

Abstract
Ribonuclease P (RNase P) is a ribonucleoprotein complex that catalyzes the cleavage of the 5′ end of precursor tRNA. To characterize the interface between the Bacillus subtilis RNA (PRNA) and protein (P protein) components, the intraholoenzyme KD is determined as a function of ionic strength using a magnetocapture-based assay. Three distinct phases are evident. At low ionic strength, the affinity of PRNA for P protein is enhanced as the ionic strength increases mainly due to stabilization of the PRNA structure by cations. Lithium substitution in lieu of potassium enhances the affinity at low ionic strength, whereas the addition of ATP, known to stabilize the structure of P protein, does not affect the affinity. At high ionic strength, the observed affinity decreases as the ionic strength increases, consistent with disruption of ionic interactions. These data indicate that three to four ions are released on formation of holoenzyme, reflecting the number of ion pairs that occur between the P protein and PRNA. At moderate ionic strength, the two effects balance so that the apparent KD is not dependent on the ionic strength. The KD between the catalytic domain (C domain) and P protein has a similar triphasic dependence on ionic strength. Furthermore, the intraholoenzyme KD is identical to or tighter than that of full-length PRNA, demonstrating that the P protein binds solely to the C domain. Finally, pre-tRNAasp (but not tRNAasp) stabilizes the PRNA•P protein complex, as predicted by the direct interaction between the P protein and pre-tRNA leader.
Keywords: ribozyme, electrostatic, magnesium, cations, ribonuclease P
INTRODUCTION
RNase P is the ubiquitous endonuclease found in prokaryotes, eukaryotes, and archaebacteria responsible for catalyzing the hydrolysis of a specific phosphodiester bond in pre-tRNA, yielding a mature tRNA with a 5′ phosphate group (Apirion and Miczak 1993). In all organisms, RNase P is a ribonucleoprotein complex (having both RNA and protein components; Xiao et al. 2002). The bacterial RNase P RNA component alone under high salt conditions is capable of catalyzing pre-tRNA maturation (Guerrier-Takada and Altman 1984); however, the single protein component is essential for in vivo catalytic activity, and enhances substrate affinity in Bacillus subtilis RNase P (Kurz et al. 1998). The eukaryotic RNase P is composed of a single RNA and several proteins, all of which are necessary for activity (Chamberlain et al. 1998; Xiao et al. 2001). Interestingly, an RNA component has not yet been identified for RNase P’s from chloroplast and mitochondria from higher eukaryotes (Gegenheimer 1995; Rossmanith and Karwan 1998; Rossmanith and Potuschak 2001).
RNase P requires both monovalent and divalent cations for stabilization of structure and enhancement of catalysis. Monovalent cations lower the dissociation constant of PRNA for both substrate (Crary et al. 1998) and product (Hardt et al. 1993; Beebe et al. 1996), but they are unable to compensate for magnesium in catalysis (Smith et al. 1992). Even though >100 magnesium ions surround PRNA (Beebe et al. 1996; Kurz et al. 1998), at least three classes of magnesium ions stabilize the tertiary structure and are needed for optimal activity (Smith and Pace 1993), at least two are necessary for E•S or E•P complex formation, and a minimum of one site stabilizes the transition state (Beebe et al. 1996). Substitution of a sulfur for an oxygen on the phosphate of the scissile bond in pre-tRNA in both prokaryotes (Chen et al. 1997; Warnecke et al. 1999) and eukaryotes (Thomas et al. 2000) decreases catalysis 104-fold, suggesting that magnesium may directly interact with the scissile phosphate. Modification of the 2′ hydroxyl at the cleavage site decreases the cleavage rate constant significantly, suggesting that this group functions as a hydrogen bond donor (Smith and Pace 1993; Persson et al. 2003). The highly conserved helix P4 of PRNA is proposed to comprise the active site of the RNase P from phosphorothioate (Harris and Pace 1995; Christian et al. 2000; Crary et al. 2002) and nucleotide analog interference mapping studies (Kaye et al. 2002).
