

Abstract
Mortalin, also known as mot2/mthsp70/GRP75/PBP74, is a member of the heat-shock protein 70 family that is heat-uninducible. It is differentially distributed in cells that have normal and immortal phenotypes, has been localized to various subcellular sites, and has several binding partners and functions. Here, we describe the construction and use of mortalin-specific conventional and hybrid ribozymes to elucidate its crucial role in cell proliferation. Whereas conventional hammerhead ribozymes did not cause any repression of endogenous mortalin expression, RNA-helicase-linked hybrid ribozymes successfully suppressed the expression of mortalin, which resulted in the growth arrest of transformed human cells. We show that, first, RNA helicase-coupled hybrid ribozymes that have a linked unwinding activity can be used to target genes for which conventional hammerhead ribozymes are ineffective; second, the targeting of mortalin by RNA-helicase-coupled hybrid ribozymes causes growth suppression of transformed human cells and could be used as a treatment for cancer.
Introduction
Ribozymes are RNA molecules that have enzymatic properties (Cech, 1986). They are naturally occurring molecules that provide fine-tuning of gene expression cascades (replication, transcription and translation) by catalysing essential steps such as cleavage and ligation (Zhang & Cech, 1997; Cech, 2000; Doudna & Cech, 2002). Hammerhead ribozymes are the smallest ribozymes, and are used as 'molecular scissors' in molecular biology and biotechnology to elucidate and eliminate gene functions. The ribozyme RNA is induced to fold into its active conformation by the binding of metal ions. It forms two domains: the scaffold (domain 2) on which the ribozyme is built, and the active centre (the catalytic domain; known as domain 1; Cech & Uhlenbeck, 1994; Hammann & Lilley, 2002). During the past two decades, the mechanism of action of hammerhead ribozymes, describing the requirement for divalent metal ions, definition of the catalytic domains, and the sequence specificity, which is usually referred to as the target site, has been shown (Kawasaki et al., 1996; Koseki et al., 1999; Takagi et al., 2001; Kato et al., 2001; Akashi et al., 2002; Hammann & Lilley, 2002; Pyle, 2002). The activity of ribozymes in vivo is dependent on the level of expression at which they are effective, their specificity, and their intracellular stability, target colocalization and the accessibility of target sites (Yarus, 1999). These technical issues have caused severe problems for the use of ribozymes in vivo. Various modifications of the ribozyme expression plasmid have therefore come into play. For example, it has been shown that ribozymes expressed under the control of the RNA polymerase III promoter (the transfer RNA (tRNA)Val promoter) are efficiently expressed, highly stable and are exported to the cytoplasm (Kuwabara et al., 2001; Miyagishi et al., 2001). Such expression increased ribozyme activity in vivo many times over. However, ribozymes that are efficiently expressed and highly stable could still lack actvity because of inaccessibility of the target sites, which is a major problem because of unforeseeable secondary and tertiary RNA structures. Recently, this obstacle was elegantly resolved by modifying ribozyme expression plasmids. A new hybrid ribozyme that combined the cleavage activity of hammerhead ribozymes with the unwinding activity of endogenous RNA helicases was developed. Such RNA-helicase-based hybrid ribozymes, expressed from the RNA polymerase III promoter (tRNAVal) were indeed shown to have substrate-unwinding activity, as well as a strong cleavage activity (Warashina et al., 2001; Kawasaki & Taira, 2002; Kuwabara et al., 2002).
Here, we show the use of conventional and RNA-helicase-linked hammerhead ribozymes to suppress mortalin expression. Mortalin (mot2/mthsp70/GRP75/PBP74) is a member of the heat-shock protein 70 family and is involved in pathways that regulate cell proliferation, tumorigenesis and the stress response (Wadhwa et al., 1993, 1998, 2002a,b). We report that whereas conventional hammerhead ribozymes were ineffective for all of the chosen target sites, some of the RNA-helicase-linked hybrid ribozymes successfully repressed mortalin expression, resulting in the growth arrest of transformed human cells.
