

Abstract
Mutations in copper-zinc superoxide dismutase (SOD1) have been linked to a subset of familial amytrophic lateral sclerosis (fALS), a fatal neurodegenerative disease characterized by progressive motor neuron death. An increasing amount of evidence supports that mitochondrial dysfunction and apoptosis activation play a critical role in the fALS etiology, but little is known about the mechanisms by which SOD1 mutants cause the mitochondrial dysfunction and apoptosis. In this study, we use proteomic approaches to identify the mitochondrial proteins that are altered in the presence of a fALS-causing mutant G93A-SOD1. A comprehensive characterization of mitochondrial proteins from NSC34 cells, a motor neuron-like cell line, was achieved by two independent proteomic approaches. Four hundred seventy unique proteins were identified in the mitochondrial fraction collectively, 75 of which are newly discovered proteins that previously had only been reported at the cDNA level. Two-dimensional gel electrophoresis was subsequently used to analyze the differences between the mitochondrial proteomes of NSC34 cells expressing wild-type and G93A-SOD1. Nine and 36 protein spots displayed elevated and suppressed abundance respectively in G93A-SOD1-expressing cells. The 45 spots were identified by MS, and they include proteins involved in mitochondrial membrane transport, apoptosis, the respiratory chain, and molecular chaperones. In particular, alterations in the post-translational modifications of voltage-dependent anion channel 2 (VDAC2) were found, and its relevance to regulating mitochondrial membrane permeability and activation of apoptotic pathways is discussed. The potential role of other proteins in the mutant SOD1-mediated fALS is also discussed. This study has produced a short list of mitochondrial proteins that may hold the key to the mechanisms by which SOD1 mutants cause mitochondrial dysfunction and neuronal death. It has laid the foundation for further detailed functional studies to elucidate the role of particular mitochondrial proteins, such as VDAC2, in the pathogenesis of familial ALS.
Amyotrophic lateral sclerosis (ALS)1 is a fatal neurodegenerative disease characterized by progressive motor neuron death. Approximately 10% of ALS patients are familial cases (fALS), and mutations in the gene encoding copper-zinc superoxide dismutase (SOD1) were linked with a subset of fALS (1, 2). To date, more than 90 mutations in SOD1 are known to be responsible for ~25% of fALS (3), most of which are point mutations that are scattered throughout the primary sequence and structure of the protein. There has been intensive research focusing on the etiology of SOD1 mutant-mediated fALS (see reviews in Refs. 4–8). It has been demonstrated that SOD1-null mice did not develop the disease (9). In addition, transgenic mice expressing the ALS-associated mutants G93A-SOD1 (10, 11), G37R-SOD1 (12), and G85R-SOD1 (13, 14) as well as transgenic rats expressing G93A-SOD1 (15, 16) and H46R-SOD1 (16) developed progressive motor neuron disease despite normal or elevated SOD1 activity. Therefore, it is believed that the ALS-linked mutants of SOD1 have acquired unknown toxic properties that eventually lead to the disease. However, the nature of the toxic “gain-of-function” of SOD1 mutations remains incompletely understood. It is neither understood what the downstream target of the toxicity associated with SOD1 mutants is, nor the mechanism by which the toxicity leads to motor neuron degeneration.
There are several hypotheses regarding the consequence of the toxic “gain-of-function” of mutant SOD1 and the mechanisms by which motor neurons die. They include mitochondrial dysfunction and activation of apoptosis (17–28), SOD1 mutant-induced protein aggregation and its cytotoxicity (29–31), glutamate transporter EAAT2-mediated excitotoxicity (15, 32–34), Fas-trigger FADD/caspase-8 apoptotic cascade activation (35), and abnormal axonal transport caused by microfilament accumulation (36–40). This study focuses on the mitochondrial dysfunction/apoptosis hypothesis and searches for mitochondrial proteins that are altered by SOD1 mutants, i.e. the potential targets of the toxicity associated with SOD1 mutants.
Mitochondria play a vital role in the cell and mitochondrial dysfunction is likely to cause cell death. In ALS-transgenic mice, degeneration of mitochondria was observed prior to disease onset (10–12, 20). Other alterations in mitochondrial properties were also observed, such as vacuolation (11), decreased activity of respiratory complexes II and IV (21, 22), and loss of mitochondrial membrane potential (19). SOD1, which is traditionally regarded as a cytosolic protein, was recently shown to be partially localized in the mitochondrial intermembrane space in yeast (41), rat liver (42), mouse neuroblastoma N2a cells (43), and spinal cord neurons of G93A-SOD1 mice (44, 45). Therefore, mitochondria could be directly involved in the mutant SOD1-mediated motor neuron death.
There has been increasing evidence that mitochondria-mediated apoptosis is involved in motor neuron degeneration. In ALS patients, pro-apoptotic proteins Bax (46) and Par4 (47) were observed to increase. In G93A-SOD1 mice, cytosolic release of cytochrome c was observed, and expression levels of pro-apoptotic proteins Bad and Bax were increased while those of anti-apoptotic proteins Bcl2, Bcl-xL, and XIAP were decreased (24, 48). Caspase-1 and caspase-3 were sequentially activated in motor neurons and astrocytes in G93A-SOD1 mice as well as in G37R-SOD1 and G85R-SOD1 mice (25–27). Another study showed that the mitochondrial localization of SOD1 mutants triggered the release of cytochrome c and activation of the caspase cascade (49). A recent study showed that SOD1 interacted directly with Bcl2 and the high-molecular-weight complexes of mutant SOD1-bound Bcl2 in mitochondria from spinal cord (50). Three strategies intervening in mitochondria-mediated apoptosis were demonstrated to be effective in delaying disease onset and progression in G93A-SOD1 mice: i) intraparitoneal administration of minocycline, which inhibits mitochondrial permeability-transition-mediated cytochrome c release, was shown to delay disease onset and extend survival (28); ii) overexpression of Bcl2, which can rescue motor neurons from apoptotic death during early development (51), could delay activation of the caspases, attenuate neuron degeneration, and delay disease onset and mortality (18, 23); and iii) intraventricular administration of N-benzyloxycarbonyl-Val-Asp-fluoromethylketone (zVAD-fmk, a broad spectrum caspase inhibitor) could delay disease onset and mortality (25). It should be noted that apoptosis activation in ALS mice was observed months prior to the disease onset. This suggests that at least in mutant SOD1-mediated familial ALS, apoptosis is not a secondary phenomenon leading to the final degeneration of motor neurons. The chronic progression of apoptosis might be enhanced by an age-related increase of oxidative stress and/or aberrant pro-oxidative properties of mutant SOD1. It remains largely unclear what factor(s) initiate the mitochondrial dysfunction and apoptosis in ALS.
In this study, we have used unbiased proteomic approaches to characterize changes in the mitochondrial proteome in the presence of mutant SOD1. We have established a cell line model of fALS by overexpressing SOD1 mutants in NSC34 cells, a mouse embryonic spinal cord neuron-neuroblastoma hybrid cell line that has many features of motor neurons. We have surveyed the mitochondrial proteome of the mouse model motor neurons and report 75 mitochondrial proteins that previously were only reported at the gene level. We have also compared the mitochondrial proteomes of NSC34 cells expressing wild-type (WT) and G93A-SOD1, respectively, and have found an array of proteins that are altered in the presence of G93A-SOD1. The relevance of these altered proteins in mitochondrial dysfunction/apoptosis and motor neuron death is discussed.
EXPERIMENTAL PROCEDURES
Reagents
IPG strips and appropriate IPG buffers were purchased from Amersham Biosciences (Piscataway, NJ). Acrylamide (40%, 29:1) was purchased from Bio-Rad (Hercules, CA). Trypsin (modified, sequencing grade, lypholized) was purchased from Promega (Madison, WI). Protease inhibitor mixture, CHAPS, and DTT were purchased from Sigma (St. Louis, MO). Other commonly used reagents were purchased from Fisher Scientific (Hampton, NJ) unless otherwise indicated.
Establishment of the Cell Line Model of fALS
The cDNAs encoding human WT and G93A-SOD1 in YEp351 yeast expression vector were a generous gift from Joan S. Valentine (University of California Los Angeles, Los Angeles, CA). The SOD1-encoding sequences were amplified by PCR using the following two primers containing BamHI and EcoRI endonuclease restriction enzyme sites: 5′-GCGAATTCCATGGCGACGAAG-3′ and 5′-GCGGATCCTTATTGGGCGATCCCAATTAC-3′. The amplified human WT- and G93A-SOD1 genes were then subcloned into the mammalian expression vector pIRESneo3 (Clontech, Palo Alto, CA), and the sequence fidelity was verified by DNA sequencing. The pIRESneo3 vector uses the cytomegalovirus promoter to control the transcription of a single mRNA containing the human SOD1-encoding sequence, the internal ribosome entry site (IRES) of the encephalomyocarditis virus and the neomycin selection marker in tandem. The advantage of this vector is that SOD1 and neomycin will be translated from one mRNA, thus nearly 100% of cells that survived neomycin selection will express SOD1. In addition, different concentrations of neomycin can select cells with different SOD1 expression levels.
