

Abstract
Light excitation powers the reversible shuttling movement of the ring component of a rotaxane between two stations located at a 1.3-nm distance on its dumbbell-shaped component. The photoinduced shuttling movement, which occurs in solution, is based on a “four-stroke” synchronized sequence of electronic and nuclear processes. At room temperature the deactivation time of the high-energy charge-transfer state obtained by light excitation is ≈10 μs, and the time period required for the ring-displacement process is on the order of 100 μs. The rotaxane behaves as an autonomous linear motor and operates with a quantum efficiency up to ≈12%. The investigated system is a unique example of an artificial linear nanomotor because it gathers together the following features: (i) it is powered by visible light (e.g., sunlight); (ii) it exhibits autonomous behavior, like motor proteins; (iii) it does not generate waste products; (iv) its operation can rely only on intramolecular processes, allowing in principle operation at the single-molecule level; (v) it can be driven at a frequency of 1 kHz; (vi) it works in mild environmental conditions (i.e., fluid solution at ambient temperature); and (vii) it is stable for at least 103 cycles.
Keywords: molecular machine, nanoscience, photochemistry, rotaxane, supramolecular chemistry
The miniaturization race is encouraging scientists to design and construct motors on the nanometer scale, that is, at the molecular level (1–5). Such a daring goal finds its scientific origin in the existence of natural molecular motors (6–9).
Natural molecular motors, however, are extremely complex, and any attempt to construct systems of such a complexity, using the bottom-up molecular approach (10), would be challenging. What can be done, at present, is to construct simple prototypes of artificial molecular motors and machines (1–5, 11–19), consisting of a few components capable of moving in a controllable way, and to investigate the associated problems posed by interfacing them with the macroscopic world (20–25), particularly as far as energy supply is concerned.
Natural motors are “autonomous”: they keep operating, in a constant environment, as long as the energy source is available. By contrast, apart from a few recent examples (26–28), the fuel-powered artificial motors described so far are not autonomous because, after the mechanical movement induced by a chemical input, they need another, opposite chemical input to reset, which also implies generation of waste products. Addition of a fuel, however, is not the only means by which energy can be supplied to a chemical system. In fact, nature shows that, in green plants, the energy needed to sustain the machinery of life is ultimately provided by sunlight. Energy inputs in the form of photons can indeed cause mechanical movements by reversible chemical reactions without formation of waste products (13, 14, 16, 17).
In a previous work (29), we reported on the rotaxane 16+ (Scheme 1) that was carefully designed and synthesized to perform as a linear molecular motor powered exclusively by visible light, with autonomous operation relying on intramolecular processes. In such a work, however, we only succeeded in demonstrating the operation of the rotaxane as a light-driven mechanical switch that needed alternate addition of two “fuels,” namely triethanolamine and oxygen, with concomitant generation of waste products. In this work, we show that 16+ does behave as an autonomous “four stroke” motor powered only by sunlight, without generation of waste products. We have measured the rate constants and the efficiencies of the electronic and nuclear processes on which the shuttling movement is based, and we also discuss the related energetic aspects.
Scheme 1.

Structural formulas of rotaxane 16+, its dumbbell-shaped component 26+, and model compound 32+.
Just like their macroscopic counterparts, molecular-level machines have to be organized structurally and work as functionally integrated multicomponent systems (1). The rotaxane 16+ consists of as many as six molecular components suitably chosen and assembled to achieve the devised function. It comprises a bis-p-phenylene-34-crown-10 electron donor macrocycle R (hereafter called the ring) and a dumbbell-shaped component that contains two electron acceptor recognition sites for the ring, namely a 4,4′-bipyridinium (A2+1) and a 3,3′-dimethyl-4,4′-bipyridinium (A2+2) units, that can play the role of “stations” for the ring R. Molecular modeling shows that the overall length of 16+ is ≈5 nm and the distance between the centers of the two stations, measured along the dumbbell, is ≈1.3 nm. Furthermore, the dumbbell-shaped component incorporates a [Ru(bpy)3]2+-type (bpy, 2,2′-bipyridine) electron transfer photosensitizer P2+ that is able to operate with visible light and also plays the role of a stopper, a p-terphenyl-type rigid spacer S that has the task of keeping the photosensitizer far from the electron acceptor units, and finally a tetraarylmethane group T as the second stopper, crucial for the template-directed slippage synthesis of the rotaxane 16+ from the ring R and its dumbbell-shaped precursor 26+.
Results and Discussion
Electrochemical Switching. Electrochemical and NMR spectroscopic data showed (29) that the stable conformation of 16+ is by far the one in which the R component is located around the better electron acceptor station, A2+1, as represented in Scheme 1. When the interaction between R and A2+1 is switched off by selective reduction of A2+1 to A+1 (–0.44 V vs. saturated calomel electrode; SCE), the ring R moves by Brownian motion (9, 18) to the alternative A2+2 station. Successive oxidation restores the electron acceptor power of the A2+1 station and therefore the ring R moves back again by Brownian motion to surround this station.
We have carried out in acetonitrile solution spectroelectrochemical experiments by exhaustive one-electron reduction of 16+ and 26+ at a potential of –0.50 V vs. SCE. The spectra so obtained (Fig. 1) display the characteristic absorption bands, at ≈390 and 600 nm, of the one-electron reduced form of 4,4′-bipyridinium (A+1) moiety. There is no evidence for the formation of the one-electron reduced form of 3,3′-dimethyl-4,4′-bipyridinium (A+2), which has a substantially different absorption spectrum.
Fig. 1.

