

Abstract
A conformational conversion of the normal, protease- sensitive prion protein (PrP-sen or PrPC) to a protease-resistant form (PrP-res or PrPSc) is commonly thought to be required in transmissible spongiform encephalopathies (TSEs). Endogenous sulfated glycosaminoglycans are associated with PrP-res deposits in vivo, suggesting that they may facilitate PrP-res formation. On the other hand, certain exogenous sulfated glycans can profoundly inhibit PrP-res accumulation and serve as prophylactic anti-TSE compounds in vivo. To investigate the seemingly paradoxical effects of sulfated glycans on PrP-res formation, we have assayed their direct effects on PrP conversion under physiologically compatible cell-free conditions. Heparan sulfate and pentosan polysulfate stimulated PrP-res formation. Conversion was stimulated further by increased temperature. Both elevated temperature and pentosan polysulfate promoted interspecies PrP conversion. Circular dichroism spectropolarimetry measurements showed that pentosan polysulfate induced a conformational change in PrP-sen that may potentiate its PrP-res-induced conversion. These results show that certain sulfated glycosaminoglycans can directly affect the PrP conversion reaction. Therefore, depending upon the circumstances, sulfated glycans may be either cofactors or inhibitors of this apparently pathogenic process.
Keywords: heparan sulfate/pentosan polysulfate/prion/scrapie/transmissible spongiform encephalopathy
Introduction
Although the etiology of transmissible spongiform encephalopathies (TSEs) has not been fully established (Chesebro, 1998), a critical event in pathogenesis appears to be the conversion of protease-sensitive prion protein (PrP-sen) to its resistant form (PrP-res). The acquisition of partial protease resistance correlates with a conformational rearrangement of PrP from high α-helix to high β-sheet content (Caughey et al., 1991b; Pan et al., 1993). One of the basic tenets of the popular ‘protein-only’ or prion hypothesis for the TSE agent is that PrP-res itself induces the conversion of PrP-sen to PrP-res and thereby propagates itself as an ‘infectious protein’ (Griffith, 1967; Prusiner, 1982; Prusiner et al., 1984). Experiments in defined cell-free systems have demonstrated that PrP-res has at least limited self-propagating activity (Kocisko et al., 1994). Whether this activity results in the production of TSE infectivity is not known. However, cell-free conversion reactions reflect other important aspects of TSE pathobiology, such as striking species and strain specificities (Bessen et al., 1995; Kocisko et al., 1995; Bossers et al., 1997; Raymond et al., 1997, 2000). Furthermore, the apparent recapitulation of the in vivo conversion of PrP-sen under defined cell-free conditions has fostered a better understanding of the potential reaction mechanism (Horiuchi et al., 1999, 2000). Cell-free conversion involves the formation of radiolabeled PrP-res by mixing radiolabeled PrP-sen with unlabeled PrP-res in vitro. Although cell-free conversion emulates several critical aspects of disease-associated PrP-sen conversion, it has been limited to substoichiometric yields relative to input PrP-res and, therefore, is not as continuous as PrP-res formation in vivo (see below). It is possible that naturally occurring compounds, acting as pathological chaperones or cofactors (Caughey and Raymond, 1993; Caughey, 1994; Caughey et al., 1994; Telling et al., 1994, 1995; DebBurman et al., 1997), may be required for continuous PrP-res formation.
Because PrP-sen is normally a glycosylphosphatidylinositol (GPI)-anchored plasma membrane protein (Stahl et al., 1987), it may make contact with extracellular matrix elements when at the cell surface. The extracellular matrix–cell surface interface is one of the sites where PrP-sen conversion may occur (Caughey and Raymond, 1991; Caughey et al., 1991a) and, therefore, a site where natural stimulators of conversion might be found. Heparan sulfate (HS), a major sulfated glycosaminoglycan constituent of the extracellular matrix, has been implicated as an influential factor in PrP-res formation (Farquhar and Dickinson, 1986; Kimberlin and Walker, 1986; Snow et al., 1990; Caughey and Raymond, 1993; Gabizon et al., 1993; Caughey, 1994; Caughey et al., 1994). However, the available evidence appears somewhat paradoxical as to whether its role is stimulatory or inhibitory. On one hand, HS is known to stimulate amyloid formation of the Alzheimer disease β peptide (Snow and Wight, 1989), and the fact that HS is associated with PrP-res amyloid deposits in vivo (Snow et al., 1990) suggests that it may play a similar role in PrP-res formation. On the other hand, HS and certain other sulfated glycan analogs are known to potently block PrP-res formation and scrapie agent propagation in animals and tissue culture cells (Ehlers and Diringer, 1984; Kimberlin and Walker, 1986; Caughey and Raymond, 1993; Caughey, 1994; Caughey et al., 1994). Other studies have shown that sulfated glycans can bind PrP-sen (Gabizon et al., 1993; Caughey et al., 1994; Shyng et al., 1995; Brimacombe et al., 1999) and influence its intracellular trafficking (Shyng et al., 1995). It is currently not clear how to reconcile these various influences of sulfated glycans.
To determine the direct effect of sulfated glycans on the PrP conversion process, we have tested HS and pentosan polysulfate (PPS), a semi-synthetic HS analog, as well as elevated temperature in cell-free conversion and found that PrP-res-induced conversion of PrP-sen was stimulated by HS, PPS and temperature.
Results
Stimulation of PrP cell-free conversion by sulfated glycans
To test for direct effects of sulfated glycans on PrP-res formation, HS was added to a modified cell-free conversion protocol. Previous conversion protocols incorporated chaotropic ions and/or detergents to stimulate PrP-sen conversion (Kocisko et al., 1994; Horiuchi et al., 1999). The current protocol, which lacks these components, allowed us to measure the effect of sulfated glycans without contributions from such non-physiological stimulants. Hamster [35S]GPI(–) PrP-sen primarily migrated as two bands at ∼28 and ∼32 kDa, corresponding to the non-glycosylated and mono-glycosylated forms of PrP-sen, respectively, on SDS–PAGE (Figure 1A, lane 1). After conversion and proteinase K (PK) digestion, the [35S]PrP-res bands migrated as 6–7 kDa smaller proteins, as expected of PrP-res and products of the cell-free conversion reaction (Kocisko et al., 1994). Maximum stimulation (∼5-fold) was reached at an HS concentration of 5 µg/ml. The EC50 (concentration of HS giving 50% maximal stimulation) was ∼100 ng/ml.
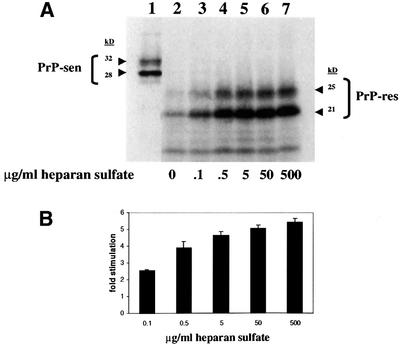
Fig. 1. HS stimulation of hamster GPI(–) PrP cell-free conversion. (A) Effect of increasing concentrations of HS on cell-free conversion. HS was added to conversion reactions at the concentrations indicated (lanes 2–7). An aliquot of the pre-incubation mixture representing 20% of the input [35S]PrP-sen is shown in lane 1. The two bands at ∼28 and ∼32 kDa show unglycosylated and partially glycosylated [35S]PrP-sen, respectively. The radiolabeled protein bands at ∼21 and ∼25 kDa after PK digestion demonstrate that the input [35S]PrP-sen had acquired limited PK resistance similar to brain-derived PrP-res (Kocisko et al., 1994). (B) Fold stimulation of cell-free conversion by increasing concentrations of HS. Error bars show the standard deviation (n = 3).
