

Abstract
The molecular code of specific DNA recognition by proteins as a paradigm in molecular biology remains an unsolved puzzle primarily because of the subtle interplay between direct protein-DNA interaction and the indirect contribution from water and ions. Transformation of the nonspecific, low affinity complex to a specific, high affinity complex is accompanied by the release of interfacial water molecules. To provide insight into the conversion from the loose to the tight form, we characterized the structure and energetics of water at the protein-DNA interface of the BamHI complex with a noncognate sequence and in the specific complex. The fully hydrated models were produced with Grand Canonical Monte Carlo simulations. Proximity analysis shows that water distributions exhibit sequence dependent variations in both complexes and, in particular, in the noncognate complex they discriminate between the correct and the star site. Variations in water distributions control the number of water molecules released from a given sequence upon transformation from the loose to the tight complex as well as the local entropy contribution to the binding free energy. We propose that interfacial waters can serve as a “hydration fingerprint” of a given DNA sequence.
INTRODUCTION
Protein binding to specific DNA sequences is a key element in various biological functions related to processing the genetic information by regulating transcription, replication, and recombination. The mechanism of DNA sequence discrimination, however, is still poorly understood. Most of our knowledge has been derived from crystal structures of specific protein-DNA complexes that revealed diverse strategies for a protein interacting with its DNA partner upon forming a high affinity complex (1,2). Besides the direct hydrogen bonds established with DNA bases, indirect interactions with phosphates and those mediated through water molecules were also found to be important determinants of selectivity. The energetic contributions of these contacts have been assessed by kinetic measurements using mutant proteins and DNA base analogs (3,4). Binding to specific sequences is associated with a negative heat capacity change that is termed the ‘thermodynamic signature’ of high affinity complex formation (5). The process of specific recognition is initiated by association of the protein with nonspecific DNA sites (6,7), which is accompanied with negligible heat capacity changes indicating that the partners are loosely bound (8–12). The protein-DNA interface remains fully hydrated (13,14), and the configurational freedom of the interacting partners is not significantly restricted (5).
Both the high mobility of the protein on the substrate and the low affinity binding of the protein are the major obstacles for structural studies of the nonspecific complexes. Only five experimental structures are proposed to represent this initial stage of protein-DNA binding (15–19). Nonspecific complexes are characterized by the lack of intimate intermolecular contacts and the excessive hydration of the protein and DNA that are held together by long-range Coulombic interactions (20,21).
The conversion of the nonspecific complexes into specific ones is accompanied by the release of water molecules from the protein-DNA interface into the bulk, which provides a favorable entropic contribution to the free energy of binding (22). The number of waters released during this transformation has been determined by osmotic stress measurements (13,14,23), although the actual values are still a matter of debate (D. Cao and L. Jen-Jacobson, personal communication, 2004). These studies can estimate the total number of waters that depart from the cognate sequence and the flanking basepairs that are required for tight binding, but they cannot give a detailed description of the process. The number of the waters released by the individual basepairs that would allow the decomposition of the binding energy and heat capacity into local contributions cannot be assessed experimentally. Since the energetics of the conversion from the loose to the tight complex is determined by the balance between the deformability (flexibility) of the given DNA sequence and the amount of waters released from the protein-DNA interface into bulk, we hypothesize that sequence dependent distribution of the interfacial water can play a role in selecting a given DNA sequence by a protein. To probe this idea we characterized the water structure and energetics at the protein-DNA interface of the nonspecific BamHI complex and compared it to the interfacial water structure in the corresponding specific complex.
BamHI is a type II restriction endonuclease that recognizes the palindromic GGATCC sequence and cleaves it with very high specificity in the presence of Mg2+ cofactors (7,24). Replacement of a single basepair, a guanine with adenine at the second position (GAATCC), decreases the KM by 3 orders of magnitude, and kcat by 6 orders of magnitude (12). A comprehensive set of BamHI structures is available: the free enzyme (1bam; 25), in complex with specific (1bhm; 26) and noncognate DNA (1esg; 18), pre- (2bam) and postreactive (3bam) complex (27) that provide snapshots along the reaction pathway.
To provide insight into water structure changes that accompany the conversion of a nonspecific to a specific complex, we provide for the first time, to our knowledge, a detailed structural and energetic analysis of the interfacial waters in a noncognate protein-DNA complex using the complex of BamHI with the noncognate GAATCC sequence. Since the solvent molecules are highly mobile, the crystal structure has an incomplete description of the waters in the interface between the protein and the DNA. To obtain a fully hydrated model of the noncognate complex, we used cavity biased grand canonical Monte Carlo (CB/GCE) simulations. We have tested the reliability of the method by comparing the observed and computed solvent sites, and we demonstrate the robustness of the CB/GCE simulations as a technique to complement crystallographic data of noncognate complexes. Based on proximity analysis of the water structure in the noncognate complex, we find that the water distribution at the protein-DNA interface of both the cognate and noncognate complexes follows a sequence specific distribution that allows for a local control of the number of waters released upon formation of the tight complex. We thus hypothesize that sequence specific structure of the water can serve as a “hydration fingerprint” of a given DNA sequence.
