

Abstract
X-ray absorption near-edge structure (XANES) spectra at the Fe K-edge have been measured and compared on solution samples of horse carbonmonoxy-myoglobin and its photoproducts, prepared by two different photolysis protocols: 1), extended illumination at low temperature (15 K) by white light; and 2), slow-cool from 140 to 10 K at a rate of 0.5 K/min while illuminating the sample with a 532-nm continuous-wave laser source. CO recombination has been followed while increasing the temperature at a rate of 1.2 K/min. After extended illumination at 15 K, a single process is observed, corresponding to CO recombination from a completely photolyzed species with CO bound to the primary docking site (formally B-state, in agreement with previous x-ray diffraction studies). The temperature peak for this single process is ∼50 K. Using slow-cool illumination, data show a two-state recombination curve, the two temperature peaks being roughly assigned to 50 K and 110 K. These results are in good agreement with previous FTIR studies using temperature-derivative spectroscopy. The XANES spectroscopic markers probe structural differences between the photoproduct induced by extended illumination at 15 K and the photoproduct induced by slow-cool illumination. These differences in the XANES data have been interpreted as due to light-induced Fe-heme relaxation that does not involve CO migration from the B-state. A quantitative description of the unrelaxed and relaxed B-states, including the measurements of the Fe-Np, Fe-NHis, and Fe-CO distances, and the out-of-plane Fe displacement, has been obtained via a procedure (MXAN) recently developed by us. This work shows that XANES, being able to extract both kinetic and structural parameters in a single experiment, is a powerful tool for structural dynamic studies of proteins.
INTRODUCTION
Myoglobin (Mb) is certainly one of the best-studied proteins. Since its structure has been revealed (Kendrew et al., 1960), the aim of many different studies has been to understand the connection between structure and function at the molecular level. According to physiology textbooks, this protein is involved in the storage of dioxygen in muscle, but its reactivity with a wide variety of small molecules such as NO and CO has long been known (Antonini and Brunori, 1971). Very recently, Mb function has been reassessed due to a large body of evidence that this protein could be involved in protecting cytochrome-c oxidase activity by scavenging potential inhibitor, especially NO (Brunori, 2001; Wittenberg and Wittenberg, 2003; and references therein). A possible allosteric functional mechanism was also hypothesized (Frauenfelder et al., 2003).
External ligands bind to the Fe-heme prosthetic group in the hydrophobic core of the protein matrix, and visible light can be used in the laboratory to photodissociate these small molecules. CO has been largely preferred as a ligand in photolysis experiments because of its quantum yield close to unity (Gibson, 1989; Chance et al., 1990; Petrich et al., 1991) and the stability of the carbonmonoxy-myoglobin derivative (MbCO).
Multiple, time-dependent energy barriers affect the motion of the CO molecule within the protein matrix, due to the presence of different intermediates between the solvent and the bound state, resulting in a non-exponential time dependence and non-Arrhenius temperature dependence of the binding kinetics (Austin et al., 1975; Scott and Gibson, 1997; Nienhaus et al., 2003a,b,c). At room temperature, the photodissociated CO molecule rebinds to the iron in a bimolecular reaction on the millisecond timescale (at 1 mM CO); at T < 160 K, CO recombination is still fast, but ligands rebind from within the protein (geminate recombination). At T < 30 K, CO rebinds very slowly (Iizuka et al., 1974) by a molecular tunneling mechanism (Alben et al., 1980), so that almost complete conversion to photoproduct can be achieved and maintained over long times. A sequential reaction model can describe the path of a CO molecule from the solvent (S) to the bound state in the active site of the protein (A),
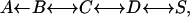 |
where the B, C, and D intermediate states involve different internal docking sites for the CO within the protein matrix. Temperature-derivative spectroscopy (TDS) is a powerful tool to study the relaxation of these intermediate states trapped at low temperature (Berendzen and Braunstein, 1990) and it has widely applied to Fourier transform infrared measurements (FTIR) using different photolysis protocols (Nienhaus et al., 1994, 1998, 2003a,b,c). This technique is very sensitive to the environment of the CO molecule and, therefore, can be used as a marker of ligand position being also sensitive to its migration, binding, and even CO internal dynamics (Mourant et al., 1993; Lamb et al., 2002; Nienhaus et al., 2002, 2003a,b,c; Kriegl et al., 2003).
Protein engineering has been a valuable tool to increase specific pathways and to selectively populate intermediate species (Scott et al., 2001; Nienhaus et al., 2002, 2003a,c). These intermediates have been characterized in recent years using room temperature time-resolved and low-temperature x-ray diffraction measurements. The low temperature images show the trapped intermediates using the same photolysis protocol used for TDS-FTIR measurements (Brunori et al., 2000; Chu et al., 2000; Ostermann et al., 2000). Consequently they have been able to match the structure and dynamics at a molecular level.
Room-temperature, time-resolved diffraction studies after pulsed laser photolysis have shown that the same intermediate states trapped at very low temperature are transiently populated along the CO rebinding pathway (Srajer et al., 1996, 2001; Schotte et al., 2003; Bourgeois et al., 2003). It is important to point out that molecular events such as dissociation and recombination of CO take place on the timescale of molecular vibrations (picoseconds), whereas most steady-state or slow kinetic studies measure a statistical average over all molecules rather than the kinetics of the individual chemical events.
X-ray absorption spectroscopy (XAS) is another, extremely sensitive technique to directly examine structural parameters such as distances and bond angles around the heme iron. It can be conveniently used to extend the relationship between structure and dynamics to a higher level of complexity. The x-ray photon absorbed excites an electron transition from a deep metal core level to final, unoccupied continuum states with dipole selected symmetry. The interference between the outgoing photoelectron wave from the metal center and the backscattering waves from neighboring atoms is recorded in a specific absorption spectrum that is consequently sensitive to the atomic environment.
In this work, we have exploited the sensitivity of XAS to study two photoproduct intermediates of horse heart myoglobin at low temperature which were prepared by different photolysis protocols. For both photoproducts, we have subsequently monitored a temperature-dependent relaxation process while increasing the temperature at a constant rate, using x-ray absorption near-edge structure (XANES) spectroscopy as a local probe. This process is different for the two different photoproducts and in agreement with FTIR-TDS results using the same protocols. This work is a first, significant step toward our goal to collect complete XANES-TDS data sets. Whereas FTIR-TDS studies have already provided valuable dynamical information on ligand migration, XANES-TDS would complement this information from the point of view of the active site of the molecule.
In previous articles, we have succeeded in extracting structural information from sperm whale MbCO crystals including photoproducts at low temperature by using XANES (Della Longa et al., 2001) that are in very good agreement with high resolution x-ray diffraction data. Moreover, we studied a thermally trapped nonequilibrium state of aquomet-myoglobin (MbMet) induced by x-rays at cryogenic temperatures and followed the temperature-dependent relaxation both in the crystal and in solution (Della Longa et al., 2003). By comparing these previous XANES results with the new results reported here, we give evidence of a light-induced Fe-heme relaxation event after ligand dissociation under prolonged illumination at very low temperature. A key step allowing the interpretation of our data is the possibility to analyze difference XANES spectra in a quantitative fashion. As we show in the present article for the first time, we have been able to improve the accuracy of our interpretation by reducing the systematic error using a modified version of the MXAN package (Benfatto and Della Longa, 2001) already applied for the analysis of XANES spectra (D'Angelo et al., 2002; Benfatto et al., 2002).
