the heterogeneity is long-lasting and the ribozyme does not bind on the membrane stably, we can now rule out the surface effect as the source for the persistent heterogeneity observed from the natural form of the hairpin ribozyme. This conclusion is likely to be true also for the minimal form because there is no reason to expect that this form lacking the junction would interact more strongly with the surface.
Finally, FRET measurements were performed on the encapsulated ribozymes with varying extravesicular ionic conditions (Fig. 6). Fig. 6 a shows the Eapp histogram of a sample encapsulated in the presence of 0.5 mM Mg2+ and measured in 0.5 mM Mg2+ outside the vesicle. Three peaks are seen, with Eapp ∼0 for the molecules without the acceptor labeling, Eapp ∼0.25 for the unfolded state, and Eapp ∼0.9 for the folded state. The low-FRET and high-FRET states coexist because the molecules fluctuate between the unfolded and folded states under these conditions (0.5 mM Mg2+). We obtained an identical histogram from ribozymes directly immobilized via biotin-streptavidin (data not shown). When the Mg2+ concentration external to the vesicle was changed to 0 mM or 1 mM, the histograms did not change (Fig. 6, b and c). This is in contrast to the data from ribozymes directly attached on the surface for the unfolded and mostly folded populations observed at 0 mM and 1 mM Mg2+, respectively. Evidently, encapsulated ribozymes are not accessible to extravesicular ions. Furthermore, the change in extravesicular Mg2+ concentration in this range was insufficient to have altered the size of the vesicle enough to induce a significant change in the effective interior Mg2+ concentration. As a comparison, when the sample cell was washed with 1 M NaCl, 0.5 mM Mg2+ buffer, the low-FRET peak disappeared. This indicates that complete folding occurred, presumably due to the increased Mg2+ concentration arising from osmotic shrinking.
FIGURE 6.

Variation of FRET efficiency with respect to the extravesicular conditions for encapsulated hairpin ribozymes. Histograms of the number of observed molecules displaying a given apparent FRET efficiency for encapsulated ribozymes in the presence of (a) 0.5 mM Mg2+; (b) 0 mM Mg2+, 1 mM EDTA; (c) 1 mM Mg2+; (d) 0.5 mM Mg2+, 1 M NaCl. For the conditions a–c, the differences in the extra- and intravesicular conditions did not induce significant size changes for the vesicles and the ribozymes were not affected by the various extravesicular Mg2+ concentrations. The FRET histograms reveal the same dynamic behavior, i.e., the molecules fluctuate between folded (Eapp ∼0.9) and unfolded (Eapp ∼0.25) states. In d, 1 M NaCl caused osmotic shrinking of the vesicles and a consequent increased concentration of Mg2+ inside the vesicle. All ribozymes adopt the folded conformation as a result, with the loss of the peak corresponding to Eapp ∼0.25.
CONCLUSION
We were able to employ the vesicle encapsulation method to show that the persistent, highly heterogeneous folding and unfolding kinetics observed from single-hairpin ribozymes are not surface-induced artifacts. Although we cannot rule out transient interactions with the inner surface of the vesicle, independent measurements showed that DNA and RNA molecules did not bind stably on the membrane and therefore membrane interaction cannot explain the observed persistent heterogeneity. A question still remains whether the heterogeneous folding kinetics are truly intrinsic to the hairpin ribozyme. For instance, the heterogeneity may arise from real chemical differences that might originate from imperfect chemical synthesis of oligonucleotides. However, we can exclude the most likely source of chemical heterogeneity, i.e., failure to remove 2′ protecting groups, since we have found previously that ribozymes synthesized using tBDMS and ACE chemistries exhibit closely similar heterogeneous dynamic properties (Tan et al., 2003). Further investigation of these properties will require new types of experiments to probe the origin of this effect.
Our data establish the vesicle encapsulation method as a viable approach in measuring the conformational dynamics of DNA and RNA, avoiding the requirement for the direct immobilization of the molecules. At the same time, our data also validate the now standard method of using biotinylated BSA and streptavidin to immobilize RNA and DNA molecules, since essentially identical kinetic parameters, including heterogeneity, were obtained from encapsulated molecules.
