

Abstract
Understanding which cytosolic domains of the dihydropyridine receptor participate in excitation-contraction (EC) coupling is critical to validate current structural models. Here we quantified the contribution to skeletal-type EC coupling of the α1S (CaV1.1) II-III loop when alone or in combination with the rest of the cytosolic domains of α1S. Chimeras consisting of α1C (CaV1.2) with α1S substitutions at each of the interrepeat loops (I-II, II-III, and III-IV loops) and N- and C-terminal domains were evaluated in dysgenic (α1S-null) myotubes for phenotypic expression of skeletal-type EC coupling. Myotubes were voltage-clamped, and Ca2+ transients were measured by confocal line-scan imaging of fluo-4 fluorescence. In agreement with previous results, the α1C/α1S II-III loop chimera, but none of the other single-loop chimeras, recovered a sigmoidal fluorescence-voltage curve indicative of skeletal-type EC coupling. To quantify Ca2+ transients in the absence of inward Ca2+ current, but without changing the external solution, a mutation, E736K, was introduced into the P-loop of repeat II of α1C. The Ca2+ transients expressed by the α1C(E736K)/α1S II-III loop chimera were ∼70% smaller than those expressed by the Ca2+-conducting α1C/α1S II-III variant. The low skeletal-type EC coupling expressed by the α1C/α1S II-III loop chimera was confirmed in the Ca2+-conducting α1C/α1S II-III loop variant using Cd2+ (10−4 M) as the Ca2+ current blocker. In contrast to the behavior of the II-III loop chimera, Ca2+ transients expressed by an α1C/α1S chimera carrying all tested skeletal α1S domains (all α1S interrepeat loops, N- and C-terminus) were similar in shape and amplitude to wild-type α1S, and did not change in the presence of the E736K mutation or in the presence of 10−4 M Cd2+. Controls indicated that similar dihydropyridine receptor charge movements were expressed by the non-Ca2+ permeant α1S(E1014K) variant, the α1C(E736K)/α1S II-III loop chimera, and the α1C(E736K)/α1S chimera carrying all tested α1S domains. The data indicate that the functional recovery produced by the α1S II-III loop is incomplete and that multiple cytosolic domains of α1S are necessary for a quantitative recovery of the EC-coupling phenotype of skeletal myotubes. Thus, despite the importance of the II-III loop there may be other critical determinants in α1S that influence the efficiency of EC coupling.
INTRODUCTION
Ca2+ signals of skeletal muscle cells are controlled by the voltage-gated L-type Ca2+ channel formed by the dihydropyridine receptor (DHPR), and by the sarcoplasmic reticulum (SR) Ca2+ release channel formed by the ryanodine receptor type 1 (RyR1). The well-established paradigm is that in response to depolarization, the DHPR produces a signal that briefly opens the RyR1 channel, leading to the release of SR-stored Ca2+. Signal transmission takes place at specialized junctions between transverse tubules and SR membranes. At these junctions, DHPRs in tetrad arrangement juxtapose a single tetrameric RyR1 channel (1–3). The narrow physical gap between the transverse tubule and the SR (∼12 nm) (2), the large protrusion of foot structure of the RyR1 channel into the gap (∼7 nm) (4), and the overall molecular dimensions of the DHPR and RyR1 complexes (5,6), all suggest that the DHPR and RyR1 channels must be in physical contact. Strong evidence for the formation of a DHPR-RyR1 complex in myotubes is provided by a recent freeze-fracture analysis of the molecular determinants that specify the arrangement of DHPRs in arrays of tetrads opposite to RyR1 (7).
The α1S subunit (CaV1.1) of the skeletal DHPR has the familiar topology of a four-repeat voltage-gated channel with five cytosolic domains adjoining the four repeats (N-terminus, I-II loop, II-III loop, III-IV loop, and C-terminus) (8–10). Reports made almost 15 years ago (11,12) and refined later (13–17), have suggested that the cytosolic loop linking repeats II and III, consisting of 132 residues, brings about the conformational change that opens the RyR1 channel under the influence of membrane depolarization. The II-III loop is commonly viewed as the cell's version of the mechanical plunger proposed by Chandler et al. (18), in which an element of the transverse tubule linked to the excitation-contraction (EC) -coupling voltage sensor exerts torque on the SR Ca2+ channel. The II-III loop model of EC coupling is simple, has intuitive appeal, and has received broad consideration. However, in reality, the signaling mechanism may be more complex. The prevailing evidence indicates that interactions between α1S and RyR1 are likely to involve the II-III loop, but also many other domains of α1S (7,19–24). Additional complexity is brought about by a deletion analysis showing that the II-III loop may not account entirely for the signaling function of the DHPR (25). Furthermore, the DHPR β1a subunit is essential for skeletal-type EC coupling (26–28), and interactions between this subunit and RyR1 are almost certain (6,29). Hence, the molecular determinants of the voltage-dependent mechanism by which the DHPR activates RyR1 may be broader than initially anticipated by the II-III loop model and are still open to debate despite unrelenting efforts.
In light of the growing multiplicity of DHPR-RyR1 interactions, here we reinvestigated the contribution of the cytosolic domains of α1S to skeletal-type EC coupling in dysgenic (α1S null) myotubes. Previous studies have focused almost exclusively on the functional identity of regions within the skeletal II-III loop (14,16,17,25,30,31). However, a voltage-clamp analysis of the EC-coupling phenotype contributed by each of the α1S interrepeat loops, as well as by the N- and C-terminal domains, has not been previously conducted. Likewise, the phenotype contributed by all the cytosolic domains together, as they would be present in the intact subunit, has not been documented. Since α1C does not express skeletal-type EC coupling (14,32), we focused on chimeras consisting of α1C (CaV1.2) with sequences from α1S (CaV1.1), substituting the interrepeat loops (I-II, II-III, or III-IV) or the N- or C-terminus . We report that Ca2+ entry-independent Ca2+ transients of a magnitude and voltage dependence similar to those expressed by wild-type (WT) α1S required a chimera with multiple α1S domains, including the II-III loop. However, the chimera with the II-III loop alone was insufficient. Some of these results have been published in abstract form (33,34).
MATERIALS AND METHODS
Identification of genotypes
Dysgenic (α1S-null mdg) mice were screened for both wild-type and mutant alleles of the DHPR α1S gene. Digestion of tail samples and subsequent verification of genotypes by the polymerase chain reaction (PCR) were described previously (27).
Primary cultures and cDNA transfection
Cultures of myotubes were prepared from hind limbs of E18 fetuses, as described previously (35). Cultures were grown at 37°C in 8% CO2 gas. After myoblast fusion (∼5 days), the medium was replaced with FBS-free medium, and CO2 was decreased to 5%. cDNA transfection was performed during the myoblast fusion stage with the polyamine LT1 (Panvera, Madison, WI). In addition to the cDNA of interest, cells were cotransfected with a plasmid encoding the T-cell protein CD8, which is used as a transfection marker. Transfected myotubes expressing CD8 were recognized by surface binding of polystyrene beads coated with anti-CD8 antibody (Dynal ASA, Oslo, Norway). Whole-cell analysis of Ca2+ currents, charge movements, and Ca2+ transients was performed 3–5 days post-transfection. The numbers of separate myotube cultures that were transfected and from which data were collected were as follows for Ca2+ current, Ca2+ transient, and charge movements when applicable: for nontransfected mdg, 10, 27, and 2; for WT α1S, 7, 3, and none; for α1S(E1014K), 3, 4, and 2; for WT α1C, 5, 3, and none; for α1C(E736K), 3, 2, and 2; for α1C/α1S N, 3, 3, and none; for α1C/α1S I-II, 4, 4, and none; for α1C/α1S II-III, 6, 4, and none; for α1C(E736K)/α1S II-III, 3, 2, and 3; for α1C/α1S III-IV, 4, 3, and none; for α1C/α1S C, 3, 2, and none; for α1C/α1S all loop, 3, 3, and none; for α1C(E736K)/α1S all loop: 3, 3, and 3; for all cadmium experiments, 2, 2, and none.
cDNA constructs
Chimeric variants were made by two-step PCR strategies using cDNAs for rabbit skeletal muscle α1S (residues 1–1873; Genbank No. M23919) and rabbit cardiac α1C (residues 1–2171; Genbank No. X15539). The PCR products were subcloned into pCR-Blunt vector (Invitrogen, Carlsbad, CA), excised by digestion with AgeI/NotI, and cloned into the pSG5 vector (Stratagene, San Diego, CA) in frame with the first 11 residues of the phage T7 gene 10 protein for antibody tagging. All constructs were sequenced twice or more at a campus facility.
Domain boundaries and nomenclature
Alignment of α1S and α1C sequences was performed with DNASTAR (Madison, WI) using the Jotun-Hein method. α1S N corresponds to residues 1–51 and replaced α1C N residues 1–154; α1S I-II loop corresponds to residues 335–432 and replaced α1C I-II loop residues 436–554; α1S II-III loop corresponds to residues 667–799 and replaced α1C II-III loop residues 789–930; α1S III-IV loop corresponds to residues 1067–1120 and replaced α1C III-IV loop residues 1188–1241; α1S C corresponds to residues 1328–1873 and replaced α1C C residues 1507–2171.