The HS analog PPS was also tested for stimulation of cell-free conversion (Figure 2). At maximum stimulation, there was a 5- to 8-fold increase in [35S]PrP-res formation. The EC50 for PPS was 50–500 ng/ml. No effect was observed when either no PPS was added or PPS was added immediately prior to PK digestion, ruling out the possibility that the increased amount of [35S]PrP-res was due to inhibition of PK activity by PPS (Figure 2C). Furthermore, no conversion was observed in the absence of input PrP-res (Figure 2D), which demonstrated that the stimulatory effect of PPS was PrP-res dependent. Results similar to those shown in Figure 2C and D were also obtained with HS (data not shown). Since PPS had similar effects to those of HS on cell-free conversion, subsequent experiments were performed with PPS.
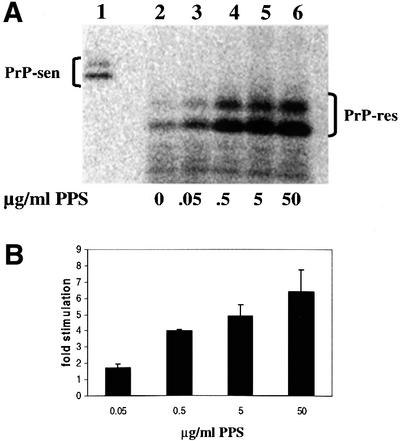
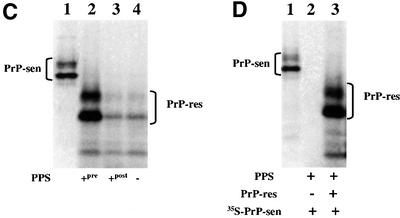
Fig. 2. PPS stimulation of hamster GPI(–) PrP cell-free conversion. (A) Effect of increasing concentrations of PPS on cell-free conversion. PPS was added to conversion reactions at the concentrations indicated (lanes 2–6). Ten percent of the input [35S]PrP-sen for each conversion reaction is shown in lane 1. (B) Fold stimulation of cell-free conversion by increasing concentrations of PPS. Error bars show the standard deviation (n = 3). The average percentage conversion at maximal stimulation (including experiments not shown) was ∼50% and could range from 15 to 80%. (C) PPS has no effect on PK digestion. PPS at 100 µg/ml was added either pre- (lane 2) or post-conversion (lane 3). No PPS was added to the control (lane 4). Twenty percent of the input [35S]PrP-sen is shown in lane 1. (D) PPS-stimulated cell-free conversion requires a PrP-res template/seed. Lane 2, PPS-stimulated cell-free conversion without input PrP-res; lane 3, PPS-stimulated cell-free conversion containing input PrP-res; lane 1, 20% of the input [35S]PrP-sen shown in lanes 2 and 3.
Because there is a wide diversity of sulfated glycosaminoglycans in vivo, other sulfated components of the extracellular matrix were tested in cell-free conversion. At the same concentrations as used with HS and PPS, chondroitin sulfate (CS) also stimulated conversion, but only to 10–20% of the effect of HS or PPS, while keratan sulfate (KS) had no stimulatory effect (data not shown). Thus, the ability to stimulate the cell-free conversion was restricted to specific sulfated glycans and was not due solely to their bulk polysulfated properties.
Stimulation of cell-free conversion by elevated temperature
In previous cell-free conversion protocols, optimum conversion occurred at an incubation temperature of 37°C (D.Kocisko, J.Chabry, S.Priola, R.Demaimay, M.Horiuchi and B.Caughey, unpublished data). To test whether the lack of chaotropic ions and/or detergents in the current protocol affected the optimum incubation temperature, we performed conversions with and without PPS at various temperatures. A temperature-dependent stimulation of [35S]PrP-res formation was found in both the presence and absence of PPS (Figure 3). The control samples had little conversion at incubation temperatures up to 42°C. However, some conversion was observed at 65°C. In contrast, the reactions with PPS had observable [35S]PrP-res formation beginning at 37°C and continued to increase to 65°C (the highest temperature tested), with the latter exceeding that obtained at 65°C alone. These results demonstrated that in the absence of chaotropic ions and detergents, conversion was stimulated by elevated temperature and further stimulated by PPS.
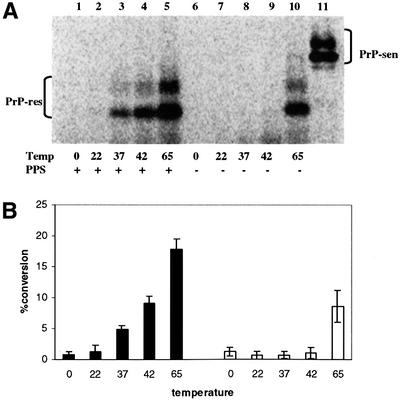
Fig. 3. Effect of temperature on hamster GPI(–) PrP cell-free conversion. (A) PPS-stimulated and non-stimulated cell-free conversion at different incubation temperatures. PPS (100 µg/ml) was included in the conversion reactions indicated in the figure. Lane 11 shows 20% of the input [35S]PrP-sen. (B) Comparison of PPS-stimulated and control cell-free conversion efficiency at different incubation temperatures. Black bars indicate samples with PPS added and white bars indicate control samples without PPS. Error bars show the range (n = 2).
PPS and/or elevated temperature promoted heterologous PrP cell-free conversion
The transmission of hamster 263K scrapie to mice is highly inefficient (Kimberlin et al., 1989; Race and Chesebro, 1998). Previous studies have shown that the transmission species barrier correlates with an inability of hamster 263K PrP-res to induce the cell-free conversion of mouse PrP-sen (Kocisko et al., 1995) and the ability of mouse PrP-sen to interfere with hamster PrP conversion reactions (Horiuchi et al., 2000). Since PPS and elevated incubation temperature stimulated conversion of hamster GPI(–) PrP-sen by hamster PrP-res, we investigated whether the same conditions could promote conversion of mouse GPI(–) PrP-sen. We found that mouse GPI(–) [35S]PrP-sen converted as efficiently as hamster GPI(–) [35S]PrP-sen in the presence of PPS at either 37 or 65°C (Figure 4). Thus, PPS apparently abrogated the species barrier-associated restriction of mouse PrP-sen conversion by hamster 263K PrP-res that has been observed under other previously described cell-free conditions.
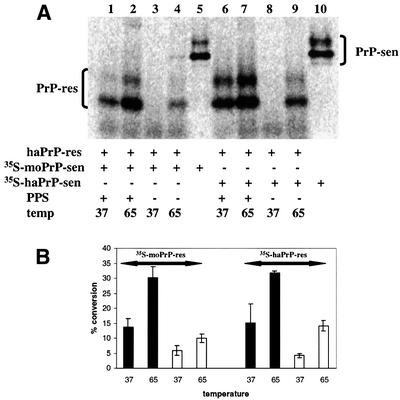
Fig. 4. Heterologous and homologous cell-free conversion with hamster PrP-res template/seed. (A) Comparison of PPS-stimulated and control [35S]PrP-res formation with mouse and hamster GPI(–) [35S]PrP-sen at 37 and 65°C. Twenty percent of mouse and hamster input [35S]PrP-sen is shown in lanes 5 and 10, respectively. (B) Percentage conversion of the PPS-stimulated and control cell-free conversion at 37 and 65°C. The solid black bars indicate PPS-stimulated conversion reactions and white bars indicate control conversions. Error bars show the standard deviation (n = 3).