NSC34, a mouse embryonic spinal cord-neuroblastoma cell line with a motor neuron phenotype (52, 53), was used as an in vitro model system to carry out this proteomic study. Cells were cultured at 37 °C under 5% CO2-95% air in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine albumin, 100 μg/ml streptomycin sulfate, and 100 U/ml penicillin (22, 52, 53). Eighty percent confluent NSC34 cells in a six-well plate were transfected with the pIRES-WT-SOD1 and pIRES-G93A-SOD1 plasmids, respectively, using the Lipofectamine protocol (Invitrogen, Carlsbad, CA). Two days after transfection, cells were transferred into a 10-cm cell culture disk and treated with 200 μg/ml neomycin for 4 wk. Medium containing 200 μg/ml neomycin was changed every 4 days during the selection period. Ten to 20 clones that survived the neomycin selection were pooled together and named NSC34WT-SOD1 and NSC34G93A-SOD1, respectively. The expression levels of human SOD1 were examined by Western blotting with SOD1 polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA).
Purification of Mitochondria from NSC34 Cells
Mitochondria purification was performed as previously described (41, 43) with slight modifications. Briefly, NSC34WT-SOD1 and NSC34G93A-SOD1 cells from four confluent 10-cm plates (~2 × 106 cells/plate) were collected and washed with ice-cold PBS. The cell pellet was resuspended in 1.8 ml of cell homogenizing medium (CHM; 10 mm Tris-Cl, 150 mm MgCl2, 10 mm KCl, 1 mm DTT, and 1% protease inhibitor mixture, pH 6.7, and homogenized with 20 strokes of the dauncer followed by adding 0.6 ml of CHM with 1 m sucrose. The homogenate was centrifuged at 1,000 × g for 5 min to remove the nucleus, plasma membrane, and unbroken cells, and this step was repeated twice. The crude mitochondrial fraction was collected from the supernatant by centrifugation at 5,000 × g for 10 min. The pellet was washed once with 1 ml of sucrose/Mg buffer (10 mm Tris-Cl, 0.15 m MgCl2, 0.25 M sucrose, 1 mm DTT, and 1% protease inhibitor mixture, pH 6.7) and resuspended with 300 μl of mannitol buffer A (5 mm HEPES, 0.25 m mannitol, 0.5 mm EGTA, 0.1% BSA, 1 mm DTT, and 1% protease inhibitor mixture, pH 7.4). Resuspended mitochondria were layered onto 3.5-ml 30% Percoll solution (30% Percoll, 25 mm HEPES, 225 mm mannitol, 1 mm EGTA, 1 mm DTT, and 1% protease inhibitor mixture, 0.1% BSA, 1 mm DTT, and 1% protease inhibitor mixture, pH 7.4) followed by centrifugation at 95,000 × g for 30 min. Approximately 400 μl of the cloudy fraction was collected and diluted with 1 ml of mannitol buffer A followed by centrifugation at 6,400 × g for 10 min. The pellet was washed three times with 1 ml of mitochondrial washing buffer (10 mm HEPES, 220 mm mannitol, 68 mm sucrose, 10 mm KCl, 1 mm EGTA, 1 mm DTT, and 1% protease inhibitor mixture, pH 7.4) and stocked at −80 °C until use.
Gel Electrophoresis of Mitochondrial Proteins
Mitochondria were subjected to both one-dimensional SDS-PAGE and two-dimensional (2D) electrophoresis. For one-dimensional gel electrophoresis, the mitochondrial pellet was resuspended and homogenized in SDS-PAGE sample buffer (~100 μg of protein in 40 μl of buffer) and centrifuged at 10,000 × g for 5 min. The supernatant was applied to 12% SDS-PAGE followed by Sypro Ruby staining.
Two-dimensional gel electrophoresis of the mitochondrial fraction isolated from NSC34WT-SOD1 and NSC34G93A-SOD1 cells was carried out as previously described (54). Briefly, the mitochondrial pellet was resuspended and homogenized in rehydration buffer (8 m urea, 2% CHAPS, 0.5% IPG buffer 3–10) (~100 to 150 μg of protein in 50 μl of buffer) and centrifuged at 10,000 × g for 5 min. The supernatant was applied to 18-cm IPG strips (pI range 3–10) through the sample loading cup located at the anode end of the strips and focused using the IPGphor protein IEF apparatus for 100,000 Vh. The strips were equilibrated and subjected to the second dimension of 12% SDS-polyacrylamide gels followed by Sypro Ruby staining. Gel images were acquired by a Storm fluorescence scanner (Amersham Pharmacia Biotech).
Analysis of the Images of 2D Electrophoresis
Two-dimensional gel images were analyzed by PDQuest software (Bio-Rad) to compare the 2D gels of the NSC34WT-SOD1 and NSC34G93A-SOD1. Spot detection and matching between six gels (three from NSC34WT-SOD1 and three from NSC34G93A-SOD1) were performed automatically, followed by manual matching. After normalization of the spot densities against the whole-gel densities, statistical analysis was performed by independent Student t test. The p values of the intensity changes of each spot among three independent experiments were calculated. The protein spots with p values less than 0.05 were considered as those displaying significant changes between NSC34WT-SOD1 and NSC34G93A-SOD1 cells.
Protein Digestion and MS Analysis
After SDS-PAGE of the mitochondrial proteins, 20 protein bands of equivalent size were sliced from the gel. After 2D gel electrophoresis of the mitochondrial proteins, 480 protein spots of greatest density were excised from the gel. All gel bands or pieces were washed and digested with trypsin as previously described (54, 55). The resulting peptides were extracted using 50 μl of 0.02% heptafluorobutylic acid (HFBA) and 50 μl of 0.02% HFBA/50% ACN and concentrated to 10 μl with a Speed Vac. The peptides from one-dimensional SDS-PAGE were subjected to LC-MS/MS analysis. Each sample was loaded to a C18 reverse-phase capillary column (75 μm × 15 cm) and eluted using a linear 1-h gradient running from 5 to 60% ACN. Peptides eluted from the column were directly sprayed into the Qstar XL Q-TOF mass spectrometer (Applied Biosystems, Foster City, CA) through a nano-electrospray source. The Qstar XL was operated in the information-dependent acquisition mode, first measuring the masses of the eluted peptides then automatically selecting the three most abundant peptide ions to perform MS/MS to obtain the sequence information. Peptides from 2D electrophoresis were desalted by Zip-tip (Millipore, Billerica, MA), spotted to MALDI plates, and analyzed by Qstar XL with the MALDI source. Both MS of all peptides and MS/MS spectra of four most abundant peptides in each sample were recorded by Qstar XL and subsequently used for protein identification.
Protein Identification and Data Analysis
The above MALDI-MS, MALDI-MS/MS, and LC-MS/MS data were all subjected to database searches for protein identification using a local MASCOT search engine (56). Multiple databases, NCBInr, Swiss-Prot, and MSDB, were searched to yield more comprehensive and complementary results. The MALDI-MS data were submitted to the MASCOT server for peptide mass fingerprint (PMF) search. The peak lists from the MALDI-MS spectra were first generated by MASCOT Wizard using the following parameters: no smoothing, signal-to-noise ratio is great than 5, and baseline is correlated by the MASCOT algorithm. The peak lists were subsequently submitted for PMF search using the following parameters: Mus musculus, maximum of one trypsin miscleavage, cysteine carbamiodomethylation, methionine oxidation, and a maximum of 100 ppm MS error tolerance. For each PMF search, a probability score is calculated by the MASCOT algorithm and is required to be higher than 62 (i.e. p < 0.05). In addition, a minimum of three matching peptides are required for positive protein identification.
The MALDI-MS/MS and LC-MS/MS data were submitted to the MASCOT server for MS/MS ion search. The peak lists from the LC-MS/MS spectra were generated by the MASCOT script embedded in the Analyst QS software using the following parameters: no smoothing, charge state determined from the MS scan, precursor ion charge states of 2+ and 3+, centroid MS/MS data, height percentage 50%, and merge distance 0.02 Da. The peak lists from the MALDI-MS/MS spectra were generated using the same parameters except using precursor ion charge states of 1+. The typical parameters used in the MASCOT MS/MS ion search are: Mus musculus, maximum of one trypsin miscleavage, cysteine carbamiodomethylation, methionine oxidation, a maximum of 100 ppm MS error tolerance, and a maximum of 0.2 Da MS/MS error tolerance. For MS/MS ion search, proteins with one peptide ion scoring higher than 45 or two peptide ions scoring higher than 30 were considered an unambiguous identification without manual inspection. All other hits were manually verified by confirming the peptide sequences from the MS/MS spectra.
RESULTS
A Cellular Model of fALS
NSC34 cells stably transfected with cDNAs of human WT- and G93A-SOD1 (designated as NSC34WT-SOD1 and NSC34G93A-SOD1, respectively) were generated as described in “Experimental Procedures.” The expression levels of SOD1 proteins were assessed by Western blotting analysis using a polyclonal antibody against both human and mouse SOD1 as shown in Fig. 1. Significant levels of human WT- and G93A-SOD1 were expressed in the stable pools. The expression levels of human SOD1 were less than the endogenous mouse SOD1, thus artifacts associated with overexpression of exogenous proteins are not a concern in these clones. Furthermore, the NSC34G93A-SOD1 cells were more susceptible to oxidative stress than the NSC34WT-SOD1 cells. Protein aggregates containing SOD1 were also observed in the NSC34G93A-SOD1 cells. A more detailed description of this cellular model of ALS will be described elsewhere (Zhang and Zhu, unpublished results).
Fig. 1. Expression levels of human SOD1 in the NSC34 cell line model of fALS.