Results and Discussion
Conventional hammerhead ribozymes for mortalin
Five mortalin-specific ribozyme (mot-Rz) expression plasmids, which were driven by the tRNAVal promoter and which targeted its five different sites, were made as described in the Methods section, and are shown schematically in Fig. 1. We first examined the catalytic activity of these ribozymes for a mortalin–luciferase reporter-construct that encodes the amino-terminal region of mortalin in-frame with the luciferase reporter protein (as described in the Methods section, and in Fig. 2). COS7 cells were used for their high transfection efficiency. These cells were transfected with the mortalin reporter plasmid and the mot-Rz plasmids, as shown in Fig. 2B. Cells were lysed and assayed for luciferase activity 48 h after transfection. Cells transfected with mot-Rz plasmids showed a significant decrease (60–80%) in the mortalin–luciferase reporter activity as compared with the activity in the vector-only-transfected cells. This was interpreted as being a result of the cleavage of the mortalin–luciferase transcript by the mot-Rzs, suggesting that the mot-Rzs directed to these target sites might be effective in vivo to suppress the expression of endogenous mortalin. A second, more direct assay, in which mortalin itself was visualized instead of the reporter protein or its activity, was performed as shown in Fig. 3. An expression plasmid that encoded a V5-epitope-tagged N-terminal region of the mortalin protein (amino-acid residues 1–435) was co-transfected with the mot-Rz constructs, as described in the Methods section. The effectiveness of the mot-Rzs was determined by the visualization of the mortalin–V5 protein by western blotting with an anti-V5 antibody (Fig. 3B). As mot-Rz 46 and mot-Rz 73 have target sites in the 5' upstream non-coding region (Fig. 1), these could not be used for this assay. mot-Rz 213 and mot-Rz 231 have overlapping targeting sites; mot-Rz 213 was more effective in luciferase assays (Figs 1,2). Thus, mot-Rz 213 and mot-Rz 272 were used for this direct assay for mortalin suppression. Both the ribozymes caused a significant reduction of mortalin–V5 protein levels. A titration experiment, in which different amounts of the mot-Rz 213 plasmid and a constant amount of the mortalin–V5 reporter-plasmid were transfected, showed a dose-dependent decrease in mortalin–V5 protein levels. Transfection efficiencies were normalized by the co-transfection of a green fluorescent protein (GFP) reporter plasmid. Protein loading was normalized by western blotting using an anti-actin antibody. These data showed that mot-Rzs have sufficient catalytic activity, and could thus be used for the suppression of the expression of endogenous mortalin in cells.
Figure 1.
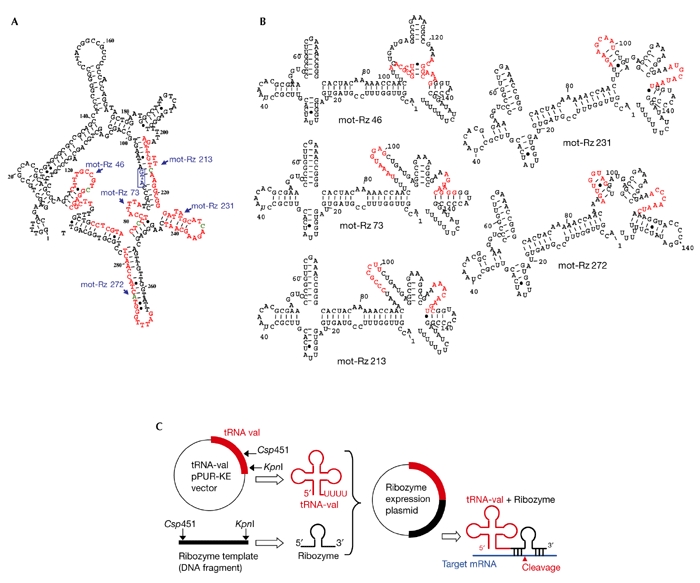
Design of mortalin ribozymes. (A) Partial secondary structure of mortalin RNA. Mortalin ribozyme (mot-Rz) target sites and cleavage sites are marked in red and green, respectively. The first ATG is shown in the blue box. (B) Partial secondary structures of each of the target sites, with the ribozyme and transfer RNA sequences (155 nucleotides) as predicted using the Mfold programme (Zuker, 1989). Target sites are marked in red. (C) Schematic representation of the construction and action of a hammerhead-ribozyme expression plasmid driven by a tRNA promoter. mRNA, messenger RNA.
Figure 2.
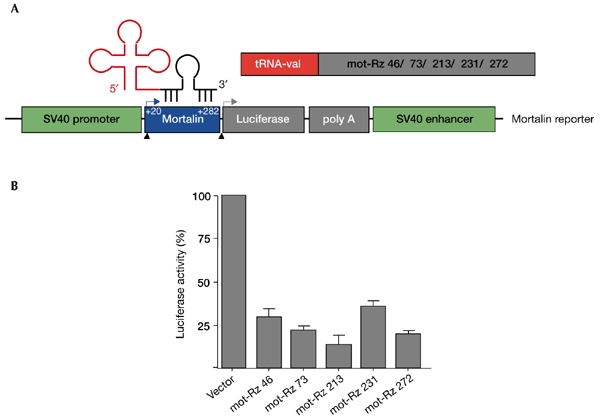
Effect of mortalin ribozymes on the expression of a mortalin–luciferase reporter. (A) Schematic representation of the mortalin–luciferase reporter expression plasmid, which is driven by the simian virus 40 (SV40) promoter. The target sites of the mortalin ribozymes (mot-Rzs) were at nucleotide residues 46, 73, 213, 231 and 272, as shown in Fig. 1. (B) Luciferase reporter activity in cells transfected with mortalin–luciferase and mot-Rzs. Cells were lysed 48 h after transfection, and were assayed as described in the Methods section. tRNA, transfer RNA.