The PrP GPI anchor interfered with PPS-stimulated cell-free conversion under conditions free of chaotropic ions and detergents
The form of [35S]PrP-sen used in the conversion experiments discussed thus far was expressed without a GPI anchor. Since cellular PrP normally contains a GPI anchor (Stahl et al., 1987), we tested the effect of PPS on the conversion of GPI(+) mouse PrP-sen with mouse and hamster PrP-res. We found that PPS (Figure 5) and HS (data not shown) either stimulated conversion poorly (2-fold) with mouse PrP-res (Figure 5A, compare lanes 10 and 11) or not at all with hamster PrP-res (compare lanes 4 and 5). In addition to the lack of a GPI anchor, the GPI(–) PrP-sen is largely unglycosylated compared with the predominantly glycosylated GPI(+) PrP-sen (Kocisko et al., 1994), as can be seen by comparing Figure 4A (lane 5) with Figure 5A (lane 6). Thus, the poor stimulation of GPI(+) PrP-sen conversion by PPS could be due to the presence of either N-linked glycans or an intact GPI anchor (Rudd et al., 1999). To address the role of the intact GPI anchor in this effect more specifically, we treated mouse wild-type, glycosylated [35S]PrP-sen-expressing cells with phosphatidylinositol phospholipase C (PIPLC), an enzyme that cleaves between the terminal phosphate and the hydrophobic diacylglycerol of the GPI anchor. The [35S]PrP-sen obtained by this procedure retained the phosphoinositol-glycan moiety, but did not have the membrane-embedded diacylglycerol component (Medof et al., 1996). PPS stimulated the conversion of this diacylglycerol-deficient mouse [35S]PrP-sen 14-fold with mouse PrP-res (Figure 5A, compare lanes 7 and 8) and 6-fold with hamster PrP-res (Figure 5A, compare lanes 1 and 2). These observations demonstrated that the presence of the diacylglycerol component of the GPI moiety inhibits full PPS stimulation under these near-physiological conditions.
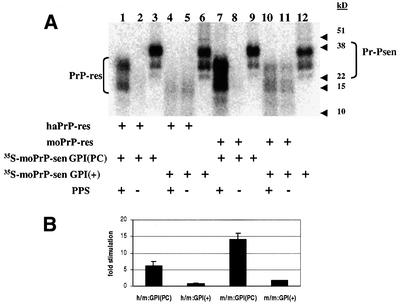
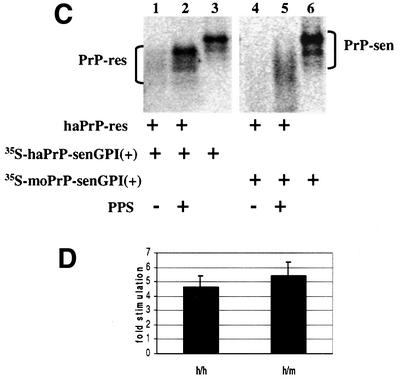
Fig. 5. Cell-free conversion of GPI(+) [35S]PrP-sen. (A) Comparison of wild-type mouse GPI(+) [35S]PrP-sen- and PIPLC-released wild-type mouse GPI(PC) [35S]PrP-sen in chaotropic ion- and detergent-free cell-free conversion. [35S]PrP-sen representing 20% of the input radioactivity is shown in lanes 3, 6, 9 and 12. (The diffused radioactive bands in lanes 4 and 5 were not identified positively as newly made [35S]PrP-res. Whether they were or not did not change the interpretation that there was little or no PPS-stimulated conversion of GPI(+) [35S]PrP-sen.) (B) Fold stimulation of GPI(+) and GPI(PC) [35S]PrP-sen by PPS. h/m:GPI(PC) = hamster PrP-res template/seed and PIPLC-released mouse [35S]PrP-sen cell-free conversion; h/m:GPI(+) = hamster PrP-res template/seed and wild-type mouse [35S]PrP-sen cell-free conversion; m/m:GPI(PC) = mouse 87V PrP-res template/seed and PIPLC-released mouse [35S]PrP-sen cell-free conversion; m/m:GPI(+) = mouse 87V PrP-res template/seed and wild-type mouse [35S]PrP-sen cell-free conversion. Error bars show the standard deviation (n = 3). (C) PPS stimulation of GPI(+) [35S]PrP-sen conversions in guanidine HCl + N-lauroyl sarkosine-based cell-free conversion. Lane 3 shows 10% of the input wild-type hamster [35S]PrP-sen of cell-free conversion shown in lanes 1 and 2. Lane 6 shows 10% of the input wild-type mouse [35S]PrP-sen of cell-free conversion shown in lanes 4 and 5. (D) Fold stimulation of GPI(+) [35S]PrP-sen cell-free conversion by PPS in guanidine HCl + N-lauroyl sarkosine-based cell-free conversion. h/h = hamster PrP-res template/seed and wild-type [35S]PrP-sen cell-free conversion; h/m = hamster PrP-res template/seed and wild-type mouse [35S]PrP-sen cell-free conversion. Error bars show the standard deviation (n = 3).
It was possible that conversion of mouse wild-type [35S]PrP-sen was blocked because the intact GPI anchor made hydrophobic interactions that limited PrP-sen interactions with PrP-res. Accordingly, we tested conversion of mouse GPI(+) [35S]PrP-sen with a protocol that included guanidine HCl and N-lauroyl sarkosine (Kocisko et al., 1994) to reduce such hydrophobic interactions. Under these conditions, PPS stimulated conversions with both mouse and hamster PrP-res (Figure 5C), supporting the hypothesis that the hydrophobic interactions of the intact GPI moiety interfered with conversion under conditions free of chaotropic ions and detergents.
PPS increased both the rate of formation and yield of [35S]PrP-res
To determine whether PPS increased the rate and/or yield of the conversion reaction, we compared the kinetics of PPS-stimulated versus non-stimulated cell-free conversion. A significant increase in the initial rate of [35S]PrP-sen conversion was seen with PPS stimulation (Figure 6). Near maximal conversion to [35S]PrP-res was reached by 3 h with PPS. In contrast, the reaction lacking PPS approached maximum only after 24–48 h and the yield of [35S]PrP-res was 3- to 4-fold less than the PPS-stimulated level. One reason for the lower maximum yield in the absence of PPS might have been the accumulation of conversion intermediates or off-pathway products that otherwise would be rescued by PPS. To test whether PPS could stimulate conversion after non-stimulated conversion had plateaued, we added PPS only after 48 h of incubation and continued the incubation for an additional 24 h (Figure 6D and E). The delayed addition of PPS stimulated [35S]PrP-res formation, but only to a level that was 63% of that achieved when PPS was added at the beginning of the incubation. Thus, PPS showed at least some ability to rescue presumably stalled conversion intermediates or off-pathway products of the conversion reaction.
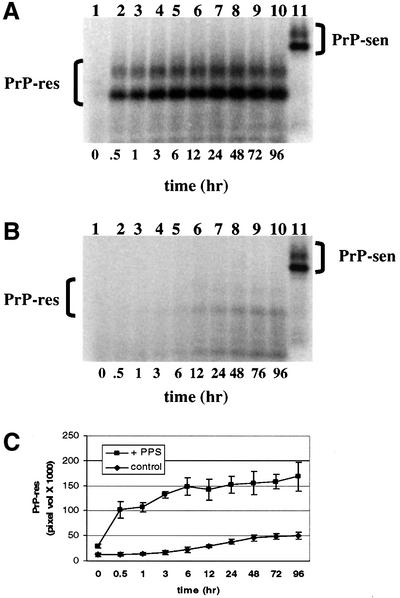
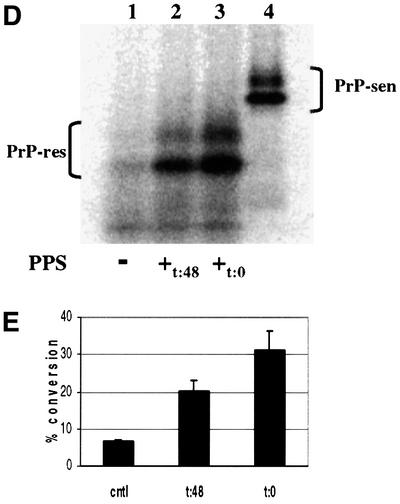
Fig. 6. Kinetics of hamster GPI(–) PrP cell-free conversion. The rate of [35S]PrP-res formation was determined for both PPS-stimulated and non-stimulated control cell-free conversion reactions over 96 h. [35S]PrP-res accumulation was determined for the time intervals indicated. [35S]PrP-sen representing 20% of the input radioactivity is shown in lane 11. (A) Kinetics of PPS-stimulated PrP cell-free conversion. (B) Kinetics of control cell-free conversion (without PPS). (C) Comparison of PPS-stimulated and control conversion reaction kinetics. Error bars show the standard deviation (n = 3). (D) Effect of adding PPS after non-stimulated cell-free conversion. Lane 1, 72 h incubation without PPS (–); lane 2, 48 h non-stimulated incubation followed by 24 h incubation with PPS (+t:48); lane 3, 72 h incubation with PPS (+t:0); lane 4, [35S]PrP-sen representing 20% of the input radioactivity shown in lanes 1, 2 and 3. (E) Percentage conversion of adding PPS after non-stimulated cell-free conversion. cntl = no added PPS; t:48 = PPS added after 48 h of non-stimulated cell-free conversion; t:0 = PPS added initially. Error bars indicate the standard deviation (n = 3).