α1C/α1S N
This chimera consists of α1S residues M1–K51 fused to the N-terminus of α1C residues P155–L2171. The N-terminus of α1S was amplified from full-length α1S and corresponds to residues 1–51. The second-step PCR product containing the α1S N-terminus and part of α1C domain I was fused to the pSG5 α1C vector using NheI/SacI sites.
α1C/α1S I-II
This chimera consists of α1C residues M1–S435 fused to the N-terminus of α1S residues G335–R432 fused to the N-terminus of α1C residues V555–L2171. The I-II loop of α1S was amplified from full-length α1S and corresponds to residues 335–432. The second-step PCR product containing the α1S I-II loop and part of α1C domains I and II was fused to the pSG5 α1C vector using BamHI/XhoI sites.
α1C/α1S II-III
This chimera consists of α1C residues M1–D788 fused to the N-terminus of α1S residues A667–T799 fused to the N-terminus of α1C residues I931–L2171. To replace the II-III loop, a HindIII site at nucleotide 2561 and a SpeI site at nucleotide 3203 were introduced into full-length α1C as silent mutations. The II-III loop of α1S was amplified from full-length α1S, and corresponds to residues 667–799. The second-step PCR product containing the α1S loop and part of α1C domain III was fused to the pSG5 α1C vector using HindIII/SpeI sites.
α1C/α1S III-IV
This chimera consists of α1C residues M1–V1187 fused to the N-terminus of α1S residues T1067–F1120 fused to the N-terminus of α1C residues E1242–L2171. The III-IV loop of α1S was amplified from full-length α1S and corresponds to residues 1067–1120. The second step PCR product containing the α1S III-IV loop and part of α1C domains III and IV was fused to the pSG5 α1C vector using SpeI/SacII sites.
α1C/α1S C
This chimera consists of α1C residues M1–M1506 fused to the N-terminus of α1S residues D1328–P1873. The C-terminus of α1S was amplified from full-length α1S using a forward primer containing a 5′ overhang of the C-terminal end of α1C domain IV up to the BclI restriction site, and a reverse primer at the stop codon of α1S. The PCR product containing the α1S C-terminus and part of α1C domain IV was fused to the pSG5 α1C vector using BclI/NotI sites.
α1C/α1S all loop (N, I-II, II-III, III-IV, C)
This chimera consists of fusions of the following peptide fragments in sequential order from N- to C-terminus: α1S(M1–K51)/α1C(P155–S435)/α1S(G335–R432)/α1C(V555–D788)/α1S(A667–T799)/α1C(I931–V1187)/α1S(T1067–F1120)/α1C(E1242–M1506)/α1S(D1382–P1873). This chimera was made by a cut-and-paste method using the chimeras and restriction sites indicated above.
α1C(E736K)
This domain II pore mutation was described elsewhere (36) and consists of a replacement of the glutamate residue at position 736 by lysine. We designed a 37-base antisense primer that introduced a mismatch at nucleotide 2397 (g2397a), and a sense primer that annealed before the BamHI site of α1C. The PCR product was cloned into the pSG5 α1C vector at BamHI/EcoRI sites.
α1S(E1014K)
This domain III pore mutation consists of replacement of a glutamate residue at position 1014 by lysine, and was previously made and described elsewhere (25).
Whole-cell voltage clamp
Whole-cell recordings were performed with an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). Effective series resistance was compensated up to the point of amplifier oscillation with the Axopatch circuit. All experiments were performed at room temperature. Patch pipettes had a resistance of 1–3 MΩ when filled with the pipette solution. To obtain Ca2+ conductance curves, cells were maintained at a holding potential of −40 mV, and depolarized in ascending order every 3 s. The pulse duration was 500 ms and was changed in 5-mV increments up to +85 mV. To obtain Ca2+ transient curves, cells were maintained at −40 mV and depolarized in descending order every 30 s to permit recovery of the resting fluorescence. The pulse duration was 200 ms and was changed in 20-mV decrements from +90 mV to –30 mV. To obtain charge movement curves, we used a P/4 protocol with a long prepulse to inactivate Na+-channel ionic and gating currents (25). The pulse protocol was as follows. The command voltage was stepped from a holding potential of −80 mV to −30 mV for 698 ms, to −50 mV for 5 ms, to the test potential for 50 ms, to −50 mV for 50 ms, and then to the −80 mV holding potential. Test potentials were applied in decreasing order every 10 mV from +100 to –80 mV. The waiting period between test pulses was 10 s.
Confocal fluorescence microscopy
Ca2+ transients were measured by confocal line scanning at room temperature. Cells were loaded with 5 μM fluo-4 acetoxymethyl ester (Molecular Probes, Eugene, OR) for ∼1 h at room temperature. Cells were viewed with an inverted IX20 Olympus microscope with a 20× (NA 1.4) objective and a Fluoview confocal attachment (Olympus, Melville, NY). The 488 nm line provided by a 5 mW Argon laser was attenuated to 6% with neutral density filters. Line scans were acquired at a speed of 2.05 ms per line. All line scans consisted of 1000 lines at a width of 512 pixels. The spatial dimension of the line scan was 30–60 μm , and covered the entire width of the myotube. Locations selected for line scans were devoid of nuclei and had a low resting fluorescence. Line scans were synchronized to start 100 ms before the onset of the depolarization for voltage-clamp experiments. The time course of the space-averaged fluorescence intensity change was estimated as described elsewhere (26,27) and is reported in ΔF/F units. The peak-to-peak noise in the baseline fluorescence averaged ∼0.1 ΔF/F units. Image analyses were performed with NIH Image software (National Institutes of Health, Bethesda, MD).
Solutions
For Ca2+ currents and Ca2+ transients, the external solution was (in mM) 130 TEA methanesulfonate, 10 CaCl2, 1 MgCl2, 10−3 TTX, and 10 HEPES titrated with TEA(OH) to pH 7.4. The pipette solution consisted of (in mM) 140 Cs aspartate, 5 MgCl2, 0.1 EGTA (when Ca2+ transients were recorded) or 5 EGTA (when only Ca2+ currents were recorded), and 10 MOPS titrated with CsOH to pH 7.2. For charge movements, the internal solution was (in mM) 120 NMG (N-methyl glucamine)-Glutamate, 10 HEPES-NMG, and 10 EGTA-NMG, pH 7.3. This solution produced a more reliable block of the outward ionic current than the internal solution used for Ca2+ currents. The external solution was supplemented with 0.5 mM CdCl2 to block background Ca2+ currents present in mdg myotubes, 0.5 mM LaCl3 to increase pipette seal resistance and 0.05 mM TTX to block residual Na+ current.
Curve fitting
The voltage dependence of the Ca2+ conductance, charge movements, and sigmoidal fluorescence-voltage relationships were fitted with a standard Boltzmann equation:
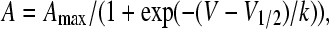 |
(1) |
where V (in mV) is the test potential, Amax is Gmax, Qmax, or ΔF/Fmax, V1/2 (in mV) is the midpoint potential, and k (in mV) is the slope factor. For bell-shaped fluorescence-voltage curves, the Boltzmann equation was modified as follows:
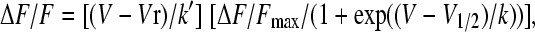 |
(2) |
where Vr (in mV) is a constant that accounts for the decrease in Ca2+ transient amplitude at positive potentials, and k′ (in mV) is an empirical scaling factor (26,27). Other parameters are the same as in Eq. 1. Parameters of a fit of averages of many cells (population average) are shown in the figures. Parameters of the fit of individual cells are shown in tables. Parameters of the fit of the population average differed slightly from the mean of the fit of individual cells. The latter parameters generated theoretical curves that were a better fit with the average data and, for that reason, were used in the figures. Analysis of variance (ANOVA) was performed with Analyze-it (Leeds, UK).