Figure 3.
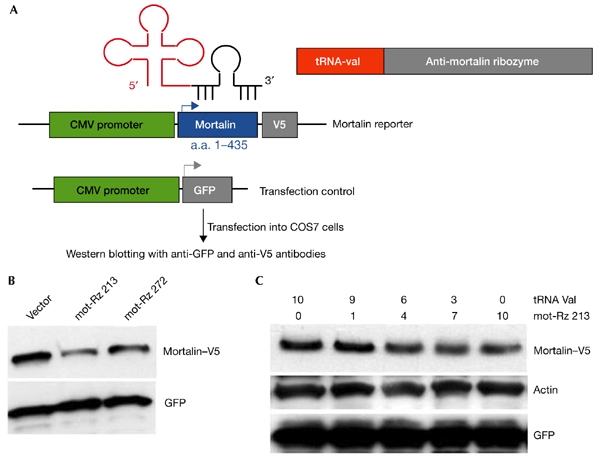
Effect of mortalin ribozymes on the expression of the mortalin–V5 reporter. (A) Schematic representation of an expression plasmid encoding a mortalin–V5 protein that is driven by the cytomegalovirus (CMV) promoter. The expression of green fluorescent protein (GFP) driven by the CMV promoter was used as a control for transfection efficiency. (B) Western blotting from cells transfected with the mortalin–V5 reporter and the indicted mortalin ribozymes (mot-Rzs). Cells were lysed 48–72 h after transfection, and the lysates were western blotted using an anti-V5 antibody. (C) Western blotting from cells transfected with the mortalin–V5 reporter and varying amounts of mot-Rz 213. GFP and actin were used as transfection and loading controls, respectively. a.a., amino acids; tRNA, transfer RNA.
Next, we transfected mot-Rzs into human fibrosarcoma (HT1080) cells. Transfected cells were selected in medium supplemented with puromycin (2.5 μg ml−1) for 3–5 days. The selected cells were maintained in medium supplemented with 0.5–1.0 μg ml−1 puromycin, and the pooled cultures were analysed for endogenous mortalin expression by northern and western blotting. Surprisingly, and in contrast to our expectations, none of the mot-Rzs caused any significant decrease of the endogenous levels of the protein (Fig. 4A); the mortalin transcript seemed to be affected only slightly. There are two possible interpretations of this data: first, the ribozymes may not have been expressed at sufficiently high levels in these cells; or second, the expressed ribozymes may not be active against the endogenous mortalin transcript, possibly due to inaccessibility of the target (Fig. 1A). To examine the first possibility, we analysed the expression of the mot-Rzs by RT–PCR (PCR after reverse transcription) and northern blotting, and found that the mot-Rzs were expressed (Fig. 4B), but at varying levels. To investigate the second possibility, we used recently developed RNA-helicase-linked hybrid ribozymes (HyRzs; Warashina et al., 2001; Kawasaki & Taira, 2002), as described below.
Figure 4.
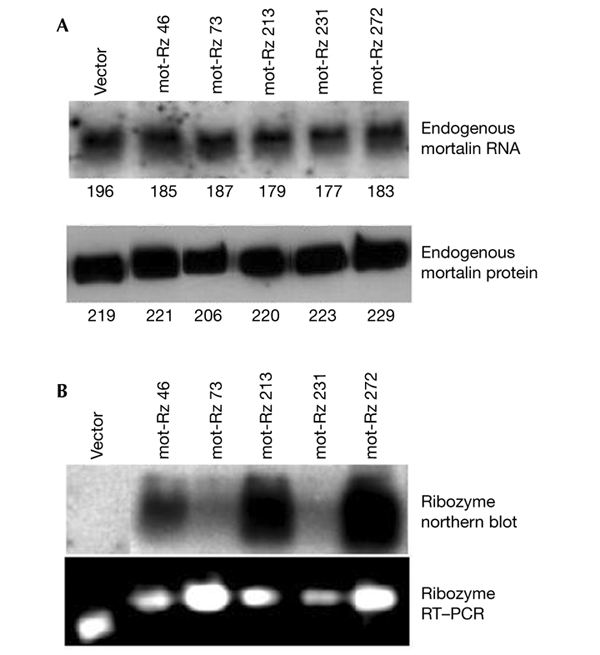
Endogenous mortalin expression in cells transfected with mortalin ribozymes. (A) Northern blot (upper panel) and western blot (lower panel) analyses of cells transfected with vector only or with the mortalin ribozyme (mot-Rz) expression plasmids indicated. The numbers below the lanes indicte relative units of mortalin expression. (B) Expression analysis of mot-Rzs by northern blotting (upper panel) and RT–PCR (PCR after reverse transcription; lower panel).