Acknowledgments
We thank Elizabeth Rhoades for useful personal communication, Evan Graves for help with FCS measurements, and Sean McKinney for providing a FRET correction program.
Funding was provided by the National Science Foundation, the National Institutes of Health, and Cancer Research-UK (Dundee).
References
- Bokinsky, G., D. Rueda, V. K. Misra, M. M. Rhodes, A. Gordus, H. P. Babcock, N. G. Walter, and X. Zhuang. 2003. Single-molecule transition-state analysis of RNA folding. Proc. Natl. Acad. Sci. USA. 100:9302–9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukobza, E., A. Sonnenfeld, and G. Haran. 2001. Immobilization in surface-tethered lipid vesicles as a new tool for single biomolecule spectroscopy. J. Phys. Chem. B. 105:12165–12170. [Google Scholar]
- Boxer, S. G. 2000. Molecular transport and organization in supported lipid membranes. Curr. Opin. Chem. Biol. 4:704–709. [DOI] [PubMed] [Google Scholar]
- Brian, A. A., and H. M. McConnell. 1984. Allogeneic stimulation of cytotoxic T cells by supported planar membranes. Proc. Natl. Acad. Sci. USA. 81:6159–6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu, D. T., C. F. Wilson, F. Ryttsen, A. Stromberg, C. Farre, A. Karlsson, S. Nordholm, A. Gaggar, B. P. Modi, A. Moscho, R. A. Garza-Lopez, O. Orwar, and R. N. Zare. 1999. Chemical transformations in individual ultrasmall biomimetic containers. Science. 283:1892–1895. [DOI] [PubMed] [Google Scholar]
- Colletier, J.-P., B. Chaize, M. Winterhalter, and D. Fournier. 2002. Protein encapsulation in liposomes: efficiency depends on interactions between protein and phospholipid bilayer. BMC Biotechnol. 2:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniz, A. A., M. Dahan, J. R. Grunwell, T. J. Ha, A. E. Faulhaber, D. S. Chemla, S. Weiss, and P. G. Schultz. 1999. Single-pair fluorescence resonance energy transfer on freely diffusing molecules: observation of Forster distance dependence and subpopulations. Proc. Natl. Acad. Sci. USA. 96:3670–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniz, A. A., T. A. Laurence, G. S. Beligere, M. Dahan, A. B. Martin, D. S. Chemla, P. E. Dawson, P. G. Schultz, and S. Weiss. 2000. Single-molecule protein folding: Diffusion fluorescence resonance energy transfer studies of the denaturation of chymotrypsin inhibitor 2. Proc. Natl. Acad. Sci. USA. 97:5179–5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniz, A. A., T. A. Laurence, M. Dahan, D. S. Chemla, P. G. Schultz, and S. Weiss. 2001. Ratiometric single-molecule studies of freely diffusing biomolecules. Annu. Rev. Phys. Chem. 52:233–253. [DOI] [PubMed] [Google Scholar]
- Dickson, R. M., A. B. Cubitt, R. Y. Tsien, and W. E. Moerner. 1997. On/off blinking and switching behaviour of single molecules of green fluorescent protein. Nature. 388:355–358. [DOI] [PubMed] [Google Scholar]
- Ha, T., I. Rasnik, W. Cheng, H. P. Babcock, G. Gauss, T. M. Lohman, and S. Chu. 2002. Initiation and re-initiation of DNA unwinding by the Escherichia coli Rep helicase. Nature. 419:638–641. [DOI] [PubMed] [Google Scholar]
- Johnson, J. M., T. Ha, S. Chu, and S. G. Boxer. 2002. Early steps of supported bilayer formation probed by single vesicle fluorescence assays. Biophys. J. 83:3371–3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, H. P., L. Y. Xun, and X. S. Xie. 1998. Single-molecule enzymatic dynamics. Science. 282:1877–1882. [DOI] [PubMed] [Google Scholar]