RESULTS
Recovery of voltage- or Ca2+ entry-dependent EC coupling in dysgenic myotubes
Studies have shown that α1C is targeted to EC-coupling junctions in cultured skeletal myotubes, and that the Ca2+ current generated by α1C can trigger Ca2+ transients by Ca2+-dependent activation of RyR1 (31,32). Chimeras with α1C in the backbone also activate Ca2+ transients by a similar mechanism when expressed in skeletal myotubes (11,14). Hence, Ca2+ release induced by the Ca2+ current is inherent to chimeras of α1C, and this component needs to be carefully separated from the release of interest triggered by mechanical DHPR-RyR1 coupling. To exclude the component of the Ca2+ transient triggered by the Ca2+ current without changing the external solution, we mutated the conserved glutamate E736 in the P-loop of repeat II of α1C, previously shown to be critical for Ca2+ permeation (36). Fig. 1 shows the EC coupling and Ca2+ current characteristics of α1C(E736K) and α1S(E1014K), localized in the P-loop of repeat III of α1S (25,37). The top traces show representative Ca2+ transients and Ca2+ currents at +30 mV, with the pore mutants depicted by shaded traces. In this and other figures, the displayed Ca2+ transients and Ca2+ currents were obtained with different protocols and different internal solutions (see Materials and Methods). For Ca2+ currents, we used 500-ms depolarizations in a highly buffered internal solution (5 mM EGTA). These conditions permitted the determination of the time course of inactivation while maintaining a low cytosolic Ca2+ at all potentials. For Ca2+ transients, we used 200-ms depolarizations in a lightly buffered internal Ca2+ solution (0.1 mM EGTA). These conditions limited SR Ca2+ release, increased the rate of resting Ca2+ recovery, and increased the intensity of the fluorescence signal. In all cases, the external Ca2+ concentration was 10 mM. WT α1S expressed slow-activating/noninactivating Ca2+ currents typical of cultured normal primary myotubes (35). In contrast, WT α1C expressed fast-activating/slow-inactivating Ca2+ currents, in agreement with previous results in mdg myotubes (11,32,38). The degree of inactivation of α1C was variable, and to some extent varied with the Ca2+ current density, consistent with Ca2+-entry-dependent inactivation of α1C investigated by others in myotubes (16). Both α1C and α1S recovered a Ca2+ current density close to that of normal myotubes reported elsewhere (35). Inward currents expressed by the pore mutants were drastically reduced. In the case of α1S(E1014K), inward current was undetectable (<0.1 pA/pF), consistent with previous results (25). In the case of α1C(E736K), the inward current at +30 mV was reduced ∼180-fold compared to WT α1C, consistent with determinations in oocytes (36). The reduction was less pronounced if calculations are based on the maximum Ca2+ conductance, Gmax, expressed by the conducting and nonconducting variants (see Tables 1 and 2). This is because the Gmax of α1S(E1014K) and α1C(E736K) is dominated by the conductance of the outward current (Fig. 1, middle panels). The latter is a mixture of outward current through the mutant Ca2+ channel and background outward currents unaffected by the mutation.
FIGURE 1.

Ca2+ currents and Ca2+ transients expressed by α1S and α1C variants. Columns show representative mdg myotubes transfected with WT α1S, WT α1C, and variants carrying pore mutations, namely α1S (E1014K) and α1C (E736K). Shaded traces and shaded symbols correspond to the pore mutant form of the variant indicated above each column. The top trace corresponds to the spatial integral of the confocal Ca2+ transient in ΔF/F units in response to a 200-ms depolarization to +30 mV from a holding potential of −40 mV. The second row of traces corresponds to the Ca2+ current elicited during a 500-ms depolarization from a holding potential of −40 mV to +30 mV. The Ca2+ transients and Ca2+ currents shown in this and other figures were obtained with different protocols (see text, especially Materials and Methods). Population averages of the Ca2+ current-voltage curves are located centrally. These were obtained by depolarizing transfected myotubes in 5-mV increments from a holding potential of −40 mV for 500 ms. At the bottom are Ca2+ transient-voltage curves for population averages in response to a 200-ms depolarization from −40 mV to the indicated potentials. The population averaged fluorescence-voltage curves for α1S and α1S(E1014K) were fit with Eq. 1, and those obtained in myotubes expressing α1C and α1C(E736K) were fit with Eq. 2. Curves were fit with the following parameters (ΔF/Fmax in ΔF/F units, V1/2 in mV, and k in mV, respectively): for α1S: 2.6, 5.3, and 11.7; for α1S(E1014K), 2.6, 6.4, and 7.8; for α1C, 1.2, 20.6, and 6.1; for α1C(E736K), 0, −19.7, and 20. For a fit of parameters by cell and number of cells included in the fit, see Table 1.
TABLE 1.
Ca2+ conductance and Ca2+ transients expressed by α1C/α1S chimeras in dysgenic (mdg) skeletal myotubes
| Gmax (pS/pF) | V1/2 (mV) | k (mV) | ΔF/Fmax | V1/2 (mV) | k (mV) | |
|---|---|---|---|---|---|---|
| NT mdg | 20 ± 3* (27) | 4 ± 5* | 19.5 ± 7.1* | —(25) | — | — |
| WT α1S | 163 ± 13 (14) | 38 ± 2 | 6.5 ± 0.7 | 2.7 ± 0.4 (5) | 7 ± 2 | 8.6 ± 1.1 |
| WT α1C | 152 ± 27 (10) | 19 ± 3 | 7.1 ± 0.7 | 1.3 ± 0.4* (6) | 24 ± 3* | 5.9 ± 1.1 |
| α1C/α1S N | 213 ± 50 (7) | 18 ± 3 | 6.3 ± 1.2 | 0.7 ± 0.2* (7) | 26 ± 10 | 11.0 ± 3.0 |
| α1C/α1S I-II | 140 ± 35 (10) | 21 ± 2 | 9.6 ± 0.5 | 1.1 ± 0.3* (8) | 19 ± 4 | 7.1 ± 1.0 |
| α1C/α1S II-III | 208 ± 29 (15) | 11 ± 2 | 5.4 ± 0.7 | 2.5 ± 0.9 (7) | 1 ± 3 | 3.3 ± 1.0* |
| α1C/α1S III-IV | 153 ± 27 (9) | 9 ± 3 | 6.8 ± 0.8 | 1.3 ± 0.4* (6) | 2 ± 2 | 4.8 ± 0.8* |
| α1C/α1S C | 151 ± 26 (12) | 13 ± 2 | 6.3 ± 0.5 | 1.0 ± 0.3* (5) | 7 ± 7 | 3.1 ± 1.5* |
| α1C/α1S all loop | 121 ± 25 (5) | 11 ± 4 | 6.2 ± 1.0 | 2.2 ± 0.6 (8) | 9 ± 5 | 8.7 ± 1.9 |
Values are mean ± SE of Boltzmann parameters fitted to each cell, with the number of cells indicated in parentheses. ΔF/Fmax, V1/2, and k are parameters of the Boltzmann fit to each cell with either Eq. 1 or Eq. 2, as indicated in the text. Changes in fluo-4 fluorescence were not detected at any test potential (−30 to +90 mV) in nontransfected dysgenic myotubes (NT mdg).
Parameters with one-way ANOVA significance p < 0.05 compared to myotubes expressing WT α1S.
TABLE 2.
Ca2+ conductance, Ca2+ transients, and charge density expressed by pore mutants in dysgenic (mdg) skeletal myotubes
| Gmax(pS/pF) | V1/2(mV) | k(mV) | ΔF/Fmax | V1/2 (mV) | K(mV) | Qmax (fC/pF) | V1/2 (mV) | k(mV) | |
|---|---|---|---|---|---|---|---|---|---|
| NT mdg | 20 ± 3 (27) | 4 ± 5 | 19.5 ± 7.1 | — (25) | — | — | 1.4 ± 0.1* (10) | −5 ± 4 | 12.0 ± 1.2 |
| α1S(E1014K) | 48 ± 12 (7) | 23 ± 14 | 16.2 ± 3.9 | 2.6 ± 0.4 (11) | 7 ± 4 | 9.3 ± 1.0 | 4.7 ± 0.8 (9) | 10 ± 2 | 17.8 ± 0.8 |
| α1C(E736K) | 25 ± 6 (7) | 21 ± 3 | 13.2 ± 1.9 | — (5) | — | — | 5.9 ± 1.3(4) | 5 ± 4 | 18.1 ± 0.8 |
| α1C(E736K)/α1S II-III | 43 ± 7 (7) | 6 ± 3 | 10.7 ± 1.5 | 0.9 ± 0.2* (7) | 20 ± 7 | 12.9 ± 2.7 | 5.8 ± 0.9 (7) | 12 ± 4 | 21.2 ± 1.9 |
| α1C(E736K)/α1S all loop | 44 ± 9 (8) | 44 ± 14 | 18.7 ± 3.8 | 2.2 ± 0.6 (8) | 11 ± 5 | 9.0 ± 2.1 | 4.5 ± 0.6 (4) | 12 ± 6 | 17.4 ± 1.8 |
Values are mean ± SEM of Boltzmann parameters fitted to each cell, with the number of cells indicated in parenthesis. Gmax, ΔF/Fmax or Qmax, V1/2, and k are parameters of the Boltzmann fit to each cell with Eq. 1. Changes in fluo-4 fluorescence were not detected at any test potential in NT mdg and α1C(E736K)-expressing myotubes. Parameters for conductance and fluorescence of NT mdg myotubes are the same as in Table 1.