RNA-helicase-linked hammerhead ribozymes for mortalin
HyRzs coupled to RNA helicase were found to have a substrate-unwinding activity that improves their ability to access targets, as shown schematically in Fig. 5A. Poly(A)-linked RNA-helicase-recruiting mortalin ribozymes were therefore constructed as described in the Methods section. For a direct comparison, the target sites used were the same as those used for the conventional mot-Rzs (Fig. 1). First, the catalytic activity of these ribozymes was examined using a mortalin–luciferase reporter plasmid construct, as described in the Methods section. All four of the mortalin HyRzs suppressed the activity of the mortalin–luciferase reporter effectively (Fig. 5B). HT1080 cells were stably transfected with these HyRzs, and were selected in medium supplemented with puromycin. Many of the colonies (60–70%) in cells transfected with HyRz 231 and HyRz 272 seemed to be growth arrested as compared with cells transfected with vector only, HyRz 73 or HyRz 213. The level of expression of endogenous mortalin was analysed in selected pooled cultures by western blotting with an anti-mortalin antibody. Cells transfected with HyRz 231 or HyRz 272 showed lower levels of mortalin expression than vector-only-transfected cells (Fig. 5C). Cells transfected with HyRz 73 or HyRz 213 showed a significant reduction in the activity of the mortalin–luciferase reporter (Fig. 5B), but there was no reduction in endogenous mortalin expression (Fig. 5C). This suggests that there is a complex mortalin mRNA structure and that the target sites are inaccessible, even in the presence of coupled helicase activity. Interestingly, cultures transfected with any of the conventional ribozymes or with HyRz 73 and HyRz 213 did not show the growth arrest observed in HyRz-231- and HyRz-272-transfected cultures. Next, we isolated colonies that seemed to be growth arrested from HT1080 cultures that were transfected with HyRz 231 and HyRz 272 (Fig. 6A). Whereas vector-only-transfected cells rapidly formed dense colonies, cells transfected with HyRz 231 or HyRz 272 had a slow growth rate and a flattened morphology (Fig. 6A). Analysis of mortalin expression in these colonies by western blotting revealed a significant decrease in expression compared with the untransfected and vector-only-transfected cells (Fig. 6B). The colonies were maintained in culture for at least nine weeks. These colonies continued to show slow growth and a rather flattened morphology compared with the untransfected and vector-only-transfected cells (Fig. 6A), suggesting that the proliferation rate of cells depends on the level of mortalin expression. These data showed that, whereas conventional mot-Rzs were ineffective, the RNA-helicase-coupled HyRzs, targeted to the same sites, caused suppression of mortalin expression. This indicates that the mortalin transcript has a structure that is not easily accessible to the conventional mot-Rzs. The novel RNA-helicase-coupled HyRzs were effective in this case, resulting in reduced mortalin expression and growth arrest of transformed cells.
Figure 5.
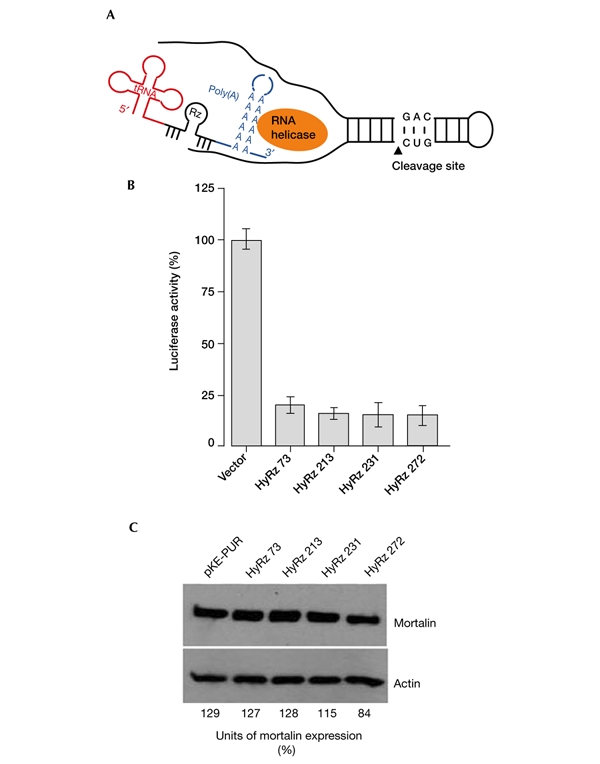
RNA-helicase-coupled ribozymes targeted against mortalin. (A) Schematic representation of RNA-helicase-coupled hammerhead ribozymes (HyRzs). The poly(A) sequence recruits RNA helicase, which uncoils the target transcript and improves the accessibility of the HyRz to the target sites. (B) Effects of mortalin HyRzs in a mortalin–luciferase reporter assay. Cells were lysed 48–72 h after transfection, and were assayed for luciferase activity as described in the Methods section. (C) Endogenous mortalin expression in cells that were transfected with mortalin HyRzs and selected in puromycin-supplemented medium. Cells transfected with HyRz 231 and HyRz 272 showed a reduction in mortalin expression. Western blots were scanned using a Micro Computer Imaging Device (MCID-M2; FUJIX). The ratios of the mortalin signals to the actin signals are shown as relative units of mortalin expression. Rz, ribozyme; tRNA, transfer RNA.