The total cell lysate of NSC34WT-SOD1 and NSC34G93A-SOD1 cells was resolved by 12% SDS-PAGE, transferred to a nitrocellulous membrane, and blotted with an SOD1 polyclonal antibody. The endogenous mouse SOD1 and human SOD1 are indicated by arrows. Significant levels of human WT- and G93A-SOD1 are expressed in NSC34WT-SOD1 and NSC34G93A-SOD1 cells, respectively.
Protein Identification from 2D Gel Electrophoresis Followed by MALDI-MS and MALDI-MS/MS Analysis
Mitochondrial proteins were separated by 2D gel electrophoresis to ~600 protein spots that were recognized by the PDQuest software (Fig. 2). Recent proteomic studies of mitochondria isolated from several tissues/organisms have reported 400–600 proteins (57, 58), similar to the number of protein spots in this study. Approximately 480 spots were excised from a 2D gel, while about 120 spots with extremely faint densities were omitted. In-gel tryptic digestion was performed, and the extracted tryptic peptides were analyzed by MALDI-MS/MS in an automated fashion. Database searching with both PMF and MS/MS spectra was also performed using the MASCOT algorithm to search the NCBInr database. In the case that a protein was not identified using the NCBInr database, the Swiss-Prot and MSDB databases were used and positive protein identification was achieved. The protein identification was positive when the following two criteria were met: 1) a probability score calculated by MASCOT is higher than 62, i.e. the probability of the random match is less than 0.05; 2) a minimum of three matching peptides are present. In addition, we took advantage of the MALDI-MS/MS capability of the Qstar XL mass spectrometer and acquired MS/MS data from the four most abundant ions. The MALDI-MS/MS data were submitted for MS/MS ion search using the parameters and criteria described in “Experimental Procedures.” In most cases, both PMF and MS/MS ion searches yielded identical protein identification results. In a few cases in which the scores of both PMF and MS/MS database searches were marginally lower than the threshold, the MS/MS spectra were manually examined to confirm the peptide sequences. Out of the 480 spots analyzed, 240 spots were positively identified. The identification rate of all 480 protein spots was ~50%, but the identification rate of ~200 protein spots with higher than moderate intensity was more than 90%. The 170 unique proteins that were identified from the 240 2D gel spots are listed in the supplemental table.
Fig. 2. A representative 2D gel electrophoresis image of the mitochondrial fraction isolated from NSC34WT-SOD1 cells.

The gel was stained by Sypro Ruby, and the image was acquired using a Storm fluorescence scanner. PDQuest software was used to analyze the image and assign the protein spots. Two hundred forty proteins spots that have been identified are indexed as numbered, and the complete list of proteins is in the supplemental table.
With a few exceptions, the positions of the protein spots in the 2D gel electrophoresis were well correlated with the theoretical values of molecular weight and pI values of the identified proteins. In the few exceptions, the protein spots might be post-translational modification products, other adducts, or degradation products of the identified proteins. It is also noted that 38 unique proteins have multiple isoforms as each of them are represented by more than two distinct 2D gel spots. For instance, 10 different 2D gel spots (spots 184, 186, 187, 188, 189, 205, 207, 221, 229, and 230 in Fig. 2) were all sub-identified as ATP synthase mitochondrial F1 complex α subunit isoform 1. These spots possibly represent the post-translational modification products of the protein. This is also the reason that 170 unique proteins were identified from 240 2D gel spots.
Protein Identification from SDS-PAGE Followed by Nano-LC-MS/MS Analysis
More than 500 MS/MS spectra were obtained from the LC-MS/MS experiment of each gel band excised from the SDS-PAGE of mitochondrial proteins. The LC-MS/MS data were subsequently subjected to MASCOT MS/MS ion search using the parameters described in “Experimental Procedures.” Proteins with one peptide ion scoring higher than 45 or two peptide ions scoring higher than 30 were considered unambiguous identification without manual inspection. All other hits were manually verified by confirming the peptide sequences from the MS/MS spectra. A total of 387 unique proteins were identified from the 20 gel bands of mitochondrial proteins, which are listed in the supplemental table.
The two independent approaches, 2D gel followed by MALDI-MS/MS and SDS-PAGE followed by LC-MS/MS, have identified 170 and 387 unique proteins, respectively, as illustrated in Fig. 3. Of these proteins, 87 proteins were commonly identified in both approaches. Eighty-three and 300 proteins were uniquely identified in the 2D gel and SDS-PAGE approach, respectively. Collectively, 470 unique proteins were identified from the mitochondrial fraction of NSC34 cells.
Fig. 3. Summary of all proteins identified by the two independent approaches: 2D gel followed by MALDI-MS/MS and SDS-PAGE followed by LC-MS/MS.
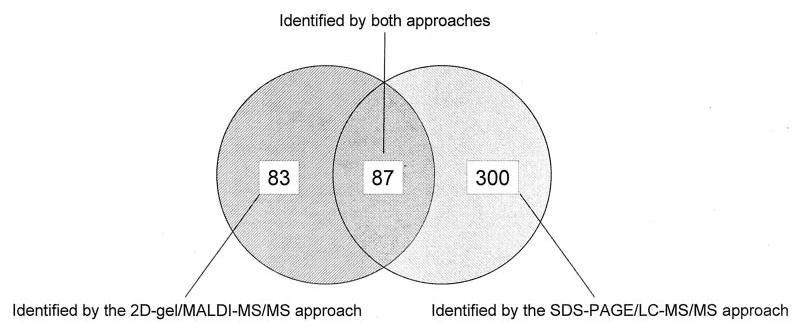
The number of proteins that have been identified by each approach is summarized here. The detailed protocols and results are described in “Experimental Procedures” and “Results.”
Two annotated protein databases, EXPASY (Swiss Institute of Bioinformatics) and Proteome BioKnowledge (Incyte), were searched to confirm the mitochondrial localization of the 470 identified proteins. Out of the 470 proteins, 171 proteins (36%) are well-characterized mitochondrial proteins, 44 proteins (9%) are shown to localize in endoplasmic reticulum (ER) and 43 proteins (9%) are cytoskeleton and related proteins. ER and cytoskeleton are commonly found to be associated with mitochondria during isolation. Sixty-nine (15%), 43 (9%), 20 (4%), 18 (4%), 6 (1%), and 1 (0.2%) proteins were shown to localize in the cytosol, membrane fraction (including plasma membrane), cytosolic ribosome, protein trafficking vesicles, nucleus, and proteasome, respectively. No subcellular localization information was available for the remaining 54 (12%) proteins. For the proteins that are reported to be localized in other subcellular compartments, it is likely that they are localized in mitochondria or associated with the mitochondrial membrane in NSC34 cells. Alternatively, they may be contaminants from the mitochondrial purification process. We have not distinguished these two possibilities.
Newly Identified Mitochondrial Proteins
Seventy-five new proteins, which were previously reported as cDNAs, have been identified in the mitochondrial fraction of NSC34 cells. All 75 proteins were submitted to the BLAST sequence similarity search against proteins from other organisms in NCBI protein database. Out of these proteins, 47 (63%) proteins display high sequence homology to the proteins of mouse or the other organisms that have been annotated in the EXPASY or Proteome BioKnowledge databases. Twenty-three (31%) proteins have sequence homology to less-characterized proteins while five (7%) proteins have no homolog to any proteins from any organisms. The 75 newly identified proteins are listed in Table I.
Table I.