MATERIALS AND METHODS
Photolysis protocols and x-ray absorption measurements
Extended illumination at 15 K
Horse heart myoglobin was purchased from Sigma and used without further purification. The lyophilized protein was dissolved in a 66% glycerol, 33% potassium phosphate buffer (v/v) (pH 7) at a final concentration of 4 mM. The sample solutions were stirred for 1 h under one atmosphere of CO. Then, the samples were reduced with twofold excess of sodium dithionite added anaerobically and stirred for another 15 min. Approximately 70 μl of the protein solution were injected in an oxygen-free sample holder having two thin Mylar windows. Immediately before measurements, another aliquot of the sample was diluted for measuring UV/V spectra to check the state of the MbCO and deoxy-Mb samples. XAS spectra of 1-mm-thick solution samples of MbCO were acquired in fluorescence mode at T = 100 K at the beam line D21 of the Laboratory for Use of Electromagnetic Radiation synchrotron facility (LURE, Orsay, France) by using an Si(111) double-crystal monochromator, an energy-resolving seven-element Ge detector, and a liquid helium cryostat. Subsequently, the sample was cooled to 15 K, and another spectrum of MbCO was taken. Thereafter, the sample was illuminated using white light from a fiber-optic illuminator for ∼3 h. The calculated optical density of the 1-mm-thick sample changes from O.D. = 192 at 420 nm, to O.D. = 14 at 540 nm, and to O.D. = 0.1 at 630 nm, so that efficient photolysis over the total thickness is induced by photons between 500 and 600 nm. With our procedure, almost complete photolysis (>95%) was achieved. In fact, at T = 15 K, due to the very low rebinding rate (by molecular tunneling; Alben et al., 1980), the steady state after saturation in photolysis kinetics corresponds to almost complete photolysis. During the illumination period, before collecting XAS spectra of the photoproduct, distinct spectral changes in the XANES spectra at E = 7124 eV were monitored until steady state was reached (Fig. 3). The kinetic curve of the x-ray absorption difference [I7124(t) − I(0)]/[I7124(∞) − I(0)] reached saturation with an integrated irradiation power density (between 500 and 600 nm) of 5 × 10−2 mW mm−2. The same saturation limit I7124(∞) at T = 15 K was obtained by changing the intensity of irradiation. The XANES spectra shown in Fig. 2 were obtained by summing, for each spectrum, two data collection frames of 330 energy points, 7 s/point acquisition time, for a total collection time of 80 min.
FIGURE 3.

Evolution of the integrated XANES signal between the A and A′ peaks as a function of the illumination time for MbCO at 15 K under extended illumination by white light. The solid line is a fit using two exponentials, (a*(1−exp(−t/t1))+(1−a)*(1−exp(−t/t2))). The faster component is assigned to Fe-CO photolysis, the slower component to light-induced Fe-heme relaxation.
FIGURE 2.

Photolysis of MbCO at 15 K under extended illumination (white light). (A) Fe K-edge XANES MbCO (dotted line) and its photoproduct Mb*CO (solid line) at 15 K after saturation of the spectral changes induced by light. The spectrum of the deoxyMb (dashed line) at T = 100 K is also shown for comparison. (B) XANES difference spectra of deoxyMb–MbCO (solid line) and Mb*CO–MbCO (dotted line).
Slow-cool illumination from 140 K
The lyophilized horse heart myoglobin was dissolved in 75% glycerol, 25% potassium phosphate buffer (v/v) (pH 8) at a final concentration of 10 mM. The sample solutions were stirred for 1 h under one atmosphere of CO. Then, the samples were reduced with twofold excess of sodium dithionite added anaerobically and stirred for another 15 min. Approximately 2 μl of the protein solution were sandwiched between a sapphire window and an aluminated Mylar foil separated by a 300-μm-thick copper spacer and mounted in an oxygen-free high conductivity copper sample cell. This sample holder design represented the best compromise to obtain both a good signal/noise ratio and a good efficiency of the photolysis protocol (∼60%). The MbCO sample chamber was filled in nitrogen atmosphere while cooling down to 140 K and evacuated below this temperature. In the slow-cool illumination protocol, the sample is cooled from 140 K to 10 K at 0.5 K/min under illumination by a continuous wave, frequency-doubled Nd-YAG laser (model Forte 530–300, Laser Quantum, Manchester, UK), emitting 300 mW of output power at 532 nm. The beam was focused on the sample. After photolysis, the temperature was raised using a gradient of 1.2 K/min, and full XANES spectra were collected every 20 K integrating over 600 s in time (12 K in temperature).
Fe K-edge x-ray absorption spectra were collected at the BM30B-FAME beam line of the European Synchrotron Radiation Facility (ESRF, Grenoble, France), in the two-third filling mode at a beam current of 150 mA. The monochromator used was an Si(111) double crystal. A high-energy resolving multielement array detector allowed collection of XAS in fluorescence mode. The number of fluorescence counts changed from ∼30 s−1 per element (before the Fe K-edge) to ∼1000 s−1 per element (after the edge), so that 3000–100,000 counts were obtained per energy point at a total collection time of 20 min for each spectrum. Sample integrity under x-ray irradiation has been checked at the end of the photolysis protocol, by looking at the final MbCO XANES spectrum at T > 160 K, which was found to be identical to the initial one.
Quantitative analysis of difference XANES spectra via the MXAN package
X-ray absorption spectroscopy calculations are carried out starting from Fermi's golden rule to obtain the photoabsorption cross section within the dipole approximation. One important step is to calculate the final state of the photoelectron transition moment, which describes the physical process of scattering the electron photoemitted by the central atom by outer atoms. This is done in the framework of multiple scattering theory (Natoli et al., 2003, and references therein). It uses a muffin-tin (MT) approximation for the shape of the potential of the cluster of atoms included. Thirty-two atoms, i.e., the porphyrin macrocycle, the histidine imidazole, and the CO molecule, were included in this case. Cluster size and the value of ℓmax (i.e., the maximum ℓ-value of the spherical harmonic expansion of the scattering path operators) were chosen on the basis of a convergence criterion; MT radii were chosen on the basis of the Norman criterion. The molecular potential for the nearest atoms, i.e., the FeN5CO cluster, were obtained by the spin-unpolarized, self-consistent field method imposing the formal valence of each atom. The exchange correlation part of the potential has been determined by the X-α approximation. Inelastic processes were taken into account by a convolution with a broad Lorentzian function, with a width given by Γ = Γc + Γ|E|. The constant part Γc includes contributions from the core hole lifetime and the experimental resolution, whereas the energy-dependent term Γ|E| represents the inelastic processes. Γ|E| is zero below an onset energy Es, and begins to increase from a value As following the universal functional form related to the mean free path in a solid (Muller et al., 1982). This method introduces three nonstructural parameters that are derived during the fit on the basis of a Monte Carlo search at each step of computation.