Figure 6.
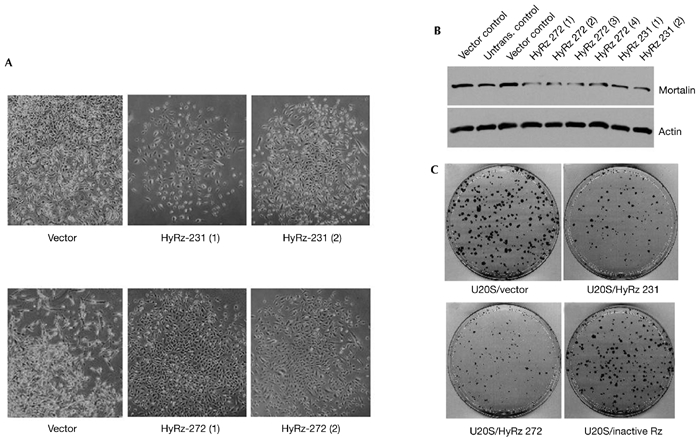
Effect of RNA-helicase-coupled ribozymes targeted against mortalin on cell growth and mortalin expression levels. (A) Phase-contrast images of human fibrosarcoma HT1080 cell clones transfected with vector only or with the RNA-helicase-coupled ribozymes (HyRzs) HyRz 231 and HyRz 272. Cells transfected with vector only grew quickly to form dense, shiny colonies. Cells transfected with HyRz 231 and HyRz 272 grew slowly, and had a flattened morphology. (B) Endogenous mortalin expression in clones selected from cells transfected with vector only, HyRz 231 or HyRz 272. HyRz-transfected clones showed low levels of mortalin expression. Actin was used as a loading control. (C) Colony-forming efficiency assay of U2OS cells transfected with empty vector, HyRz 231, HyRz 272 or an inactive, unrelated ribozyme. Suppression of colony formation was seen only in cells transfected with HyRz 231 and HyRz 272. Untrans., untransfected.
Many immortal and transformed human cell lines have higher levels of mortalin expression than their normal counterparts (Takahashi et al., 1994; Bini et al., 1997; Kaul et al., 1998). The exogenous expression of human mortalin (MOT2) caused the malignant transformation of NIH 3T3 cells (Kaul et al., 1998) and lifespan extension of normal human fibroblasts (Kaul et al., 2000), possibly by mechanisms involving the inactivation of the tumour suppressor protein p53 (Kaul et al., 2001; Wadhwa et al., 1998, 2002c) and other proteins that are thought to involve its mitochondrial import and chaperone functions (Liu et al., 2001; Wadhwa et al., 2002a,b). To determine whether the growth arrest of transformed cells by the targeting of mortalin was caused by the abrogation of its p53-inactivation function, we performed p53-dependent reporter assays in U2OS/PG13Luc cells (U2OS cells that are stably transfected with a p53-dependent luciferase reporter) that express wild-type p53. Cells transfected with HyRz 231 and HyRz 272 showed a 2–3-fold higher level of p53 activity than vector-only-transfected and HyRz-272-transfected cells, suggesting that p53 function is activated as a result of targeting mortalin. Such activation of p53 function may account for the growth arrest of cells expressing wild-type p53. To examine further the effect of mortalin targeting on the long-term proliferative ability of cells, we performed colony-forming-efficiency assays. The following cells, which have variable statuses with respect to wild-type p53, were used: HT1080 (partial loss of p53 expression); U2OS (wild-type p53); Saos-2 (complete loss of p53); and MCF7 (wild-type p53). These were transfected with vector only, HyRz 231, HyRz 272 and an inactive version of an unrelated ribozyme, which was made against the peroxisomal protein Pex19; Wadhwa et al., 2002d). Mortalinspecific HyRzs decreased colony-forming efficiency as compared with the vector and the inactive ribozyme (Fig. 6C; and data not shown). Furthermore, cells that expressed wild-type p53 (HT1080, U2OS and MCF7) showed stronger suppression of colony-forming efficiency than the p53-null (Saos-2) cells (data not shown). This data suggests that the growth arrest involves the activation of p53 and p53-independent pathways. This may involve mitochondrial import and chaperone functions of mortalin, as suggested by other studies (Liu et al., 2001; Wadhwa et al., 2002a,b). On the basis of these data, the targeting of mortalin could be used as a tool for cancer therapy. We have shown here for the first time that such targeting is possible using RNA-helicase-coupled hybrid ribozymes, a powerful tool for targeting gene expression in vivo.