PPS altered the circular dichroism spectrum of PrP-sen
One possible mechanism by which PPS stimulated cell-free conversion is by binding and altering the PrP-sen structure such that the energy barrier of the PrP-sen to PrP-res transition is reduced. Several studies have already demonstrated that PPS binds to PrP-sen (Caughey and Raymond, 1993; Caughey et al., 1994; Shyng et al., 1995; Brimacombe et al., 1999). To determine whether protein structure was altered by PPS, we performed circular dichroism (CD) spectroscopy on purified hamster GPI(–) PrP-sen (Figure 7A). To measure the CD spectrum below 200 nm, we used a buffer comprised of 10 mM sodium phosphate pH 6.5, 400 mM potassium fluoride. This buffer was transparent below 200 nm but supported PPS-stimulated conversion as well as the standard citrate/KCl conversion buffer (data not shown). The PrP-sen CD spectrum was comparable with previously published CD spectra (Swietnicki et al., 1997; Safar et al., 1998) (Figure 7B) with a prominent positive CD maximum at 192 nm and two negative CD maxima centered at 212 and 222 nm, which are diagnostic of proteins with high α-helix content (Venyaminov and Yang, 1996). Upon PPS addition, there was an immediate reduction of CD signal at 192, 212 and 222 nm. The reduction in CD signal continued slowly over 44 h. In contrast, the CD spectrum of PrP-sen incubated alone or with CS did not change over 48 h (Figure 7C). PPS or CS alone exhibited very little CD at the applicable concentration and wavelengths examined (data not shown). Nevertheless, they were subtracted along with the contribution of the buffer from the PrP-sen spectrum when appropriate. The PPS-induced change in the CD spectrum was not due to loss of protein through precipitation. At the concentrations of PrP-sen and PPS used, we found no detectable aggregation by centrifugation or change in optical density at 280 nm. In addition, there was no increase in absorbance between 400 and 300 nm, which would be expected if there were increased turbidity due to aggregation (data not shown). The change in CD signal of PrP-sen upon co-incubation with PPS was consistent with a decrease in α-helix content (Venyaminov and Yang, 1996), which supported the hypothesis that PPS stimulated conversion by altering PrP-sen structure, presumably in a way that favors [35S]PrP-res formation. To determine whether PPS affected other proteins in the same way that it affects PrP-sen, we determined the CD spectrum of chicken egg white lysozyme, a protein with high α-helical content similar to PrP-sen. We found no shift of the lysozyme CD spectrum by PPS under identical conditions (data not shown), indicating that the PPS effect on PrP-sen is not common to all proteins.
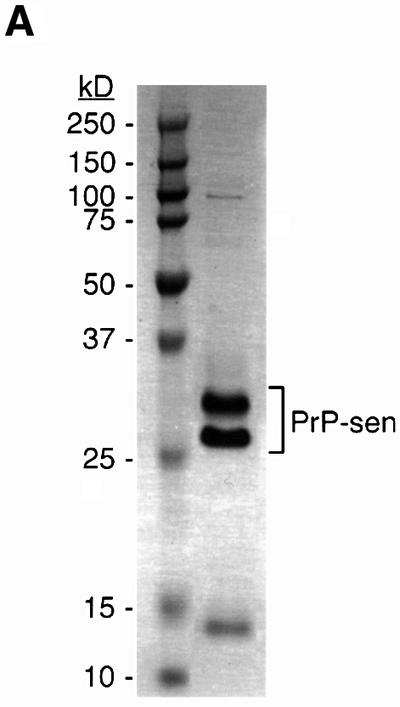
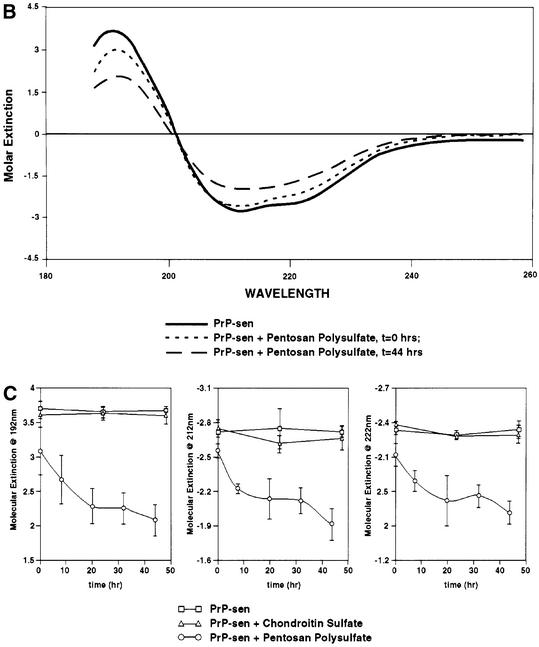
Fig. 7. Effect of PPS on the hamster PrP-sen CD spectrum. (A) Coomassie Blue-stained SDS–PAGE of purified hamster GPI(–) PrP-sen. (B) PPS-induced changes to the PrP-sen CD spectrum. Each spectrum shown is the average of three separately prepared samples. (C) PPS-induced change of molar extinction of PrP-sen at 192, 212 and 222 nm over 48 h. Error bars show the standard deviation (n = 3).
Discussion
Given that HS, PPS and certain other sulfated glycans are known to prevent PrP-res formation in scrapie-infected rodents (Beringue et al., 2000) and tissue culture cells (Caughey and Raymond, 1993), it seems paradoxical that these compounds would stimulate PrP-res formation in the cell-free conversion reaction. One possible explanation for these observations might be that sulfated glycans have a physiological effect on cells that overrides their direct stimulatory effects on PrP-res formation. For instance, when PPS is added to cells in tissue culture, the cell surface PrP-sen is shunted to endocytic compartments (Shyng et al., 1995). These results led to suggestions that either the sequestered PrP-sen is no longer in physical contact with PrP-res or the endocytic compartments are not conducive to PrP conversion. Alternatively, free exogenous sulfated glycans may compete for, and interfere with, the interaction of PrP-sen with an endogenous glycosaminoglycan or other cofactor that is necessary for PrP conversion in infected cells (Figure 8) (Caughey and Raymond, 1993; Caughey, 1994; Caughey et al., 1994). For instance, if PrP interactions with an endogenous membrane-bound glycosaminoglycan are required for both the appropriate subcellular trafficking and the PrP-res-induced conformational change, then the presence of free sulfated glycans could block PrP-res formation within cells despite the fact that they can promote the conversion in the cell-free reaction. In the cell-free conversion reaction, intermolecular contacts are likely to be much less constrained than in a membrane-bound milieu. This might allow a complex sequence of interactions to occur that might not be possible in intact cells if interactions between PrP molecules and an endogenous cofactor are blocked by free sulfated glycans (Caughey, 1993).