UV-visible difference absorption spectra obtained upon electrochemical reduction of 16+ (solid line) and 26+ (dashed line) at –0.50 V vs. SCE in a spectroelectrochemical thin-layer cell (5.0 × 10–4 mol·liter–1 acetonitrile solution at room temperature). The spectra were normalized to the intense band peaking at 392 nm for 16+.(Inset) Detail of the spectra in the 370–405 nm region.
Photochemical Switching. The [Ru(bpy)3]2+-type unit P2+ (Scheme 1) is capable of playing the role of a photosensitizer for ring shuttling because visible light excitation leads to the formation of a long-lived strongly reducing excited state. Except for luminescence quantum yield and lifetime (see below), the photophysical and electrochemical properties of the photosensitizer P2+ unit contained in 16+ and 26+ are the same as those exhibited by the model compound 32+: absorption, λmax = 458 nm, εmax = 14,800 liters·mol–1·cm–1; emission, λmax = 618 nm, τ = 880 ns; Φem = 0.065; excited-state energy ≈2.1 eV (1 eV = 1.602 × 10–19 J); E1/2(P2+/P+) =+1.15 V vs. SCE. It is worthwhile recalling that the P2+ unit can harvest sunlight quite efficiently because it displays a broad and intense absorption band in the visible region.
The overall mechanism devised to perform the light-driven shuttling process in the rotaxane 16+ is based on the following four phases (Scheme 2a).
Scheme 2.

Mechanisms of the photochemically driven ring shuttling in 16+. (a) Intramolecular mechanism. (b) Shuttling assisted by an electron relay. (c) Graphical representation of how the energy profile of shuttling changes upon reduction of 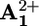 and the location of the ring at
and the location of the ring at 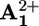 (blue) or
(blue) or 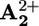 (magenta) for each molecular structure illustrated.
(magenta) for each molecular structure illustrated.
(a) Destabilization of the stable conformation. Light excitation of the photoactive unit P2+ (step 1) is followed by the transfer of an electron from the *P2+ excited state to the A2+1 station, which is encircled by the ring R (step 2), with the consequent “deactivation” of this station; such a photoinduced electron-transfer process has to compete with the intrinsic decay of *P2+ (step 3).
(b) Ring displacement. After reduction (“deactivation”) of the 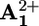 station to
station to 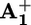 , the ring moves by Brownian motion to
, the ring moves by Brownian motion to 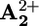 (step 4), a step that has to compete with back electron-transfer from
(step 4), a step that has to compete with back electron-transfer from 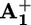 to the oxidized photoactive unit P3+ (step 5). This requirement is the most difficult one to meet because step 4 involves only slightly exergonic nuclear motions, whereas step 5 is an exergonic outer-sphere electron transfer process.
to the oxidized photoactive unit P3+ (step 5). This requirement is the most difficult one to meet because step 4 involves only slightly exergonic nuclear motions, whereas step 5 is an exergonic outer-sphere electron transfer process.
(c) Electronic reset. A back electron-transfer process from the “free” reduced station 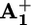 to P3+ (step 6) restores the electron acceptor power of the
to P3+ (step 6) restores the electron acceptor power of the 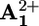 station.
station.
(d) Nuclear reset. As a consequence of the electronic reset, the ring moves back again by Brownian motion from 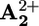 to
to 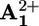 (step 7).
(step 7).
To verify whether such a devised mechanism works, we have performed luminescence quenching and flash photolysis experiments both in the absence and presence of phenothiazine as an electron relay.
Luminescence quenching. In deaerated acetonitrile solution containing the rotaxane 16+ or the dumbbell-shaped compound 26+, the emission lifetimes and intensities of the P2+ units are quenched when compared with the values obtained for the model compound 32+. The occurrence of dynamic quenching can be ruled out because of the high dilution of the solution. As will be clear from the transient absorption data, the luminescence quenching is due to an electron-transfer process from the *P2+ excited state to the 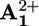 unit. The luminescence lifetimes and quantum yields for the electron transfer process at different temperatures are gathered in Table 3, which is published as supporting information on the PNAS web site. The values for the electron transfer rate constant ket are reported in Table 1. Flash photolysis. Upon flash excitation of 16+, transient absorption spectral changes were observed (see Fig. 5, which is published as supporting information on the PNAS web site). The transient spectrum 6 μs after light excitation displayed the features of the monoreduced 4,4′-bipyridinium unit
unit. The luminescence lifetimes and quantum yields for the electron transfer process at different temperatures are gathered in Table 3, which is published as supporting information on the PNAS web site. The values for the electron transfer rate constant ket are reported in Table 1. Flash photolysis. Upon flash excitation of 16+, transient absorption spectral changes were observed (see Fig. 5, which is published as supporting information on the PNAS web site). The transient spectrum 6 μs after light excitation displayed the features of the monoreduced 4,4′-bipyridinium unit 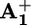 (for the monoreduced form of the 1,1′-dibenzyl-4,4′-bipyridinium model compound in acetonitrile, λmax = 402 nm and εmax = 35,000 liters·mol–1·cm–1) and some residual bleaching in the spectral region of the ground state absorption of P2+. There is no evidence of formation of the monoreduced 3,3′-dimethyl-4,4′-bipyridinium unit, A+2, whose spectrum is substantially different from that of
(for the monoreduced form of the 1,1′-dibenzyl-4,4′-bipyridinium model compound in acetonitrile, λmax = 402 nm and εmax = 35,000 liters·mol–1·cm–1) and some residual bleaching in the spectral region of the ground state absorption of P2+. There is no evidence of formation of the monoreduced 3,3′-dimethyl-4,4′-bipyridinium unit, A+2, whose spectrum is substantially different from that of 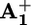 . These results indicate that the species formed upon light excitation of 16+ is a compound in which an electron has been transferred from the *P2+ excited state to the
. These results indicate that the species formed upon light excitation of 16+ is a compound in which an electron has been transferred from the *P2+ excited state to the 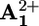 unit. The same results have been obtained for dumbbell 26+.
unit. The same results have been obtained for dumbbell 26+.