Methods
Construction of RNA-polymerase-III-driven hammerhead-ribozyme expression plasmids.
Target sites flanking the ten putative ribozyme cleavage sites (GUC, GUA, CUC and CUA) in the 5' region of the mortalin complementary DNA sequence were selected. A partial secondary structure of mortalin RNA (Fig. 1A), and structures of each of the target sites, the ribozyme and the transfer-RNA sequence (155 nucleotides; Fig. 1B) were predicted using the Mfold programme (ftp://iubio.bio.indiana.edu/molbio/mac/mulfold.hqx; Zuker, 1989). Five target sites (with cleavage sites at nucleotides 46, 73, 213, 231 and 272; Fig 1B, shown in red) that had at least a 60% open structure (Fig. 1B) when embedded in ribozyme and tRNA sequences were selected, and were constructed in the pKE-PUR vector, as described in Koseki et al. (1999; Fig. 1C). The DNA oligonucleotide (TCCCCGGTTCGAAACCGGGCACTACAAAAACCAACTTTXXXXXXXXX CTGATGAGGCCGAAAGGCCGAAXXXXXXXXXGGTACCCCGGATATCTTTTTTT), which comprises the tRNA sequence, the ribozyme sequence (flanked by the gene-targeting sequence (X9)), and the linker sequence, was synthesized. This oligonucleotide was made by PCR, using the upperstrand primer 5'-TCCCCGGTTCGAAACCGGGCACTAC-3' and the lower-strand primer 5'-AGAAAAAAAGATATCCGGGGTACC-3', with 25 cycles of 94 °C for 30 s, 58 °C for 30 s, and 72 ° C for 45 s. The product was purified, cut with Csp45I and KpnI, and ligated into the pPUR-KE vector (Kato et al., 2001). The empty vector, which contained the tRNA sequence but no ribozyme, was used as a negative control.
Construction of mortalin reporter plasmids.
Two kinds of mortalin reporter plasmids were constructed. The first consisted of a mortalin–luciferase reporter driven by the simian virus 40 promoter. Mortalin amino-acid residues 20–282 were fused to the N-terminus of luciferase (Fig. 2A). The reporter activity was determined by luciferase assays. The second mortalin reporter plasmid encoded the N-terminal region of mortalin tagged with a V5 epitope at its carboxyl terminus (Fig. 3A) under the control of the cytomegalovirus promoter (provided by the pCDNA3.1 vector, Invitrogen). The reporter activity was determined by western blotting using an anti-V5 antibody (Invitrogen).
Cell culture, transfections and colony-forming assays.
HT1080, osteosarcoma (U2OS and Saos-2), breast carcinoma (MCF7) and monkey kidney (COS7) cells were cultured in a humidified incubator in DMEM supplemented with 10% FBS, penicillin, streptomycin and fungizone (Life Technologies, Inc.) at 37 °C in the presence of 5% CO2.
Transfections were performed using Lipofectamine PLUS (Invitrogen). Usually, 3 or 8 μg of plasmid DNA was used for each 6-cm or 10-cm dish, which contained cells at 60% confluence. Cells were selected in medium supplemented with puromycin (2.5 μg ml−1) for 3–5 days, and were then cultured in the presence of 0.5–1 μg ml−1 puromycin. For colony-forming assays, 1,000 cells were plated in 10-cm dishes and were monitored until the appearance of colonies (10–14 days). Colonies were washed with PBS, fixed with methanol:acetone (1:1) and stained with 0.1% aqueous crystal violet (Sigma).
Luciferase reporter assay.
Cells were transfected with the mortalin–luciferase reporter plasmid and the mot-Rzs. Cells were then selected in medium supplemented with puromycin (2.5 μg ml−1) for 2–4 days, and were then maintained in medium lacking puromycin for at least 24–48 h. Luciferase assays were performed using a Dual-Luciferase Reporter Assay System (Promega).
Northern blotting.