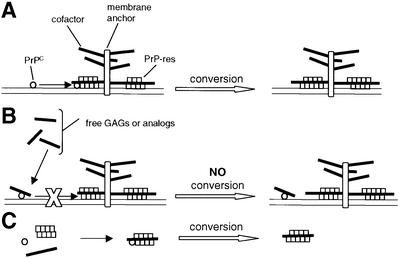
Fig. 8. Model consistent with the paradoxical effects of glycosaminoglycans (GAGs) and GAG analogs in PrP-res formation. (A) A cofactor in PrP-res formation is proposed that is either a membrane (diagrammed) or extracellular matrix-associated GAG moiety, or a different molecule whose influence can be mimicked by GAGs and GAG analogs. PrP-sen must interact with both the cofactor and PrP-res for efficient conversion. This is consistent with the association of GAGs with PrP-res deposits in vivo (Snow et al., 1989). (B) Exogenous free GAGs or GAG analogs can inhibit PrP-res formation by binding to PrP-sen at the cofactor binding site and interfering with its interaction with the cofactor and PrP-res. This is consistent with the ability of such compounds to bind PrP-sen (Gabizon et al., 1993; Caughey et al., 1994; Shyng et al., 1995; Brimacombe et al., 1999) and potently inhibit PrP-res formation in scrapie-infected cells and animals (Ehlers and Diringer, 1984; Kimberlin and Walker, 1986; Caughey and Raymond, 1993; Caughey, 1994; Caughey et al., 1994; Brimacombe et al., 1999). (C) In cell-free reactions between PrP-sen and purified PrP-res that is stripped of cofactor(s) during purification, all three elements can freely associate to allow GAGs or GAG analogs to substitute for the endogenous GAG in stimulating conversion.
Stimulation of cell-free conversion by elevated temperature
We have shown in this study that elevated temperature promoted cell-free conversion of PrP-sen. The cell-free conversion reaction is comprised of at least two steps: the rapid initial binding of [35S]PrP-sen to PrP-res and the rate-limiting conversion of the bound [35S-PrP]sen to [35S]PrP-res (Horiuchi et al., 1999). The fact that the conversion step is slow and rate limiting suggests that there is a significant energy barrier to the conformational rearrangement. Increasing the incubation temperature may increase the probability of overcoming this energy barrier to conversion. By combining elevated temperature with PPS, we found an additive effect that demonstrated that the mechanism(s) of stimulation by elevated temperature and PPS are complementary.
The stimulation of cell-free conversion by elevated incubation temperatures above 37°C was not seen previously in conditions containing guanidine HCl and/or N-lauroyl sarkosine. However, in preliminary experiments, a short pulse of 70°C after 37°C incubation and dilution of guanidine HCl were found to enhance conversion (L.Herrmann, S.Priola and B.Caughey, unpublished results). By eliminating these destabilizing reagents during the conversion reaction, we were able to raise the incubation temperature and demonstrate an extended temperature-dependent increase in [35S]PrP-res formation. When denaturants are present, higher temperatures may cause irreversible denaturation of the PrP-res, which then prevents conversion (Caughey et al., 1997).
PPS-stimulated heterologous PrP cell-free conversion
The hamster and mouse scrapie models are commonly used for studying species barriers in TSE transmissions (Kimberlin and Walker, 1978; Kimberlin et al., 1987, 1989). Specific amino acid sequence differences between the prion proteins of hamsters and mice have been shown to be critical in maintaining the species barrier to hamster scrapie in mice (Prusiner et al., 1990; Priola et al., 1994; Priola and Chesebro, 1995). Our results demonstrate that the barrier to conversion reactions between hamster and mouse PrP isoforms is reduced or eliminated with PPS in cell-free conversion. These results raise the possibility that endogenous sulfated glycosaminoglycans may also aid in overcoming barriers to interspecies PrP conversion when it occurs in vivo (e.g. Race and Chesebro, 1998; Hill et al., 2000). PrP-res formation in vivo is likely to be influenced by a complex interplay of positive and negative effects of sulfated glycosaminoglycans and/or other functionally related molecules. It is clear, however, that certain exogenous sulfated glycans block PrP-res formation in vivo and in vitro, and these negative effects may supersede any positive effects of such compounds on heterologous PrP conversion.
The GPI anchor and PPS-stimulated cell-free conversion
The fact that PPS stimulated GPI(+) PrP-sen conversions only in the presence of guanidine HCl and N-lauroyl sarkosine suggests that the lipophilic GPI anchor interfered with conversion through hydrophobic interactions. GPI-linked proteins are known to form pseudomicelles in minimal detergent solutions (Medof et al., 1996). Such micelles of PrP-sen might block interactions with PrP-res that occur via surfaces near the C-terminus where the GPI anchor is attached (Horiuchi et al., 1999). Since the GPI anchor of PrP-sen is usually sequestered in a membrane, its presence may not limit accessibility to PrP-res (which may also be membrane bound) to the same extent. Conversion studies with membrane-bound GPI(+) PrP-sen are in progress to test this hypothesis.
Alteration of PrP-sen secondary structure by PPS
The CD spectra showed that PPS induced a loss of α-helical content in PrP-sen. In contrast, there was no detectable change in the PrP-sen CD spectrum when it was incubated alone or co-incubated with CS. The latter result was consistent with the observation that CS does not bind PrP-sen as well as PPS or HS (Caughey et al., 1994; Shyng et al., 1995) and is a poor stimulator of cell-free conversion (this study). With the exception of a small β-sheet comprised of two strands of four amino acid residues, the stable secondary structure of native PrP-sen is predominantly α-helix (Riek et al., 1996; Donne et al., 1997; James et al., 1997; Riek et al., 1997). It is reasonable to speculate that a reduction in the α-helical content of PrP-sen by PPS reflects a relaxation of the constraints between the PrP conformers and thus a lowering of the activation energy for the PrP-sen to PrP-res transition.
The potential relaxation of the PrP-sen structure by PPS could also apply to PPS stimulation of heterologous PrP-sen conversion. As discussed above, there is probably a significant energy barrier between PrP-sen and PrP-res conformers. Critical amino acid sequence mismatches could reduce the ability of hamster PrP-res to make the contacts necessary to catalyze the conversion of mouse PrP-sen. A relaxation of the PrP-sen conformation by PPS and/or elevated temperature may potentiate such intermolecular contacts and allow PrP-res to induce the heterologous conversion reaction.
Potential mechanisms of sulfated glycan stimulation of cell-free conversion
There are several conceivable mechanisms by which PPS and HS enhance PrP-res formation in cell-free conversion. One possibility is that [35S]PrP-sen availability is increased. For instance, the soluble [35S]PrP-sen concentration can be reduced by non-specific binding to the plastic tubes used for conversion reactions. However, we found that PPS did not affect the recovery of [35S]PrP-sen from the reaction solution (data not shown). A second possibility is that PPS and HS increase the effective [35S]PrP-sen concentration by ‘molecular crowding’ via sequestration of water, a well-known property of glycosaminoglycans. However, this possibility is unlikely because the effective concentrations of HS and PPS were much too low to sequester a significant proportion of the water. Furthermore, although KS is an authentic glycosaminoglycan with presumably a similar ability to bind water, it did not stimulate conversion even at 105 times the effective HS and PPS concentrations. A third possibility might be that PPS and HS provide a stabilizing scaffold for the newly formed [35S]PrP-res (Figure 8) (Caughey and Raymond, 1993; Caughey, 1994), similar to a role proposed for the high molecular weight glucose polymer component of brain-derived PrP-res (Appel et al., 1999). A fourth possibility is that PPS acts catalytically on [35S]PrP-sen, as proposed above. However, PrP-sen conversion appears to be more complex than a simple bimolecular reaction with a single equilibrium. If that were the case, the level of [35S]PrP-res formation in the absence of PPS would eventually have reached the level of the PPS-stimulated conversion. However, for reasons that are still not clear, cell-free conversion stalls even when [35S]PrP-sen is not limiting (see legend to Figure 2B). One explanation could be that trapped intermediates or off-pathway conformers of [35S-PrP]sen block the seeding site and interfere with further conversion (Horiuchi et al., 2000). By interacting with these trapped or off-pathway conformers and relaxing their structure, PPS might remove or ‘repair’ these blockages. Thus, PPS may perform an ‘editing’ function on partially converted [35S]PrP molecules. This hypothesis is consistent with the observation that PPS stimulates conversion even when added after reactions lacking PPS had stalled (Figure 6D and E). Since this stimulation was less than that observed when PPS was present from the beginning of the reaction, there appear to be some off-pathway events that PPS cannot reverse. This is also consistent with our finding that PPS itself does not promote continuous cell-free conversion (data not shown). We hypothesize that PPS and HS facilitate the refolding of native [35S]PrP-sen as well as some kinetically trapped or off-pathway [35S]PrP conformers. Thus, in the conversion reaction, these sulfated glycans may interact with PrP-sen in a manner similar to that proposed for chaperone proteins such as GroEL or HSP104 in cell-free conversion (DebBurman et al., 1997).