Parameters with one-way ANOVA significance p < 0.05 compared to myotubes expressing α1S(E1014K). Using the mean Qmax provided in the table, the ratio (ΔF/Fmax)/Qmax was 0.55 ± 0.08 for α1S(E1014K) (n = 11); 0.16 ± 0.04 for α1C(E736K)/α1S II-III (n = 7); and 0.45 ± 0.13 for α1C/α1S all loop (n = 8). The ratio for α1C(E736K)/α1S II-III was significantly lower (t-test significance p < 0.05) than that of α1S(E1014K) or that of α1C(E736K)/α1S all loop.
Ca2+ transients expressed by the conducting and nonconducting variants were obtained by line-scan integration of confocal fluo-4 fluorescence in myotubes held under voltage-clamp conditions. As indicated by the superimposed traces at the top of Fig. 1, the pore mutation α1S(E1014K) had a minimal effect on the time course of the Ca2+ transient compared to WT α1S. This result is in agreement with previous observations (25,37), and confirms expression of a bona fide skeletal-type EC-coupling phenotype. In contrast, the amplitude of the Ca2+ transient evoked by α1C(E736K) was severely depressed compared to WT α1C. The bottom graphs of Fig. 1 show the relationship between the amplitude of the Ca2+ transient and the magnitude of the depolarization, in the presence and absence of pore mutations. The Ca2+ transient amplitude in these plots was measured just before the end of a 200-ms depolarization, and thus excluded Ca2+ release triggered by the tail current, which was prominent in some myotubes. For α1S and α1S(E1014K), the expressed Ca2+ transients had a sigmoidal voltage-dependence with saturation at potentials more positive than +30 mV. Furthermore, there was good agreement between the two sets of data. We interpreted this result as an indication that the EC coupling expressed by α1S or α1S(E1014K) was entirely Ca2+-entry-independent. In contrast, Ca2+ transients recovered by α1C were smaller in magnitude, reached a maximum at ∼+30 mV, and decreased at more positive potentials. This biphasic behavior gives rise to a bell-shaped fluorescence-voltage curve that mirrors the bell-shaped dependence of the Ca2+ current-voltage curve (see Fig. 1, middle panel). This result agrees with previous determinations in dysgenic myotubes (32), and suggests that Ca2+ transients may be triggered by the Ca2+ current via a Ca2+-dependent mechanism. Consistent with this interpretation, the pore mutant α1C(E736K) failed to express a detectable Ca2+ transient. The changes in fluorescence in myotubes expressing WT α1C may also reflect changes in cytosolic Ca2+ produced directly by the Ca2+ current. The contribution of the Ca2+ current to the cell fluorescence was determined previously in myotubes treated with ryanodine (27). For cells with a comparable Ca2+ current density, we found that the contribution was <0.25 ΔF/F at +30 mV (27). In this study, the Ca2+ transient expressed by WT α1C at +30 mV was ∼1.3 ΔF/F (Table 1). For this reason, we believe that the Ca2+ current expressed by WT α1C contributed only modestly to the overall cell fluorescence.
Incomplete recovery of skeletal-type EC coupling by the α1S II-III loop revealed by pore mutations that eliminate inward Ca2+ current
Ca2+ currents and transients expressed by chimeras consisting of α1C carrying each of the cytosolic domains of α1S, namely the N-terminal, I-II loop, II-III loop, III-IV loop, or C-terminal domains, are shown in Fig. 2. Coordinates of skeletal domains donated to α1C, and of homologous regions removed from α1C, are described in Materials and Methods. The top traces correspond to inward Ca2+ currents for depolarizations to –30 mV and +30 mV in dysgenic myotubes expressing each of the five indicated chimeras. All chimeras expressed high-density, fast-activating, and slow-inactivating Ca2+ currents typical of WT α1C when expressed in myotubes. Furthermore, the fitted Gmax was statistically similar to that of α1C or α1S (Table 1). Two chimeras, that carrying the N-terminal domain (labeled α1C N) and that carrying the II-III loop (labeled α1C/α1S II-III), expressed Ca2+ current at a density higher than that of WT α1C, although the difference was not significant. Representative Ca2+ transients at +30 mV, and the voltage dependence of the Ca2+ transient amplitude measured at the end of a 200-ms depolarization, are shown in the bottom graphs of Fig. 2. For reference, the shaded lines in the plots indicate the fitted fluorescence-voltage relationship of myotubes expressing WT α1S and WT α1C described above. The II-III loop chimera consistently expressed Ca2+ transients that were much larger in amplitude than those expressed by the other chimeras. Furthermore, only the II-III loop chimera expressed a fluorescence-voltage relationship that was sigmoidal like that of WT α1S. All other chimeras expressed fluorescence-voltage relationships that were bell-shaped like that of WT α1C, indicative of a Ca2+-dependent mechanism. The bell-shaped voltage dependence is particularly prominent for the III-IV loop and the C-terminal domain chimeras. Furthermore, in these cases there was a significant negative shift in midpoint potential compared to WT α1C. The shifts were consistent with differences in midpoint potential found for the Ca2+ conductance described in Table 1. In summary, only the α1C/α1S II-III loop chimera expressed Ca2+ transients with a sigmoidal fluorescence-voltage curve indicative of skeletal-type EC coupling. This result is consistent with previous observations, which have suggested that the II-III loop is a critical domain for skeletal-type EC coupling (11,13–17,30,32). Other domains of α1S, namely the III-IV loop and the C-terminal domain, affected channel gating by shifting Ca2+ current activation to more negative potentials, resulting in a concurrent shift in the voltage dependence of the Ca2+ transient. Furthermore, the skeletal N-terminal domain and the I-II loop do not appear to influence the Ca2+-dependent EC-coupling phenotype in a significant manner.
FIGURE 2.

Ca2+ currents and Ca2+ transients expressed by α1C/α1S chimeras. Columns show representative mdg myotubes transfected with the identified α1C/α1S chimeras. Nomenclature and domain boundaries for the chimeras are described in Materials and Methods. The top traces correspond to Ca2+ currents from the same cell in response to a 500-ms depolarization from a holding potential of −40 mV to –30 mV (no current) and to +30 mV (near maximum inward current). Population averages of the Ca2+ current-voltage curves are located centrally. These were obtained by depolarizing transfected myotubes in 5-mV increments from a holding potential of −40 mV for 500 ms. The traces in the middle correspond to confocal Ca2+ transients in mdg myotubes expressing each of the chimeras in ΔF/F units in response to a 200-ms depolarization to +30 mV from a holding potential of −40 mV. At the bottom are Ca2+ transient-voltage curves for population averages in response to a 200-ms depolarization from −40 mV to the indicated potentials. The shaded traces without data in each graph correspond to the fitted voltage dependence of WT α1S and WT α1C from Fig. 1. The fluorescence-voltage curves for all α1C/α1S chimeras were fit with Eq. 2, except that obtained in myotubes expressing α1C/α1S II-III, which was fit with Eq. 1. Curves (black) were fit with the following parameters (ΔF/Fmax in ΔF/F units, V1/2 in mV, and k in mV, respectively): for α1C/α1S N, 0.5, 16.7, and 10.0; for α1C/α1S I-II, 1.3, 20.0, and 9.9; for α1C/α1S II-III, 2.4, 2.8, and 4.9. For α1C/α1S III-IV, 1.0, 1.2, and 4.5; and for α1C/α1S C, 0.8, 4.9, and 3.9.
To examine the extent of recovery of the skeletal phenotype by the II-III loop chimera, we investigated Ca2+ transients elicited by the II-III loop chimera in the presence of the α1C(E736K) pore mutation. For the sake of clarity, we refer to this mutation as E736K in all cases even though the actual coordinate will differ for some chimeras. The left panel of Fig. 3 shows representative Ca2+ transients and Ca2+ currents expressed by the α1C(E736K)/α1S II-III loop chimera compared to the Ca2+ conducting variant at +30 mV. Current-voltage relationships and fluorescence-voltage relationships are shown immediately below. Shaded areas are reserved for the pore mutant. Inward Ca2+ currents were severely curtailed in the α1C(E736K)/α1S II-III chimera compared to the Ca2+-conducting variant. Furthermore, inward current was blocked to the same extent as with α1C(E736K). This can be readily appreciated by comparing current-voltage curves in Figs. 1 and 3, and by the parameters of the conductance fit. Table 1 shows data for WT α1C and α1C/α1S II-III, and Table 2 for α1C(E736K) and α1C(E736K)/α1S II-III. Based on these results, the mutation was deemed useful for removing a component of the Ca2+ transient of the II-III loop chimera triggered by the Ca2+ current, if such a component was present. Quite surprisingly, Ca2+ transients expressed by the α1C(E736K)/α1S II-III loop chimera were severely depressed relative to those expressed by the Ca2+-conducting α1C/α1S II-III loop variant. This can be appreciated in the noticeably small Ca2+ transient shown at the top of Fig. 3 for a representative myotube expressing α1C(E736K)/α1S II-III, and by the fluorescence-voltage relationship at the bottom of Fig. 3. At face value, the data suggests that the II-III loop chimera relies heavily on external Ca2+ for activation of SR Ca2+ release, and that the voltage-dependent skeletal-type component is minor. However, other explanations must be considered also. It is possible that α1C/α1S chimeras are only partially functional, and, as a consequence, none of them are able to rescue the skeletal phenotype to the same extent as WT α1S. Also, the charge movement density expressed by the II-III chimera may be inherently low, and hence unable to rescue a substantial skeletal-type component. Finally, the E736K mutation in repeat II may have adversely affected skeletal-type EC coupling. The latter is troublesome since it is known that the E1014K charge reversal eliminates high-affinity Ca2+ binding to the α1S subunit (39). The Ca2+-binding site in the pore region could be the Ca2+-binding site identified as critical for the DHPR voltage sensor (40,41). In what follows, we tested these alternatives.