In vivo, RNase P recognizes all pre-tRNAs with different primary sequences; consequently, the characteristic secondary and tertiary structures of tRNA are essential molecular recognition elements (Tobian et al. 1985; Krupp et al. 1991; Pan and Jakacka 1996; Lee et al. 1997). Previous work has focused on the characterization of the interactions between substrate and PRNA in the ribozyme reaction (Christian et al. 1998; Loria and Pan 1998; Christian and Harris 1999; Siew et al. 1999; Zahler et al. 2003) and demonstrated that the substrate directly contacts the P protein in RNase P (Crary et al. 1998; Niranjanakumari et al. 1998b). However, less is known about the interactions between the PRNA and the P protein within the holoenzyme complex.
In the absence of substrate at low ionic strength, small-angle X-ray scattering studies have shown that the P protein•PRNA aggregates to form dimers (PRNA2/P protein2; Fang et al. 2001), but the holoenzyme complex is monomeric in the presence of substrate or at high ionic strength (Barrera et al. 2002). The recent X-ray crystal structure of the S domain of the B. subtilis PRNA suggests that the dimer interface is adjacent to the putative pre-tRNA binding site in the S domain (Krasilnikov et al. 2003). X-ray crystal structures of P protein from B. subtilis (Stams et al. 1998) and Thermotoga maritima (Kazantsev et al. 2003), and the structure of P protein from Staphylococcus aureus solved by NMR techniques (Spitzfaden et al. 2000), suggest regions of the protein that could potentially bind RNA. To date, no crystallographic or NMR studies on the holoenzyme structure have been reported; however, biochemical structure-probing studies have been performed to produce models of the ribonucleoprotein and enzyme–substrate complexes (Rox et al. 2002; Tsai et al. 2003; S. Niranjanakumari, J.J. Day-Storms, R.A. Venters, M. Ahmed, and C.A. Fierke, in prep.).
To investigate further the nature of the interaction between the PRNA and P protein of B. subtilis RNase P, we developed a magnetocapture-based assay to determine the intraholoenzyme KD using P protein with a His-tag on either the C or the N terminus. In monitoring the apparent intraholoenzyme KD as a function of ionic strength, three phases are observed for both full-length PRNA and the C domain only. At low ionic strength, the apparent affinity between the protein and RNA increases as the ionic strength increases, as previously demonstrated (Talbot and Altman 1994a,b). The addition of 1 mM ATP has little effect on this phenomenon, whereas the substitution of Li+ for K+ or C domain for wild-type PRNA shifts this phase to lower ionic strength. Therefore, the enhancement of binding affinity at low ionic strength is likely due to structural stabilization or rearrangement of the PRNA. At high ionic strength, the apparent intraholoenzyme KD increases as a function of ionic strength, suggesting important ionic interactions. The magnitude of this effect indicates that a net formation of three to four ionic pairs stabilize the PRNA/P protein interaction. Finally, at intermediate ionic strength conditions, these two phenomena cancel each other so that the KD is independent of ionic strength. These data demonstrate that electrostatic interactions between PRNA and P protein, likely between the highly conserved P4 helix of PRNA and the positively charged RNR motif of P protein, are important for formation of the holoenzyme complex.