Materials and methods
PrP-res and PrP-sen
Brain-derived PrP-res was prepared from hamsters infected with scrapie strain 263K and from mice infected with scrapie strain 87V, as described previously (Kocisko et al., 1994; Caughey et al., 1999). Six different radiolabeled PrP-sen proteins were used in this study. The [35S]methionine-labeled hamster and mouse 3F4 (Kascsak et al., 1987) epitope-tagged PrP-sen with and without the GPI anchor were expressed in mixed cultures of Psi-2 and PA317 mouse retroviral packaging cells as described (Robertson et al., 1992; Priola et al., 1994). Also used in this study was radiolabeled mouse wild-type PrP-sen immunoprecipitated with rabbit R30 antibody from lysates of mouse neuroblastoma cells (clone 5E4E), which overexpressed mouse PrP after being transduced with a murine retrovirus-expressing mouse PrP (S7 allele) in the pSFF vector as described (Robertson et al., 1992; Priola et al., 1994). To obtain the mouse wild-type [35S]PrP-sen without an intact GPI moiety, the PrP-sen was enzymatically released from the cells with PIPLC and purified from the medium by immunoprecipitation with antibody R30 as described (Horiuchi et al., 1999).
In this study, we refer to cell-free conversion reactions where the [35S]PrP-sen was from the same species as the input PrP-res as ‘homologous’. Reactions between [35S]PrP-sen and PrP-res of different species are referred to as ‘heterologous’. However, it should be noted that the wild-type mouse and 3F4 epitope-tagged mouse PrP-sen molecules differ at two amino acid residues, Leu108 and Val111, both of which are replaced with methionines. Furthermore, the wild-type mouse S7 allele [35S]PrP-sen differs at two residues from the P7 allotype of the mouse 87V strain PrP-res. The mouse S7 allele has a leucine at 108 and a threonine at 189, which are phenylalanine and valine, respectively, in the P7 allele.
Pentosan polysulfate and glycosaminoglycans
PPS (catalog #P8275), HS (catalog #H5393), chondroitin sulfate A (CS; catalog #C8529) and KS (catalog #2876) were purchased from Sigma. These reagents were initially dissolved in water at 10–20 mg/ml and stored at –20°C. The concentrated solutions were diluted to a final concentration in conversion buffer as described for the experiment.
Cell-free conversion
Two cell-free conversion protocols were used in this study. The guanidine HCl/N-lauroyl sarkosine-based conversion was performed as described (Caughey et al., 1999). The second protocol was a modification of that of Horiuchi et al. (1999) and was performed as follows. The stock PrP-res was diluted to 50 ng/µl in water. [35S]PrP-sen was diluted to 10 000–30 000 c.p.m./µl with 0.1 M acetic acid. For each conversion reaction, 2 µl of the 50 ng/µl PrP-res were mixed with 2 µl of [35S]PrP-sen in 6 µl of 100 mM sodium citrate pH 6.0, 400 mM KCl, and, except where noted, pre-incubated at 0°C for 3–12 h. For the standard conversion reaction, 10 µl of the pre-incubation mixture were transferred to a fresh tube containing either 10 µl of PPS (200 µg/ml) or water (PPS diluent) and incubated at 37°C for 24 h. Samples were either frozen at –20°C for subsequent PK (Calbiochem) digestion or digested with PK immediately as outlined below. Experiments testing the effect of HS, CS and KS on cell-free conversion were performed identically to those outlined for PPS. For kinetic experiments, samples were prepared as above, incubated at 37°C and at the specified time points rapidly frozen at –20°C to stop further conversion. For testing the effect of incubation temperature in the experiment shown in Figure 3, the samples were prepared as above, pre-incubated at 0°C for 30 min, mixed with PPS and incubated at the temperatures indicated in the experiment for 6 h. For testing the effect of incubation temperature on homologous and heterologous conversion shown in Figure 4, the samples were prepared as above, pre-incubated at 0°C, mixed with PPS and incubated at either 37 or 65°C for 24 h. After incubation under the conditions required for the experiment, the samples were digested with PK as follows: 32 µl of 23.4 ng/µl PK in 150 mM Tris pH 8.0, 450 mM NaCl were added to each conversion reaction and incubated at 37°C for 60 min. The PK was quenched by addition of 4 µl of 5 mg/ml thyroglobulin (Sigma), 4 µl of 0.1 M Pefabloc (Boehringer Mannheim) and 200 µl of cold methanol per reaction. The samples were chilled at –20°C to precipitate the proteins. The samples were centrifuged and the pellets resuspended in SDS–PAGE sample buffer and heated at 100°C for 5 min. The samples were resolved on pre-cast 16% acrylamide–Tris–glycine gels (Novex). The SDS–PAGE gels were dried and exposed to Storage Phosphor Screens (Molecular Dynamics) for visualization of the radioactive proteins. IMAGEQUANT (Molecular Dynamics) software was used for digital quantitation of the radioactive bands. Determination of the amount of newly converted [35S]PrP-res was based on the intensity of the [35S]PrP-res bands at ∼21 and ∼25 kDa, and the fold stimulation was calculated as the ratio of the [integrated intensity of [35S]PrP-res (stimulated)/integrated intensity of [35S]PrP-res (control)]. For determination of the efficiency of the conversion reaction, samples representing 10% of the input radiolabeled PrP-sen were removed from the post-incubation conversion mixtures or samples representing 20% of the input [35S]PrP-sen from the pre-incubation mixture were mixed with 20 µg of thyroglobulin and precipitated immediately with cold methanol. These samples were resolved by SDS–PAGE along with the conversion samples. Calculations of the percentage conversion were performed as follows: [integrated image intensity of PrP-res bands/integrated image intensity of the input [35S]PrP-sen] × 10 or 20 depending on the sampling percentage used in the experiment. There are no methionines in [35S]PrP-res within the region trimmed by PK, so no additional correction factor was necessary. For the kinetics experiment shown in Figure 6, the radioactivity of the [35S]PrP-res bands was compared by their integrated image intensity and expressed as a pixel volume.