Table 1. Kinetic data for photoinduced processes of 16+ in deaerated acetonitrile solution in the temperature range 284–303 K.
| T, K | ket, s-1 | kbet, s-1 | kptz, liter·mol-1·s-1 | krd, s-1 | τbet, μs | τrd, μs |
|---|---|---|---|---|---|---|
| 284 | 1.5 × 105 (1.8 × 105) | 1.4 × 105 (5.4 × 105) | 7.9 × 108 (5.0 × 108) | 8.1 × 102 | 7.4 (1.8) | 1,230 |
| 288 | 1.7 × 105 (2.0 × 105) | 1.4 × 105 (5.9 × 105) | 8.4 × 108 (5.0 × 108) | 1.8 × 103 | 7.1 (1.7) | 550 |
| 293 | 1.7 × 105 (2.1 × 105) | 1.4 × 105 (6.7 × 105) | 7.6 × 108 (4.9 × 108) | 3.5 × 103 | 7.0 (1.5) | 285 |
| 299 | 1.8 × 105 (2.4 × 105) | 1.4 × 105 (7.0 × 105) | 7.8 × 108 (4.8 × 108) | 1.3 × 104 | 7.1 (1.4) | 78 |
| 303 | 2.5 × 105 (3.1 × 105) | 1.5 × 105 (6.9 × 105) | 8.1 × 108 (4.8 × 108) | 2.1 × 104 | 6.7 (1.4) | 47 |
The corresponding data for 26+ are reported in parentheses. Subscripts are defined as follows: et, photoinduced electron transfer (Scheme 2a, step 2); bet, back electron transfer (Scheme 2a, steps 5 and 6); ptz, bimolecular electron transfer between the A1+ unit and ptz+ (Scheme 2b, steps 9 and 10); rd, displacement of the ring from A1+ to the A22+ station corresponding to the shift of the transient absorption maximum of the A1+ unit (Scheme 2 a and b, step 4).
In the case of model compound 32+, where photoinduced electron transfer cannot take place because it does not contain any electron acceptor unit, the observed spectral changes were consistent with the formation of the excited state of the P2+ unit (see Fig. 6, which is published as supporting information on the PNAS web site). The decay time of the transient absorption spectrum was in full agreement with the luminescence decay. An isosbestic point between the absorption spectra of the ground and excited states of the P2+ unit in such a model compound was found at λiso = 398 nm. This observation and the fact that the spectrum of 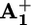 possesses a very intense band peaking in the same region (Fig. 1) allowed us to monitor the formation and decay of the
possesses a very intense band peaking in the same region (Fig. 1) allowed us to monitor the formation and decay of the 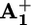 unit in the case of 16+ and 26+, and hence to measure the rate constants for the forward (*P2+ to
unit in the case of 16+ and 26+, and hence to measure the rate constants for the forward (*P2+ to 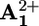 , step 2; Scheme 2a) and the back (
, step 2; Scheme 2a) and the back (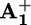 to P3+, steps 5 and 6; see below) electron transfer processes. Typical transient absorption kinetics recorded at λiso are shown in Fig. 7, which is published as supporting information on the PNAS web site.
to P3+, steps 5 and 6; see below) electron transfer processes. Typical transient absorption kinetics recorded at λiso are shown in Fig. 7, which is published as supporting information on the PNAS web site.
The changes of the transient absorption, monitored at λiso, as a function of time were measured at five different temperatures (the exact position of λiso was determined on model compound 32+ at each temperature). In all cases, the rise time of the transient absorption was in agreement, within experimental error, with the lifetime of the excited state of the P2+ component (Table 3), and the transient absorption signal completely disappeared with the first-order rate constants, kbet reported in Table 1. We have also observed that both 16+ and 26+ do not undergo any photodegradation after 103 light pulses. The quantum yields of the photoinduced electron transfer processes obtained from transient absorption data are in agreement, within experimental error, with those found from luminescence measurements.
In summary, the luminescence and transient absorption data demonstrate that visible excitation of the P2+ component in 16+ and 26+ causes an electron transfer process (ket) to the 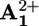 unit, followed by a back electron transfer (kbet) leading to the ground state according to a monoexponential law (Table 2).
unit, followed by a back electron transfer (kbet) leading to the ground state according to a monoexponential law (Table 2).
Table 2. Values of the efficiency of ring displacement, ηrd, and quantum yield of ring shuttling, Φsh, for 16+ in the temperature range 284–303 K.
| Intramolecular mechanism (Scheme 2a)
|
Mechanism with assistance of phenothiazine (Scheme 2b)
|
||||||
|---|---|---|---|---|---|---|---|
| T, K | k4, s-1 | k5, s-1 | ηrd | Φsh | k9[ptz+]0, s-1 | ηrd | Φsh |
| 284 | 8.1 × 102 | 1.4 × 105 | <0.01 | <0.001 | 2.5 × 103 | 0.25 | 0.04 |
| 288 | 1.8 × 103 | 1.4 × 105 | 0.01 | 0.002 | 3.2 × 103 | 0.36 | 0.06 |
| 293 | 3.5 × 103 | 1.4 × 105 | 0.025 | 0.004 | 3.3 × 103 | 0.51 | 0.08 |
| 299 | 1.3 × 104 | 1.3 × 105 | 0.09 | 0.015 | 5.1 × 103 | 0.72 | 0.12 |
| 303 | 2.1 × 104 | 1.3 × 105 | 0.14 | 0.02 | 6.7 × 103 | 0.76 | 0.12 |
Conditions were as follows: deaerated acetonitrile solution, and, for the assisted mechanism of Scheme 2b, [ptz] = 5.0 × 10-5 mol·liter-1.