Newly identified proteins in the mitochondrial fraction of NSC34 cells
| Proteins | Accession no. | Homolog of BLAST search | Sequence similarity percentage | Annotated localization of the homologs |
|---|---|---|---|---|
| Hypothetical protein | 2137411 | No homolog | N/A | Unknown |
| Hypothetical protein Xp_158679 | 20912531 | No homolog | N/A | Unknown |
| RIKEN cDNA 0610008F14 | 21536220 | Mitochondrial F1 ATP synthase δ subunit (rat) | 95 | Mitochondria |
| RIKEN cDNA 0610009D10 | 16741459 | ATP synthase subunit d (rat) | 89 | Mitochondria |
| RIKEN cDNA 0610040B21 | 13384700 | Thioredoxin superfamily member 18 kDa (human) | 93 | ER |
| RIKEN cDNA 0610041L09 | 21313618 | FLJ20420 (human) | 90 | Unknown |
| RIKEN cDNA 1110055E19 | 38083712 | Filamin C γ (human) | 96 | Cytoskeleton |
| RIKEN cDNA 1300006L01 | 21311845 | Solute carrier family 25 member 22 (human) | 96 | Mitochondria |
| RIKEN cDNA 1300007C21 | 26334035 | LOC207121 (rat) | 59 | Unknown |
| RIKEN cDNA 1700007H16 | 13386272 | Citrate synthase (mouse) | 92 | Mitochondria |
| RIKEN cDNA 1700034J06 | 30409998 | Solute carrier family 25 member 4 (mouse) | 72 | Mitochondria |
| RIKEN cDNA 1700041C02 | 12839878 | No homolog | N/A | Unknown |
| RIKEN cDNA 1810020E01 | 13384870 | DUF1370 domain of unknown function family (human) | 71 | Unknown |
| RIKEN cDNA 1810047C23 | 20149754 | FLJ11200 (human) | 84 | Unknown |
| RIKEN cDNA 2010203J19 | 13385408 | Ribosomal protein L11 (human) | 100 | Ribosome |
| RIKEN cDNA 2010310D06 | 29789387 | TMPIT (human) | 95 | Unknown |
| RIKEN cDNA 2210402A09 | 13399310 | Ribosomal protein S10 (human) | 99 | Ribosome |
| RIKEN cDNA 2300002G02 | 27370092 | Tu translation elongation factor, mitochondrial (human) | 94 | Mitochondria |
| RIKEN cDNA 2310008M10 | 13384930 | DC2 (human) | 99 | Unknown |
| RIKEN cDNA 2310016N05 | 21312676 | FLJ10511 (human) | 98 | Unknown |
| RIKEN cDNA 2310050B20 | 21313468 | Ts translation elongation factor mitochondrial (human) | 80 | Mitochondria |
| RIKEN cDNA 2310057H16 | 27754056 | Tubulin β 3 (mouse) | 93 | Cytoskeleton |
| RIKEN cDNA 2410002K23 | 13385998 | Heat shock protein 75 (human) | 88 | Cytosolic |
| RIKEN cDNA 2410003K15 | 20820749 | C7ORF30 (human) | 72 | Unknown |
| RIKEN cDNA 2610028H14 | 30424601 | Mitochondrial ribosomal protein S27 (human) | 77 | Mitochondria |
| RIKEN cDNA 2700060E02 | 13386026 | C14ORF166 (human) | 97 | Unknown |
| RIKEN cDNA 2700067E09 | 30424792 | Hydroxysteroid dehydrogenase-like 1 (human) | 88 | Unknown |
| RIKEN cDNA 2810403L02 | 22094989 | TIMM50 (human) | 92 | Mitochondria |
| RIKEN cDNA 2810442I22 | 22137680 | ATPase Ca2+ transporting plasma membrane 1 (human) | 99 | Membrane |
| RIKEN cDNA 2900024O10 | 28526420 | No homolog | N/A | Unknown |
| RIKEN cDNA 2900053E13 | 13195624 | NADH dehydrogenase (ubiquinone) 1 α subcomplex, 10, 42 kDa (human) | 77 | Mitochondria |
| RIKEN cDNA 3110052F15 | 12963643 | Mitochondrial ribosomal protein L46; chromosome 15 open reading frame 4 (human) | 81 | Mitochondria |
| RIKEN cDNA 4430402G14 | 13385168 | Ubiquinol-cytochrome c reductase Rieske iron-sulfur polypeptide (rat) | 96 | Mitochondria |
| RIKEN cDNA 4631422C05 | 21707978 | Glutamic pyruvate transaminase 2 (human) | 93 | Cytosolic |
| RIKEN cDNA 4733401H18 | 12963603 | LOC56769 (rat) | 91 | Unknown |
| RIKEN cDNA 4933421G18 | 38076907 | No homolog | N/A | Unknown |
| RIKEN cDNA 4933435E07 | 27502349 | Peptidase (mitochondrial processing) alpha (human) | 91 | Mitochondria |
| RIKEN cDNA 5630400A | 19343750 | Cytoskeleton-associated protein 4 (human) | 79 | Cytoskeleton |
| RIKEN cDNA 5630400A09 | 19343750 | Cytoskeleton-associated protein 4 (human) | 79 | Cytoskeleton |
| RIKEN cDNA 5730438N18 | 15030091 | Bone marrow stromal cell mitochondrial carrier protein (human) | 91 | Mitochondria |
| RIKEN cDNA 5730466P16 | 20859189 | PMI protein (human) | 97 | Cytosolic |
| RIKEN cDNA 6720485C15 | 27754054 | Dephospho-CoA kinase (human) | 91 | Cytosolic |
| RIKEN cDNA 9430016H08 | 19353157 | FLJ22555 (human) | 77 | Unknown |
| RIKEN cDNA A230072I16 | 28510843 | CGI-147 (human) | 79 | Unknown |
| RIKEN cDNA C230096C10 | 31542273 | KIAA0090 (human) | 93 | Unknown |
| RIKEN cDNA C820010P03 | 38074235 | UDP-glucose glycoprotein (human) | 91 | ER |
| RIKEN cDNA D330038I09 | 23956316 | 10-formyltetrahydrofolate dehydrogenase (mouse) | 75 | Mitochondria |
| RIKEN cDNA G431004K08 | 26006117 | General control of amino-acid synthesis 1-like (human) | 95 | Cytosolic |
| RIKEN cDNA5730568A12 | 21313384 | FLJ12436 (human) | 83 | Unknown |
| Unnamed protein product | 12848167 | Lyric (rat) | 96 | Unknown |
| Unnamed protein product | 26354076 | DKFZp761D221 (human) | 92 | Unknown |
| Unnamed protein product | 26331048 | FLJ20793 (human) | 92 | Unknown |
| Unnamed protein product | 12859602 | LOC56926 (human) | 96 | Unknown |
| Unnamed protein product | 26344842 | KIAA0152 (human) | 93 | Unknown |
| Unnamed protein product | 12839842 | CHCHD3 (human) | 90 | Unknown |
| Unnamed protein product | 12838089 | FLJ10525 (human) | 98 | Unknown |
| Unnamed protein product | 12834738 | MGC4825 (human) | 82 | Unknown |
| Unnamed protein product | 26336489 | Alanyl-tRNA synthetase (human) | 95 | Unknown |
| Unnamed protein product | 12855874 | IQ motif-containing GTPase activating protein 1 (human) | 96 | Cytoskeleton |
| Unnamed protein product | 7670472 | α-1,3-mannosyltransferase (human) | 80 | ER |
| Unnamed protein product | 12833038 | Stomatin-like 2 (human) | 94 | Membrane fraction |
| Unnamed protein product | 26378399 | Thioredoxin domain containing disulfide oxidoreductase (human) | 76 | Membrane fraction |
| Unnamed protein product | 26344894 | Acyl-Coenzyme A dehydrogenase family member 9 (human) | 86 | Mitochondria |
| Unnamed protein product | 12846081 | Ubiquinol-cytochrome c reductase core protein I human (human) | 88 | Mitochondria |
| Unnamed protein product | 26331164 | 4-Aminobutyrate aminotransferase (rat) | 97 | Mitochondria |
| Unnamed protein product | 26339056 | Isocitrate dehydrogenase 3 (NAD+) α (rat) | 99 | Mitochondria |
| Unnamed protein product | 26342525 | NADH dehydrogenase (ubiquinone) 1 α subcomplex 10 (human) | 77 | Mitochondria |
| Unnamed protein product | 12832967 | Succinyl-CoA synthetase α subunit (rat) | 95 | Mitochondria |
| Unnamed protein product | 26337655 | Mitochondrial carrier family protein (human) | 84 | Mitochondria |
| Unnamed protein product | 12833077 | Cytochrome c1 (human) | 91 | Mitochondria |
| Unnamed protein product | 12832971 | 2,4-Dienoyl-CoA reductase (rat) | 94 | Mitochondria |
| Unnamed protein product | 12833468 | Succinate dehydrogenase complex iron sulfur subunit B (human) | 91 | Mitochondria |
| Unnamed protein product | 26352986 | NADH dehydrogenase (ubiquinone) 1 β subcomplex 5 (human) | 76 | Mitochondria |
| Unnamed protein product | 12847456 | ATP synthase H+ transporting mitochondrial F1 complex δ subunit (rat) | 95 | Mitochondria |
| Unnamed protein product | 26336675 | Phosphoinositide-3-kinase class 2 β (human) | 87 | Membrane fraction |
Out of the 47 new proteins that have sequence similarities to other annotated proteins, 18 (38%) proteins are known to be localized in mitochondria. Three (6%) and two (4%) proteins are homologous to proteins associated with cytoskeleton and ER, respectively. The subcellular localization information for 16 (34%) other proteins was not found.
Function of the Identified Proteins
The functions of the 171 mitochondrial proteins that have been annotated in either EXPASY or Proteome BioKnowledge databases are illustrated in Fig. 4. The largest percentage of proteins (92 proteins, 54%) are those with catalytic activities related to metabolism and energy production. More than half of the metabolic proteins are enzymes involved in the TCA cycle or the mitochondrial respiratory chain. These findings are consistent with the fact that the mitochondrion is the main compartment responsible for energy generation. Mitochondrial ribosomal proteins, membrane transporters, heat shock proteins/chaperones, and proteins with redox function consist of 18, 14, 2, and 2% of these proteins, respectively. The functions of a small subset of the proteins (1%) are not yet well defined.
Fig. 4. Summary of the functions of the mitochondrial proteins that have been annotated in the EXPASY or BioKnowledge databases.
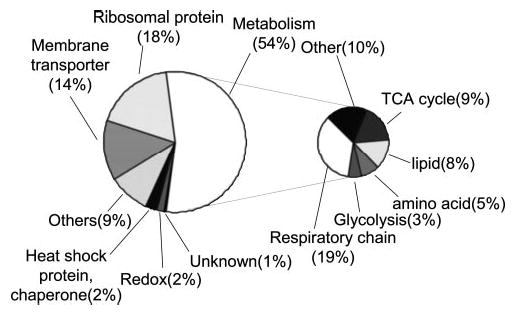
One hundred seventy-one mitochondrial proteins were annotated in the databases, and the functions of other identified mitochondrial proteins need to be elucidated.