Total RNA was prepared from cells at 70% confluence in 10-cm dishes using Trizol (Life Technologies, Inc.). The RNA was denatured and fractionated according to size using 1% agarose gels containing 2.2 M formaldehyde, and was then transferred to a Hybond N+ membrane (Amersham Biosciences). 1.6-kb fragments of the human mortalin cDNA or the ribozyme sequence were used as probes for determining the expression levels of mortalin and ribozyme, respectively. Hybridization was performed at 65 °C in express hybridization buffer (Clontech). The membrane was washed for 10 min in each of 2 × SSC and 2 × SSC, 0.1% SDS, and was then washed twice in 1 × SSC and 0.1% SDS, followed by autoradiography. The amount of RNA loaded was determined by hybridization of the blots with an actin probe.
Detection of ribozyme expression by RT–PCR.
RT–PCR was carried out on total RNA (2 μg) prepared from cells transfected with the ribozyme plasmids or with empty vector (as a control). The RNA was reverse-transcribed using the lower-strand primer described below and Moloney murine leukaemia virus reverse transcriptase (at 42 °C for 90 min). The reverse-transcribed product was then amplified by PCR using the upper-strand primer 5'-TCCCCGGTTCGAAACCGGGCA-3' and the lower-strand primer 5'-GCTTGCATGCCTGCAGGTCGACGCGATAGAAAAAAAGATATCCGGGGT-3' for 20 cycles at 94 °C, 52 °C and 72 °C for 30 s each. The amplified product (100 bp) was run on a 2.6% agarose gel and visualized by ethidium bromide staining.
Western blot analysis.
Protein samples (10–20 μg) were separated on SDS–polyacrylamide gels and electroblotted onto nylon membranes (Millipore) using a semi-dry-transfer apparatus. Immunoassays were performed with polyclonal anti-mortalin (Wadhwa et al., 1993) or anti-actin (MAB1501R; Chemicon) antibodies. The immunocomplexes formed were visualized with horseradish-peroxidase-conjugated secondary antibodies using an enhanced chemiluminescence kit (Amersham Biosciences).
Acknowledgments
We thank T. Yaguchi for excellent technical assistance. The study was partly supported by NEDO Research Grants, Japan.
References
- Akashi H., Kawasaki H., Kim W.J., Akaike T., Taira K. & Maruyama A. (2002) Enhancement in the cleavage activity of a hammerhead ribozyme by cationic comb-type polymers and an RNA helicase in vitro. J. Biochem., 131, 687–692. [DOI] [PubMed] [Google Scholar]
- Bini L. et al. (1997) Protein expression profiles in human breast ductal carcinoma and histologically normal tissue. Electrophoresis, 18, 2832–2841. [DOI] [PubMed] [Google Scholar]
- Cech T.R. (1986) Biologic catalysis by RNA. Harvey Lect., 82, 123–144. [PubMed] [Google Scholar]
- Cech T.R. (2000) Structural biology. The ribosome is a ribozyme. Science, 289, 878–879. [DOI] [PubMed] [Google Scholar]
- Cech T.R. & Uhlenbeck O.C. (1994) Ribozymes. Hammerhead nailed down. Nature, 372, 39–40. [DOI] [PubMed] [Google Scholar]
- Doudna J.A. & Cech T.R. (2002) The chemical repertoire of natural ribozymes. Nature, 418, 222–228. [DOI] [PubMed] [Google Scholar]
- Hammann C. & Lilley D.M. (2002) Folding and activity of the hammerhead ribozyme. Chembiochem, 3, 690–700. [DOI] [PubMed] [Google Scholar]
- Kato Y., Kuwabara T., Warashina M., Toda H. & Taira K. (2001) Relationships between the activities in vitro and in vivo of various kinds of ribozyme and their intracellular localization in mammalian cells. J. Biol. Chem., 276, 15378–15385. [DOI] [PubMed] [Google Scholar]
- Kaul S.C., Duncan E.L., Englezou A., Takano S., Reddel R.R., Mitsui Y. & Wadhwa R. (1998) Malignant transformation of NIH3T3 cells by overexpression of mot-2 protein. Oncogene, 17, 907–911. [DOI] [PubMed] [Google Scholar]
- Kaul S.C., Reddel R.R., Sugihara T., Mitsui Y. & Wadhwa R. (2000) Inactivation of p53 and life span extension of human diploid fibroblasts by mot-2. FEBS Lett., 474, 159–164. [DOI] [PubMed] [Google Scholar]