METHODS
Models
In choosing the two crystal structures for this study, our sole criterion was to select structures representing the different binding modes. For studies of the enzymatic mechanism, these structures may be less than ideal. Based on energetic considerations, the noncognate complex (1esg) is considered as an intermediate in course of the transition from the loose, nonspecific to the tight, specific complex (28), rather than a snapshot of nonspecific binding that occurs during linear diffusion of proteins on DNA. Also, the relevance of the asymmetric contacts in the minor groove was questioned based on kinetic studies using modified oligonucleotides (29). In our study, we assume that this problem does not affect the structure of the protein facing the cognate sequence since assuming otherwise would imply that the cognate sequence can be recognized in different ways.
The noncognate model ESG has been derived from the crystal structure of BamHI with the noncognate TGAATCCA sequence (the star site is displayed in italics; PDB code: 1esg (18)), whereas the corresponding specific model BHM was constructed from the complex with the cognate TATGGATCCATA sequence without bivalent metal ions (PDB code 1bhm (26)). Hydrogens were built by the HBUILD module of the program CHARMM version 23 (30). For the GCE simulations, all crystallographic water molecules were removed from both complexes. The generated water sites of the noncognate and specific complexes were collected into the ESG_GS and BHM_GS models. Numbering of the phosphates corresponds to base numbers of the shortened substrates; from P2-P7 in the first strand and P10-P15 in the second strand. The scissile groups are P3 and P11, respectively.
Grand canonical Monte Carlo simulations
To obtain a description of the solvent molecules at the protein-DNA interface in the noncognate complex of BamHI, the CB/GCE method has been applied (31,32). The CB/GCE technique has demonstrated its robustness in modeling solvent molecules at crystal hydrates and protein active sites (32,33). In this approach the insertion of a molecule is attempted if a cavity of an appropriate radius is found and the insertion is accepted with a probability:
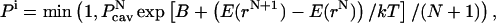 |
where 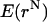 is the potential energy of the system of N particles at configuration rN, and
is the potential energy of the system of N particles at configuration rN, and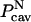 is the probability of finding a cavity of a specific size. To maintain microscopic reversibility, the probability of a deletion of a particle is given by
is the probability of finding a cavity of a specific size. To maintain microscopic reversibility, the probability of a deletion of a particle is given by
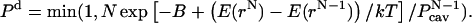 |
The parameter B is related to the excess chemical potential μ′ as
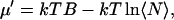 |
where 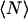 is the average number of particles.
is the average number of particles.
This method overcomes the problem of particle generation in a condensed phase at random positions by first calculating the probability Pcav based on a grid scan of the system and then choosing randomly from the available cavities. Simulations in the grand canonical ensemble result in an excess chemical potential at a density which is obtained after the equilibrium has been reached. The density of the bulk phase (i.e., far from solute) is regulated by adjusting the B parameter. After equilibrium has been reached, the B parameter is modified according to the deviations of the bulk phase density from the target density until the target bulk density is obtained.
Water site definition
The water positions from the simulation were determined by the generic solvent site (GSS) approach developed by Mezei and Beveridge (34). For each configuration of the trajectory, waters are assigned to generic sites (GSs) based on a procedure using the so-called Hungarian method of graph theory (35). Here, the maximum deviation is minimized between the GS and the water assigned to it in each snapshot. If the water-site distance exceeds 3.5 Å from the GS, a new site is added at the position of the water site. This process is iterated until convergence. Since the GSs do not carry labels, water molecules can exchange between GSs. The GSs are defined by the mean oxygen positions and characterized by the mean square deviation of the position and by the occupancy of the site. The occupancy is computed as the number of configurations in which a water molecule is assigned to the GS divided by the total number of configurations.
Proximity analysis
The water structure at the protein-DNA interface was determined using the method of proximity analysis (36,37). Proximity analysis assigns a proximal region to each solute atom defined by Voronoi tessellation of the space generated by the solute atoms. This is equivalent to partitioning the space by the bisector planes of neighboring atoms. The proximity regions of solute groups are the unions of the proximity regions of solute atoms forming the group. The definition of the solute groups is flexible; they can be residues, basepairs, grooves, or phosphate groups. This algorithm is based on calculating quasicomponent distribution functions of solvent molecules belonging to the proximity region of each group of the solute. The distribution functions are computed from the snapshots generated by the simulation of a given ensemble. The proximity radial distribution function gi(r) is defined as the quasicomponent correlation function at distance r between solute atom i and the solvents as,
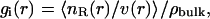 |
where nR(r) gives the number of waters whose distance r from the nearest solute atom i falls into the interval [r, r + dr], v(r) is the volume of the shell containing points nearest to solute atom i and falling into the interval [r, r + dr], and the symbol <…> signifies an average over the snapshots of the trajectory. The running coordination number is defined as
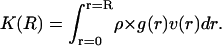 |
Energy of specific water sites
The energy associated with a site was evaluated as the average of the interaction energies between the water and the protein-DNA complex calculated for all the waters contributing to that site. The energy associated with a given region (e.g., minor groove) was calculated as the average of the energies of all sites in that region, weighted by their occupancies.