Differentially Expressed Proteins in Mitochondria of NSC34WT-SOD1 and NSC34G93A-SOD1
Mitochondrial dysfunction and apoptosis have been demonstrated in fALS but the mechanisms by which SOD1 mutations cause these mitochondrial abnormalities are not clear. After surveying the mitochondrial proteome of NSC34 cells, we used the 2D gel electrophoresis approach to examine the differentially expressed mitochondrial proteins between NSC34 cells expressing WT and G93A mutant SOD1. The mitochondrial fractions were isolated from NSC34WT-SOD1 to NSC34G93A-SOD1 cells, respectively, and subjected to 2D gel electrophoresis as described earlier. The mitochondrial protein expression profiles of NSC34WT-SOD1 to NSC34G93A-SOD1 cells were analyzed by PDQuest software. The density of each spot was determined by the software and normalized against the total gel density that represents the total protein quantity in the mitochondrial fraction. The experiment was carried out three times independently, and the Student t test was performed to determine the statistically significant alterations. Nine and 36 protein spots were found to have increased and decreased abundance, respectively, in NSC34G93A-SOD1 cells compared with those in NSC34WT-SOD1 cells. The p values for these spots in three independent experiments were less than 0.05. The 45 protein spots that display altered abundances are shown in Table II, and representative images of two such spots are shown in Fig. 5.
Table II.
Protein spots with differential abundances in NSC34G93A
| Spot no. | NSC34G93A/NSC34WT | p value | Proteins | Accession no. | Spots of the protein |
|---|---|---|---|---|---|
| Increased abundances in NSC34G93A | |||||
| 36 | 3.431 | 0.009 | NADH dehydrogenase (ubiquinone) Fe-S protein 8 | 21450107 | 35, 36 |
| 63 | 2.799 | 0.025 | Peripherin | 2253159 | 63 |
| 133 | 2.571 | 0.027 | Septin 5 | 6685763 | 133 |
| 217 | 2.476 | 0.007 | Aldolase 1, A isoform | 6671539 | 217, 218, 219 |
| 203 | 2.381 | 0.028 | Aconitase 2 | 18079339 | 202, 203 |
| 125 | 1.969 | 0.019 | Similar to septin6 type II | 26324430 | 125, 126 |
| 197 | 1.539 | 0.041 | Retinal pigment (fragment) | 30691136 | 197 |
| 146 | 1.500 | 0.023 | RIKEN cDNA 2810403L02 | 22094989 | 146 |
| 156 | 1.361 | 0.023 | Pyrroline-5-carboxylate reductase 1 | 13879494 | 156 |
| Decreased abundances in NSC34G93A | |||||
| 20 | 0.774 | 0.031 | γ-Actin | 809561 | 20 |
| 150 | 0.740 | 0.041 | RIKEN cDNA 2310050B20 | 21313468 | 150 |
| 164 | 0.676 | 0.023 | VDAC2 | 6755965 | 152, 157, 158, 164, 165 |
| 227 | 0.653 | 0.035 | VDAC1 | 6755963 | 173, 227, 228, 231 |
| 135 | 0.636 | 0.012 | Proline-rich protein | 284995 | 135 |
| 187 | 0.614 | 0.023 | ATP synthase, H+ transporting, mitochondrial F1 complex, α subunit, isoform 1 | 6680748 | 184, 186, 187, 188, 189, 205, 207, 221, 229, 230 |
| 189 | 0.675 | 0.012 | ATP synthase, H+ transporting, mitochondrial F1 complex, α subunit, isoform 1 | 6680748 | 184, 186, 187, 188, 189, 205, 207, 221, 229, 230 |
| 205 | 0.264 | 0.014 | ATP synthase, H+ transporting, mitochondrial F1 complex, α subunit, isoform 1 | 6680748 | 184, 186, 187, 188, 189, 205, 207, 221, 229, 230 |
| 230 | 0.672 | 0.034 | ATP synthase, H+ transporting, mitochondrial F1 complex, α subunit, isoform 1 | 6680748 | 184, 186, 187, 188, 189, 205, 207, 221, 229, 230 |
| 213 | 0.612 | 0.035 | Acetyl-Coenzyme A acetyltransferase 1 precursor | 21450129 | 213, 214, 215 |
| 115 | 0.578 | 0.038 | Tyrosyl-tRNA synthetase | 15488844 | 115 |
| 64 | 0.578 | 0.038 | Heat shock 70-kDa protein 5 | 31981722 | 5, 64, 75 |
| 75 | 0.522 | 0.043 | Heat shock 70-kDa protein 5 | 31981722 | 5, 64, 75 |
| 212 | 0.545 | 0.016 | Acetyl-Coenzyme A dehydrogenase, medium chain | 6680618 | 212 |
| 109 | 0.517 | 0.037 | Dnaj (Hsp40) homolog subfamily B member 11 | 20892117 | 109, 137 |
| 96 | 0.496 | 0.030 | Chaperonin subunit 5 (ε) | 6671702 | 96 |
| 106 | 0.485 | 0.021 | Ornithine aminotransferase | 8393866 | 106 |
| 74 | 0.473 | 0.013 | Tubulin α-2 chain | 135412 | 11, 74 |
| 116 | 0.470 | 0.047 | Chaperonin subunit 6A (ζ) | 6753324 | 116 |
| 110 | 0.445 | 0.007 | Stomatin | 12963591 | 110, 144 |
| 142 | 0.434 | 0.035 | Serine hydroxymethyl transferase 2 | 21312298 | 142, 206, 208 |
| 8 | 0.432 | 0.001 | ATP synthase, H+ transporting mitochondrial F1 complex, β subunit | 25052136 | 8, 10 |
| 103 | 0.421 | 0.004 | Dihydrolipoamide S-succinyltransferase | 21313536 | 102, 103 |
| 198 | 0.396 | 0.018 | Cytochrome c oxidase, subunit Va | 21707954 | 198 |
| 77 | 0.391 | 0.002 | Tubulin, β 5 | 7106439 | 77 |
| 204 | 0.389 | 0.001 | RIKEN cDNA 4631422C05 | 21707978 | 204 |
| 179 | 0.378 | 0.008 | Adenylate kinase 2 | 34328230 | 179 |
| 90 | 0.354 | 0.004 | Mitochondrial ATP-dependent protease Lon | 26984237 | 90 |
| 216 | 0.336 | 0.013 | Isocitrate dehydrogenase 3, β subunit | 18700024 | 216 |
| 91 | 0.283 | 0.049 | Mitochondrial inner membrane protein (Mitofilin) | 29427692 | 91, 92, 121 |
| 85 | 0.270 | 0.016 | Peroxiredoxin 3 | 6680690 | 85, 181 |
| 191 | 0.235 | 0.009 | RIKEN cDNA 4430402G14 | 13385168 | 191 |
| 235 | 0.226 | 0.028 | NADH dehydrogenase (Ubiquinone) 1 β subcomplex, 10 | 20899100 | 235 |
| 131 | 0.002 | 0.001 | Pyruvate dehydrogenase E1 α 1 | 6679261 | 131, 132 |
| 178 | 0.001 | 0.000 | RIKEN cDNA 0610041L09 | 21313618 | 176, 178, 180 |
| 202 | 0.001 | 0.002 | Aconitase 2 | 18079339 | 202, 203 |
Fig. 5. Representative 2D gel protein spots that displayed differential levels between NSC34WT-SOD1 and NSC34G93A-SOD1 cells.

A, the protein named “Similar to septin6 type II” (spot 125 in Fig. 2) has increased abundance in NSC34G93A-SOD1 cells (NSC34G93A-SOD1/ NSC34WT-SOD1 = 1.97, p = 0.019). B, the protein “Heat shock 70-kDa protein 5” (spot 64 in Fig. 2) has decreased abundance in NSC34G93A-SOD1 cells (NSC34G93A-SOD1/NSC34WT-SOD1 = 0.58, p = 0.038). Forty-five 2D gel spots display altered abundance between the mitochondrial fractions isolated from NSC34WT-SOD1 and NSC34G93A-SOD1 cells. These 45 protein spots were identified as 40 unique proteins, and they are listed in Table II.
As noted earlier, post-translational modification occurs in many mitochondrial proteins, resulting in the fact that multiple proteins spots in the 2D gel are identified as the same protein. Out of 45 differentially expressed protein spots listed in Table II, 24 spots were identified to be 19 unique proteins that have multiple spots in 2D gel. In other words, more than half of the protein spots that are changed in the mitochondria of NSC34G93A-SOD1 cells are those representing protein post-translational modification isoforms. The results suggest that the fALS mutant G93A-SOD1 often causes alterations in protein post-translational modifications.
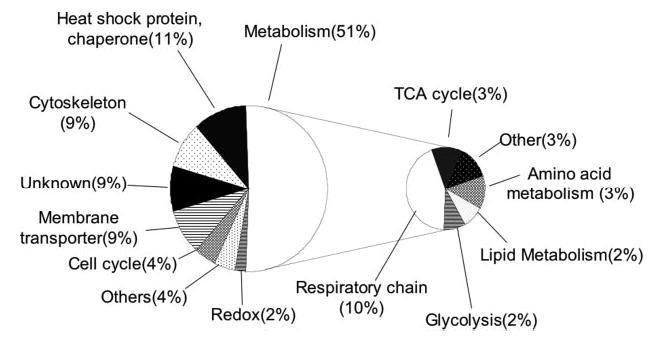
Functions of the proteins in Table II, which are changed in the presence of G93A-SOD1 in NSC34 cells, are related to many essential cellular functions as shown in Fig. 6. Half of them have the metabolic functions, mainly involved in the mitochondrial respiratory chain including the subunits of complex I, III, IV, and V. Heat shock proteins/molecular chaperones were also identified from five spots consisting of 11% of the total proteins altered: chaperonin subunit 5 (ε), chaperonin subunit 6a (ζ), DnaJ (Hsp40) homolog subfamily B member 11, and two spots of heat shock 70-kDa protein 5. It is noted that all molecular chaperone proteins displayed decreased abundances in NSC34G93A-SOD1. Mitochondrial membrane proteins, voltage-dependent anion channel 1 (VDAC1) and VDAC2, are affected by G93A-SOD1 as well. VDACs are components of mitochondrial membrane permeability transition pore and are known to regulate the mitochondrial membrane permeability and the cytochrome c release from mitochondria. The modification isoforms of VDAC1 and VDAC2 showed decreased levels in NSC34G93A-SOD1 cells, which may lead to deregulation of cytochrome c release and activation of apoptosis. The significance of the alterations in these proteins will be discussed, and this study will lay ground for further studies.