In the rotaxane 16+ the back electron transfer can take place by either step 5 or 6 (Scheme 2a), depending on the rate of ring displacement (step 4). If step 5 is much faster than step 4, the photoinduced electron transfer is simply followed by a back electron transfer, without any ring motion. If, on the contrary, step 4 is much faster than step 5, the photoinduced electron transfer causes the displacement of the ring R from the 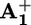 station to
station to 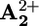 . In such a case, back electron transfer (now step 6) leads subsequently to the unstable isomer of 16+ (D in Scheme 2a) which relaxes thermally (step 7) to give back the starting conformation. If the rates of steps 4 and 5 are not much different from each other, both steps 5 and 6 contribute to the back electron transfer process. The results discussed above showed that steps 1, 2, 3, and 5 and/or 6 take place, but we were not able to establish whether the photoinduced ring displacement, step 4, occurs (Scheme 2a). This problem has been tackled as discussed below.
. In such a case, back electron transfer (now step 6) leads subsequently to the unstable isomer of 16+ (D in Scheme 2a) which relaxes thermally (step 7) to give back the starting conformation. If the rates of steps 4 and 5 are not much different from each other, both steps 5 and 6 contribute to the back electron transfer process. The results discussed above showed that steps 1, 2, 3, and 5 and/or 6 take place, but we were not able to establish whether the photoinduced ring displacement, step 4, occurs (Scheme 2a). This problem has been tackled as discussed below.
Experiments in the presence of phenothiazine as an electron relay. We have used an electron relay to slow down the back electron transfer process (step 5) to observe the ring displacement (step 4) and measure its rate.
A compound X capable of playing the role of an electron relay in our system must satisfy several requirements: (i) it should not quench the *P2+ excited state by energy transfer; (ii) its oxidation process must be chemically reversible; (iii) its X+/X redox potential must be less positive than that of P3+/P2+ (+1.15 V vs. SCE) to scavenge P3+; and (iv) both X and X+ should not absorb the light used to cause the photoreaction and to monitor its occurrence. After trying and discarding several compounds (DABCO, diphenylamine and triphenylamine), we chose phenothiazine, ptz, which is reversibly oxidized [E1/2(ptz+/ptz) = +0.60 V vs. SCE] (30) and does not show absorption bands in the visible region. Furthermore, its radical cation ptz+ exhibits a band at 520 nm (31) that does not interfere with the spectrum of 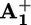 and also can be useful for monitoring the behavior of the electron relay during the process.
and also can be useful for monitoring the behavior of the electron relay during the process.
When photoexcitation of 16+ is performed in the presence of ptz (Scheme 2b), the photoinduced electron transfer (step 2) is expected to be followed by a diffusion-controlled reaction between ptz and P3+ with formation of ptz+ and regeneration of P2+ (step 8). Therefore, intramolecular back electron transfer (step 5) should no longer occur, and ring displacement (step 4) should successfully compete with the much slower intermolecular back electron transfer reaction between ptz+ and 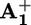 (step 9).
(step 9).
Flash-photolysis experiments of solutions containing 16+ or 26+ (1.0 × 10–4 mol·liter–1) and phenothiazine (5.0 × 10–5 mol·liter–1) at all of the examined temperatures showed the presence of the characteristic bands of 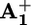 at ≈390 and 600 nm and of ptz+ at 520 nm (Fig. 2). The transient absorption signals at 600, 520, and 390 nm completely disappeared according to identical second-order processes occurring in the millisecond time scale (Table 1 and Fig. 2 Inset). Therefore, under such conditions the back electron transfer reaction is much slower (from 2 to 3 orders of magnitude) than in the absence of ptz, thereby leaving more time for ring displacement.
at ≈390 and 600 nm and of ptz+ at 520 nm (Fig. 2). The transient absorption signals at 600, 520, and 390 nm completely disappeared according to identical second-order processes occurring in the millisecond time scale (Table 1 and Fig. 2 Inset). Therefore, under such conditions the back electron transfer reaction is much slower (from 2 to 3 orders of magnitude) than in the absence of ptz, thereby leaving more time for ring displacement.
Fig. 2.

Transient UV-visible absorption difference spectrum of a deaerated acetonitrile solution containing 1.0 × 10–4 mol·liter–1 16+ and 5.0 × 10–5 mol·liter–1 ptz, recorded 50 μs after 532-nm laser excitation at room temperature. (Inset) Normalized transient absorption decays monitored at 520 (solid line) and 600 (dotted line) nm, which correspond to signals assigned to ptz+ and 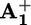 , respectively.
, respectively.