One expects the accuracy of the structural parameters extracted by solution samples to be worse than those extracted by the P21 MbCO single crystals studied previously (Della Longa et al., 2001), due to the convolution of the polarized contributions in the solution spectrum. Another difficulty concerns the use of the Γ(E) function: due to strong asymmetry of the Fe-heme site, the function Γ(E) used to fit unpolarized spectra of a system with approximately C4v symmetry has a vectorial form with different values of the Es and As parameters for the two polarized components of the spectrum. In total, five nonstructural parameters, Γc, 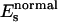 ,
, 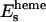 ,
, 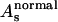 , and
, and 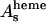 are used in the fitting procedure. Theoretically it is justified, considering that all physical quantities involved, in particular the dielectric function associated to the calculation of the self-energy of the system, must have the same symmetry of the geometrical cluster. As a consequence, the mean free path term is largely anisotropic, as is evident by looking at the systematic difference in the values Es found for the ɛ//c and ɛ//a* polarized spectra of the MbCO crystal.
are used in the fitting procedure. Theoretically it is justified, considering that all physical quantities involved, in particular the dielectric function associated to the calculation of the self-energy of the system, must have the same symmetry of the geometrical cluster. As a consequence, the mean free path term is largely anisotropic, as is evident by looking at the systematic difference in the values Es found for the ɛ//c and ɛ//a* polarized spectra of the MbCO crystal.
A constant experimental error corresponding to a noise/signal ratio of 0.0025 normalized units (i.e., units for which the absorption jump is equal to 1) was chosen. The minimization of the χ2 function was performed in the space of the n selected structural parameters (the χ2/n value is reported in Table 1 to quantify the fit goodness). They are the core size, the Fe-heme displacement, d|Fe-C|, d(Fe-Nhis|, the tilting angle α between the Fe-C vector and the heme normal, the bending angle β between the Fe-C vector and the C-O bond, and the C-O bond length. Doming of the heme in addition to Fe-heme displacement was not considered here, but could be included in a further study. During the fit, the outer atoms of the imidazole ring of the histidine and of the pyrrole rings of the porphyrin rigidly followed the motion of the Nhis and Np atoms, respectively. In Fig. 1, the sensitivity of the calculated XANES difference spectrum for each of these parameters is shown, keeping all other parameters constant. It is evident here that the sensitivity of the Mb*CO XANES spectrum in solution to the angular parameters (tilting and bending CO angles) and to small variations of the distance d(C-O) is very low. This result is different from the case of the solution spectrum of MbCO, where sensitivity arises from the tight proximity of the CO scatterer molecule to the Fe center, and also different from the case of the(ɛ//heme_normal) spectrum of single crystal Mb*CO (Della Longa et al., 2001), where sensitivity arises essentially from the polarization factors. For this reason, these parameters are omitted from Table 1.
TABLE 1.
Structural result of the fit of XANES difference spectra
| Fit | State | Chemsh(eV) | %Photolysis | χ2/n | ΔCore(Å) | Fe-heme Displ. (Å) | d(Fe-C)(Å) | d(Fe-NHis) (Å) | d(Fe-Np)(Å) |
|---|---|---|---|---|---|---|---|---|---|
| MbCO (x-ray diffraction: PDB code 1A6G) | |||||||||
| − | A | − | − | − | 0.00 | 0.05 | 1.83 | 2.06 | 1.98 |
| Photoproduct (XANES diff: extended illumination at 15 K) | |||||||||
| 1 | B relaxed | 1.25 | 100% | 4.27 | −0.01(4) | 0.50(5) | 3.18(11) | 2.06(3) | 2.03(2) |
| 2 | B relaxed | 0.3 | 100% | 4.65 | +0.02 | 0.40 | 3.23 | 2.07 | 2.04 |
| 3 | B relaxed | 0.0 | 100% | 5.37 | +0.04 | 0.31 | 3.24 | 2.09 | 2.06 |
| Photoproduct (XANES diff: slow-cool illumination from 140 K) | |||||||||
| 4 | B | 0.0 | 30% | 2.58 | +0.01 | 0.07 | 3.22 | 2.01 | 2.00 |
| 5 | B | 0.0 | 60% | 2.74 | +0.01 | 0.09 | 3.23 | 2.01 | 2.00 |
| 6 | B | 0.0 | 100% | 3.74 | 0.00 | 0.2 | 3.28 | 2.01 | 1.99 |
Chemsh is the chemical shift assumed for the B-relaxed state, i.e., a rigid shift of the Mb*CO spectrum with respect to the MbCO spectrum according to partial reduction of the net charge at the iron site. %Photolysis is the photolysis percentage assumed for the slow-cool illumination protocol. Statistical errors on the last digits are reported in parentheses, only in the case of Fit 1.
FIGURE 1.

Sensitivity of the simulated XANES difference spectrum to the important structural features of the Mb*CO spectrum. Parameters have been varied around a hypothetical structure intermediate between MbCO and the best-fit structure of Mb*CO.
Alignment of the theoretical and experimental spectra was obtained with a rigid energy shift of 7115.5 eV. A value of Γc = 1.7 ± 0.2 eV results from the fit procedure, fully consistent with the sum of the known value of the core-hole lifetime (1.2 eV) and the experimental resolution (∼0.5 eV). The ranges of the values found for the Es and As parameters of the Γ|E) function are 10 ± 4 eV and 12 ± 2 eV, respectively.
The fits of the XANES difference spectra Mb*CO–MbCO were obtained by modifying the MXAN program already used in our previous studies, for the following procedure:
The theoretical XANES spectrum of MbCO was calculated as a reference, based on the structural coordinates of the PDB file 1A6G (Kachalova et al., 1999), using the real part of the Hedin-Lundqvist potential without including any damping factor.
At each step of the fitting procedure, the MXAN package calculated the undamped Mb*CO theoretical spectrum, taking the coordinates from the PDB file 1ABS (Schlichting et al., 1994) as starting parameters and varying the selected coordinate parameters shown in Table 1.
The undamped difference spectrum Mb*CO–MbCO was calculated.
Finally, the damping factors were convoluted directly to the difference spectrum and fitted to the experimental difference spectrum.
This procedure assumes that the damping parameters are not correlated to the structural parameters, and that identical (or at least very similar) values pertain to the Mb*CO and MbCO spectrum. The number of parameters used by this modified MXAN procedure is the same as in previous studies of XANES spectra of heme proteins.
Fitting difference spectra has the important advantage that some systematic errors of the XANES theory can be reduced. As observed during the simulation of polarized spectra of single crystal MbCO and Mb*CO, respectively, for a chosen experimental error of 0.01 normalized units, the accuracy of the fit is much better for the (ɛ//heme normal) polarization, for which we obtained χ2/n values of 2.09 and 1.85, than for the (ɛ//heme plane) polarization, with χ2/n values of 3.46 and 3.08. We believe that our systematic errors are mostly due to the poor approximation used for the phenomenological broadening function Γ|E) that mimics the electronic damping. In all cases studied until now (Della Longa et al., 2001, 2003; Benfatto et al., 2002; D'Angelo et al., 2002; Hayakawa et al., 2004), systematic errors did not appreciably affect the structural results, confirming how this spectroscopy is dominated by the geometry of the atomic cluster rather than its electronic structure. Systematic errors linked to the (ɛ//heme plane) polarized component give more problems for the extraction of the axial parameters of heme proteins in solution. As the majority of the signal in solution is due to the (ɛ//heme plane) scattering, these errors affect the measurement of axial parameters more seriously and render them less accurate. However, as was observed in the crystal, the (ɛ//heme plane) MbCO and Mb*CO spectra are very similar, so that these systematic errors affect both spectra in a very similar way and are mostly eliminated in the difference spectra. In the present study, we observe rather large deviations between theory and experiment in the range from 15 to 30 eV of the XANES difference spectra. Therefore, we cannot exclude other sources of systematic errors that can also substantially affect the final values of the structural fits. As discussed below, various assumptions were required to be able to apply the XANES analysis (e.g., no contribution from D-states, no doming effects). However, we believe that they do not affect the overall conclusion from the quantitative analysis, i.e., the ability to distinguish between relaxed and unrelaxed Fe-heme conformations, and to quantify the degree of relaxation, which is the main goal of this work.