- Kaul S.C., Reddel R.R., Mitsui Y. & Wadhwa R. (2001) An N-terminal region of mot-2 binds to p53 in vitro. Neoplasia, 3, 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki H. & Taira K. (2002) Identification of genes by hybrid ribozymes that couple cleavage activity with the unwinding activity of an endogenous RNA helicase. EMBO Rep., 3, 443–450. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kawasaki H., Ohkawa J., Tanishige N., Yoshinari K., Murata T., Yokoyama K.K. & Taira K. (1996) Selection of the best target site for ribozyme-mediated cleavage within a fusion gene for adenovirus E1A-associated 300 kDa protein (p300) and luciferase. Nucleic Acids Res., 24, 3010–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koseki S., Tanabe T., Tani K., Asano S., Shioda T., Nagai Y., Shimada T., Ohkawa J. & Taira K. (1999) Factors governing the activity in vivo of ribozymes transcribed by RNA polymerase III. J. Virol., 73, 1868–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwabara T., Warashina M., Koseki S., Sano M., Ohkawa J., Nakayama K. & Taira K. (2001) Significantly higher activity of a cytoplasmic hammerhead ribozyme than a corresponding nuclear counterpart: engineered tRNAs with an extended 3' end can be exported efficiently and specifically to the cytoplasm in mammalian cells. Nucleic Acids Res., 29, 2780–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwabara T., Warashina M. & Taira K. (2002) Cleavage of an inaccessible site by the maxizyme with two independent binding arms: an alternative approach to the recruitment of RNA helicases. J. Biochem., 132, 149–155. [DOI] [PubMed] [Google Scholar]
- Liu Q., Krzewska J., Liberek K. & Craig E.A. (2001) Mitochondrial Hsp70 Ssc1: role in protein folding. J. Biol. Chem., 276, 6112–6118. [DOI] [PubMed] [Google Scholar]
- Miyagishi M., Kuwabara T. & Taira K. (2001) Transport of intracellularly active ribozymes to the cytoplasm. Cancer Chemother. Pharmacol., 48, S96–S101. [DOI] [PubMed] [Google Scholar]
- Pyle A.M. (2002) Metal ions in the structure and function of RNA. J. Biol. Inorg. Chem., 7, 679–690. [DOI] [PubMed] [Google Scholar]
- Takagi Y., Warashina M., Stec W.J., Yoshinari K. & Taira K. (2001) Survey and summary: recent advances in the elucidation of the mechanisms of action of ribozymes. Nucleic Acids Res., 29, 1815–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S. et al. (1994) Correlation of heat shock protein 70 expression with estrogen receptor levels in invasive human breast cancer. Am. J. Clin. Pathol., 101, 519–525. [DOI] [PubMed] [Google Scholar]
- Wadhwa R., Kaul S.C., Ikawa Y. & Sugimoto Y. (1993) Identification of a novel member of mouse hsp70 family. Its association with cellular mortal phenotype. J. Biol. Chem., 268, 6615–6621. [PubMed] [Google Scholar]
- Wadhwa R., Takano S., Robert M., Yoshida A., Reddel R.R., Nomura H., Mitsui Y. & Kaul S.C. (1998) Inactivation of tumor suppressor p53 by mot-2, an hsp70 family member. J. Biol. Chem., 273, 29586–29591. [DOI] [PubMed] [Google Scholar]
- Wadhwa R., Taira K. & Kaul S.C. (2002a) An hsp70 family chaperone, mortalin/mthsp70/PBP74/Grp75: what, when and where? Cell Stress Chaperones, 7, 309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadhwa R., Taira K. & Kaul S.C. (2002b) Mortalin: a potential candidate for biotechnology and biomedicine. Histochem. Histopathol., 17, 1173–1177. [DOI] [PubMed] [Google Scholar]
- Wadhwa R., Yaguchi T., Hasan M.K., Mitsui Y., Reddel R.R. & Kaul S.C. (2002c) Hsp70 family member, mot-2/mthsp70/GRP75, binds to the cytoplasmic sequestration domain of the p53 protein. Exp. Cell Res., 274, 246–253. [DOI] [PubMed] [Google Scholar]
- Wadhwa R., Sugihara T., Hasan M.K., Taira K., Reddel R.R. & Kaul S.C. (2002d) A major functional difference between the mouse and human ARF tumor suppressor proteins. J. Biol. Chem., 277, 36665–36670. [DOI] [PubMed] [Google Scholar]
- Warashina M., Kuwabara T., Kato Y., Sano M. & Taira K. (2001) RNA–protein hybrid ribozymes that efficiently cleave any mRNA independently of the structure of the target RNA. Proc. Natl Acad. Sci. USA, 98, 5572–5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarus M. (1999) Boundaries for an RNA world. Curr. Opin. Chem. Biol., 3, 260–267. [DOI] [PubMed] [Google Scholar]
- Zhang B. & Cech T.R. (1997) Peptide bond formation by in vitro selected ribozymes. Nature, 390, 96–100. [DOI] [PubMed] [Google Scholar]
- Zuker M. (1989) On finding all suboptimal foldings of an RNA molecule. Science, 244, 48–52. [DOI] [PubMed] [Google Scholar]