To measure the rate of ring displacement, we carefully analyzed the time evolution of the shape of the 390-nm transient absorption band of the reduced station 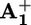 (see Supporting Text and Figs. 8–10, which are published as supporting information on the PNAS web site). We found that the maximum of such a band in 15+ undergoes a slight red shift upon decay, whereas it does not change for 25+ (Fig. 3), which has the same structural elements of the rotaxane, except for the “movable” ring, and features all of the processes that allow the operation of the motor but the motion of the ring itself. The shift of the maximum of the
(see Supporting Text and Figs. 8–10, which are published as supporting information on the PNAS web site). We found that the maximum of such a band in 15+ undergoes a slight red shift upon decay, whereas it does not change for 25+ (Fig. 3), which has the same structural elements of the rotaxane, except for the “movable” ring, and features all of the processes that allow the operation of the motor but the motion of the ring itself. The shift of the maximum of the 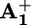 band in 15+ that accompanies the transient signal decay thus can be related to the displacement of the ring from
band in 15+ that accompanies the transient signal decay thus can be related to the displacement of the ring from 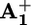 to
to 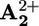 (krd, step 4 in Scheme 2a). This assumption is supported by the fact that, 850 μs after light excitation (303 K), the transient UV band in the case of 16+ is slightly red-shifted compared with that observed for 26+ (Fig. 3) as observed in the spectroelectrochemical experiments between the one electron reduced form 15+ (in which the ring encircles
(krd, step 4 in Scheme 2a). This assumption is supported by the fact that, 850 μs after light excitation (303 K), the transient UV band in the case of 16+ is slightly red-shifted compared with that observed for 26+ (Fig. 3) as observed in the spectroelectrochemical experiments between the one electron reduced form 15+ (in which the ring encircles 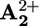 ) and 25+ (Fig. 1).
) and 25+ (Fig. 1).
Fig. 3.

Time dependence of the wavelength of the absorption maximum (λmax) of the UV band of photogenerated 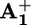 for 16+ (filled circles) and 26+ (open circles) at 303 K. The full line represents the monoexponential data fit used to obtain krd for 16+.
for 16+ (filled circles) and 26+ (open circles) at 303 K. The full line represents the monoexponential data fit used to obtain krd for 16+.
A kinetic analysis (see ref. 14 and Supporting Text) has shown that the shift of the band maximum (i.e., the ring displacement) takes place according to a monoexponential law (Fig. 3) with a rate constant, krd, that rises with increasing temperature (Table 1; see also Fig. 11, which is published as supporting information on the PNAS web site). The free energy of activation for the ring displacement is ≈50 kJ·mol–1 at 298 K. The relatively small values of the ring displacement rate constants suggest that such a movement requires considerable structural rearrangements, possibly including unfolding processes. Under the experimental conditions used, both 16+ or 26+ showed a reversible behavior, with no change in the steady-state absorption and luminescence spectra after 103 laser pulses.
In conclusion, in the presence of ptz the photoinduced electron transfer reaction (step 2) is unaffected (for more details, see Supporting Text; see also Fig. 12, which is published as supporting information on the PNAS web site), P3+ is completely scavenged by ptz (step 8), ring displacement takes place (step 4), and both electron (steps 9 and/or 10) and nuclear (step 7) reset processes occur quantitatively because the system can undergo at least 103 cycles. Therefore 16+ behaves as an autonomous molecular motor that consumes only photons of visible light (Scheme 2b). The role played by ptz is that of an electron relay with a “negative” kinetic effect.
Intramolecular mechanism. As reported above, by using ptz as an electron relay we have obtained the rate (krd; Table 2) of ring displacement (step 4) from the photogenerated 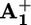 station to the
station to the 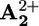 station. Therefore, we can go back to the discussion about what happens in the absence of ptz (Scheme 2a). To understand whether ring displacement takes place also in the case of the intramolecular mechanism, we must compare the values of k4 (i.e., krd) and k5, the rate of the back electron-transfer process from the
station. Therefore, we can go back to the discussion about what happens in the absence of ptz (Scheme 2a). To understand whether ring displacement takes place also in the case of the intramolecular mechanism, we must compare the values of k4 (i.e., krd) and k5, the rate of the back electron-transfer process from the 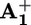 station, still encircled by the ring, to P3+. The kinetic treatment and more details are given in Supporting Text and Schemes 3 and 4, which are published as supporting information on the PNAS web site. Contrary to what happens when ptz is present, in the case of the intramolecular mechanism the exact position of
station, still encircled by the ring, to P3+. The kinetic treatment and more details are given in Supporting Text and Schemes 3 and 4, which are published as supporting information on the PNAS web site. Contrary to what happens when ptz is present, in the case of the intramolecular mechanism the exact position of 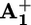 absorption maximum and, if any, its shift during the signal decay cannot be established because of interference by the absorption spectra of *P2+ and P3+. However, we can estimate the rate constant of step 6 from the experiments performed on the dumbbell component 26+ (kbet = k6), and we can reasonably assume that krd is the same as that observed for the ptz-assisted mechanism. From Table 2 we can see that k6 (i.e., kbet for 26+) is at least 35 times larger than krd, so that conformer C has to disappear (step 6) as soon as it is formed (step 4). Therefore, in 16+ kbet = k5 + krd. At 303 K (Table 1) k5 = 1.5 × 105 – 2.1 × 104 = 1.3 × 105 s–1, which means that the displacement of the ring (step 4) competes (14%) with the intramolecular back electron transfer (step 5, Scheme 2a). In conclusion, also in the absence of ptz the shuttling process does take place with appreciable efficiency at 303 K. The drawback of the low efficiency is compensated by the fact that the operation of the system relies exclusively on intramolecular processes. This artificial molecular motor does not rely on consumption or even assistance of external species; it is stable for at least 103 cycles, and it operates by simply exposing the solution to sunlight.
absorption maximum and, if any, its shift during the signal decay cannot be established because of interference by the absorption spectra of *P2+ and P3+. However, we can estimate the rate constant of step 6 from the experiments performed on the dumbbell component 26+ (kbet = k6), and we can reasonably assume that krd is the same as that observed for the ptz-assisted mechanism. From Table 2 we can see that k6 (i.e., kbet for 26+) is at least 35 times larger than krd, so that conformer C has to disappear (step 6) as soon as it is formed (step 4). Therefore, in 16+ kbet = k5 + krd. At 303 K (Table 1) k5 = 1.5 × 105 – 2.1 × 104 = 1.3 × 105 s–1, which means that the displacement of the ring (step 4) competes (14%) with the intramolecular back electron transfer (step 5, Scheme 2a). In conclusion, also in the absence of ptz the shuttling process does take place with appreciable efficiency at 303 K. The drawback of the low efficiency is compensated by the fact that the operation of the system relies exclusively on intramolecular processes. This artificial molecular motor does not rely on consumption or even assistance of external species; it is stable for at least 103 cycles, and it operates by simply exposing the solution to sunlight.