Circular dichroism
The PrP-sen protein used in CD experiments was recombinant hamster (23–231) PrP that had a stop codon inserted after amino acid 231 so that it was expressed without the GPI anchor (Kocisko et al., 1994). The protein was isolated from mixed cultures of Psi2 and PA317 mouse retroviral packaging cells (Robertson et al., 1992; Priola et al., 1994). Because the PrP protein lacked a GPI anchor, the expressed protein was secreted into the medium, from where it was purified. The expressed PrP protein bound to nickel-charged NTA–agarose beads (Qiagen), which were added to the medium (1 ml of a 50% slurry/200 ml medium) and agitated at room temperature for 1–2 h. The beads were collected by centrifugation and washed with 100 mM NaPO4 pH 8.0, 150 mM NaCl. The PrP protein was eluted with 100 mM NaPO4 pH 8.0, 150 mM NaCl, 50 mM l-histidine. The pH of the eluant was adjusted to 6.5 and it was applied to an SP Sepharose (Pharmacia) column equilibrated with 100 mM NaPO4 pH 6.5, 150 mM NaCl. The column was washed with 10 column volumes of equilibration buffer, five column volumes of 100 mM NaPO4 pH 6.5, 300 mM NaCl. The PrP protein was eluted with 100 mM NaPO4 pH 6.5, 1 M NaCl. The column fractions were assayed by western blotting with 3F4 monoclonal antibody (Kascsak et al., 1987) and the peak PrP fractions were diluted to <300 mM NaCl and reapplied to the SP Sepharose column, washed of remaining NaCl and eluted with 20 mM NaPO4 pH 6.5, 800 mM KF. The buffer was adjusted to 10 mM NaPO4 pH 6.5, 400 mM KF for all CD determinations.
CD spectra were recorded with an OLIS 16 CD spectropolarimeter using a 1 mm cylindrical quartz cell (Starna) at 37°C. For each spectrum, a minimum of nine scans were collected from 260 to 188 nm with a step resolution of 1 nm under constant nitrogen gas purge. All spectra were verified by three separately prepared samples. Baseline spectra of the buffer, buffer + PPS and buffer + CS were collected and subtracted from the experimental spectra where appropriate. PrP-sen spectra were determined at 70 µg/ml protein. When PPS or CS were used, they were added to a final concentration of 60 µg/ml. The spectropolarimeter was calibrated with (1S)-(+)-camphorsulfonic acid.
Acknowledgments
Acknowledgements
The authors wish to thank Gregory Raymond for providing brain-derived PrP-res, Suzette Priola and Kim Hasenkrug for critical reading of the manuscript prior to publication, and Gary Hettrick and Anita Mora for graphics in Figure 7.
REFERENCES
- Appel T.R., Dumpitak,C., Matthiesen,U. and Riesner,D. (1999) Prion rods contain an inert polysaccharide scaffold. Biol. Chem., 380, 1295–1306. [DOI] [PubMed] [Google Scholar]
- Beringue V., Adjou,K.T., Lamoury,F., Maignien,T., Deslys,J.P., Race,R. and Dormont,D. (2000) Opposite effects of dextran sulfate 500, the polyene antibiotic MS-8209 and Congo red on accumulation of the protease-resistant isoform of PrP in the spleens of mice inoculated intraperitoneally with the scrapie agent. J. Virol., 74, 5432–5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessen R.A., Kocisko,D.A., Raymond,G.J., Nandan,S., Lansbury,P.T.,Jr and Caughey,B. (1995) Nongenetic propagation of strain-specific phenotypes of scrapie prion protein. Nature, 375, 698–700. [DOI] [PubMed] [Google Scholar]
- Bossers A., Belt,P.B.G.M., Raymond,G.J., Caughey,B., de Vries,R. and Smits,M.A. (1997) Scrapie susceptibility-linked polymorphisms modulate the in vitro conversion of sheep prion protein to protease-resistant forms. Proc. Natl Acad. Sci. USA, 94, 4931–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brimacombe D.B., Bennett,A.D., Wusteman,F.S., Gill,A.C., Dann,J.C. and Bostock,C.J. (1999) Characterization and polyanion-binding properties of purified recombinant prion protein. Biochem. J., 342, 605–613. [PMC free article] [PubMed] [Google Scholar]
- Caughey B. (1993) Scrapie associated PrP accumulation and its prevention: insights from cell culture. Br. Med. Bull., 49, 860–872. [DOI] [PubMed] [Google Scholar]
- Caughey B. (1994) Scrapie-associated PrP accumulation and agent replication: effects of sulphated glycosaminoglycan analogues. Philos. Trans. R. Soc. Lond. Ser. B, 343, 399–404. [DOI] [PubMed] [Google Scholar]
- Caughey B. and Raymond,G.J. (1991) The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J. Biol. Chem., 266, 18217–18223. [PubMed] [Google Scholar]
- Caughey B. and Raymond,G.J. (1993) Sulfated polyanion inhibition of scrapie-associated PrP accumulation in cultured cells. J. Virol., 67, 643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B., Raymond,G.J., Ernst,D. and Race,R.E. (1991a) N-terminal truncation of the scrapie-associated form of PrP by lysosomal protease(s): implications regarding the site of conversion of PrP to the protease-resistant state. J. Virol., 65, 6597–6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B.W., Dong,A., Bhat,K.S., Ernst,D., Hayes,S.F. and Caughey,W.S. (1991b) Secondary structure analysis of the scrapie-associated protein PrP 27–30 in water by infrared spectroscopy. Biochemistry, 30, 7672–7680. [DOI] [PubMed] [Google Scholar]
- Caughey B., Brown,K., Raymond,G.J., Katzenstien,G.E. and Thresher,W. (1994) Binding of the protease-sensitive form of PrP (prion protein) to sulfated glycosaminoglycan and Congo red. J. Virol., 68, 2135–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B., Raymond,G.J., Kocisko,D.A. and Lansbury,P.T.,Jr (1997) Scrapie infectivity correlates with converting activity, protease resistance and aggregation of scrapie-associated prion protein in guanidine denaturation studies. J. Virol., 71, 4107–4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B., Horiuchi,M., Demaimay,R. and Raymond,G.J. (1999) Assays of protease-resistant prion protein and its formation. Methods Enzymol., 309, 122–133. [DOI] [PubMed] [Google Scholar]
- Chesebro B. (1998) Prion diseases—BSE and prions: uncertainties about the agent. Science, 279, 42–43. [DOI] [PubMed] [Google Scholar]
- DebBurman S.K., Raymond,G.J., Caughey,B. and Lindquist,S. (1997) Chaperone-supervised conversion of prion protein to its protease-resistant form. Proc. Natl Acad. Sci. USA, 94, 13938–13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donne D.G., Viles,J.H., Groth,D., Mehlhorn,I., James,T.L., Cohen,F.E., Prusiner,S.B., Wright,P.E. and Dyson,H.J. (1997) Structure of the recombinant full-length hamster prion protein PrP(29–231): the N terminus is highly flexible. Proc. Natl Acad. Sci. USA, 94, 13452–13457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers B. and Diringer,H. (1984) Dextran sulphate 500 delays and prevents mouse scrapie by impairment of agent replication in spleen. J. Gen. Virol., 65, 1325–1330. [DOI] [PubMed] [Google Scholar]
- Farquhar C.F. and Dickinson,A.G. (1986) Prolongation of scrapie incubation period by an injection of dextran sulphate 500 within the month before or after infection. J. Gen. Virol., 67, 463–473. [DOI] [PubMed] [Google Scholar]
- Gabizon R., Meiner,Z., Halimi,M. and Bensasson,S.A. (1993) Heparin-like molecules bind differentially to prion proteins and change their intracellular metabolic fate. J. Cell Physiol., 157, 319–325. [DOI] [PubMed] [Google Scholar]
- Griffith J.S. (1967) Self-replication and scrapie. Nature, 215, 1043–1044. [DOI] [PubMed] [Google Scholar]