FIGURE 3.

Full restoration of skeletal-type EC coupling by the all loop chimera and partial restoration by the II-III loop chimera. Columns show representative mdg myotubes transfected with the α1C/α1S II-III loop chimera and α1C/α1S all loop chimera, both in the presence and absence of the pore mutation E736K. Shaded traces and shaded symbols correspond to pore mutants. The top traces correspond to the spatial integral of the confocal Ca2+ transient in ΔF/F units in response to a 200-ms depolarization to +30 mV from a holding potential of −40 mV. The second row of traces corresponds to the Ca2+ current elicited during a 500-ms depolarization from −40 mV to +30 mV. Population averages of the Ca2+ current-voltage curves are located centrally. These were obtained by depolarizing transfected myotubes in 5-mV increments from a holding potential of −40 mV for 500 ms. At the bottom are Ca2+ transient-voltage curves for population averages in response to a 200-ms depolarization from −40 mV to the indicated potentials. All fluorescence-voltage curves were fit with Eq. 1. Curves were fit with the following parameters (ΔF/Fmax in ΔF/F units, V1/2 in mV, and k in mV, respectively): for α1C/α1S II-III, 2.4, 2.8, and 4.9; for α1C(E736K)/α1S II-III, 0.9, 17.1, and 15.5; for α1C/α1S all loop, 2.1, 1.7, and 10.9; for α1C(E736K)/α1S all loop: 2.1, 14.3, and 8.
To determine if the tested α1C/α1S chimeras were broadly EC-coupling-defective, we investigated the behavior of α1C/α1S all loop, a chimera consisting of α1C and all the cytoplasmic elements of α1S characterized in Fig. 2, namely the N-terminal, I-II loop, II-III loop, III-IV loop, and C-terminal domains. If chimeras are defective in general, such a defect should also be present in a chimera containing the II-III loop and the rest of the skeletal cytoplasmic elements. Alternatively, if the α1C/α1S II-III loop chimera was partially defective due to the absence of critical skeletal elements, the α1C/α1S all loop chimera should recover the skeletal phenotype more effectively. These results are shown in the right panel of Fig. 3. The α1C/α1S all loop chimera expressed Ca2+ currents with a fast activation and slow inactivation, typical of WT α1C and all tested α1C/α1S chimeras. Furthermore, the Ca2+ current density was not significantly different from that of the other chimeras (Table 2), and as shown by the current-voltage curves, the E736K pore mutation curtailed the bulk of the inward Ca2+ current. Significantly, the voltage dependence of the Ca2+ transients was sigmoidal for both the Ca2+-conducting α1C/α1S all loop and for the poorly conducting α1C(E736K)/α1S all loop chimeras. The maximum fluorescence at large positive potentials was also the same. This is indicated by the traces of cell fluorescence at +30 mV in separate representative myotubes shown at the top of Fig. 3, and by the fluorescence-voltage relationships at the bottom of Fig. 3. The fitted Boltzmann parameters indicated a small difference in midpoint potential; however, the difference was not significant. Parameters of the fit of the voltage dependence of Ca2+ transients are shown in Table 1 for α1C/α1S all loop and in Table 2 for α1C(E736K)/α1S all loop. From the similarity in shape and maximum Ca2+ fluorescence at large potentials, we concluded that this pair of chimeras recapitulated well the behavior of WT α1S and α1S(E1014K) described above. To determine effectiveness of recovery of the skeletal-type EC-coupling components by the II-III loop and all loop chimeras, we compared the behavior of α1C(E736K)/α1S all loop and α1C(E736K)/α1S II-III loop in terms of the maximum amplitude of the Ca2+ transients elicited at large positive potentials (Table 2). The all-skeletal loop chimera recovered a ΔF/Fmax statistically similar to that of α1S(E1014K), and ∼2.4-fold higher than that of the II-III loop chimera (t-test significance <0.05). Hence the all loop chimera was more effective than the II-III loop chimera in recovery of a skeletal EC-coupling component. The results show that it is possible to design an α1C/α1S chimera with a phenotype similar to WT α1S, and that α1C/α1S chimeras are not inherently EC-coupling-defective. Evidently, several cytoplasmic loops of α1S, not only the II-III loop, are required for a quantitative recovery of the EC-coupling phenotype.
Fig. 4 shows normalized charge movement density (fC/pF) expressed by the II-III loop chimera, the all loop chimera, and the two WT constructs. We carried out a standard gating current protocol (25) in myotubes expressing variants carrying the pore mutation. This reduced the ionic current, and hence increased the success of the measurement. The traces in the insets show the transient outward ON current at the start of a 50-ms test voltage step to +60 mV followed by inward OFF current at the end of the voltage step as charges return to the original position. The complete pulse protocol (see Materials and Methods) included a 698-ms prepulse depolarization to −30 mV to remove immobilization-sensitive components of the gating current unrelated to the DHPR, and pretest P/4 pulses to eliminate linear components unrelated to voltage sensors. The graphs show normalized charge density-voltage relationships obtained by integration of the OFF component which, in our hands, was less contaminated by residual ionic current than the ON component. The main contaminant of the ON component was an outward current, presumably from a K+ channel, that was not always blocked by the pipette solution. In all cases, the OFF charge increased in a sigmoidal fashion starting at ∼−40 mV and saturated at potentials more positive than +60 mV. Data were adequately fit by a Boltzmann equation (Eq. 1). In nontransfected myotubes, we detected a small background charge with a maximum density of ∼1 fC/pF, similar to that reported previously (25). The average charge movement density of nontransfected cells is shown by the line without data, and the parameters of the fit are shown in Table 2. The maximum charge-movement densities expressed by the α1C(E736K)/α1S II-III loop chimera, the α1C(E736K)/α1S all loop chimera, α1C(E736K), and α1S(E1014K) were statistically similar (Table 2). These values agreed with previous determinations in dysgenic myotubes expressing WT α1C and WT α1S (12,25). The results show that DHPR voltage-sensor density was not a confounding factor in our studies .
FIGURE 4.

Charge movement density expressed α1C/α1S chimeras carrying pore mutations. Graphs show the population-averaged voltage dependence of the OFF charge in femto-Coulombs normalized to the linear membrane capacitance of each cell in pico-Farads. The OFF charge was obtained by integration of the OFF charge movement current at the end of a 50-ms test pulse after on-line subtraction of the linear component. Measurements were made in mdg myotubes transfected with the indicated constructs using a pulse protocol and external and pipette solutions described in Materials and Methods. The shaded trace in each graph corresponds to the fit of the population averaged charge movement of nontransfected dysgenic myotubes. Insets are representative traces of charge movement currents during the test-pulse phase of the pulse protocol elicited from a holding potential of –80 mV to a test potential of +60 mV for 50 ms. Charge movement versus voltage curves were fit with Eq. 1 with the following parameters (Qmax in fC/pF, V1/2 in mV, and k in mV, respectively); for α1S(E1014K), 5.3, 8.2, and 19.1; for α1C(E736K), 5.9, 2.7, and 17.6; for α1C(E736K)/α1S II-III, 5.7, 10.6, and 20.1; for α1C(E736K)/α1S all loop, 4.4, 12.0, and 17.6; and for nontransfected dysgenic myotubes (shaded traces): 1.4, −7.0, and 12.9. For a fit of parameters by cell and number of cells included in the fit, see Table 2.