RESULTS
Temperature-dependent XANES under different photolysis protocols
In Fig. 2 A, the Fe K-edge XANES spectra are shown for horse MbCO (dotted line) and Mb*CO (solid line) after photolysis under extended illumination at 15 K. The spectrum of deoxyMb (dashed line) obtained by reduction of the aquomet adduct by sodium dithionite, taken at T = 100 K, is also shown for comparison. The most prominent changes observed are: 1), a red-shift of the rising absorption edge (the region between the A and A′ labels); 2), an increase and a fine modulation of the C, D, and D′ features; 3), a disappearing of the C2 bump; and 4), an increase of the E feature. The Mb*CO spectrum obtained by this protocol is very similar to that of deoxyMb, but the red-shift of the latter (2.5–3 eV) is, however, slightly larger than that of the former (2–2.5 eV).
Fig. 2 B shows the difference spectra Mb*CO–MbCO (dotted curve) and deoxyMb–MbCO (solid curve). The spectra differ markedly in the features P, A, and A′, and it is not possible to superimpose them by simple scaling as would be the case if the difference between them resulted from the extent of photolysis.
The evolution of the XANES difference spectrum as a function of time, leading to the Mb*CO spectrum in Fig. 2, is presented in Fig. 3. Each point on the graph represents an integrated intensity signal between 7118 eV and 7124 eV at the boundaries of the rising edge region (A′, A region in Fig. 2 B. This time course is biphasic and within the accuracy of the data can be fitted by a sum of two exponentials (solid line), and even the slower one was completed when we acquired the spectrum of Mb*CO. As we will discuss later, this XANES signal is extremely sensitive both to the breaking of the Fe-CO bond and to the heme-plane fine-structure changes. A structural explanation will be discussed in the next section according to a possible decoupling of the photodissociation of the CO molecule and the following relaxation of the heme structure. Further evidences will be presented to address this point. At the moment we want only to show that the time dependence of the photolysis protocol is pointing in the direction of a species that is fully relaxed(a B-relaxed state) even at this very low temperature. The differences between the deoxyMb-MbCO and Mb*CO-MbCO spectra may be interpreted as due to: 1), a reduction of the net charge at the iron site in deoxyMb relative to the B-relaxed state, induced by the reducing agent, resulting in a nearly rigid spectral shift of the features P and of the rising edge; and 2), the absence of a CO molecule in the B-site, and the presence of a water molecule in the distal pocket, that can influence the fine structure of the C, D, D′ features in deoxyMb with respect to Mb*CO.
A completely different photolysis protocol has been used to trap the CO molecule in another site in the protein matrix. This second protocol was originally introduced to study light-induced relaxation process at low temperatures (Nienhaus et al., 1994) both in wild-type horse heart and wild-type sperm whale myoglobin. Later on, an intermediate state with CO trapped in the proximal side of the heme plane of wild-type horse heart myoglobin was identified due to cryocrystallography technique using this same protocol (Chu et al., 2000). The same result was obtained for the L29W mutant of sperm whale myoglobin under the same experimental condition (Ostermann et al., 2000). The CO molecule was found in the so-called Xe1 cavity; this intermediate state was denoted as D or Mb**CO. Rebinding from this second docking site requires overcoming a higher energetic barrier as compared with rebinding from docking site B.
Using a continuous-wave 532-nm laser, we illuminated the sample while cooling slowly from 140 to 10 K at a constant rate of 0.5 K/min. With this second illumination protocol, we were able to obtain only partial photolysis, so that the final spectrum at 10 K is a superposition of spectra of MbCO and the two different photoproducts denoted as Mb*CO (B) and Mb**CO (D). In Fig. 4, we show the MbCO XANES spectrum (dotted line) and the spectrum obtained after slow-cool illumination, at 10 K (solid line). To estimate precisely the percentage of photolysis is difficult; however, by the analysis of the difference spectra shown later, we assume that the photolysis in the second protocol is in the range of 60% of the total.
FIGURE 4.

Photolysis of MbCO by slow-cool illumination. Fe K-edge XANES spectra of horse MbCO (dotted line), and the spectrum obtained at 10 K after slow-cool illumination from 140 K (solid line). The estimated fraction of photolysis is ∼60%. According to previous studies (Nienhaus et al., 1994; Chu et al., 2000; Ostermann et al., 2000), the photoproduct contains both the B- and D-states.
In Fig. 5 A, the temperature dependence of the integrated XANES absorption difference between the peaks A and A′, ΔAA<′(T), is plotted while the sample temperature was ramped up by 1.2 K/min. The signal represents CO rebinding for the two different trapped photoproducts after extended illumination at 15 K (open circles), and after slow-cool illumination (solid squares). Data are plotted as fractions of the maximum difference observed, ΔAA′(T)/ΔAA′max. In the former case, we only acquired the few points in the energy range between the features A and A′ so that we obtained a large number of points with a time resolution faster than the temperature gradient. In the latter case, we collected full XANES spectra that represent averages over 10 K along the temperature gradient, so that we lost time (temperature) resolution but were able to obtain the full spectra of the trapped intermediate species. In Fig. 5 B, we have plotted the consecutive difference spectra obtained under the temperature gradient after slow-cool illumination. Two distinct rebinding processes are well separated, and there is an intermediate region (80–100 K) where the XANES spectrum does not show any detectable rebinding feature. Therefore, even though there are only a few points with a larger temperature average for this second protocol, they contain more information as it is possible to plot the entire XANES spectrum for the associated temperature interval. Finally, the same MbCO XANES spectrum is recovered at higher temperature for both protocols.
FIGURE 5.

(A) CO recombination after preparation of different photoproducts while ramping the temperature up at 1.2 K/min, as measured from the XANES signal ΔAA′(T) integrated between 7.12 and 7.125 keV. Each point in the panel was calculated as ΔAA′(T)/ΔAA′max. (Open circles) Recombination after extended illumination at T = 15 K; (solid squares) recombination after slow-cool illumination. (B) XANES difference spectra (XANES-TDS) upon recombination after slow-cool illumination, from 10 K to 160 K.