RESULTS
Magnetocapture assay to measure the affinity of PRNA for P protein
To determine the affinity of the B. subtilis PRNA for P protein, henceforth termed the intraholoenzyme affinity, we developed a magnetocapture assay (Fig. 1 ▶) to separate PRNA from the PRNA•P protein holoenzyme complex. For this assay, we first prepared B. subtilis P protein containing a 6×His-tag incorporated onto either the C terminus or the N terminus. The His-tagged P protein•PRNA holoenzyme complex was formed by incubating varying concentrations of His-tagged P protein with trace concentrations of re-folded PRNA containing a radioactive label at the 5′ end for 30 min at 25°C (Fig. 1 ▶, step A). Varying the incubation time from 30 to 60 min did not affect the amount of complex formed, indicating that equilibrium was achieved. The His-tagged P protein•PRNA complex was separated from free PRNA by incubation with magnetic agarose beads containing nickel coordinated by NTA (Ni-NTA; Fig. 1 ▶, step B). In the absence of the His-tagged P protein, typically <5% of the radioactivity in the PRNA was retained on either the magnetic beads or on the sides of the microfuge tube. At saturating concentrations of P protein, 70%–90% of the radiolabeled PRNA was routinely retained on the magnetic beads. Therefore, the formation of the RNase P holoenzyme could be quantified from the bound radioactivity indicating the concentration of PRNA bound to the beads. The fraction of the total PRNA that binds to the beads has a hyperbolic dependence on the concentration of P protein, as expected for a binding isotherm (Fig. 2 ▶). The values for the apparent intraholoenzyme KD were calculated from a fit of a binding isotherm (Equation 2) to these data. The location of the His-tag does not affect the protein affinity because the values for the intraholoenzyme KD are identical for the C-terminal and the N-terminal His-tagged P proteins at 15 ± 2 nM and 15 ± 4 nM, respectively (Fig. 2 ▶). These values are higher than those observed previously using a gel retardation assay (0.4 nM) under different ionic conditions, temperature, protein (Escherichia coli C5) and RNA components (Talbot and Altman 1994a).
FIGURE 1.

Schematic of magnetocapture assay. (A) Radiolabeled PRNA (gray, helical) and P protein (black, with His-tag labeled H) are incubated to form equilibrium concentrations of free PRNA, free P protein, and holoenzyme complex. (B) Magnetic agarose Ni-NTA beads are added to bind the free P protein and the holoenzyme complex. (C) An external magnetic strip is applied to pull the magnetic beads with the bound holoenzyme complex and noncomplexed His-tagged P protein from the bulk solution where the free PRNA is located. (D) The free radiolabeled PRNA is removed in the wash and counted in a scintillation counter. (E) The bound complexes and proteins are removed from the beads and quantitated by Cerenkov scintillation counting.
FIGURE 2.

Measurement of the intraholoenzyme KD for His-tagged P protein and PRNA. The observed KD was determined using trace concentrations (<0.1 nM) of refolded, 32P-5′ end-labeled PRNA and varying concentrations (0.5 nM–1 μM) of either C-terminal His-tagged (•) or N-terminal His-tagged (○) B. subtilis P protein in 50 mM Tris-HCl (pH 8), 10 mM imidazole, 10 mM MgCl2, and 100 mM KCl. The dissociation constants were determined from a fit of Equation 1 to these data, giving values of 15 ± 4 nM and 15 ± 2 nM, respectively, for the N-terminal His-tagged P protein and C-terminal His-tagged P protein. (Inset) Measurement of the intraholoenzyme KD between the C-terminal His-tagged B. subtilis P protein (•) or E. coli P protein (▵) for the B. subtilis PRNA or the C-terminal His-tagged B. subtilis P protein and the S. cerevisiae RPR1 RNA (□) in higher salt (50 mM Tris-HCl at pH 8, 10 mM imidazole, 10 mM MgCl2, and 400 mM NH4Cl). The affinity of the B. subtilis P protein•PRNA complex is 40 ± 10 nM, whereas the affinities for the E. coli P protein•B. subtilis PRNA complex and the B. subtilis P protein•S. cerevisiae RPR1 RNA complex are 400 ± 200 nM and 1500 ± 500 nM, respectively.
To investigate the specificity of the PRNA/P protein interaction, we compared the affinity of the B. subtilis PRNA for the C-terminal His-tagged B. subtilis and E. coli P proteins (Fig. 2 ▶, inset; Rivera-Leon et al. 1995); at 400 mM ammonium chloride, the value for the KD for the B. subtilis P protein of 40 ± 10 nM is 10-fold lower than that of the E. coli P protein (400 ± 200 nM). Under similar conditions, the Saccharomyces cerevisiae RPR1 RNA binds the B. subtilis protein >30-fold less tightly than does the B. subtilis PRNA (1500 ± 500 nM), which is similar to affinities measured for nonspecific RNA–protein interactions at moderate ionic strength (Ucci and Cole 2004). These data suggest that the P protein recognizes regions of PRNA that are conserved between the E. coli and B. subtilis species, such as the conserved P4 helix that contains 11 of the 21 invariant nucleotides found in all cellular PRNAs (Frank and Pace 1998). Consistent with this, previous studies have demonstrated that the B. subtilis P protein activates the catalytic activity of the E. coli PRNA significantly (Guerrier-Takada et al. 1983) and the archaeal PRNA modestly (Pannucci et al. 1999). However, differences between the PRNAs, including the presence or absence of helices and the elaboration of helices such as helix P3 in both E. coli and S. cerevisiae (Frank and Pace 1998; Hsieh et al. 2004), could account for the decreased affinity of the P protein for heterologous PRNAs.