Fig. 6. Summary of the annotated functions of the 40 differentially expressed mitochondrial proteins between NSC34WT-SOD1 and NSC-34G93A-SOD1 cells.
DISCUSSION
Despite increasing evidence suggesting that mitochondrial dysfunction and apoptosis play a role in the pathogenesis of mutant SOD1-mediated fALS, little is known about the mechanisms by which mutant SOD1 causes mitochondrial dysfunction in motor neurons that are specifically damaged in the disease. A comprehensive analysis of the mitochondrial protein expression profile in the presence and absence of fALS-linked SOD1 mutant can provide important insight into the role of mitochondria in the disease. We have used two independent approaches, 2D gel electrophoresis followed by MALDI-MS/MS and SDS-PAGE followed by LC-MS/MS, to characterize the mitochondrial proteome of motor neuron-like NSC34 cells as well as the alterations in the mitochondrial proteome in the presence of fALS-linked mutant G93A-SOD1.
The first achievement of this study is the comprehensive survey of mitochondrial proteins in NSC34 cells, a mouse embryonic spinal cord-neuroblastoma hybrid cell line. Trans-genic mice are powerful tools in biomedical research, providing animal models for many diseases. The identification of mitochondrial proteins in mouse neuronal cells is not only essential to the comparative analysis in this study but also invaluable to the scientific community, particularly those studying the role of mitochondria in various diseases using mice as animal models.
Two-dimensional gel electrophoresis separation of the isolated mitochondrial fraction of NSC34 cells yielded ~600 protein spots, which is close to the number of mitochondrial proteins isolated from the human heart and other sources (52, 53). Subsequent MALDI-MS/MS analysis identified 170 unique proteins from 240 2D gel spots. A complimentary approach, SDS-PAGE fractionation of mitochondrial proteins followed by LC-MS/MS analysis, has identified 383 unique proteins. Eighty-seven unique proteins were commonly identified by both approaches (see Fig. 3). The LC-MS/MS technique using extremely slow flow-rate HPLC (150–200 nl/min) coupled with nanoelectrospray MS/MS is more sensitive, so more proteins are identified by this approach. For instance, many mitochondrial ribosomal proteins were only identified by the SDS-PAGE followed by LC-MS/MS approach. In addition, many proteins identified only in the SDS-PAGE followed by LC-MS/MS approach have isoelectric point values higher than 9, i.e. basic proteins. This reflects the difficulties in analyzing proteins with very low or very high isoelectric point values by 2D gel electrophoresis. Combining results from both approaches, 470 unique proteins have been identified from mitochondria of NSC34 cells, and the complete list is shown in the supplemental table.
This study has discovered 75 mouse mitochondrial proteins that previously had only been reported at the cDNA level. The newly discovered mouse mitochondrial proteins are listed in Table I. Though this study is not designed to study the functions of these novel mouse mitochondrial proteins, which need to be further investigated by other groups in the field, it provides initial evidence that these genes are expressed in mouse neuronal cells and isolated in the mitochondrial fraction.
The functions of the mitochondrial proteins that have been annotated in the EXPASY and Proteome BioKnowledge databases include metabolism (TCA cycle, respiratory chain, amino acid metabolism, lipid metabolism, etc.), membrane transport, mitochondrial ribosome, heat shock/molecular chaperone, redox, and antioxidant proteins, among others. This finding is consistent with the fact that the mitochondrion is the main organelle responsible for energy generation.
Many identified proteins in this study are well-characterized mitochondrial proteins as noted in two annotated protein databases, EXPASY and Proteome BioKnowledge. Other proteins are not annotated as mitochondrial proteins in the above two databases. Most of those are known as ER and nucleus proteins. We used the Percoll gradient ultracentrifugation technique to isolate the mitochondrial fraction, which is widely recognized as the technique producing least contamination. However, it remains possible that other proteins, particularly ER proteins, may be co-localized in the mitochondrial fraction as contaminants. It is also possible that some of the “contaminating” proteins are partially localized in mitochondria or interact with other mitochondrial proteins. More detailed studies such as immuno-colocalization experiments are needed to distinguish these different possibilities, which are beyond the scope of this study.
To better understand the mechanisms underlying mitochondrial dysfunction in mutant SOD1-mediated fALS, the mitochondrial proteomes from NSC34WT-SOD1 and NSC34G93A-SOD1 were compared by 2D gel electrophoresis in this study. Nine and 36 spots displayed increased and decreased levels, respectively. These 45 spots represent 40 distinct proteins. Out of the 40 unique proteins that displayed differential levels in the mitochondrial fractions, 27 proteins have been clearly described as mitochondrial proteins, such as VDAC1, VDAC2, aconitase, ATP synthase, etc. Five of the differentially expressed proteins (RIKEN cDNA 2810403L02, RIKEN cDNA 2310050B20, RIKEN cDNA 4631422C05, RIKEN cDNA 4430402G14, and RIKEN cDNA 0610041L09) were solely reported at the DNA level previously, thus their localizations were not clearly defined in literature. However, our BLAST search has demonstrated that four out of five of these proteins have high sequence similarity to well-defined mitochondrial proteins (see Table I). For example, RIKEN cDNA 4430402G14 has 96% sequence similarity to rat Ubiquinol-cytochrome c reductase Rieske iron-sulfur polypeptide.
Twenty-four of 45 2D gel spots that displayed differential abundances are proteins that have multiple 2D gel spots, suggesting that G93A-SOD1 often causes alterations in protein post-translational modifications. Modifications in a particular protein named VDAC2 is discussed below. It is worth noting that an advantage of the 2D gel electrophoresis approach is its ability of detecting such differences in the modification states of a protein.
The main hypothesis driving this study is that the mitochondrial dysfunction and activation of apoptosis play a role in the motor neuron death in fALS, which has been supported by various results from different laboratories. One of the differentially expressed protein spots we have identified is a mitochondrial outer membrane protein named VDAC2 (see Table II). VDAC2 is one of three mammalian VDACs and shows 75 and 73% sequence identity to VDAC1 and VDAC3, respectively. VDACs are mitochondrial outer membrane proteins that can form membrane permeability transition pores, and their common functions are to control ADP translocation into mitochondria and the mitochondrial outer membrane potential (59, 60). Thus, VDAC2 could be directly involved in abnormal mitochondrial morphology observed in the ALS-transgenic mice. It is well documented that VDAC1 is involved in apoptosis activation by interacting with bcl-2 family members (60–63). VDAC2 is recently reported to be a specific inhibitor of BAK-dependent mitochondrial apoptosis by interacting with the inactive form of BAK and preventing BAK from being activated (64). Consequently, VDAC2 deficiency is lethal to mouse embryos (64). Thus, it is reasonable to speculate that VDAC2 can be directly involved in regulating apoptosis as has been implicated in the ALS etiology.
We observed two lines of evidence supporting the potential role of VDAC2 in ALS (1). Five distinct 2D gel spots (spots 152, 157, 158, 164, and 165 in Fig. 2) with the same molecular mass (~33 kDa) but different pI values (ranging from 4 to 8) were all identified as VDAC2. Equivalent molecular mass of the five spots suggests that they are not likely to be different splicing products. Rather, the 2D gel pattern suggests that they are modified isoforms of VDAC2. The nature of the modification has not been determined in this study yet, but is speculated to phosphorylation or oxidation, which can change the pI values (2). The abundance of one particular isoform (spot 164) decreased ~2-fold in NSC34G93A-SOD1 cells compared with that in NSC34WT-SOD1, while other spots (spots 152, 157, 158, and 165) were not significantly changed. Little is known about the post-translational modifications of VDAC2 or the mechanisms by which VDAC2 regulates apoptosis. It is not uncommon that specific modification events, particularly protein phosphorylation, can modulate protein function and signaling pathways. Thus, it is conceivable that the change in the specific modification state of VDAC2 is likely to be involved in mutant SOD1-mediated toxicity and the subsequent mitochondrial dysfunction and apoptosis activation. Future studies of VDAC2 modifications and its role in regulating apoptosis are necessary to elucidate its relevance to the pathogenesis of fALS.