Computational details
The noncognate and cognate models were placed in a rectangular cell with the dimensions 84 Å × 84 Å × 84 Å for ESG and 93 Å × 62 Å × 75 Å for BHM, respectively. The protein-DNA systems were equilibrated for 107 Monte Carlo steps by adjusting the B parameter to reach an average density in the outer 5 Å layer (considered to be bulk phase) comparable to that of liquid water. Then a production run for 107 Monte Carlo steps was conducted for both complexes. During the production run, the bulk phase density was found to vary around 1.001 ± 0.003 g/ml for the noncognate complex and 0.998 ± 0.002 g/ml for the cognate complex. The protein and DNA conformations were kept fixed. The CB/GCE simulations were performed with the MMC program (http://fulcrum.physiobio.mssm.edu/∼mezei/mmc) using the force field of CHARMM, version 22 (38). Waters were represented by the TIP3P model (39). Nonbonded interactions were treated under the minimum image convention for the solute-water interactions, based on distances from the residue centers to the water oxygen, and water-water interactions were cut off at 10.0 Å.
RESULTS
Comparison of simulated and crystallographic water positions
To assess the reliability of our calculations, water positions generated by CB/GCE simulations were compared to the water sites observed in the crystal structure of cognate and noncognate BamHI complexes. In the crystal structure of the BHM model with the specific GGATCC sequence, 97 water molecules out of the total 215 are found in the interface region, defined as a box of 35 Å × 40 Å × 30 Å centered at the protein-DNA interface that could accommodate the central eight basepairs and the active site. All of the 97 waters have been successfully located by the CB/GCE calculations with an RMS deviation of 1.2 Å. Unlike for the noncognate complex (vide infra) the simulation has found no additional sites. This is consistent with the tight packing between the protein and DNA in the specific complex and the low thermal factors of the observed waters.
In the noncognate complex (ESG), 579 fully occupied crystallographic water positions were observed. We extracted water molecules from the 107 configurations collected during the simulation and filtered those that belong to the protein-DNA complex using the recently developed circular variance criteria (40). This analysis produced 4795 GSs of which 2603 sites were fully occupied and 1338 additional sites had occupancy above 0.5. GSs are found at 578 out of the 579 crystallographically observed water sites. The GSs are in excellent agreement with the crystallographic water positions with an RMS deviation of 1.3 Å. The convergence of the simulations was tested by locating GSs using the waters in the interface region (within a box of 35 Å × 40 Å × 30 Å) independently for the two sets of 5 × 106 configurations collected during the simulation. In either set, 787 GSs could be determined with an average RMS of 0.9 Å. If all 107 configurations are analyzed, 795 water sites could be located. These waters sites are quite stable; 786 positions are occupied in more than half of the configurations. These GSs correspond to 149 out of the 150 water positions in the same region of the crystal structure with RMS deviation of 1.4 Å.
The good agreement between the computed and the experimental water positions in this complex and also in previous studies (32,33,41) demonstrate the robustness of the CB/CGE technique in generating fully hydrated models of crystallographic structures. Application of CB/CGE simulations to noncognate protein-DNA complexes is particularly useful due to the large number of solvent molecules that cannot be located at the protein-DNA interface due to their high mobility.
Water structure around noncognate and cognate substrates in complex with BamHI
To investigate sequence effects on local water distribution and discern its possible role in sequence discrimination, hydration patterns around specific and noncognate complexes have been analyzed. To this end, the proximity radial distribution functions and the corresponding running coordination numbers of the interfacial waters obtained in CB/GCE simulations were computed in both noncognate and specific complexes (Fig. 1). For g(R) around the phosphates, see Supplementary Material.
FIGURE 1.

Radial distribution functions (g(R)) and running coordination numbers (K(R)) of waters around the major and minor groove of the recognition sequence of specific (bold line) and noncognate (thin line) substrates in complex with BamHI.
The radial distribution functions in the grooves of the specific BHM_GS complex are mostly limited to the first two hydration shells, whereas in the noncognate ESG_GS complex they extend almost to a distance of 10 Å due to the larger separation between the protein and the noncognate sequence. The GC3 and CG6/TA6 basepairs neighboring the scissile phosphate group are exceptions because the minor groove faces the bulk, resulting in a continuous radial distribution function till 10 Å. In both the specific and the noncognate complexes, the phosphates pointing toward the protein (P2, P3, and P10–P12) are surrounded by a single hydration shell. For the phosphates facing the solvent (P6, P7, P14, P15), the g(R)s show several additional hydration layers, which are not well structured with the exception of P13 in the middle of the cognate sequence.
The running coordination numbers of the water molecules computed for the first hydration shell (up to 3.5 Å) along the six basepairs of the recognition sequence in BHM_GS and ESG_GS models are presented in Table 1. The coordination numbers in the first two hydration shells are summarized in the Supplementary Material. The noncognate ESG_GS complex retains a full hydration layer around the recognition sequence with the exception of the phosphates, whereas the tight BHM_GS complex is significantly dehydrated especially at the major groove that contacts the protein. The major groove of the noncognate complex is also more dehydrated than the minor groove by nine water molecules.