The interaction between the B. subtilis P protein and PRNA is ionic in nature
Interactions between RNA and RNA binding proteins frequently include important ionic contributions. For example, ionic interactions are important for RNA recognition in the HIV Tat protein (Weeks and Crothers 1992), the phage R17 coat protein (Carey and Uhlenbeck 1983), the E. coli ribosomal L11 protein (Ryan and Draper 1989), and Bacillus stearothermophilus ribosomal S4 protein (Gerstner et al. 2001). However, previous studies using a gel retardation assay to monitor the affinity of the E. coli PRNA (M1 RNA) for the C5 protein indicated that the apparent KD decreases as the ionic strength increases for all concentrations of NH4Cl measured. These results, combined with enthalpy measurements, suggested that the RNA/protein contact in E. coli RNase P holoenzyme was a novel, hydrophobic interaction (Talbot and Altman 1994b). However, recent models of the structure of the RNase P holoenzyme suggest that the positively charged “RNR” motif of the P protein interacts with PRNA (Tsai et al. 2003; S. Niranjanakumari, J.J. Day-Storms, R.A. Venters, M. Ahmed, and C.A. Fierke, in prep.).
We probed the importance of ionic interactions in the formation of the B. subtilis RNase P holoenzyme by determining the apparent intraholoenzyme KD as a function of the ionic strength at 10 mM MgCl2 and varying concentrations of KCl (0.1–800 mM). Three distinct phases are clearly evident in a plot of the log KD,app as a function of the log of the ionic strength (Fig. 3 ▶). At low ionic strength, the KD decreases as the ionic strength increases, as observed previously (Talbot and Altman 1994b), whereas at high ionic strength the KD increases as the ionic strength increases, as is often the norm for nucleic acid binding proteins (Vartikar and Draper 1989; Weeks and Crothers 1992). At moderate ionic strengths, these two effects are balanced so that the KD appears to be independent of the ionic strength.
FIGURE 3.

Measurement of the intraholoenzyme KD for His-tagged P protein and PRNA (or C domain) as a function of ionic strength. The observed KD for wild-type holoenzyme complex (•) and the C domain–P protein complex (○) were determined in 50 mM Tris-HCl (pH 8), 10 mM MgCl2, 10 mM imidazole, and 0.1–800 mM KCl at room temperature. The ionic strength was determined as described in Equation 3. Equation 1, derived from the model shown in Scheme 1 ▶, was fit to the data. The results of these fits are listed in Table 1 ▶.
The multiple effects of ionic strength on the intraholoenzyme KD are consistent with Scheme 1 ▶, suggesting two classes of ion binding sites that affect P protein affinity, where one class enhances affinity (Zn1) and the second class decreases affinity (Zn3). This model assumes complete cooperativity in binding each group of ions. Equation 1, derived from this model,
SCHEME 1.
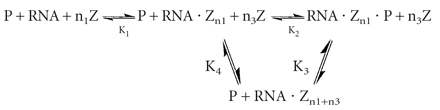
Model describing the dependence of the intraholoenzyme KD on ionic strength.