In addition, it is noted that fALS is a complex disease and the SOD1 mutants may disturb other mitochondrial functions. Findings in this comprehensive proteomic study can explain several other observations in fALS. First, five heat shock proteins/chaperones, chaperonin subunit 5 (ε), chaperonin subunit 6a (ζ), DnaJ (Hsp40) homolog subfamily B member 11, and two spots of heat shock 70-kDa protein 5, were down-regulated in NSC34G93A-SOD1. It has been proposed that the availability of heat shock proteins (HSPs) is reduced by the misfolded and aggregated mutant SOD1 (8, 31), leading to the compromised molecular chaperone activities and cellular damages. The levels of HSPs in motor neurons were shown to be lower than those in astrocytes (65). Beneficial effects of elevated levels of HSPs were also demonstrated in cell line and transgenic mouse models (66, 67). Moreover, HSPs should play a role in protein translocation into mitochondria because proteins need to be at least partially unfolded to pass through the mitochondrial membrane pore and subsequently need to refold. In fact, cytosolic HSPs were shown to partially block the mitochondrial entry of mutant SOD1 but not WT-SOD1 (43). Recent studies showed that mutant SOD1 were present in mitochondria from motor neurons but WT SOD1 were not (45, 50). We observed both WT and G93A-SOD1 in the mitochondrial fraction isolated from NSC34 cells by Western blotting (data not shown). We identified manganese SOD (SOD2, a typical mitochondrial matrix protein, see the supplemental table) but not SOD1 using the proteomic approaches in this study, probably because the level of SOD1 was below the detection limit of the approaches. Though little is known about the detailed mechanisms by which a fraction of SOD1 enters mitochondria, it is likely that mitochondrial HSPs can also affect the entry of mutant SOD1 into mitochondria, thus playing a role in the mutant SOD1-mediated mitochondrial abnormalities. Thus, the finding of this study, i.e. the decreased levels of mitochondrial HSPs in NSC34G93A-SOD1 cells, can be related with the entry of mutant SOD1 into mitochondria and the subsequent activation of apoptosis.
Second, half of the differentially expressed proteins are involved in various metabolic pathways, mainly the mitochondrial respiratory chain including the subunits of complex I, III, IV, and V. It was previously reported that the activity of complexes II and IV were suppressed in a similar cell line model of fALS (21, 22). The findings of this study may underline the mechanisms of such suppressed respiratory activities.
The findings in this study provide comprehensive knowledge of the mitochondrial proteome in mouse neuronal cells, which has broad impact on the research of mitochondrial diseases using transgenic mouse models. This study has yielded a short list of mitochondrial proteins that may hold the key to the mechanisms by which SOD1 mutants cause abnormal mitochondrial morphology and apoptosis activation. It has laid ground for future detailed functional studies to elucidate the role of particular mitochondrial proteins, such as VDAC2, in the pathogenesis of fALS.
Supplementary Material
Acknowledgments
We are grateful to Joan S. Valentine (University of California Los Angeles, Los Angeles, CA) for providing the human SOD1 cDNA.
Footnotes
This study is partly supported by the University of Kentucky College of Medicine start-up funds and National Institutes of Health Grants (R21ES12025 and R01NS49126, to H. Z.). Purchase of the QSTAR XL mass spectrometer was made possible by a grant from the National Science Foundation Experimental Program to Stimulate Competitive Research program and matching funds from the University of Kentucky.
The on-line version of this manuscript (available at http://www.mcponline.org) contains supplemental material.
The abbreviations used are: ALS, amyotrophic lateral sclerosis; SOD1, copper zinc superoxide dismutase; fALS, familial amyotrophic lateral sclerosis; WT, wild-type; VDAC, voltage-dependent anion channel; IRES, internal ribosome entry site; CHM, cell homogenizing medium; HFBA, heptafluorobutylic acid; PMF, peptide mass fingerprinting; 2D, two-dimensional; ER, endoplasmic reticulum; HSP, heat shock protein.
References
- 1.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 2.Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, Getzoff ED, Hu P, Herzfeldt B, Roos RP, et al. Amyotrophic lateral sclerosis and structural defects in Cu, Zn superoxide dismutase. Science. 1993;261:1047–1051. doi: 10.1126/science.8351519. [DOI] [PubMed] [Google Scholar]
- 3.Gaudette M, Hirano M, Siddique T. Current status of SOD1 mutations in familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:83–89. doi: 10.1080/14660820050515377. [DOI] [PubMed] [Google Scholar]
- 4.Brown RH., Jr SOD1 aggregates in ALS: Cause, correlate or consequence? Nat Med. 1998;4:1362–1364. doi: 10.1038/3945. [DOI] [PubMed] [Google Scholar]
- 5.Cleveland DW, Liu J. Oxidation versus aggregation—How do SOD1 mutants cause ALS? Nat Med. 2000;6:1320–1321. doi: 10.1038/82122. [DOI] [PubMed] [Google Scholar]
- 6.Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: Deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- 7.Julien JP. Amyotrophic lateral sclerosis. Unfolding the toxicity of the misfolded. Cell. 2001;104:581–591. doi: 10.1016/s0092-8674(01)00244-6. [DOI] [PubMed] [Google Scholar]
- 8.Valentine JS, Hart PJ. Misfolded CuZnSOD and amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2003;100:3617–3622. doi: 10.1073/pnas.0730423100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH, Jr, Scott RW, Snider WD. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13:43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- 10.Gurney ME. Transgenic-mouse model of amyotrophic lateral sclerosis. N Engl J Med. 1994;331:1721–1722. doi: 10.1056/NEJM199412223312516. [DOI] [PubMed] [Google Scholar]
- 11.Chiu AY, Zhai P, Dal Canto MC, Peters TM, Kwon YW, Prattis SM, Gurney ME. Age-dependent penetrance of disease in a transgenic mouse model of familial amyotrophic lateral sclerosis. Mol Cell Neurosci. 1995;6:349–362. doi: 10.1006/mcne.1995.1027. [DOI] [PubMed] [Google Scholar]
- 12.Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- 13.Ripps ME, Huntley GW, Hof PR, Morrison JH, Gordon JW. Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 1995;92:689–693. doi: 10.1073/pnas.92.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 15.Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc Natl Acad Sci U S A. 2002;99:1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagai M, Aoki M, Miyoshi I, Kato M, Pasinelli P, Kasai N, Brown RH, Jr, Itoyama Y. Rats expressing human cytosolic copper-zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: Associated mutations develop motor neuron disease. J Neurosci. 2001;21:9246–9254. doi: 10.1523/JNEUROSCI.21-23-09246.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rabizadeh S, Gralla EB, Borchelt DR, Gwinn R, Valentine JS, Sisodia S, Wong P, Lee M, Hahn H, Bredesen DE. Mutations associated with amyotrophic lateral sclerosis convert superoxide dismutase from an antiapoptotic gene to a proapoptotic gene: Studies in yeast and neural cells. Proc Natl Acad Sci U S A. 1995;92:3024–3028. doi: 10.1073/pnas.92.7.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kostic V, Jackson-Lewis V, de Bilbao F, Dubois-Dauphin M, Przedborski S. Bcl-2: Prolonging life in a transgenic mouse model of familial amyotrophic lateral sclerosis. Science. 1997;277:559–563. doi: 10.1126/science.277.5325.559. [DOI] [PubMed] [Google Scholar]
- 19.Carri MT, Ferri A, Battistoni A, Famhy L, Gabbianelli R, Poccia F, Rotilio G. Expression of a Cu, Zn superoxide dismutase typical of familial amyotrophic lateral sclerosis induces mitochondrial alteration and increase of cytosolic Ca2+ concentration in transfected neuroblastoma SH-SY5Y cells. FEBS Lett. 1997;414:365–368. doi: 10.1016/s0014-5793(97)01051-x. [DOI] [PubMed] [Google Scholar]
- 20.Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci. 1998;18:3241–3250. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borthwick GM, Johnson MA, Ince PG, Shaw PJ, Turnbull DM. Mitochondrial enzyme activity in amyotrophic lateral sclerosis: Implications for the role of mitochondria in neuronal cell death. Ann Neurol. 1999;46:787–790. doi: 10.1002/1531-8249(199911)46:5<787::aid-ana17>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 22.Menzies FM, Cookson MR, Taylor RW, Turnbull DM, Chrzanowska-Lightowlers ZMA, Dong L, Figlewicz DA, Shaw PJ. Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain. 2002;125:1522–1533. doi: 10.1093/brain/awf167. [DOI] [PubMed] [Google Scholar]
- 23.Vukosavic S, Stefanis L, Jackson-Lewis V, Guegan C, Romero N, Chen C, Dubois-Dauphin M, Przedborski S. Delaying caspase activation by Bcl-2: A clue to disease retardation in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci. 2000;20:9119–9125. doi: 10.1523/JNEUROSCI.20-24-09119.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guegan C, Vila M, Rosoklija G, Hays AP, Przedborski S. Recruitment of the mitochondrial-dependent apoptotic pathway in amyotrophic lateral sclerosis. J Neurosci. 2001;21:6569–6576. doi: 10.1523/JNEUROSCI.21-17-06569.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li M, Ona VO, Guegan C, Chen M, Jackson-Lewis V, Andrews LJ, Olszewski AJ, Stieg PE, Lee JP, Przedborski S, Friedlander RM. Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science. 2000;288:335–339. doi: 10.1126/science.288.5464.335. [DOI] [PubMed] [Google Scholar]