TABLE 1.
Number of water molecules around the minor groove, major groove, and phosphates along the DNA basepairs of the cognate (BHM_GS) and noncognate (ESG_GS) complex in the first solvation shell (up to 3.5 Å)
| Major grove
|
Minor groove
|
Phosphate strand 1
|
Phosphate strand 2
|
Total
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| BHM_GS | ESG_GS | BHM_GS | ESG_GS | BHM_GS | ESG_GS | BHM_GS | ESG_GS | Σ(BHM) | Σ(ESG) | |
| GC2 (P2,P15) | 1.7 | 7.1 | 6.2 | 8.8 | 0.0 | 2.8 | 4.6 | 3.8 | 12.5 | 22.5 |
| GC3 (P3,P14) | 2.1 | 5.0 | 4.4 | 6.2 | 1.5 | 2.1 | 5.4 | 4.5 | 13.4 | 17.8 |
| AT4 (P4,P13) | 2.0 | 6.6 | 1.0 | 7.2 | 1.0 | 0.8 | 1.9 | 5.5 | 5.9 | 20.1 |
| TA5 (P5,P12) | 0.1 | 7.0 | 2.3 | 8.3 | 0.2 | 1.6 | 0.0 | 1.1 | 2.6 | 18.0 |
| CG6,TA6 (P6,P11) | 3.2 | 5.4 | 6.4 | 7.9 | 1.9 | 3.8 | 0.8 | 2.3 | 12.3 | 19.4 |
| CG7 (P7,P10) | 2.0 | 5.8 | 3.7 | 7.7 | 4.0 | 4.3 | 0.7 | 3.4 | 10.4 | 21.2 |
| Σ | 11.1 | 36.9 | 24.0 | 46.1 | 8.6 | 15.4 | 13.4 | 20.6 | 57.1 | 119.0 |
The total coordination number computed for the individual basepairs shows variations along the recognition sequence in both complexes, although in opposite directions. In the specific complex, a maximal hydration is observed for the basepairs 3′ to the scissile bond (GC3 and CG6), whereas the corresponding correct site in the noncognate structure (GC3) exhibits a minimal coordination number. The hydration pattern is perturbed around the star site (TA6); the running coordination number is greater by 1.6 water molecules than that of the correct site. This suggests that water distribution at the protein-DNA interface may be dependent on the actual DNA sequence.
Water structure in both the minor and major grooves shows sequence dependent variations (Fig. 2). It suggests that in a loosely associated protein-DNA complex, the water distribution is determined by the DNA sequence even in the groove that faces the protein. In the major groove of the specific complex, the basepairs neighboring the scissile phosphates from the 3′ side (GC3 and CG6) are the most solvent exposed, whereas in the noncognate complex the corresponding basepairs (GC3 and TA6) are the least hydrated. In the specific complex, waters in the major groove next to the scissile phosphate occupy the position of the catalytically essential metal ion cofactor and also fill the space that is required for the conformational changes during the catalytic reaction. Partial dehydration of the basepairs 3′ to the cleavage site suggests that GC3 and TA6 are the most exposed to form contacts with the protein.
FIGURE 2.

Coordination numbers (K(R)) of water molecules in the first (up to 3.5 Å, ▪) and the first two hydration shells (up to 5.5 Å, ▴) (A) in the major groove and (B) in the minor groove along the basepairs of the recognition sequence. Values referring to the specific sequence are connected by straight lines, whereas those computed for the noncognate sequence are connected by dash-dotted lines.
Since the protein and DNA coordinates were kept fixed in the CB/GCE run, the asymmetry of the two subunits in the specific complex is also reflected in the water structure obtained from the simulation. Due to the contact of the DNA to Asp-196 of the R subunit, the minor groove of the first half-site is less hydrated in the specific complex and the maximum hydration is shifted from the GC3 to GC2. We expect that in the fully functional state of BamHI, where no such minor groove contact is present in either subunit (29), the minor groove of both basepairs 3′ to the scissile phosphates has the highest number of coordinated water molecules.
Indirect interactions with the phosphates are important for stability of the specific complex, the formation of which results in exclusion of most of the waters from the hydration shells of those phosphates that contact the protein (P2–P5 of the first strand and P13–P15 of the second strand). The few remaining water molecules form a single hydration shell with a low (0–2) coordination number around the phosphates, which participate in the indirect interactions. In the noncognate complex the contacts between the protein and DNA are restricted to water mediated interactions with the phosphates, mostly at the correct first half-site. Therefore P2–P5 and P13–P15 are also less hydrated than the other phosphates facing the solvent, but their hydration is increased compared to that in the specific complex (Table 1). The difference between the specific and noncognate complex is most pronounced at the star site with the G→A substitution (1.5 water molecules).