Shuttling Efficiency. As we have seen above, the results obtained have allowed us to establish the following relationship among the rate constants of the steps involved in the intramolecular and ptz-assisted mechanisms (Scheme 2) and the experimentally determined rate constants (Tables 1, 2, 3): k2 = ket (Table 2); k3 = 1/τ0 (Table 1); k4 = krd (Table 2); k5 = kbet – krd (Table 3); k6 and k10 are obtained from the experimentally measured kbet of 26+ (Table 1); k9 is obtained from Eq. S13 in Supporting Text (Table 2, in which the values of k9[ptz+]0, more easily comparable with k4, are reported). Concerning step 8, its rate constant is diffusion controlled, k8 ≈ 1 × 1010 liters·mol–1·s–1, and under our experimental conditions it is fast enough to fully prevent intramolecular back electron transfer, thereby activating the sequence of processes illustrated in Scheme 2b. Finally, step 7 is thermodynamically allowed, and it does not compete with any other step; the value of its rate constant, k7, which is not relevant for the discussion on quantum yields and efficiencies, is estimated to be 103 s–1 (see Supporting Text).
We can now discuss the performance of 16+ as a photochemically driven molecular motor when it operates according to the mechanisms illustrated in Scheme 2. The quantum yield of the shuttling process (number of ring shuttling events divided by number of absorbed photons) amounts to Φsh = Φet× ηrd× ηer × ηnr, where Φet is the quantum yield of the photoinduced electron transfer process that causes the destabilization of the stable conformation A, ηrd is the efficiency of ring displacement, ηer is the efficiency of the electronic reset, and ηnr is the efficiency of the nuclear reset.
The quantum yield of the photoinduced electron transfer process is the same, regardless of the working mechanism, Φet = k2/(k2+k3), e.g., 0.16 at 303 K (Scheme 2 and Table 3). The efficiency of the electronic reset, ηer, is equal to unity in both mechanisms because back electron transfer takes place in any case (via steps 5 and 6 in the intramolecular mechanism and via steps 9 and 10 in the ptz-assisted one), as shown by lack of any degradation after 103 cycles. The efficiency of the nuclear reset, ηnr, is equal to 1 in all cases because k7 does not compete with any other step. Therefore, the expression of the quantum yield of ring shuttling reduces to Φsh = Φet× ηrd. The efficiency ηrd depends on the operating mechanism and the experimental conditions, as shown in Table 2. The values of Φsh at the various temperatures examined also are gathered in Table 2.
When 16+ works according to the intramolecular mechanism (Scheme 2a), at 303 K ηrd = 0.14, and therefore the quantum yield for the shuttling process, Φsh, is ≈2%. On decreasing temperature, the quantum yield decreases because k4 (i.e., krd) refers to a strongly activated process, whereas (k5 + k4) (i.e., kbet) is almost temperature independent.
When 16+ works with assistance of ptz (Scheme 2b), at 303 K ηrd = 0.76 and Φsh = 12%. On decreasing temperature ηrd decreases, but Φsh is still appreciable (4%) at 284 K. On changing the ptz+ concentration, the values of ηrd and Φsh would of course change.
Energetics. The energy available (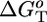 ) for the shuttling, which could, in principle, be used to do mechanical work, can be estimated on the basis of thermodynamic considerations (see Scheme 5, which is published as supporting information on the PNAS web site). The energy levels involved in the photoinduced ring shuttling of 16+ at room temperature are shown in Fig. 4. Altogether, the fraction F of the excited-state energy used for the motion of the ring amounts to ≈10%, and the overall efficiency of light-to-mechanical energy conversion, ηen.conv. = Φsh× F, is ≈1% for the kinetically assisted system and ≈0.2% in the case of the intramolecular mechanism (at 303 K). Considering that the nuclear reset (step 7 in Scheme 2a) is 1 ms, the motor can be driven at a frequency of 103 Hz, potentially generating a power of 3 × 10–17 W per molecule.
) for the shuttling, which could, in principle, be used to do mechanical work, can be estimated on the basis of thermodynamic considerations (see Scheme 5, which is published as supporting information on the PNAS web site). The energy levels involved in the photoinduced ring shuttling of 16+ at room temperature are shown in Fig. 4. Altogether, the fraction F of the excited-state energy used for the motion of the ring amounts to ≈10%, and the overall efficiency of light-to-mechanical energy conversion, ηen.conv. = Φsh× F, is ≈1% for the kinetically assisted system and ≈0.2% in the case of the intramolecular mechanism (at 303 K). Considering that the nuclear reset (step 7 in Scheme 2a) is 1 ms, the motor can be driven at a frequency of 103 Hz, potentially generating a power of 3 × 10–17 W per molecule.
Fig. 4.