- Hill A.F., Joiner,S., Linehan,J., Desbruslais,M., Lantos,P.L. and Collinge,J. (2000) Species-barrier-independent prion replication in apparently resistant species. Proc. Natl Acad. Sci. USA, 97, 10248–10253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi M., Chabry,J. and Caughey,B. (1999) Specific binding of normal prion protein to the scrapie form via a localized domain initiates its conversion to the protease-resistant state. EMBO J., 18, 3193–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi M., Priola,S.A., Chabry,J. and Caughey,B. (2000) Interactions between heterologous forms of prion protein: binding, inhibition of conversion and species barriers. Proc. Natl Acad. Sci. USA, 97, 5836–5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James T.L. et al. (1997) Solution structure of a 142-residue recombinant prion protein corresponding to the infectious fragment of the scrapie isoform. Proc. Natl Acad. Sci. USA, 94, 10086–10091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kascsak R.J., Rubenstein,R., Merz,P.A., Tonna-DeMasi,M., Fersko,R., Carp,R.I., Wisniewski,H.M. and Diringer,H. (1987) Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J. Virol., 61, 3688–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimberlin R.H. and Walker,C.A. (1978) Evidence that the transmission of one source of scrapie agent to hamsters involves separation of agent strains from a mixture. J. Gen. Virol., 39, 487–496. [DOI] [PubMed] [Google Scholar]
- Kimberlin R.H. and Walker,C.A. (1986) Suppression of scrapie infection in mice by heteropolyanion 23, dextran sulfate and some other polyanions. Antimicrob. Agents Chemother., 30, 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimberlin R.H., Cole,S. and Walker,C.A. (1987) Temporary and permanent modifications to a single strain of mouse scrapie on transmission to rats and hamsters. J. Gen. Virol., 68, 1875–1881. [DOI] [PubMed] [Google Scholar]
- Kimberlin R.H., Walker,C.A. and Fraser,H. (1989) The genomic identity of different strains of mouse scrapie is expressed in hamsters and preserved on reisolation in mice. J. Gen. Virol., 70, 2017–2025. [DOI] [PubMed] [Google Scholar]
- Kocisko D.A., Come,J.H., Priola,S.A., Chesebro,B., Raymond,G.J., Lansbury,P.T. and Caughey,B. (1994) Cell-free formation of protease-resistant prion protein. Nature, 370, 471–474. [DOI] [PubMed] [Google Scholar]
- Kocisko D.A., Priola,S.A., Raymond,G.J., Chesebro,B., Lansbury,P.T.,Jr and Caughey,B. (1995) Species specificity in the cell-free conversion of prion protein to protease-resistant forms: a model for the scrapie species barrier. Proc. Natl Acad. Sci. USA, 92, 3923–3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medof M.E., Nagarajan,S. and Tykocinski,M.L. (1996) Cell-surface engineering with GPI-anchored proteins. FASEB J., 10, 574–586. [DOI] [PubMed] [Google Scholar]
- Pan K.-M. et al. (1993) Conversion of α-helices into β-sheets features in the formation of the scrapie prion protein. Proc. Natl Acad. Sci. USA, 90, 10962–10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priola S.A. and Chesebro,B. (1995) A single hamster amino acid blocks conversion to protease-resistant PrP in scrapie-infected mouse neuroblastoma cells. J. Virol., 69, 7754–7758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priola S.A., Caughey,B., Race,R.E. and Chesebro,B. (1994) Heterologous PrP molecules interfere with accumulation of protease-resistant PrP in scrapie-infected murine neuroblastoma cells. J. Virol., 68, 4873–4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner S.B. (1982) Novel proteinaceous infectious particles cause scrapie. Science, 216, 136–144. [DOI] [PubMed] [Google Scholar]
- Prusiner S.B., Groth,D.F., Bolton,D.C., Kent,S.B. and Hood,L.E. (1984) Purification and structural studies of a major scrapie prion protein. Cell, 38, 127–134. [DOI] [PubMed] [Google Scholar]
- Prusiner S.B. et al. (1990) Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell, 63, 673–686. [DOI] [PubMed] [Google Scholar]
- Race R. and Chesebro,B. (1998) Scrapie infectivity found in resistant species. Nature, 392, 770. [DOI] [PubMed] [Google Scholar]
- Raymond G.J. et al. (1997) Molecular assessment of the transmissibilities of BSE and scrapie to humans. Nature, 388, 285–288. [DOI] [PubMed] [Google Scholar]
- Raymond G.J. et al. (2000) Evidence of a molecular barrier limiting susceptibility of humans, cattle and sheep to chronic wasting disease. EMBO J., 19, 4425–4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riek R., Hornemann,S., Wider,G., Billeter,M., Glockshuber,R. and Wuthrich,K. (1996) NMR structure of the mouse prion protein domain PrP(121–231). Nature, 382, 180–182. [DOI] [PubMed] [Google Scholar]
- Riek R., Hornemann,S., Wider,G., Glockshuber,R. and Wuthrich,K. (1997) NMR characterization of the full-length recombinant murine prion protein, mPrP(23–21). FEBS Lett., 413, 282–288. [DOI] [PubMed] [Google Scholar]
- Robertson M.N., Spangrude,G.J., Hasenkrug,K., Perry,L., Nishio,J., Wehrly,K. and Chesebro,B. (1992) Role and specificity of T-cell subsets in spontaneous recovery from Friend virus-induced leukemia in mice. J. Virol., 66, 3271–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd P.M. et al. (1999) Glycosylation differences between the normal and pathogenic prion protein isoforms. Proc. Natl Acad. Sci. USA, 96, 13044–13049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safar J., Wille,H., Itri,V., Groth,D., Serban,H., Torchia,M., Cohen,F.E. and Prusiner,S.B. (1998) Eight prion strains have PrP(Sc) molecules with different conformations. Nature Med., 4, 1157–1165. [DOI] [PubMed] [Google Scholar]
- Shyng S.L., Lehmann,S., Moulder,K.L. and Harris,D.A. (1995) Sulfated glycans stimulate endocytosis of the cellular isoform of the prion protein, PrPC, in cultured cells. J. Biol. Chem., 270, 30221–30229. [DOI] [PubMed] [Google Scholar]
- Snow A.D. and Wight,T.N. (1989) Proteoglycans in the pathogenesis of Alzheimer’s disease and other amyloidoses. Neurobiol. Aging, 10, 481–497. [DOI] [PubMed] [Google Scholar]
- Snow A.D., Kisilevsky,R., Willmer,J., Prusiner,S.B. and DeArmond,S.J. (1989) Sulfated glycosaminoglycans in amyloid plaques of prion diseases. Acta Neuropathol. (Berl.), 77, 337–342. [DOI] [PubMed] [Google Scholar]
- Snow A.D., Wight,T.N., Nochlin,D., Koike,Y., Kimata,K., DeArmond,S.J. and Prusiner,S.B. (1990) Immunolocalization of heparan sulfate proteoglycans to the prion protein amyloid plaques of Gerstmann–Straussler syndrome, Creutzfeldt–Jakob disease and scrapie. Lab. Invest., 63, 601–611. [PubMed] [Google Scholar]
- Stahl N., Borchelt,D.R., Hsiao,K. and Prusiner,S.B. (1987) Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell, 51, 229–240. [DOI] [PubMed] [Google Scholar]
- Swietnicki W., Petersen,R., Gambetti,P. and Surewicz,W.K. (1997) pH-dependent stability and conformation of the recombinant human prion protein PrP(90–231). J. Biol. Chem., 272, 27517–27520. [DOI] [PubMed] [Google Scholar]
- Telling G.C. et al. (1994) Transmission of Creutzfeldt–Jakob disease from humans to transgenic mice expressing chimeric human–mouse prion protein. Proc. Natl Acad. Sci. USA, 91, 9936–9940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telling G.C., Scott,M., Mastrianni,J., Gabizon,R., Torchia,M., Cohen,F.E., DeArmond,S.J. and Prusiner,S.B. (1995) Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell, 83, 79–90. [DOI] [PubMed] [Google Scholar]
- Venyaminov S.Y. and Yang,J.T. (1996) Determination of protein secondary structure. In Fasman,G.D. (ed.), Circular Dichroism and the Conformational Analysis of Biomolecules. Plenum Press, New York, NY, pp. 69–108.