Differences in hydration of the recognition sequence between the cognate and noncognate complex
Water release is one of the major driving forces of specific complex formation between the protein and a DNA, and the concomitant entropy increase makes a major contribution to the free energy of binding (42,43). We found that local water structure is influenced by a single basepair substitution in the recognition sequence. We analyze the effect of this substitution on the amount of released water in the formation of the specific complex.
Running coordination numbers (presented in Table 1) computed for the first and the first two solvation shells of the specific BHM_GS complex have been subtracted from those obtained for the noncognate ESG_GS complex. In total, the first solvation shell of the noncognate complex contains 62 waters more than that of the specific complex, whereas including the second solvation shell, 97 more waters can be found. We propose that the additional water molecules are released into the bulk in the process of specific complex formation, i.e., upon transition from the noncognate to the cognate structure. During this transition, 26 water molecules are displaced from the first hydration shell of the major groove and 22 from the minor groove. Phosphates of the two strands contribute to water release almost an equal amount of seven waters each. Water release from the minor groove might be overestimated due to the contact between the R subunit of BamHI and the first half-site of the substrate in the specific complex.
Due to the perturbation in local water structure, the amount of water released shows considerable variations along the recognition sequence (Fig. 3). Largest hydration changes of 14–15 water molecules are associated with the middle two basepairs (AT4 and TA5), whereas the smallest difference of 4.5 water molecules can be observed at the correct scissile site (GC3). Interestingly, on the corresponding star site of the sequence (TA6), the hydration difference is larger by 2.5 water molecules than at the correct site, indicating that water release is affected at single basepair level. Sequence dependent variations in hydration are more pronounced when the second solvation shell is also taken into account (Fig. 3 B); changes in solvation around the correct and the star site increase to 3.5 water molecules.
FIGURE 3.

Difference between the coordination numbers computed for the major groove (▪), minor groove (•), first strand phosphates (▴), second strand phosphates (▾), and total (♦) of each basepair of the recognition sequence (A) in the first hydration shell (up to 3.5 Å) and (B) in the second hydration shell (up to 5.5 Å).
Energetics of interfacial water molecules in the noncognate complex
We have shown that the formation of a high affinity complex requires the displacement of more water molecules from the incorrect site of the recognition sequence than from the correct site. However, energetic requirements of replacing water molecules by protein groups are determined by the interaction energies of the waters with the specific site to which they are coordinated. In an attempt to elucidate whether interaction energies can discriminate between the correct and the star site, we have computed the solute-solvent interaction energies of water molecules around the grooves and phosphates of the recognition sequence (Table 2). Clearly, solute-solvent energies are dependent on the sequence and thus they might serve as the energetic basis for sequence discrimination. In the major groove, interaction energies of the waters are strongest with the middle basepair AT4, predicting the importance of this basepair for stability of the complex, but are almost equal for the correct and the star site (GC3 and AT6, respectively). Solute-solvent energies of the scissile phosphates of the correct and the star site clearly show a difference: the interaction energy of P11 is lower by 2.9 kcal/mol than that of the corresponding phosphate on the first half-strand (P3), suggesting that it is more difficult to replace the water molecules around the scissile phosphate of the star site than around the correct site. Interestingly, the interaction energies of the opposite phosphates (P6 and P14) with the surrounding water molecules also differ from each other by 1.6 kcal/mol. Phosphates of the central basepairs (P4 and P12) that contact BamHI have lowest interaction energies in agreement with their role as clamps in the high affinity complex with a full 12 basepair DNA (29).
TABLE 2.
Average solute-solvent energies (kcal/mol) of water molecules bound in the minor groove, major groove, and phosphates of the noncognate recognition sequence (ESG_GS)
| Major groove | Minor groove | Phosphate I | Phosphate II | |
|---|---|---|---|---|
| GC2 (P2,P15) | −7.55 ± 0.55 | −13.85 ± 0.95 | −13.27 ± 0.42 | −16.43 ± 0.43 |
| GC3 (P3,P14) | −7.58 ± 0.43 | −9.78 ± 0.47 | −12.65 ± 0.45 | −15.1 ± 0.37 |
| AT4 (P4,P13) | −12.74 ± 0.50 | −9.30 ± 0.32 | −16.81 ± 0.97 | −14.01 ± 0.32 |
| TA5 (P5,P12) | −7.03 ± 0.36 | −10.65 ± 0.44 | −15.63 ± 0.64 | −17.02 ± 0.85 |
| CG6,TA6 (P6,P11) | −7.32 ± 0.41 | −11.33 ± 0.52 | −13.51 ± 0.36 | −15.55 ± 0.53 |
| CG7 (P7,P10) | −7.16 ± 3.1 | −13.51 ± 0.71 | −17.02 ± 0.43 | −12.28 ± 0.34 |
DISCUSSION
Water is an essential participant in macromolecular binding and it can contribute to recognition in several ways. Complex formation is initiated by interactions between partners of protein-protein or protein-DNA molecules with fully hydrated surfaces. During the process, specific bound waters are expelled from the interface leading to burial of the contact surfaces. Several water molecules, however, may remain trapped at the interface and serve to mediate interactions between the macromolecules. Interfacial water molecules in specific complexes not only act as linkers, but they have also been shown to buffer electrostatic repulsion between negatively charged groups of protein and DNA (44).