Energy level diagram for the processes taking place in rotaxane 16+ when it works as a light-driven molecular motor based on intramolecular processes at room temperature (see Scheme 2a). The energy of a 532-nm photon (225 kJ·mol–1) is used to produce the long-lived excited state of the P2+ component (205 kJ·mol–1, E), which decays by electron transfer to the 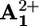 station (B) with 14% yield. An energy of 10 kJ·mol–1 is then released in the Brownian motion of the ring R over the 50 kJ·mol–1 barrier (forward stroke) from
station (B) with 14% yield. An energy of 10 kJ·mol–1 is then released in the Brownian motion of the ring R over the 50 kJ·mol–1 barrier (forward stroke) from 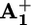 to
to 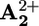 (C). A further 145 kJ·mol–1 is used to transfer the electron from
(C). A further 145 kJ·mol–1 is used to transfer the electron from 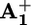 back to P3+ (D), and the remaining 8 kJ·mol–1 is released in the motion of the ring (backward stroke) from
back to P3+ (D), and the remaining 8 kJ·mol–1 is released in the motion of the ring (backward stroke) from 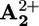 to
to 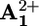 (A).
(A).
Conclusions
We have shown that in rotaxane 16+ displacement of the ring for 1.3 nm between the two stations A2+ and 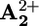 , that are placed along the dumbbell-shaped component, can be obtained by light energy inputs without consumption of chemical fuels and formation of waste products. Such photoinduced ring displacement can occur by (i) a purely intramolecular mechanism and (ii) an intermolecular mechanism, involving the assistance of an external electron relay. The system behaves as an autonomous linear motor powered by visible light, and its operation takes place in four strokes: destabilization of the stable conformation, displacement of the ring, electronic reset, and nuclear reset.
, that are placed along the dumbbell-shaped component, can be obtained by light energy inputs without consumption of chemical fuels and formation of waste products. Such photoinduced ring displacement can occur by (i) a purely intramolecular mechanism and (ii) an intermolecular mechanism, involving the assistance of an external electron relay. The system behaves as an autonomous linear motor powered by visible light, and its operation takes place in four strokes: destabilization of the stable conformation, displacement of the ring, electronic reset, and nuclear reset.
Each phase (Scheme 2a) corresponds, in kind, to the fuel injection and combustion (A), piston displacement (B), exhaust removal (C), and piston replacement (D) of a four-stroke engine. The timing of the strokes is intrinsic to the molecular motor and cannot be tuned up externally as in fuel-injected car engines. The mechanism relies indeed on the competition between the motion of the ring (step 4) and the back electron transfer (step 5). If the latter process is much faster than the former, the photoinduced electron transfer (step 2) is simply followed by a back electron transfer (step 5), with no relative motion involving the ring. Such a situation would be equivalent to injection of fuel and removal before combustion. In the present case, at 303 K, the back electron transfer is 7 times faster than ring displacement, leading to an overall quantum efficiency of the motor of 2%. When the ring shuttling is assisted by the electron relay (Scheme 2b), the “negative” kinetic effect exerted by ptz on the back electron transfer increases the overall quantum efficiency of the motor to 12%.
The low efficiency of this motor when it works in an autonomous way with the assistance of an external relay, and even more so when it works purely with an intramolecular mechanism, may seem disappointing, but it should be noted that the fuel (sunlight) is free. Besides being powered by sunlight and operating as an autonomous motor, the investigated system shows other quite interesting properties: it works in mild environmental conditions, it is remarkably stable, and it can be driven at high frequency (kilohertz). In principle, when working by intramolecular mechanism, it is also suitable for operation at the single-molecule level.
The field of artificial molecular motors is still in its infancy. The construction of an autonomous, sunlight-driven molecular motor was a very ambitious task from the outset. Our results show that the structural and functional integration of suitably selected modular units into carefully designed multicomponent molecular systems is indeed a valuable approach.
Materials and Methods
The syntheses of 16+, 26+, and 32+ have been previously reported (29).
Spectroelectrochemical measurements were performed in situ with a custom-made optically transparent thin-layer electrochemical cell by using a diode array spectrophotometer. The working and counter electrodes were Pt minigrids, and the quasi-reference electrode was an Ag wire; all three electrodes were melt-sealed into a polyethylene spacer.
Luminescence decay and transient absorption experiments were performed in degassed acetonitrile solutions by exciting the sample with 10 ns (full width at half maximum) pulses from the second harmonic (532 nm) of a Nd:YAG laser. In the transient absorption studies, a Xe lamp perpendicular to the laser beam was used as a probing light. The absorption transient decays were plotted as ΔA = log(I0/It) vs. time, where I0 and It were the probing light intensity before the laser pulse and after delay t, respectively. Transient absorption spectra were obtained from the decays measured at various wavelengths (5-nm increment), by sampling the absorbance changes at constant delay time. In the determination of the shift of λmax, a wavelength increment of 1 nm in the 350–440 nm region was adopted; to obtain a significant comparison between the bands of the rotaxane and of its dumbbell-shaped component, the decay at each wavelength was measured in parallel for otherwise identical solutions of 16+ and 26+. The same laser/monochromator/phototube setup was used to measure luminescence lifetimes. The accuracy on the wavelength values was estimated to be ±2 nm. The experimental error was 4% for the lifetime values and 20% for the quantum yield of the photoinduced electron transfer processes. More details can be found in Supporting Text.
Supplementary Material
Acknowledgments
We thank Dr. Lucia Flamigni for useful discussion. This work was supported by the Ministry of Education, University and Research in Italy and by the National Science Foundation. The European Union provided support under the auspices of the Biomach project and a Marie Curie Individual Fellowship (to B.F.).
Author contributions: V.B., A.C., M.V., and J.F.S. designed research; M.C.-L. and B.F. performed research; V.B., A.C., B.F., M.V., and A.H.F. analyzed data; and V.B. and A.C. wrote the paper.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: SCE, saturated calomel electrode.