Incomplete recovery of skeletal-type EC coupling by the α1S II-III loop revealed by Cd2+ block of inward Ca2+ current
Is the reduced activity of the α1C(E736K)/α1S II-III loop chimera a consequence of the E736K mutation? To answer this critical question, in Fig. 5 we compared Ca2+ transients expressed by WT α1S and the Ca2+-conducting II-III loop and all loop chimeras in the presence of external Cd2+ to block the inward Ca2+ current. Titrations indicated that 10−4 M, but not 10−6 M Cd2+, blocked the Ca2+ conductance to the same extent as the pore mutations. This is shown in Table 3, with control data in the absence of Cd2+ in Table 1. Ca2+ currents and Ca2+ transients in control external solution and in external solution supplemented with 10−4 M Cd2+ are shown for WT α1S, α1C/α1S II-III loop and α1C/α1S all loop in left, center, and right panels as indicated. Shaded traces and symbols are reserved for myotubes in external solution containing Cd2+. Inward Ca2+ currents were severely curtailed by 10−4 M Cd2+ in all cases. This can be readily appreciated from the insets of Ca2+ currents at +30 mV, and by comparing current-voltage curves in the presence and absence of the blocker. It is significant to observe that Ca2+ transients in the presence of Cd2+ reproduced the same phenotype as those seen in the presence of the pore mutations. The Ca2+ transients expressed by the α1C/α1S II-III loop chimera were severely depressed in the presence of the Ca2+ current blocker. However, the Ca2+ transients expressed by WT α1S and the α1C/α1S all loop chimera were not depressed by 10−4 M Cd2+. This can be appreciated in the Ca2+ transients shown as insets for a representative myotube expressing the indicated construct, and by the fluorescence-voltage relationship in the center of each panel in Fig. 5. The results are further confirmed by the correlation presented in the histograms at the bottom of each panel. Ca2+ current density and Ca2+ transient amplitude at +30 mV, at two Cd2+ concentrations, were normalized to the mean Ca2+ current and Ca2+ transient amplitude in control solution. In all cases, there was a significant decrease in fractional Ca2+ current density at 10−4 M Cd2+. However, only in the case of the α1C/α1S II-III loop chimera did we observe a concomitant decrease in fractional Ca2+ transient amplitude. This was not the case for WT α1S or the α1C/α1S all loop chimera. A statistical analysis of the parameters of Ca2+ conductance and cell fluorescence in the presence of Cd2+ are described in Table 3. The Ca2+ fluorescence data in Table 3 are in agreement with the statistical analysis of the data at a single potential presented in histogram form at the bottom of Fig. 5. The ΔF/Fmax fitted to cells expressing the II-III loop chimera in 10−4 M Cd2+, but not that of the all loop chimera, was significantly lower relative to controls in external solution with 10−6 M Ca2+ or external solution without Cd2+. The results confirmed that the E736K mutation was not responsible for the weak EC coupling recovered by the II-III loop chimera. Rather, the behavior of the all loop chimera emphasized that a quantitative recovery of the skeletal phenotype could not be achieved unless all the cytosolic elements of α1S were present. It remains to be determined in future work which cytosolic domains in addition to the II-III loop are required for a quantitative functional recovery.
FIGURE 5.

Cd2+ block of Ca2+-dependent EC coupling promoted by the II-III loop chimera. Columns show representative mdg myotubes expressing the II-III loop and all loop chimeras. The left panel shows myotubes expressing WT α1S used as control. The top graphs correspond to population average current-voltage curves for depolarizations of 500 ms from a holding potential of −40 mV. Insets show representative Ca2+ currents elicited at +30 mV from a holding potential of −40 mV. With the exception of the bar graphs at the bottom, all shaded traces and shaded symbols are in 10−4 M CdCl2 added to the bath solution. The middle graphs show population-averaged Ca2+ transient-voltage relationships for depolarizations of 200 ms from a holding potential of −40 mV. Insets correspond to the spatial integral of the confocal fluorescence in ΔF/F units in response to a 200-ms depolarization to +30 mV from a holding potential of −40 mV. Fluorescence-voltage curves were fit with Eq. 1. Curves were fit with the following parameters (Fmax in ΔF/F units, V1/2 in mV, and k in mV, respectively): for WT α1S in 0 Cd2+, 2.5, 13.7, and 14.1; for WT α1S in 10−4 Cd2+, 2.2, 3.0, and 8.2; for α1C/α1S II-III in 0 Cd2+, 2.8, 2.8, and 4.7; for α1C/α1S II-III in 10−4 Cd2+, 0.7, 17.0, and 13.5; for α1C/α1S all loop in 0 Cd2+, 2.1, 1.7, and 10.9; for α1C/α1S all loop in 10−4 Cd2+, 2.7, 15.6, and 9.3. For a fit of parameters by cell and number of cells included in the fit, see Table 3. The bottom bar graphs correspond to normalized Ca2+ currents (shaded bars) and normalized ΔF/Fmax (black) in control solution without Cd2+, external solution with 10−6 M CdCl2, and external solution with 10−4 M CdCl2. Ca2+ currents at +30 mV were normalized to the population averaged Ca2+ current at +30 mV in the absence of Cd2+. ΔF/Fmax from a fit of individual cells was normalized to ΔF/Fmax from a fit of the population average in the absence of Cd2+.
TABLE 3.
Ca2+ conductance and Ca2+ transients expressed by α1C/α1S chimeras in presence of extracellular cadmium
| Gmax(pS/pF) | V1/2(mV) | k (mV) | ΔF/Fmax | V1/2 (mV) | k (mV) | |
|---|---|---|---|---|---|---|
| WT α1S + 10−4 Cd2+ | 43 ± 4 (7) | 18 ± 4 | 6.7 ± 2.0 | 2.2 ± 0.5 (9) | 0 ± 3 | 6.9 ± 1.7 |
| WT α1S + 10−6 Cd2+ | 158 ± 15* (6) | 11 ± 2 | 3.8 ± 0.3 | 3.4 ± 0.6 (7) | 3 ± 3 | 5.8 ± 0.4 |
| α1C/α1S II-III + 10−4 Cd2+ | 53 ± 8 (5) | 14 ± 4 | 10.1 ± 3.6 | 0.7 ± 0.3* (7) | 16 ± 3* | 13.0 ± 2.0* |
| α1C/α1S II-III + 10−6 Cd2+ | 211 ± 9* (4) | 2 ± 6 | 5.4 ± 3.3 | 1.4 ± 0.6 (4) | 13 ± 9 | 5.1 ± 0.7 |
| α1C/α1S all loop + 10−4 Cd2+ | 29 ± 8 (6) | 38 ± 14 | 20.1 ± 5.8* | 2.8 ± 0.7 (6) | 18 ± 4* | 10.6 ± 2.2 |
| α1C/α1S all loop + 10−6 Cd2+ | 104 ± 9* (5) | 6 ± 3 | 5.2 ± 1.6 | 2.4 ± 0.4 (5) | 5 ± 4 | 6.9 ± 1.9 |
Values are mean ± SEM of Boltzmann parameters fitted to each cell, with the number of cells indicated in parentheses. Gmax or ΔF/Fmax, V1/2, and k are parameters of the Boltzmann fit to each cell with Eq. 1.
Parameters with one-way ANOVA significance p < 0.05 compared to WT α1S + 10−4 Cd2+.
DISCUSSION
Studies in mdg myotubes designed to investigate the contribution of α1S to skeletal-type EC coupling have concluded that the II-III loop is functionally unique (11,12,14,16,17,25,30,32). Our results confirmed this observation. However, it was not previously known if the functional recovery produced by the α1S II-III loop was comparable to that produced by WT α1S, or if multiple cytosolic domains of α1S were necessary for a quantitative recovery of the EC-coupling phenotype. Earlier studies (11) did not provide a voltage-clamp analysis of the contribution of the skeletal loops alone or in combination, and the more recent studies have focused exclusively on domains within the α1S II-III loop (16,17,25). The efficiency of EC-coupling recovery by the α1S II-III loop is a significant issue since structural models suggest that the area of juxtaposition between the α1S subunit and RyR1 is large, and therefore the number of interaction sites is potentially large (5,6). The EC-coupling triggering mechanism could thus be much more complex than a single DHPR loop orchestrating SR Ca2+ release. Sequence divergence between α1S and α1C is mostly confined to the long cytoplasmic domains rather than the transmembrane repeats. Hence, it was reasonable to expect that a α1C/α1S chimera carrying all the cytosolic domains of α1S could recover the skeletal EC-coupling phenotype quantitatively. We found that a large fraction of the Ca2+ transient expressed by the α1C/α1S II-III loop chimera was eliminated when the pore mutation α1C(E736K) was introduced to reduce the inward Ca2+ current. This was not the case for the α1C/α1S chimera carrying all the cytosolic domains of α1S. Furthermore, the results were confirmed using Ca2+-conducting variants of the same chimeras in which the inward Ca2+ current was blocked by a low concentration of Cd2+. The α1C/α1S all loop chimera, unlike the α1C/α1S II-III loop chimera, rescued Ca2+ transients of the same ΔF/Fmax regardless of the presence of the pore mutation. When normalized by the DHPR charge movement and compared to α1C/α1S all loop and WT α1S (see Table 2 legend), the maximum Ca2+ fluorescence expressed by the II-III loop chimera was 0.16 ± 0.04 Fmax/Qmax, compared to 0.45 ± 0.13 Fmax/Qmax for α1C/α1S all loop, and 0.55 ± 0.08 Fmax/Qmax for α1S(E1014K), or ∼3-fold lower (t-test significance, p < 0.05). These ratios only have comparative value since it is unlikely that all the DHPR charge movement is involved in EC coupling (42). The observations lend themselves to the conclusion that a conglomerate of cytoplasmic domains, rather than the II-III loop alone, is necessary to restore normal EC-coupling function.