Both crystallographic and computational evidences show that hydration around the free DNA is mostly local and correlates with the groove width (45–49). Thus water distribution is sequence dependent: in general CG basepairs are more hydrated than AT basepairs. Major groove hydration patterns were proposed to offer the possibility for sequence recognition (45–49). Since the protein-DNA interface in nonspecific complexes is almost fully hydrated, we hypothesize that water structure around the DNA in such complexes is also determined by its sequence. This could offer a sequence dependent control of the amount of water released during transformation of the loose (nonspecific) to the tight (specific) form and thus regulate the local entropic contribution of a given DNA sequence to the binding energy. The presence of a protein, however, can perturb the water structure around the DNA. The extent of the perturbation of the water structure around the DNA by BamHI in noncognate sequences could be characterized by comparing the hydration pattern found in this work with the hydration pattern around uncomplexed DNA—this comparison is the subject of future work.
Interfacial water structure in noncognate complexes has not been characterized so far. To deduce the role of local water structure in sequence discrimination, we have compared the water distribution around cognate and noncognate sequences in complex with the BamHI protein. The question we focused on was whether local solvent structure around the individual basepairs in the loose protein-DNA complex can reflect perturbations in the recognition sequence. To this end, fully hydrated models of both complexes were generated using CB/GCE simulations, and water distributions around grooves and phosphates have been compared for the specific and noncognate substrates. Since neither in the specific nor in the noncognate complex does BamHI bend the DNA upon binding, conformational effects on the hydration pattern were not analyzed.
We demonstrated the robustness of the CB/GCE technique by reproducing all but one of the experimentally determined waters and showed the usefulness of the technique for locating highly mobile water molecules that cannot be resolved in the electron density map, thus complementing crystallographic data on noncognate complexes. The reason for missing the last crystallographic site could be either a minor shortcoming of the potential parameter set or a minor error in some of the heavy atom positions in the crystal structure. Indeed, an additional use of the CB/GCE technique could be to help refining both experimental structures and potential parametrizations through the analysis of such ‘missed’ experimental sites.
Interfacial water structure around individual basepairs was found to follow a sequence dependent distribution in both specific and noncognate BamHI complexes. This suggests that DNA hydration in a loosely associated noncognate complex is in principle determined by the basepair series, thus serving as a “fingerprint” of the given sequence. Variations along the grooves of the six basepairs of the recognition sequence are in opposite directions in the two complexes: although basepairs 3′ to the scissile phosphates (GC3 and CG6) have the highest number of coordinated water molecules in the specific complex, they are least hydrated in the noncognate complex. Since the noncognate complex represents an intermediate during the conversion from the nonspecific to the tight complex (18), the partial dehydration of the basepairs 3′ to the scissile phosphates in the noncognate complex can indicate their role as first contact points in the formation of the tight complex (50).
Sequence dependent variations of the water coordination numbers in both the specific and noncognate complexes result in sequence dependent modulation of the number of water molecules that are released between the loose and tight complex forms. Most waters are released from the middle basepairs and thus will provide the largest entropy contribution to the free energy of binding in agreement with their role as clamps in the high affinity complex with a full 12 basepair DNA (29). We also found that waters are most strongly associated with the major groove and phosphate of these sites. Basepairs 3′ to the scissile phosphates release the smallest number of water molecules, thus they are not likely a key factor in stabilizing the specific complex. We must note, however, that the presence of metal ions can change this observation, although specific binding by BamHI can be achieved even in the absence of metals. There is a clear difference of 2.5 water molecules in the first hydration shell and 3.5 waters in the second hydration shell between the water release from the correct and the star site. We can conclude that sequence dependence of interfacial water structure can locally control the number of released water molecules and can be used for discriminating between correct and star sites. Generalization of this “fingerprint” hypothesis would require analysis of more such nonspecific complexes. This is currently in progress in our laboratory.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
This work was supported by grants F 046164 and T 34131 from the Hungarian Scientific Research Fund, Public Health Service CA 63317 (R.O.), and Bolyai János fellowships for M.F.