The two time courses under temperature gradient reported in Fig. 5 A show clear differences: using the extended illumination at 15 K, a single process is observed so that it can be interpreted as simple rebinding from photoproduct state B in which the ligand is located in the primary docking site. A single temperature peak for this process is obtained from a derivative of the curve at ∼T = 50 K in complete agreement with the experiment using FTIR-TDS (Nienhaus et al., 1994; Chu et al., 1995). Using slow-cool illumination, data show a two-state recombination curve being strongly evident as a second process at higher temperature. The two temperature peaks in the derivative of the curve have been roughly assigned to T = 50 K and T = 110 K so that even these data are in good agreement with previous FTIR studies using temperature-derivative spectroscopy (Nienhaus et al., 1994; Chu et al., 1995). According to those previous results we have assigned the first recombination event to the CO rebinding process from the B-state and the second recombination event to the rebinding process of a fraction of CO molecules populating the higher energy D-state (formally assigned to the Xe1 pocket).
Comparison of low temperature XANESdifference spectra
Fig. 6 A shows the XANES difference spectrum obtained under the temperature gradient after slow-cool illumination (10–80 K, solid line). For comparison, we show the difference spectrum obtained by extended illumination at 15 K (dotted curve), scaled to account for partial photolysis in the slow-cool illumination protocol. Both difference spectra are interpreted as resulting from CO rebinding from the B-state. However, it is evident that there are large differences, especially around the XANES features A, C, and D, suggesting structural dissimilarities in the environment of the heme iron.
FIGURE 6.

Rebinding of CO after slow-cool illumination, obtained raising temperature from 10 to 80 K. (A) XANES difference spectrum (T = 10–80 K) compared with that obtained upon extended illumination at 15 K, rescaled for comparison (dotted line). (B) The same spectrum is compared with the (ɛ//heme normal) polarized XANES difference spectrum of a sperm whale MbCO single crystal under extended illumination at 15 K (Della Longa et al., 1999), also rescaled for comparison.
For comparison, we plot the slow-cool difference spectrum (10–80 K, solid line) with previously published results on single sperm whale MbCO crystals (Della Longa et al., 1999) in Fig. 6 B. The (ɛ//heme normal) polarized XANES difference taken after photolysis of the crystal under extended illumination at 15 K is shown as a dotted line. Also this difference spectrum has been rescaled to account for partial photolysis. The strong similarity between the spectra suggests that the B-state photoproduct obtained by slow-cool illumination has structural parameters of the heme plane very close to that of the bound state, so that their contribution is eliminated in the difference spectrum as well as by taking only (ɛ//heme normal) polarized spectra. In other words, contrary to the experiment done by extended illumination at 15 K, involving a B-relaxed state, this photoproduct involves a very small Fe-heme relaxation. This hypothesis is based on the qualitative comparison of the two difference spectra obtained but has been confirmed by the quantitative XANES analysis. According to this interpretation, in the following we will refer to these photoproducts as B-relaxed (extended illumination at 15 K) and B (slow-cool illumination, CO rebinding up to 80 K).
Quantitative analysis of XANES differencespectra using the MXAN program
Fig. 7 presents a comparison between the theoretical best fits obtained via the MXAN package (solid line) and the experimental XANES difference spectra (open circles) obtained by the protocol of extended illumination at 15 K (Fig. 7 A) and by slow-cool illumination, CO rebinding up to 80 K (Fig. 7 B). The structural parameters extracted by the quantitative analysis are listed in Table 1, and a sketch of the structural states B and B-relaxed obtained is depicted in Fig. 8. The theoretical calculations fully confirm the experimental observations: the XANES difference spectrum obtained by extended illumination at 15 K (B-relaxed state) is explained by an almost complete relaxation of the Fe-heme toward the structure assumed in deoxy-Mb with the CO molecule in the primary docking site B. Three best-fit procedures were performed, the first one without assuming any chemical shift (that is related to the reduction of the net charge at the Fe site), the second one assuming a −0.3 eV chemical shift upon going from MbCO to Mb*CO, the third one assuming a −1.25 eV chemical shift. There is statistical correlation between the chemical shift assumed, and the first shell distances found by MXAN, as they both concur to the relative position of the absorption edge. This correlation is not included in our error evaluation. However, the chemical shift for Mb*CO should be less than about −0.3 eV, as confirmed by the almost complete absence of red-shift of the P features in the pre-edge region going from MbCO to Mb*CO. By contrast, a shift of the P features of about −1 eV is visible upon going from MbCO to deoxy-Mb. A chemical shift of −0.3 eV does not significantly change the structural parameters, so that for the photoproduct after extended illumination at 15 K we have obtained a slightly expanded core size, Δcore = +0.02(4) Å, an almost complete Fe-heme displacement= 0.40(5) Å, and a d(Fe-Np) = 2.04(2) Å, which is close to the distance found for deoxyMb; moreover, d(Fe-NHis) = 2.07(3) Å and d(Fe-CO) = 3.23(10) Å, according to the XRD studies locating the CO at the B docking site (Schlichting et al., 1994). As already discussed in Materials and Methods, above, the error found on the angular parameters of Mb*CO is much larger than in the case of the single crystal measurements (Della Longa et al., 2001), because their influence on the multiple scattering terms without considering the polarization factors, is minimal (see Fig. 1); for this reason they are not reported in Table 1.
FIGURE 7.

Theoretical XANES fits (solid lines) of the experimental difference spectra (open circles) obtained as follows: (A) before and after extended illumination at 15 K (B-relaxed state, Fit 2 of Table 1) and (B) The photoproduct at 10 K, and after rising temperature at 80 K in the dark, after slow-cool illumination (B state, Fit 5 of Table 1). The extracted structural parameters are depicted for clarity in Fig. 8.
FIGURE 8.

Sketch of the structure of the B-state obtained by slow-cool illumination, CO rebinding up to 80 K (upper frame), and the B-relaxed state obtained by extended illumination at 15 K (lower frame), according to the MXAN fits of the XANES difference spectra shown in Fig. 7.
The MXAN procedure has been performed in the same way for the XANES difference spectrum obtained by slow-cool illumination, CO rebinding up to 80 K (B-state). In this experiment the percentage of photolysis is uncertain. As stated above, by comparison with the (ɛ//heme normal) polarized XANES difference spectrum observed in a single crystal, the photolyzed fraction is estimated to be 60%. The XANES fit has been carried out by assuming that the bound states A of reassociated MbCO at 80 K (and also the residual MbCO kept frozen at 10 K at the end of the slow-cool illumination protocol, due to partial photolysis) have the same structure reported by XRD for MbCO at 100 K (Kachalova et al., 1999), and in turn, the same XANES spectrum, which is reported in Fig. 1. A second assumption is that the reassociation process observed between 10 K and 80 K is a two-state process B → A, i.e., the fraction of D-states that recombine at ∼110 K has no structural dynamics detectable by our technique at lower temperature. These two assumptions represent the simplest starting approximation allowing XANES analysis to get a first insight to MbCO protein dynamics.
We have repeated the fitting procedure assuming a photolysis fraction between 30 and 100%, without finding any relevant statistical correlation with the structural parameters obtained. For 60% photolysis, the photodissociated heme that participates to the rebinding process at T < 80 K is characterized, with respect to MbCO, by an almost irrelevant change of the core size, Δcore = 0.01(4) Å, Fe-heme displacement = 0.09(5) Å, d(Fe-Np) = 2.00(5) Å, d(Fe-NHis) = 2.01(4) Å. Even in this case, we have measured d(Fe-CO) = 3.23(11) Å, locating the CO in the same B docking site.