The Ca2+ transient expressed by CSk3, the α1C/α1S II-III loop chimera described by Tanabe et al. (11) was substantially reduced in the presence of inorganic blockers of the inward Ca2+ current. Ca2+ fluorescence-voltage relationships for CSk3 have not been described (11,14); therefore, it is difficult to compare the two sets of results. However, qualitatively the results are similar and indicate that both II-III loop chimeras have a sizable EC-coupling component unrelated to the skeletal phenotype. CSk3 consists of α1C(1–787)/α1S(666–791)/α1C(923–2171), whereas our chimera included eight additional residues, namely α1S(1–788)/α1S(667–799)/α1C(931–2171). These differences may not be significant since critical determinants within the α1S II-III loop have been mapped to the segment of residues 671–690 and 720–765 present in both chimeras (14,25). Furthermore, charge movements expressed by both chimeras in dysgenic myotubes are comparable (12) (Table 2). Hence, possible differences in voltage-sensor densities, which could conceivably affect the density of mechanically coupled RyR1 channels, are nonexistent. Interestingly, Ca2+ fluorescence-voltage relationships have been published for GFP-CSk53, an α1C/α1S chimera carrying the α1S II-III loop residues 720–765 deemed critical for skeletal-type EC coupling. This chimera also has an N-terminal fusion of the green fluorescent protein (GFP). The Fmax expressed by GFP-CSk53 was considerably lower than that expressed by the GFP-WT α1S fusion even in external solution containing the standard 10 mM Ca2+ (31). Thus, the low Fmax of a II-III loop chimera may not be related in all instances to the presence of an enlarged Ca2+-dependent EC-coupling component, which is removed when the inward Ca2+ current is blocked. It could be that in this case, the GFP moiety hinders EC coupling in the CSk53 chimera, or that residues 720–765 are insufficient for a quantitative EC-coupling recovery comparable to that of WT α1S. In support of the latter, it is important to note that the identification of critical α1S II-III loop residues 720–765 in chimeras without GFP was achieved at the expense of a substantial loss in Ca2+ transient amplitude relative to the Ca2+ transient expressed by the parent CSk3 chimera carrying the complete α1S II-III loop (14). Thus, an appropriate structural context is essential for a quantitative recovery of EC-coupling function.
It was intriguing to find that, on average, the shape of the fluorescence-voltage curve for the Ca2+-conducting and pore-mutant variants of the α1C/α1S II-III loop chimera were both sigmoidal (Fig. 3). A somewhat bell-shaped curve was expected for the former based on the significant dependence on the Ca2+ current for triggering release. We considered the possibility that the reduction in ΔF/Fmax observed in the pore-mutant variant of the II-III loop chimera reflected inactivation of the voltage sensor due to the elimination of a critical Ca2+-binding site (41). Several reasons argue against this possibility. First, the pore mutation α1S(E1014K) was shown to eliminate high-affinity Ca2+ binding to the subunit (39). Yet, WT α1S and α1S(E1014K) had fluorescence-voltage characteristics that were superimposable (Fig. 1). Second, the Ca2+-conducting and pore-mutant variants of the α1C/α1S all loop chimera expressed skeletal-type EC coupling with a similar ΔF/Fmax (Tables 1 and 2). This was not expected under the assumption that the pore mutation inactivated the voltage sensor. Finally, and most significantly, a reduction in ΔF/Fmax was observed in the Ca2+-conducting α1C/α1S II-III loop chimera when the Ca2+ current was blocked by Cd2+. This was not observed for WT α1S, nor for the α1C/α1S all loop chimera (Fig. 5). Hence, it could not be argued that Cd2+ block limited access of Ca2+ to the binding site on the voltage sensor. We further searched for an explanation in the data itself. A cell-by-cell fit of the fluorescence-voltage curve indicated that some cells expressing the Ca2+-conducting II-III loop chimera displayed a slight bell-shaped voltage dependence (three out of seven) whereas others did not (four out of seven). This heterogeneity contributed to the much larger variance of the data collected for the II-III loop chimera relative to other single-loop chimeras (see Fig. 2). A freeze-fracture analysis of mdg myotubes expressing the CSk3 chimera revealed that only a few of the chimeric DHPR particles present at a DHPR-RyR1 junction were arranged as tetrads (43). DHPR tetrad formation is a precondition for skeletal-type EC coupling (1). Hence, junctions with tetrads are more likely to trigger Ca2+ release by a skeletal-type mechanism whereas junctions without tetrads, or with “incomplete” tetrads, are more likely to trigger Ca2+ release via the Ca2+ current. We suggest that in myotubes expressing the II-III loop chimera, the EC-coupling mechanism itself may be heterogeneous due to a variable density of tetrads in each DHPR-RyR1 junction. The voltage dependence of the Ca2+ transient may approximate a sigmoidal relationship when the number of skeletal-like junctions in a muscle cell is high and bell-shaped when it is low. In addition, it is necessary to realize that the Ca2+-entry dependent component, in the best of circumstances, is small. This can be appreciated by comparing the maximum Ca2+ transient amplitudes expressed by WT α1S and WT α1C in Fig. 1. Such a component may not be always possible to detect, particularly when the voltage-dependent component is much larger.
The presence of a Ca2+-entry-dependent EC-coupling component in the α1S II-III loop chimera and not in the chimera carrying all the cytosolic domains of α1S, strongly suggests that structural determinants unknown at this point contribute to the stability of the DHPR-RyR1 complex. The mechanism by which the II-III loop in combination with other cytoplasmic domains of α1S enable the formation of a stable DHPR-RyR1 complex remains to be understood.
Acknowledgments
This work was supported by National Institutes of Health grants AR46448, HL47053, and T32 HL07936, a Training Grant predoctoral fellowship to L.C., a Cremer Fellowship to L.C., and a predoctoral fellowship from the Wisconsin Heart Association to D.C.S.
References
- 1.Flucher, B. E., and C. Franzini-Armstrong. 1996. Formation of junctions involved in excitation-contraction coupling in skeletal and cardiac muscle. Proc. Natl. Acad. Sci. USA. 93:8101–8106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Franzini-Armstrong, C., and F. Protasi. 1997. Ryanodine receptors of striated muscles: a complex channel capable of multiple interactions. Physiol. Rev. 77:699–729. [DOI] [PubMed] [Google Scholar]
- 3.Protasi, F., C. Franzini-Armstrong, and P. D. Allen. 1998. Role of ryanodine receptors in the assembly of calcium release units. J. Cell Biol. 140:831–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu, Z., J. Zhang, P. Li, S. R. W. Chen, and T. Wagenknecht. 2002. Three-dimensional reconstruction of the recombinant type 2 ryanodine receptor and localization of its divergent region 1. J. Biol. Chem. 277:46712–46719. [DOI] [PubMed] [Google Scholar]
- 5.Sherysheva, I. I., S. J. Ludtke, M. R. Backer, W. Chui, and S. L. Hamilton. 2002. 3D structure of the voltage-gated L-type Ca2+ channel by electron cryomicroscopy. Proc. Natl. Acad. Sci. USA. 99:10370–10375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolf, M., A. Eberhart, H. Glossmann, J. Striessnig, and N. Grigorieff. 2003. Visualization of the domain structure of an L-type Ca2+ channel using electron cryo-microscopy. J. Mol. Biol. 332:171–182. [DOI] [PubMed] [Google Scholar]
- 7.Takekura, H., C. Paolini, C. Franzini-Armstrong, G. Kugler, M. Grabner, and B. E. Flucher. 2004. Differential contribution of skeletal and cardiac II–III loop sequences to the assembly of dihydropyridine- receptor arrays in skeletal muscle. Mol. Biol. Cell. 15:5408–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perez-Reyes, E., and T. Schneider. 1994. Calcium channels: structure, function, and classification. Drug Dev. Res. 33:295–318. [Google Scholar]
- 9.De Ward, M., C. A. Gurnett, and P. K. Campbell. Structure and diversity of voltage-gated Ca2+ channels. 1996. In Ion Channels. T. Narahashi, editor. Plenum Press, New York. 41–87. [DOI] [PubMed]
- 10.Bezanilla, F. 2000. The voltage sensor in voltage-dependent ion channels. Physiol. Rev. 80:555–592. [DOI] [PubMed] [Google Scholar]
- 11.Tanabe, T., K. G. Beam, B. A. Adams, T. Niidome, and S. Numa. 1990. Regions of the skeletal muscle dihydropyridine receptor critical for excitation-contraction coupling. Nature. 346:567–572. [DOI] [PubMed] [Google Scholar]