References
- 1.Jones, S., P. van Heyningen, H. M. Berman, and J. M. Thornton. 1999. Protein-DNA interactions: a structural analysis. J. Mol. Biol. 287:877–896. [DOI] [PubMed] [Google Scholar]
- 2.Nadassy, K., S. J. Wodak, and J. Janin. 1999. Structural features of protein-nucleic acid recognition sites. Biochemistry. 38:1999–2017. [DOI] [PubMed] [Google Scholar]
- 3.Jen-Jacobson, L. 1997. Protein-DNA recognition complexes: conservation of structure and binding energy in the transition state. Biopolymers. 44:153–180. [DOI] [PubMed] [Google Scholar]
- 4.Oda, M., and H. Nakamura. 2000. Thermodynamic and kinetic analyses for understanding sequence-specific DNA recognition. Genes Cells. 5:319–326. [DOI] [PubMed] [Google Scholar]
- 5.Jen-Jacobson, L., L. E. Engler, J. T. Ames, M. R. Kurpiewski, and A. Grigorescu. 2000. Thermodynamic parameters of specific and non-specific protein-DNA binding. Supramol. Chem. 12:143–160. [Google Scholar]
- 6.von Hippel, P. H. 1994. Protein-DNA recognition: new perspectives and underlying themes. Science. 263:769–770. [DOI] [PubMed] [Google Scholar]
- 7.Pingoud, A., and A. Jeltsch. 2001. Structure and function of type II restriction endonucleases. Nucleic Acids Res. 29:3705–3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takeda, Y., P. D. Ross, and C. P. Mudd. 1992. Thermodynamics of Cro protein-DNA interactions. Proc. Natl. Acad. Sci. USA. 89:8180–8184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ladbury, J. E., J. G. Wright, J. M. Sturtevant, and P. B. Sigler. 1994. A thermodynamic study of the trp repressor-operator interaction. J. Mol. Biol. 238:669–681. [DOI] [PubMed] [Google Scholar]
- 10.Merabet, E., and G. K. Ackers. 1995. Calorimetric analysis of lambda cI repressor binding to DNA operator sites. Biochemistry. 34:8554–8563. [DOI] [PubMed] [Google Scholar]
- 11.Frank, D. E., R. M. Saecker, J. P. Bond, M. W. Capp, O. V. Tsodikov, S. E. Melcher, M. M. Levandoski, and M. T. Record Jr. 1997. Thermodynamics of the interactions of lac repressor with variants of the symmetric lac operator: effects of converting a consensus site to a non-specific site. J. Mol. Biol. 267:1186–1206. [DOI] [PubMed] [Google Scholar]
- 12.Engler, L. E. 1998. Specificity determinants in the BamHI endonuclease-DNA interaction. PhD thesis. University of Pittsburgh, Pittsburgh, PA.
- 13.Garner, M. M., and D. C. Rau. 1995. Water release associated with specific binding of gal repressor. EMBO J. 14:1257–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sidorova, N. Y., and D. C. Rau. 1996. Differences in water release for the binding of EcoRI to specific and nonspecific DNA sequences. Proc. Natl. Acad. Sci. USA. 93:12272–12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luisi, B. F., W. X. Xu, Z. Otwinowski, L. P. Freedman, K. R. Yamamoto, and P. B. Sigler. 1991. Crystallographic analysis of the interaction of the glucocorticoid receptor with DNA. Nature. 352:497–505. [DOI] [PubMed] [Google Scholar]
- 16.Winkler, F. K., D. W. Banner, C. Oefner, D. Tsernoglou, R. S. Brown, S. P. Heathman, R. K. Bryan, P. D. Martin, K. Petratos, and K. S. Wilson. 1993. The crystal structure of EcoRV endonuclease and of its complexes with cognate and non-cognate DNA fragments. EMBO J. 12:1781–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Albright, R. A., M. C. Mossing, and B. W. Matthews. 1998. Crystal structure of an engineered Cro monomer bound nonspecifically to DNA: possible implications for nonspecific binding by the wild-type protein. Protein Sci. 7:1485–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Viadiu, H., and A. K. Aggarwal. 2000. Structure of BamHI bound to nonspecific DNA: a model for DNA sliding. Mol. Cell. 5:889–895. [DOI] [PubMed] [Google Scholar]
- 19.Kalodimos, C. G., N. Biris, A. M. Bonvin, M. M. Levandoski, M. Guennuegues, R. Boelens, and R. Kaptein. 2004. Structure and flexibility adaptation in nonspecific and specific protein-DNA complexes. Science. 305:386–389. [DOI] [PubMed] [Google Scholar]
- 20.Record, M. T. Jr., J. H. Ha, and M. A. Fisher. 1991. Analysis of equilibrium and kinetic measurements to determine thermodynamic origins of stability and specificity and mechanism of formation of site-specific complexes between proteins and helical DNA. Methods Enzymol. 208:291–343. [DOI] [PubMed] [Google Scholar]
- 21.Breyer, W. A., and B. W. Matthews. 2001. A structural basis for processivity. Protein Sci. 10:1699–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lundback, T., and T. Hard. 1996. Sequence-specific DNA-binding dominated by dehydration. Proc. Natl. Acad. Sci. USA. 93:4754–4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robinson, C. R., and S. G. Sligar. 1998. Changes in solvation during DNA binding and cleavage are critical to altered specificity of the EcoRI endonuclease. Proc. Natl. Acad. Sci. USA. 95:2186–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pingoud, A., and A. Jeltsch. 1997. Recognition and cleavage of DNA by type-II restriction endonucleases. Eur. J. Biochem. 246:1–22. [DOI] [PubMed] [Google Scholar]