DISCUSSION
According to our experimental evidence and theoretical calculations, the XANES spectrum of the photoproduct obtained under extended illumination at 15 K corresponds to a relaxed Fe-heme structure (B-relaxed state), very similar to deoxyMb. However, the rather complete relaxation of the Fe-heme observed in this experiment is not in agreement with other diffraction (Schlichting et al., 1994; Teng et al., 1997) and EXAFS (Teng et al., 1987) studies reporting essentially a constrained relaxation (30–70%) of the Fe-heme, quantified from the Fe-heme displacement and/or from the Fe-Np distance increase. Moreover, even the previous XANES experiment of photolysis under extended illumination at 15 K on single crystal MbCO (Della Longa et al., 1999, 2001) gave evidence of a constrained relaxation.
We interpret the variations in the results reported from these different experiments as due to light-induced Fe-heme relaxation at low temperature, depending essentially on the net amount of incident photons on the sample. At the same time, we interpret the biphasic behavior of the photolysis kinetics curve shown in Fig. 3 as due to two structural determinants. The faster component of the XANES kinetics is determined by CO dissociation, inducing a red-shift of the (ɛ//heme normal) polarized XANES component, whereas the slower component probes the light-induced Fe-heme relaxation, giving in turn the red-shift of the (ɛ//heme plane) polarized XANES component.
In the following, we give a summary of the experimental evidence and of the theoretical results supporting this conclusion.
Comparison between the XANES photolysis experiment performed in solution and previous experiment with single crystal
The experimental results shown in Fig. 2 for horse MbCO (and similar results for sperm whale MbCO, data not shown) are in partial disagreement with previous results obtained by polarized XANES on single crystal sperm whale MbCO (Della Longa et al., 1999). In the crystal we have observed dramatic changes, including a 2 eV red-shift of the rising edge, for the polarization vector of the x-ray photons oriented along the Fe-CO bond in the (ɛ//heme normal) polarization. On the other hand, very small changes and no edge shift were observed in the (ɛ//heme plane) polarization.
The overall behavior was accurately reproduced by quantitative XANES analysis (Della Longa et al., 2001), and the edge red-shift observed in the (ɛ//heme normal) polarization was interpreted as due to energy relaxation of the empty states of the photoelectron of pz symmetry, mainly arising from a decrease of the ligand field caused by the CO molecule migrating away from the Fe coordination environment. The spectral changes were then quantified in structural terms: the XANES quantitative analysis provided a picture of the structural dynamics accompanying photolysis, including the CO dissociation and accommodation in the B docking site and a very small relaxation of the Fe-heme parameters, namely the Fe-Np distance and Fe-heme displacement. However, the experimental changes observed for the crystal are small compared with the changes observed in solution (see Fig. 2). In particular, the 2 eV edge red-shift observed in the (ɛ//heme normal) polarization would appear much smaller in a solution spectrum, i.e., <1 eV, due to the 1:2 average of the (ɛ//heme normal) and (ɛ//heme plane) signals, in contrast with the large red-shift of ∼2–2.5 eV seen in Fig. 1. So our explanation for such discrepancy between the results obtained in solution and those obtained in the crystal is that, in solution we have been able to obtain a fully relaxed form of the Fe-heme, whereas in the crystal this relaxation was incomplete.
Comparison with previous experiments on the thermally induced water dissociation in photoreduced aquomet-Mb
Even though there was only negligible spectral relaxation in the (ɛ//heme plane) polarization due to extended illumination at 15 K of single crystal MbCO, a substantial relaxation of this component was indeed observed in the temperature-induced dissociation of water from Fe-heme in photoreduced aquomet-myoglobin at low temperature (Della Longa et al., 2003). In this experiment, x-ray photoreduction below 70 K does not detect any water dissociation; this occurs only at T > 180 K when collective motions of the protein become thermally activated. This observation is consistent with the finding that photoreduced aquomet-myoglobin binds water at the sixth coordination, where it can be removed by photolysis (Lamb et al., 1998, 2000). In the XANES experiment on photoreduced aquomet-myoglobin, both the (ɛ//heme normal) and (ɛ//heme plane) XANES spectra do exhibit big changes at T > 180 K, both including edge red-shifts when the water dissociates. These changes are also reproduced in the analogous experiment in solution and the spectrum of the photoreduced species can be overlapped with the expected combination 2:1 of the (ɛ//heme plane) and (ɛ//heme normal) XANES spectra (Della Longa et al., 2003). These differences are also in agreement with the well-known differences between aquomet-myoglobin and dithionite-reduced deoxyMb. The observed relaxation of the (ɛ//heme plane) XANES spectrum is easily interpreted and reproduced by XANES theory as due mainly to an increase of the first shell average distance, i.e., the Fe-Np distance. The red-shift of XANES features with the increase of the first shell distance is reproduced by (ɛ//heme plane) XANES calculations, and can also be approximately calculated as a generalization of a rule (Natoli, 1983) already applied to predict the energy position of the main XANES peaks in diatomic hydrocarbon compounds (Hitchcock et al., 1984).
Consequently, comparing the experiments performed with crystals of MbCO and aquomet-Mb, with the corresponding experiments in the same condition in solution, it seems evident that the structural parameters related to ligand dissociation and Fe-heme relaxation can be decoupled in the polarized experiments on the crystals, but they have to be reproduced under the same condition in solution. Polarized XANES can probe and resolve their dynamics with unique sensitivity; both these parameters determine the edge red-shift observed in solution going from ligated to unligated states of the Fe-heme. The missing edge red-shift that is observed in the low-temperature photolysis on the crystal of MbCO is therefore due to an incomplete relaxation of this species. Possible explanations for this difference under the same photolysis protocol is that either crystal forces contribute to restrain the Fe-heme relaxation at very low temperature, even under extended illumination, or the efficiency of photolysis was decreased in the case of the crystal due to scattering of the light induced by the quartz capillary within which the crystal was mounted.
Our observation of the B-relaxed structure under extended illumination at 15 K is well in agreement with the evidence of light-induced relaxation of the CO rebinding barrier reported by IR-TDS after prolonged illumination at 25 K on sperm whale myoglobin (Nienhaus et al., 1994), an effect that has nothing to do with CO migration. At this temperature, barrier changes occur while the CO remains in the photolyzed state B, resulting in a shift of the related peak of absorbance difference ΔA(TR) from T = 43 K to T = 48 K and an increase of the calculated enthalpy barrier of 1.2 kJ/mol, that according to our results could be considered as the proximal work necessary for the readjustment of the Fe-heme-proximal histidine geometry from the B-relaxed state to the bound state.
Finally, the XANES difference spectrum between 10 and 20 K in Fig. 5 B exhibits extended features that may be associated to the exchange process between the two infrared CO stretching bands of the B-state observed in the TDS contour maps (Nienhaus et al., 1994, 2002; Chu et al., 1995). The XANES changes extend up to 7.16 keV, corresponding to a photoelectron energy of ∼40 eV. At this energy the absorption changes are almost certainly related to structural rearrangements around the Fe coordination sphere. Further experimental and theoretical work will reveal if these XANES changes are related to the readjustment of the CO molecule (flip-flop) within the B-site at such a low temperature and/or to further Fe-heme dynamics.