References
- 1.Balzani, V., Credi, A., Raymo, F. M. & Stoddart, J. F. (2000) Angew. Chem. Int. Ed. 39, 3348–3391. [DOI] [PubMed] [Google Scholar]
- 2.Stoddart, J. F., ed. (2001) Acc. Chem. Res. 34 (6), special issue, 409–522. [Google Scholar]
- 3.Sauvage, J.-P., ed. (2001) Struct. Bond. 99, special volume, 1–281. [Google Scholar]
- 4.Balzani, V., Credi, A. & Venturi, M. (2003) Molecular Devices and Machines: A Journey into the Nano World (Wiley-VCH, Weinheim, Germany).
- 5.Kelly, T. R., ed. (2005) Top. Curr. Chem. 262, special volume, 1–227. [Google Scholar]
- 6.Schliwa, M., ed. (2002) Molecular Motors (Wiley-VCH, Weinheim, Germany).
- 7.Oster, G. & Wang, H. (2003) Trends Cell Biol. 13, 114–121. [DOI] [PubMed] [Google Scholar]
- 8.Goodsell, D. S. (2004) Bionanotechnology: Lessons from Nature (Wiley, New York).
- 9.Jones, R. A. L. (2005) Soft Machines: Nanotechnology and Life (Oxford Univ. Press, Oxford).
- 10.Lehn, J.-M. (2002) Proc. Natl. Acad. Sci. USA 99, 4763–4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steinberg-Yfrach, G., Rigaud, J.-L., Durantini, E. N., Moore, A. L., Gust, D. & Moore, T. A. (1998) Nature 392, 479–482. [DOI] [PubMed] [Google Scholar]
- 12.Kelly, T. R., De Silva, H. & Silva, R. A. (1999) Nature 401, 150–152. [DOI] [PubMed] [Google Scholar]
- 13.Koumura, N., Zijlstra, R. W., van Delden, R. A., Harada, N. & Feringa, B. L. (1999) Nature 401, 152–155. [DOI] [PubMed] [Google Scholar]
- 14.Brouwer, A. M. Frochot, C., Gatti, F. G., Leigh, D. A., Mottier, L., Paolucci, F., Roffia, S. & Wurpel, G. W. H. (2001) Science 291, 2124–2128. [DOI] [PubMed] [Google Scholar]
- 15.Badjic, J. D., Balzani, V., Credi, A., Silvi, S. & Stoddart, J. F. (2004) Science 303, 1845–1849. [DOI] [PubMed] [Google Scholar]
- 16.Hernandez, J. V., Kay, E. R. & Leigh, D. A. (2004) Science 306, 1532–1537. [DOI] [PubMed] [Google Scholar]
- 17.Mobian, P., Kern, J.-M. & Sauvage, J.-P. (2004) Angew. Chem. Int. Ed. 43, 2392–2395. [DOI] [PubMed] [Google Scholar]
- 18.Sherman, W. B. & Seeman, N. C. (2004) Nano Lett. 4, 1203–1207. [Google Scholar]
- 19.Astumian, R. D. (2005) Proc. Natl. Acad. Sci. USA, 102, 1843–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng, X. L. Mulcahy, M. E., Horinek, D., Galeotti, F., Magnera, T. F. & Michl, J. (2004) J. Am. Chem. Soc. 126, 4540–4542. [DOI] [PubMed] [Google Scholar]
- 21.Katz, E., Lioubashevsky, O. & Willner, I. (2004) J. Am. Chem. Soc. 126, 15520–15532. [DOI] [PubMed] [Google Scholar]
- 22.van Delden, R. A., ter Wiel, M. K. J., Pollard, M. M., Vicario, J., Koumura, N. & Feringa, B. L. (2005) Nature 437, 1337–1340. [DOI] [PubMed] [Google Scholar]
- 23.Liu, Y., Flood, A. H., Bonvallet, P. A., Vignon, S. A., Northrop, B. H., Tseng, H.-R., Jeppesen, J. O., Huang, T. J., Brough, B., Baller, M., et al. (2005) J. Am. Chem. Soc. 127, 9745–9759. [DOI] [PubMed] [Google Scholar]
- 24.Nguyen, T. D., Tseng, H.-R., Celestre, P. C., Flood, A. H., Liu, Y., Stoddart, J. F. & Zink, J. I. (2005) Proc. Natl. Acad. Sci. USA 102, 10029–10034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berná, J., Leigh, D. A., Lubomska, M., Mendoza, S. M., Pérez, E. M., Rudolf, P., Teobaldi, G. & Zerbetto, F. (2005) Nat. Mater. 4, 704–710. [DOI] [PubMed] [Google Scholar]
- 26.Tian, Y. & Mao, C. D. (2004) J. Am. Chem. Soc. 126, 11410–11411. [DOI] [PubMed] [Google Scholar]
- 27.Tian, Y., He, Y., Chen, Y., Yin, P. & Mao, C. D. (2005) Angew. Chem. Int. Ed. 44, 4355–4358. [DOI] [PubMed] [Google Scholar]
- 28.Bath, J., Green, S. J. & Turberfield, A. J. (2005) Angew. Chem. Int. Ed. 44, 4358–4361. [DOI] [PubMed] [Google Scholar]
- 29.Ashton, P. R., Ballardini, R., Balzani, V., Credi, A., Dress, K. R., Ishow, E., Kleverlaan, C. J., Kocian, O., Preece, J. A., Spencer, N., et al. (2000) Chem. Eur. J. 6, 3558–3574. [DOI] [PubMed] [Google Scholar]
- 30.Kowert, B. A., Marcoux, L. & Bard, A. J. (1972) J. Am. Chem. Soc. 94, 5538–5550. [Google Scholar]
- 31.Hanson, P. & Norman, R. O. C. (1973) J. Chem. Soc. Perkin Trans. 2, 264–271. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.