- 26.Pasinelli P, Borchelt DR, Houseweart MK, Cleveland DW, Brown RH., Jr Caspase-1 is activated in neural cells and tissue with amyotrophic lateral sclerosis-associated mutations in copper-zinc superoxide dismutase. Proc Natl Acad Sci U S A. 1998;95:15763–15768. doi: 10.1073/pnas.95.26.15763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pasinelli P, Houseweart MK, Brown RH, Jr, Cleveland DW. Caspase-1 and -3 are sequentially activated in motor neuron death in Cu, Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2000;97:13901–13906. doi: 10.1073/pnas.240305897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, Sarang S, Liu AS, Hartley DM, Wu du C, Gullans S, Ferrante RJ, Przedborski S, Kristal BS, Friedlander RM. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]
- 29.Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- 30.Johnston JA, Dalton MJ, Gurney ME, Kopito RR. Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2000;97:12571–12576. doi: 10.1073/pnas.220417997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watanabe M, Dykes-Hoberg M, Cizewski Culotta V, Price DL, Wong PC, Rothstein JD. Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol Dis. 2001;8:933–941. doi: 10.1006/nbdi.2001.0443. [DOI] [PubMed] [Google Scholar]
- 32.Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- 33.Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 34.Honig LS, Chambliss DD, Bigio EH, Carroll SL, Elliott JL. Glutamate transporter EAAT2 splice variants occur not only in ALS, but also in AD and controls. Neurology. 2000;55:1082–1088. doi: 10.1212/wnl.55.8.1082. [DOI] [PubMed] [Google Scholar]
- 35.Raoul C, Estevez AG, Nishimune H, Cleveland DW, deLapeyriere O, Henderson CE, Haase G, Pettmann B. Motoneuron death triggered by a specific pathway downstream of Fas. Potentiation by ALS-linked SOD1 mutations. Neuron. 2002;35:1067–1083. doi: 10.1016/s0896-6273(02)00905-4. [DOI] [PubMed] [Google Scholar]
- 36.Williamson TL, Bruijn LI, Zhu Q, Anderson KL, Anderson SD, Julien JP, Cleveland DW. Absence of neurofilaments reduces the selective vulnerability of motor neurons and slows disease caused by a familial amyotrophic lateral sclerosis-linked superoxide dismutase 1 mutant. Proc Natl Acad Sci U S A. 1998;95:9631–9636. doi: 10.1073/pnas.95.16.9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999;2:50–56. doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]
- 38.Kong J, Xu Z. Overexpression of neurofilament subunit NF-L and NF-H extends survival of a mouse model for amyotrophic lateral sclerosis. Neurosci Lett. 2000;281:72–74. doi: 10.1016/s0304-3940(00)00808-9. [DOI] [PubMed] [Google Scholar]
- 39.Couillard-Despres S, Zhu Q, Wong PC, Price DL, Cleveland DW, Julien JP. Protective effect of neurofilament heavy gene overexpression in motor neuron disease induced by mutant superoxide dismutase. Proc Natl Acad Sci U S A. 1998;95:9626–9630. doi: 10.1073/pnas.95.16.9626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown RH, Jr, Ludlow CL, Fischbeck KH. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33:455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- 41.Sturtz LA, Diekert K, Jensen LT, Lill R, Culotta VC. A fraction of yeast Cu, Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J Biol Chem. 2001;276:38084–38089. doi: 10.1074/jbc.M105296200. [DOI] [PubMed] [Google Scholar]
- 42.Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu, Zn-SOD in mitochondria. J Biol Chem. 2001;276:38388–38393. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- 43.Okado-Matsumoto A, Fridovich I. Amyotrophic lateral sclerosis: A proposed mechanism. Proc Natl Acad Sci U S A. 2002;99:9010–9014. doi: 10.1073/pnas.132260399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Higgins CM, Jung C, Ding H, Xu Z. Mutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS. J Neurosci. 2002;22:RC215. doi: 10.1523/JNEUROSCI.22-06-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu J, Lillo C, Jonsson PA, Velde CV, Ward CM, Miller TM, Subramaniam JR, Rothstein JD, Marklund S, Andersen PM, Brannstrom T, Gredal O, Wong PC, Williams DS, Cleveland DW. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43:5–17. doi: 10.1016/j.neuron.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 46.Martin LJ. Neuronal death in amyotrophic lateral sclerosis is apoptosis: Possible contribution of a programmed cell death mechanism. J Neuropathol Exp Neurol. 1999;58:459–471. doi: 10.1097/00005072-199905000-00005. [DOI] [PubMed] [Google Scholar]
- 47.Pedersen WA, Luo H, Kruman I, Kasarskis E, Mattson MP. The prostate apoptosis response-4 protein participates in motor neuron degeneration in amyotrophic lateral sclerosis. FASEB J. 2000;14:913–924. doi: 10.1096/fasebj.14.7.913. [DOI] [PubMed] [Google Scholar]
- 48.Ishigaki S, Liang Y, Yamamoto M, Niwa J, Ando Y, Yoshihara T, Takeuchi H, Doyu M, Sobue G. X-Linked inhibitor of apoptosis protein is involved in mutant SOD1-mediated neuronal degeneration. J Neurochem. 2002;82:576–584. doi: 10.1046/j.1471-4159.2002.00998.x. [DOI] [PubMed] [Google Scholar]
- 49.Takeuchi H, Kobayashi Y, Ishigaki S, Doyu M, Sobue G. Mitochondrial localization of mutant superoxide dismutase 1 triggers caspase-dependent cell death in a cellular model of familial amyotrophic lateral sclerosis. J Biol Chem. 2002;277:50966–50972. doi: 10.1074/jbc.M209356200. [DOI] [PubMed] [Google Scholar]
- 50.Pasinelli P, Belford ME, Lennon N, Bacskai BJ, Hyman BT, Trotti D, Brown RH., Jr Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron. 2004;43:19–30. doi: 10.1016/j.neuron.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 51.Sato N, Sakuma C, Kato H, Milligan CE, Oppenheim RW, Yaginuma H. Bcl-2 rescues motoneurons from early cell death in the cervical spinal cord of the chicken embryo. J Neurobiol. 2002;53:381–390. doi: 10.1002/neu.10108. [DOI] [PubMed] [Google Scholar]
- 52.Cashman NR, Durham HD, Blusztajn JK, Oda K, Tabira T, Shaw IT, Dahrouge S, Antel JP. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev Dyn. 1992;194:209–21. doi: 10.1002/aja.1001940306. [DOI] [PubMed] [Google Scholar]
- 53.Durham HD, Dahrouge S, Cashman NR. Evaluation of the spinal cord neuron X neuroblastoma hybrid cell line NSC-34 as a model for neurotoxicity testing. Neurotoxicology. 1993;14:387–395. [PubMed] [Google Scholar]
- 54.Hunter TC, Yang L, Zhu H, Majidi V, Bradbury EM, Chen X. Peptide mass mapping constrained with stable isotope-tagged peptides for identification of protein mixtures. Anal Chem. 2001;73:4891–4902. doi: 10.1021/ac0103322. [DOI] [PubMed] [Google Scholar]
- 55.Zhu H, Hunter TC, Pan S, Bradbury EM, Chen X. Residue-specific mass signatures for the efficient identification of protein modifications by mass spectrometry. Anal Chem. 2002;74:1687–1694. doi: 10.1021/ac010853p. [DOI] [PubMed] [Google Scholar]
- 56.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 57.Taylor SW, Fahy E, Zhang B, Glenn GM, Warnock DE, Wiley S, Murphy AN, Gaucher SP, Capaldi RA, Gibson BW, Ghosh SS. Characterization of the human heart mitochondrial proteome. Nat Biotechnol. 2003;21:281–286. doi: 10.1038/nbt793. [DOI] [PubMed] [Google Scholar]
- 58.Mootha VK, Bunkenborg J, Olsen JV, Hjerrild M, Wisniewski JR, Stahl E, Bolouri MS, Ray HN, Sihag S, Kamal M, Patterson N, Lander ES, Mann M. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell. 2003;115:629–640. doi: 10.1016/s0092-8674(03)00926-7. [DOI] [PubMed] [Google Scholar]
- 59.Colombini M. Regulation of the mitochondrial outer membrane channel, VDAC. J Bioenerg Biomembr. 1987;19:309–20. doi: 10.1007/BF00768534. [DOI] [PubMed] [Google Scholar]
- 60.Zamzami N, Kroemer G. The mitochondrion in apoptosis: How Pandora’s box opens. Nat Rev Mol Cell Biol. 2001;2:67–71. doi: 10.1038/35048073. [DOI] [PubMed] [Google Scholar]
- 61.Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- 62.Tsujimoto Y, Shimizu S. VDAC regulation by the Bcl-2 family of proteins. Cell Death Differ. 2000;7:1174–1181. doi: 10.1038/sj.cdd.4400780. [DOI] [PubMed] [Google Scholar]
- 63.Tsujimoto Y, Shimizu S. The voltage-dependent anion channel: An essential player in apoptosis. Biochimie. 2002;84:187–193. doi: 10.1016/s0300-9084(02)01370-6. [DOI] [PubMed] [Google Scholar]
- 64.Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science. 2003;301:513–517. doi: 10.1126/science.1083995. [DOI] [PubMed] [Google Scholar]
- 65.Batulan Z, Shinder GA, Minotti S, He BP, Doroudchi MM, Nalbantoglu J, Strong MJ, Durham HD. High threshold for induction of the stress response in motor neurons is associated with failure to activate HSF1. J Neurosci. 2003;23:5789–5798. doi: 10.1523/JNEUROSCI.23-13-05789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bruening W, Roy J, Giasson B, Figlewicz DA, Mushynski WE, Durham HD. Up-regulation of protein chaperones preserves viability of cells expressing toxic Cu/Zn-superoxide dismutase mutants associated with amyotrophic lateral sclerosis. J Neurochem. 1999;72:693–699. doi: 10.1046/j.1471-4159.1999.0720693.x. [DOI] [PubMed] [Google Scholar]
- 67.Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med. 2004;10:402–405. doi: 10.1038/nm1021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.