CONCLUDING REMARKS
The present work shows the ability of XAS to provide both structural and dynamical information, and to connect a large body of different experiments, both on crystal and solution samples. We have been able for the first time to quantitatively analyze XANES difference spectra, a key-step to interpret our experiments and to relate them to the results reported by both FTIR-TDS and x-ray diffraction experiments. Our XANES analysis allows us to quantify the degree of relaxation of the Fe-heme and eventually distinguish the combined effects of heme-core size change and Fe-heme displacement.
Acknowledgments
A.A. is thankful to both Stiftelsen Wenner-Grenska Samfundet and Wenner-Gren Stiftelserna for the grant obtained to work at the Stockholm University. G.U.N. was supported by the Deutsche Forschungsgemeinschaft (SFB 569, GRK 328, and Ni-291/3) and the “Fonds der Chemischen Industrie”.
References
- Alben, J. O., D. Beece, S. F. Bowne, L. Eisenstein, H. Frauenfelder, D. Good, M. C. Marden, P. P. Moh, L. Reinisch, A. H. Reynolds, and K. T. Yue. 1980. Isotope effect in molecular tunnelling. Phys. Rev. Lett. 44:1157–1160. [Google Scholar]
- Antonini, E., and M. Brunori. 1971. Hemoglobin and Myoglobin in their Reactions with Ligands. North-Holland, Amsterdam, the Netherlands.
- Austin, R. H., K. W. Benson, L. Eisenstein, H. Frauenfelder, and I. C. Gunsalus. 1975. Dynamics of ligand binding to myoglobin. Biochemistry. 14:5355–5373. [DOI] [PubMed] [Google Scholar]
- Benfatto, M., and S. Della Longa. 2001. Geometrical fitting of experimental XANES spectra by a full multiple-scattering procedure. J. Synchr. Rad. 8:1087–1094. [DOI] [PubMed] [Google Scholar]
- Benfatto, M., P. D'Angelo, S. Della Longa, and N. V. Pavel. 2002. Evidence of distorted fivefold coordination of the Cu2+ aqua ion from a XAS quantitative analysis. Phys. Rev. B. 65:174205-1–174205-5. [Google Scholar]
- Berendzen, J., and D. Braunstein. 1990. Temperature-derivative spectroscopy: a tool for protein dynamics. Proc. Natl. Acad. Sci. USA. 87:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeois, D., B. Vallone, F. Schotte, A. Arcovito, A. E. Miele, G. Sciara, M. Wulff, P. Anfinrud, and M. Brunori. 2003. Complex landscape of protein structural dynamics unveiled by nanosecond Laue crystallography. Proc. Natl. Acad. Sci. USA. 22:8704–8709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunori, M., B. Vallone, F. Cutruzzola, C. Travaglini-Allocatelli, J. Berendzen, K. Chu, R. M. Sweet, and I. Schlichting. 2000. The role of cavities in protein dynamics: crystal structure of a photolytic intermediate of a mutant myoglobin. Proc. Natl. Acad. Sci. USA. 97:2058–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunori, M. 2001. Nitric oxide, cytochrome-c oxidase and myoglobin. Trends Biochem. Sci. 26:21–23. [DOI] [PubMed] [Google Scholar]
- Chance, M. R., S. H. Courtney, M. D. Chavez, M. R. Ondrias, and J. M. Friedman. 1990. O2 and CO reactions with heme proteins: quantum yields and geminate recombination on picosecond time scales. Biochemistry. 29:5537–5545. [DOI] [PubMed] [Google Scholar]
- Chu, K., R. M. Ernst, H. Frauenfelder, J. R. Mourant, G. U. Nienhaus, and R. Philipp. 1995. Light-induced and thermal relaxation in a protein. Phys. Rev. Lett. 74:2607–2610. [DOI] [PubMed] [Google Scholar]
- Chu, K., J. Vojtechovsky, B. H. McMahon, R. M. Sweet, J. Berendzen, and I. Schlichting. 2000. Structure of a ligand-binding intermediate in wild-type carbonmonoxy-myoglobin. Nature. 403:921–923. [DOI] [PubMed] [Google Scholar]
- D'Angelo, P., M. Benfatto, S. Della Longa, and N. V. Pavel. 2002. Combined XANES and EXAFS analysis of Co2+, Ni2+ and Zn2+ aqueous solutions. Phys. Rev. B. 66:064209-1–064209-7. [Google Scholar]
- Della Longa, S., A. Arcovito, B. Vallone, A. Congiu Castellano, Y. Soldo, and J. L. Hazemann. 1999. Polarized x-ray absorption spectroscopy (P-XAS) of photolyzed carbonmonoxy-myoglobin. J. Synchr. Rad. 6:1138–1147. [Google Scholar]
- Della Longa, S., A. Arcovito, M. Girasole, J. L. Hazemann, and M. Benfatto. 2001. Quantitative analysis of x-ray absorption near edge structure data by a full multiple scattering procedure: the Fe-CO geometry in photolyzed carbonmonoxy-myoglobin single crystal. Phys. Rev. Lett. 87:15550–15551. [DOI] [PubMed] [Google Scholar]
- Della Longa, S., A. Arcovito, M. Benfatto, A. Congiu-Castellano, M. Girasole, J. L. Hazemann, and A. Lo Bosco. 2003. Redox-induced structural dynamics of Fe-heme-ligand in myoglobin by x-ray absorption spectroscopy. Biophys. J. 85:549–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauenfelder, H., B. H. McMahon, R. H. Austin, K. Chu, and J. T. Groves. 2003. The role of structure, energy landscape, dynamics, and allostery in the enzymatic function of myoglobin. Proc. Natl. Acad. Sci. USA. 98:2370–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson, Q. H. 1989. Hemoproteins, ligands and quanta. J. Biol. Chem. 264:20155–20158. [PubMed] [Google Scholar]
- Hayakawa, K., K. Hatada, P. D'Angelo, S. Della Longa, C. R. Natoli, and M. Benfatto. 2004. Full quantitative multiple-scattering analysis of x-ray absorption spectra: application to potassium ferro- and ferricyanide complexes. J. Am. Chem. Soc. 26:5618–5623. [DOI] [PubMed] [Google Scholar]
- Hitchcock, A. P., S. Beaulieu, T. Steel, J. Stohr, and F. Sette. 1984. Carbon K-shell electron energy loss spectra of 1- and 2-butenes, trans-1,3-butadiene and perfluoro-2-butene: bond lengths from continuum shape resonances. J. Chem. Phys. 80:3927–3935. [Google Scholar]
- Iizuka, T., H. Yamamoto, M. Kotani, and T. Yonetani. 1974. Low temperature photodissociation of hemoproteins: carbon monoxide complex of myoglobin and hemoglobin. Biochim. Biophys. Acta. 371:126–139. [DOI] [PubMed] [Google Scholar]
- Kachalova, G. S., A. N. Popov, and H. D. Bartunik. 1999. A steric mechanism for inhibition of CO binding to heme proteins. Science. 284:473–476. [DOI] [PubMed] [Google Scholar]
- Kendrew, J. C., R. E. Dickerson, B. E. Strandberg, R. G. Hart, and D. R. Davies. 1960. Structure of myoglobin: a three-dimensional Fourier synthesis at 2 Å resolution. Nature. 185:422–427. [DOI] [PubMed] [Google Scholar]
- Kriegl, J. M., K. Nienhaus, P. Deng, J. Fuchs, and G. U. Nienhaus. 2003. Ligand dynamics in a protein internal cavity. Proc. Natl. Acad. Sci. USA. 100:7069–7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb, D. C., V. Prusakov, N. Engler, A. Ostermann, P. Schellenberg, F. G. Parak, and G. U. Nienhaus. 1998. Photodissociation and rebinding of H2O to ferrous sperm whale myoglobin. J. Am. Chem. Soc. 120:2981–2982. [Google Scholar]
- Lamb, D. C., J. Kriegl, K. Kastens, and G. U. Nienhaus. 2000. Quantum-mechanical tunneling of water in heme proteins. J. Phys. Org. Chem. 13:659–663. [Google Scholar]
- Lamb, D. C., K. Nienhaus, A. Arcovito, F. Draghi, A. E. Miele, M. Brunori, and G. U. Nienhaus. 2002. Structural dynamics of myoglobin: ligand migration among protein cavities studied by Fourier transform infrared/temperature derivative spectroscopy. J. Biol. Chem. 277:11636–11644. [DOI] [PubMed] [Google Scholar]
- Mourant, J. R., D. P. Braunstein, K. Chu, H. Frauenfelder, G. U. Nienhaus, P. Ormos, and R. D. Young. 1993. Ligand binding to heme proteins. II. Transitions in the heme pocket of myoglobin. Biophys. J. 65:1496–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, J. E., O. Jepsen, and J. W. Wilkins. 1982. X-ray absorption spectra: K-edges of 3D transition metals, L-edges of 3D and 4D metals and M-edges of palladium. Solid State Commun. 42:365–368. [Google Scholar]
- Natoli, C. R. 1983. EXAFS and Near Edge Structure. A. Bianconi, L. Incoccia, and S. Stipcich, editors. Springer, Berlin, Germany. 43–56.