- Maiti, S., U. Haupts, and W. W. Webb. 1997. Fluorescence correlation spectroscopy: diagnostics for sparse molecules. Proc. Natl. Acad. Sci. USA. 94:11753–11757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murchie, A. I. H., J. B. Thomson, F. Walter, and D. M. J. Lilley. 1998. Folding of the hairpin ribozyme in its natural conformation achieves close physical proximity of the loops. Mol. Cell. 1:873–881. [DOI] [PubMed] [Google Scholar]
- Murphy, M. C., I. Rasnik, W. Cheng, T. M. Lohman, and T. Ha. 2004. Biophys. J. 86:2530–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoades, E., E. Gussakovsky, and G. Haran. 2003. Watching proteins fold one molecule at a time. Proc. Natl. Acad. Sci. USA. 100:3197–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell, P. J., S. Berger, O. Kensch, S. Felekyan, M. Antonik, B. M. Wohrl, T. Restle, R. S. Goody, and C. A. Seidel. 2003. Multiparameter single-molecule fluorescence spectroscopy reveals heterogeneity of HIV-1 reverse transcriptase:primer/template complexes. Proc. Natl. Acad. Sci. USA. 100:1655–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupert, P. B., and A. R. Ferre-D'Amare. 2001. Crystal structure of a hairpin ribozyme-inhibitor complex with implications for catalysis. Nature. 410:780–786. [DOI] [PubMed] [Google Scholar]
- Schuler, B., E. A. Lipman, and W. A. Eaton. 2002. Probing the free-energy surface for protein folding with single-molecule fluorescence spectroscopy. Nature. 419:743–747. [DOI] [PubMed] [Google Scholar]
- Talaga, D. S., W. L. Lau, H. Roder, J. Y. Tang, Y. W. Jia, W. F. DeGrado, and R. M. Hochstrasser. 2000. Dynamics and folding of single two-stranded coiled-coil peptides studied by fluorescent energy transfer confocal microscopy. Proc. Natl. Acad. Sci. USA. 97:13021–13026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, E., T. J. Wilson, M. K. Nahas, R. M. Clegg, D. M. J. Lilley, and T. Ha. 2003. A four way junction accelerates hairpin ribozyme folding via a discrete intermediate. Proc. Natl. Acad. Sci. USA. 100:9308–9313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walde, P., and S. Ichikawa. 2001. Enzymes inside lipid vesicles: preparation, reactivity and applications. Biomol. Eng. 18:143–177. [DOI] [PubMed] [Google Scholar]
- Walter, N. G., K. J. Hampel, K. M. Brown, and J. M. Burke. 1998. Tertiary structure formation in the hairpin ribozyme monitored by fluorescence resonance energy transfer. EMBO J. 17:2378–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss, S. 1999. Fluorescence spectroscopy of single biomolecules. Science. 283:1676–1683. [DOI] [PubMed] [Google Scholar]
- Wennmalm, S., L. Edman, and R. Rigler. 1997. Conformational fluctuations in single DNA molecules. Proc. Natl. Acad. Sci. USA. 94:10641–10646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda, R., T. Masaike, K. Adachi, H. Noji, H. Itoh, and K. Kinosita, Jr. 2003. The ATP-waiting conformation of rotating F1-ATPase revealed by single-pair fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA. 100:9314–9318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz, A., J. N. Forkey, S. A. McKinney, T. Ha, Y. E. Goldman, and P. R. Selvin. 2003. Myosin V walks hand-over-hand: single fluorophore imaging with 1.5-nm localization. Science. 300:2061–2065. [DOI] [PubMed] [Google Scholar]
- Zhuang, X. W., L. E. Bartley, H. P. Babcock, R. Russell, T. J. Ha, D. Herschlag, and S. Chu. 2000. A single-molecule study of RNA catalysis and folding. Science. 288:2048–2051. [DOI] [PubMed] [Google Scholar]
- Zhuang, X. W., H. Kim, M. J. B. Pereira, H. P. Babcock, N. G. Walter, and S. Chu. 2002. Correlating structural dynamics and function in single ribozyme molecules. Science. 296:1473–1476. [DOI] [PubMed] [Google Scholar]