- 12.Adams, B. A., T. Tanabe, A. Mikami, S. Numa, and K. G. Beam. 1990. Intramembrane charge movement restored in dysgenic skeletal muscle by injection of dihydropyridine receptor cDNAs. Nature. 346:569–572. [DOI] [PubMed] [Google Scholar]
- 13.Casarotto, M. G., F. Gibson, S. M. Pace, S. M. Curtis, M. Mulcair, and A. F. Dulhunty. 2000. A structural requirement for activation of skeletal ryanodine receptors by peptides of the dihydropyridine receptor II–III loop. J. Biol. Chem. 275:11631–11637. [DOI] [PubMed] [Google Scholar]
- 14.Nakai, J., T. Tanabe, T. Konno, B. Adams, and K. G. Beam. 1998. Localization in the II–III loop of the dihydropyridine receptor of a sequence critical for excitation-contraction coupling. J. Biol. Chem. 273:24983–24986. [DOI] [PubMed] [Google Scholar]
- 15.Saiki, Y., R. El-Hayek, and N. Ikemoto. 1999. Involvement of the Glu724-Pro760 region of the dihydropyridine receptor II–III loop in skeletal muscle-type excitation-contraction coupling. J. Biol. Chem. 274:7825–7832. [DOI] [PubMed] [Google Scholar]
- 16.Wilkens, C. M., N. Kasielke, B. E. Flucher, K. G. Beam, and M. Grabner. 2001. Excitation-contraction coupling is unaffected by drastic alteration of the sequence surrounding residues L720–L764 of the α1S II–III loop. Proc. Natl. Acad. Sci. USA. 98:5892–5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kugler, G., R. G. Weiss, B. E. Flucher, and M. Grabner. 2004. Structural requirements of the dihydropyridine receptor α1S II–III loop for skeletal-type excitation-contraction coupling. J. Biol. Chem. 279:4721–4728. [DOI] [PubMed] [Google Scholar]
- 18.Chandler, W. K., R. F. Rakowski, and M. F. Schneider. 1976. Effects of glycerol treatment and maintained depolarization on charge movement in skeletal muscle. J. Physiol. 254:285–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leong, P., and D. H. MacLennan. 1998. A 37-amino acid sequence in the skeletal muscle ryanodine receptor interacts with the cytoplasmic loop between domain II and domain III in the skeletal muscle dihydropyridine receptor. J. Biol. Chem. 273:7791–7794. [DOI] [PubMed] [Google Scholar]
- 20.Leong, P., and D. H. MacLennan. 1998. The cytoplasmic loops between domains II and III and domains III and IV in the skeletal muscle dihydropyridine receptor bind to a contiguous site in the skeletal muscle ryanodine receptors. J. Biol. Chem. 273:29958–29964. [DOI] [PubMed] [Google Scholar]
- 21.Leong, P., and D. H. MacLennan. 1998. Complex interactions between skeletal muscle ryanodine receptor and dihydropyridine receptor. Biochem. Cell Biol. 76:681–694. [DOI] [PubMed] [Google Scholar]
- 22.Sencer, S., R. V. L. Papineni, D. B. Halling, P. Pate, J. Krol, J.-Z. Zhang, and S. L. Hamilton. 2001. Coupling of RyR1 and L-type calcium channels via calmodulin binding domains. J. Biol. Chem. 276:38237–38241. [DOI] [PubMed] [Google Scholar]
- 23.Proenza, C., J. O'Brien, J. Nakai, S. Mukherjee, P. D. Allen, and K. G. Beam. 2002. Identification of a region of RyR1 that participates in allosteric coupling with the α1S (CaV1.1) II-III loop. J. Biol. Chem. 277:6530–6535. [DOI] [PubMed] [Google Scholar]
- 24.Papadopoulos, S., V. Leuranguer, R. A. Bannister, and K. G. Beam. 2004. Mapping sites of potential proximity between the dihydropyridine receptor and RyR1 using a cyan fluorescent protein-yellow fluorescent protein tandem as a fluorescent resonance energy transfer probe. J. Biol. Chem. 279:44046–44056. [DOI] [PubMed] [Google Scholar]
- 25.Ahern, C. A., D. Bhattacharya, L. Mortenson, and R. Coronado. 2001. A component of excitation-contraction coupling triggered in the absence of the T671–L690 and L720–Q765 regions of the II–III loop of the dihydropyridine receptor α1S pore subunit. Biophys. J. 81:3294–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sheridan, D. C., W. Cheng, C. A. Ahern, L. Mortenson, D. Alsammarae, P. Vallejo, and R. Coronado. 2003. Truncation of the carboxyl terminus of the dihydropyridine receptor β subunit promotes Ca2+ dependent excitation-contraction coupling in skeletal myotubes. Biophys. J. 84:220–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sheridan, D. C., L. Carbonneau, C. A. Ahern, P. Nataraj, and R. Coronado. 2003. Ca2+-dependent excitation-contraction coupling triggered by the heterologous cardiac/brain DHPR β2a-subunit in skeletal myotubes. Biophys. J. 85:3739–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sheridan, D. C., W. Cheng, L. Carbonneau, C. Ahern, and R. Coronado. 2004. Involvement of a heptad repeat in the carboxyl terminus of the dihydropyridine receptor β1a subunit in the mechanism of excitation-contraction coupling in skeletal muscle. Biophys. J. 87:929–942 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen, L., E. Esteves, J. M. Sabatier, M. Ronjat, M. De Waard, P. D. Allen, and I. N. Pessah. 2003. Maurocalcine and peptide A stabilize distinct subconductance states of the ryanodine receptor type 1, revealing a proportional gating mechanism. J. Biol. Chem. 278:16095–16106. [DOI] [PubMed] [Google Scholar]
- 30.Grabner, M., R. T. Dirksen, N. Suda, and K. G. Beam. 1999. The II–III loop of the skeletal muscle dihydropyridine receptor is responsible for the bi-directional coupling with the ryanodine receptor. J. Biol. Chem. 274:21913–21919. [DOI] [PubMed] [Google Scholar]
- 31.Kasielke, N., G. J. Obermair, G. Kugler, M. Grabner, and B. E. Flucher. 2003. Cardiac-type EC-coupling in dysgenic myotubes restored with Ca2+ channel subunit isoforms α1C and α1D does not correlate with current density. Biophys. J. 84:3816–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garcia, J., T. Tanabe, and K. G. Beam. 1994. Relationship of calcium transients to calcium currents and charge movements in myotubes expressing skeletal and cardiac dihydropyridine receptors. J. Gen. Physiol. 103:125–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carbonneau, L., D. Bhattacharya, and R. Coronado. 2004. Limited role of cytoplasmic loops of the skeletal muscle DHPR pore subunit alpha-1S in excitation-contraction coupling. Biophys. J. 86:219a (Abstr.). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carbonneau, L., D. Bhattacharya, W. Cheng, D. C. Sheridan, and R. Coronado. 2005. Limited role of the II–III loop of the skeletal dihydropyridine receptor α1S pore subunit in excitation-contraction coupling. Biophys. J. 88:640a (Abstr.). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beurg, M., M. Sukhareva, C. Strube, P. A. Powers, R. Gregg, and R. Coronado. 1997. Recovery of Ca2+ current, charge movements, and Ca2+ transients in myotubes deficient in dihydropyridine receptor beta 1 subunit transfected with beta 1 cDNA. Biophys. J. 73:807–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang, J., P. T. Ellinor, W. A. Sather, J.-F. Zhang, and R. W. Tsien. 1993. Molecular determinants of Ca2+ selectivity and ion permeation in L-type Ca2+ channels. Nature. 366:158–161. [DOI] [PubMed] [Google Scholar]
- 37.Dirksen, R. T., and K. G. Beam. 1999. Role of calcium permeation in dihydropyiridine receptor function. Insights into channel gating and excitation-contraction coupling. J. Gen. Physiol. 114:393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tanabe, T., B. A. Adams, S. Numa, and K. G. Beam. 1991. Repeat I of the dihydropyridine receptor is critical in determining calcium channel activation kinetics. Nature. 352:800–803. [DOI] [PubMed] [Google Scholar]
- 39.Peterson, B. Z., and W. A. Catterall. 1995. Calcium binding in the pore of L-type calcium channels modulates high affinity dihydropyridine binding. J. Biol. Chem. 270:18201–18204. [DOI] [PubMed] [Google Scholar]
- 40.Luttgau, H. C., and W. Spiecker. 1979. The effects of calcium deprivation upon mechanical and electrophysiological parameters in skeletal muscle fibers of the frog. J. Physiol. (Lond.). 296:411–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brum, G., R. Fitts, G. Pizarro, and E. Rios. 1988. Voltage sensors of the frog skeletal muscle membrane require calcium to function in excitation-contraction coupling. J. Physiol. (Lond.). 398:475–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Melzer, W., M. F. Schneider, B. J. Simon, and G. Szucs. 1986. Intramembrane charge movement and calcium release in frog skeletal muscle. J. Physiol. 373:481–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paolini, C. C. Franzini-Armstrong, M. Grabner, P. D. Allen, and F. Protasi. 2003. CSk3 and R9/R16 restore tetrads in α1S DHPR/RyR1 double knock-out myotubes. Biophys. J. 8417a (Abstr.).