- Natoli, C. R., M. Benfatto, S. Della Longa, and K. Hatada. 2003. X-ray absorption spectroscopy in biological systems: state-of-the-art analysis J. Synchr. Rad. 10:26–42. [DOI] [PubMed] [Google Scholar]
- Nienhaus, G. U., J. R. Mourant, K. Chu, and H. Frauenfelder. 1994. Ligand binding to heme proteins: the effect of light on ligand binding in myoglobin. Biochemistry. 33:13413–13430. [DOI] [PubMed] [Google Scholar]
- Nienhaus, G. U., K. Chu, and K. Jesse. 1998. Structural heterogeneity and ligand binding in carbonmonoxy myoglobin crystals at cryogenic temperatures. Biochemistry. 37:6819–6823. [DOI] [PubMed] [Google Scholar]
- Nienhaus, K., D. C. Lamb, P. Deng, and G. U. Nienhaus. 2002. The effect of ligand dynamics on heme electronic transition band III in myoglobin. Biophys. J. 82:1059–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nienhaus, K., P. Deng, J. M. Kriegl, and G. U. Nienhaus. 2003a. Structural dynamics of myoglobin: spectroscopic and structural characterization of ligand docking sites in myoglobin mutant L29W. Biochemistry. 42:9633–9646. [DOI] [PubMed] [Google Scholar]
- Nienhaus, K., P. Deng, J. M. Kriegl, and G. U. Nienhaus. 2003b. Structural dynamics of myoglobin: the effect of internal cavities on ligand migration and binding. Biochemistry. 42:9647–9658. [DOI] [PubMed] [Google Scholar]
- Nienhaus, K., P. Deng, J. S. Olson, J. J. Warren, and G. U. Nienhaus. 2003c. Structural dynamics of myoglobin: ligand migration and binding in Valine-68 mutants. J. Biol. Chem. 278:42532–42544. [DOI] [PubMed] [Google Scholar]
- Ostermann, A., R. Waschipky, F. G. Parak, and G. U. Nienhaus. 2000. Ligand binding and conformational motions in myoglobin. Nature. 404:205–208. [DOI] [PubMed] [Google Scholar]
- Petrich, J. W., J. C. Lambry, K. Kuczera, M. Karplus, C. Poyart, and J. L. Martin. 1991. Ligand binding and protein relaxation in heme proteins: a room temperature analysis of NO geminate recombination. Biochemistry. 30:3975–3987. [DOI] [PubMed] [Google Scholar]
- Schlichting, I., J. Berendzen, G. N. Phillips, Jr., and R. M. Sweet. 1994. Crystal structure of photolysed carbonmonoxy-myoglobin. Nature. 371:808–812. [DOI] [PubMed] [Google Scholar]
- Schotte, F., M. Lim, T. A. Jackson, A. V. Smirnov, J. Soman, J. S. Olson, G. N. Phillips, Jr., M. Wulff, and P. A. Anfinrud. 2003. Watching a protein as it functions with 150-ps time-resolved x-ray crystallography. Science. 300:1944–1947. [DOI] [PubMed] [Google Scholar]
- Scott, E. E., and Q. H. Gibson. 1997. Ligand migration in sperm whale myoglobin. Biochemistry. 36:11909–11917. [DOI] [PubMed] [Google Scholar]
- Scott, E. E., Q. H. Gibson, and J. S. Olson. 2001. Mapping the pathways for O2 entry into and exit from myoglobin. J. Biol. Chem. 276:5177–5188. [DOI] [PubMed] [Google Scholar]
- Srajer, V., T. Y. Teng, T. Ursby, C. Pradervand, Z. Ren, S. I. Adachi,W. Schildkamp, D. Bourgeois, M. Wulff, and K. Moffat. 1996. Photolysis of the carbon monoxide complex of myoglobin: nanosecond time-resolved crystallography. Science. 274:1726–1729. [DOI] [PubMed] [Google Scholar]
- Srajer, V., Z. Ren, T. Y. Teng, M. Schmidt, T. Ursby, D. Bourgeois, C. Pradervand, W. Schildkamp, M. Wulff, and K. Moffat. 2001. Protein conformational relaxation and ligand migration in myoglobin: a nanosecond to millisecond molecular movie from time-resolved Laue x-ray diffraction. Biochemistry. 40:13802–13815. [DOI] [PubMed] [Google Scholar]
- Teng, T.-Y., H. W. Huang, and G. A. Olah. 1987. 5 K extended x-ray absorption fine structure and 40 K 10-s resolved extended x-ray absorption fine structure studies of photolyzed carboxymyoglobin. Biochemistry. 26:8066–8077. [DOI] [PubMed] [Google Scholar]
- Teng, T.-Y., V. Srajer, and K. Moffat. 1997. Initial trajectory of carbon monoxide after photodissociation from myoglobin at cryogenic temperatures. Biochemistry. 36:12087–12100. [DOI] [PubMed] [Google Scholar]
- Wittenberg, J. B., and B. A. Wittenberg. 2003. Myoglobin function reassessed. J. Exp. Biol. 206:2011–2020. [DOI] [PubMed] [Google Scholar]