- 25.Newman, M., T. Strzelecka, L. F. Dorner, I. Schildkraut, and A. K. Aggarwal. 1994. Structure of restriction endonuclease BamHI and its relationship to EcoRI. Nature. 368:660–664. [DOI] [PubMed] [Google Scholar]
- 26.Newman, M., T. Strzelecka, L. F. Dorner, I. Schildkraut, and A. K. Aggarwal. 1995. Structure of BamHI endonuclease bound to DNA: partial folding and unfolding on DNA binding. Science. 269:656–663. [DOI] [PubMed] [Google Scholar]
- 27.Viadiu, H., and A. K. Aggarwal. 1998. The role of metals in catalysis by the restriction endonuclease BamHI. Nat. Struct. Biol. 5:910–916. [DOI] [PubMed] [Google Scholar]
- 28.Sun, J., H. Viadiu, A. K. Aggarwal, and H. Weinstein. 2003. Energetic and structural considerations for the mechanism of protein sliding along DNA in the nonspecific BamHI-DNA complex. Biophys. J. 84:3317–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engler, L. E., P. Sapienza, L. F. Dorner, R. Kucera, I. Schildkraut, and L. Jen-Jacobson. 2001. The energetics of the interaction of BamHI endonuclease with its recognition site GGATCC. J. Mol. Biol. 307:619–636. [DOI] [PubMed] [Google Scholar]
- 30.Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy, minimization and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- 31.Mezei, M. 1987. Grand canonical Monte Carlo study of dense liquid Lenard-Jones, soft spheres and water. Mol. Phys. 61:565–582. [Google Scholar]
- 32.Resat, H., and M. Mezei. 1994. Grand canonical Monte Carlo simulation of water position in crystal hydrates. J. Am. Chem. Soc. 116:7451–7452. [Google Scholar]
- 33.Resat, H., and M. Mezei. 1996. Grand canonical ensemble Monte Carlo simulation of the dCpG/proflavine crystal hydrate. Biophys. J. 71:1179–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mezei, M., and D. L. Beveridge. 1984. Generic solvation sites in a crystal. J. Comput. Chem. 6:523–527. [Google Scholar]
- 35.Berge, C. 1962. The Theory of Graphs. John Wiley and Sons Inc., New York.
- 36.Mezei, M., and D. L. Beveridge. 1986. Structural chemistry of biomolecular hydration via computer simulation: the proximity criterion. Methods Enzymol. 127:21–47. [DOI] [PubMed] [Google Scholar]
- 37.Mezei, M. 1988. Modified proximity criterion for the analysis of the solvation environment of a polyfunctional solute. Mol. Simul. 1:327–332. [Google Scholar]
- 38.MacKerell, A. D., Jr., D. Bashford, R. L. Bellot, R. L. Dunbrack Jr., J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnik, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher 3rd, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, and M. Karplus. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- 39.Jorgensen, W. L., J. Chandrashekar, J. D. Madura, R. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 40.Mezei, M. 2003. A new method for mapping macromolecular topography. J. Mol. Graph. Model. 21:463–472. [DOI] [PubMed] [Google Scholar]
- 41.Marrone, T. J., H. Resat, C. N. Hodge, C. H. Chang, and J. A. McCammon. 1998. Solvation studies of DMP323 and A76928 bound to HIV protease: analysis of water sites using grand canonical Monte Carlo simulations. Protein Sci. 7:573–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spolar, R. S., and M. T. Record Jr. 1994. Coupling of local folding to site-specific binding of proteins to DNA. Science. 263:777–784. [DOI] [PubMed] [Google Scholar]
- 43.Ha, J. H., R. S. Spolar, and M. T. J. Record. 1989. Role of hydrophobic effect in the stability of site-specific protein-DNA complexes. J. Mol. Biol. 209:801–816. [DOI] [PubMed] [Google Scholar]
- 44.Reddy, C. K., A. Das, and B. Jayaram. 2001. Do water molecules mediate protein-DNA recognition? J. Mol. Biol. 314:619–632. [DOI] [PubMed] [Google Scholar]
- 45.Kopka, M. L., A. V. Fratini, H. R. Drew, and R. E. Dickerson. 1983. Ordered water structure around a B-DNA dodecamer. A quantitative study. J. Mol. Biol. 163:129–146. [DOI] [PubMed] [Google Scholar]
- 46.Quintana, J. R., K. Grzeskowiak, K. Yanagi, and R. E. Dickerson. 1992. Structure of a B-DNA decamer with a central T-A step: C-G-A-T-T-A-A-T-C-G. J. Mol. Biol. 225:379–395. [DOI] [PubMed] [Google Scholar]
- 47.Schneider, B., and H. M. Berman. 1995. Hydration of the DNA bases is local. Biophys. J. 69:2661–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saenger, W. 1987. Structure and dynamics of water surrounding biomolecules. Annu. Rev. Biophys. Biophys. Chem. 16:93–114. [DOI] [PubMed] [Google Scholar]
- 49.Feig, M., and B. M. Pettitt. 1999. Modeling high-resolution hydration patterns in correlation with DNA sequence and conformation. J. Mol. Biol. 286:1075–1095. [DOI] [PubMed] [Google Scholar]
- 50.Shoemaker, B. A., J. J. Portman, and P. G. Wolynes. 2000. Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc. Natl. Acad. Sci. USA. 97:8868–8873. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.