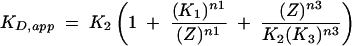 |
(1) |
provides a good description of the ionic strength dependence of KD (Fig. 3 ▶). At very low salt concentration, the apparent KD decreases, as seen in the previous studies (Talbot and Altman 1994b). This enhanced affinity could be explained by stabilization of the native structure of either the PRNA by bound cations (Pan 1995) or the P protein by bound anions (Stams et al. 1998; Henkels et al. 2001), or a combination of both. These possibilities will be discussed in detail later; Scheme 1 ▶ illustrates a mechanism where cations bind to PRNA to stabilize a conformation with high affinity for the P protein (RNA•Zn1). We will refer to this portion of the curve as “activation” by ions. Under these conditions, the apparent dissociation constant (as described by Equation 1) extrapolates to K2K1n1 / [Z]n1, reflecting the equilibrium constants for both structure stabilization by the ions (K1) and affinity of PRNA for P protein (K2). Because of the innate difficulty of obtaining very low (i.e., submillimolar concentration) ionic strength while maintaining the magnesium concentration at 10 mM, KD measurements at low salt have the largest degree of error. A fit of Equation 1 to the data in Figure 3 ▶ suggests approximate values for K1 and n1 of 0.09 ± 0.01 M and 4.4 ± 0.9, respectively (Table 1 ▶). At intermediate concentrations, there is a region that is relatively independent of ionic strength that can be described by the binding of P protein to PRNA•Zn1 to form holoenzyme, K2 (13 ± 2 nM), with no net uptake or release of ions. A similar plateau region was observed in the salt dependence of the affinity of E. coli ribosomal S4 protein for rRNA (Vartikar and Draper 1989). Finally, at high ionic strength the apparent intraholoenzyme KD increases as the ionic strength increases. Decreased affinity for nucleic acids at high salt is observed for many nucleic acid binding proteins, including ribosomal proteins and the HIV Tat protein (Carey and Uhlenbeck 1983; Ryan and Draper 1989; Weeks and Crothers 1992; Gerstner et al. 2001). In this region, binding affinity is decreased because cations in solution are competing with the P protein for interactions with specific sites on the nucleic acid and solution anions are competing for the RNA binding regions of the P protein. In Scheme 1 ▶, this is illustrated by the equilibrium K3 where the P protein displaces a class of cations (n3) when it binds to PRNA. We call this the “destabilization” portion of the curve. The apparent dissociation constant at high salt concentrations can be approximated by [Z]n3/K3n3 (Equation 1). A fit of Equation 1 to the data in Figure 3 ▶ indicates that K4 (where K4n3 = K2K3n3) equals 0.38 ± 0.04 M with a value for n3 of 4.0 ± 0.5. This portion of the curve will be discussed in greater detail later.
TABLE 1.
Analysis of ion dependence of intraholoenzyme KD,appa
| KClb | KCl (+ATP)c | LiCld | KCl (+pre-tRNAAsp)e | C domainf | |
| K1 (M) | 0.09 ± 0.01 | 0.07 ± 0.01 | 0.03 ± 0.02 | 0.15 ± 0.05 | 0.06 ± 0.01 |
| n1 | 4.4 ± 0.9 | >5 | ≫5 | 5 ± 2 | ≫5 |
| K2 (nM) | 13 ± 2 | 12 ± 3 | 8 ± 6 | 1.3 ± 1 | 12.0 ± 0.8 |
| K4g (M) | 0.38 ± 0.04 | 0.5 ± 0.1 | 0.2 ± 0.2 | 0.4 ± 0.2 | 0.44 ± 0.02 |
| n3h | 4.0 ± 0.5 | 3.8 ± 1.4 | 1 ± 0.7 | 3.7 ± 1.4 | 3.7 ± 0.3 |
| n3logK3h | 6.2 ± 0.1 | 6.7 ± 0.2 | 7.3 ± 0.1i | 7.2 ± 0.2i | 6.6 ± 0.1 |
aThe apparent intraholoenzyme KDs were measured at varying ionic conditions as described in the legends of Figures 3 ▶–5 ▶ and modeled using equation 1 derived for scheme 1 ▶.
bVarying the concentration of KCl.
cVarying the concentration of KCl in the presence of 1 mM ATP.
dVarying the concentration of LiCl.
eVarying the concentration of KCl in the presence of 0.1 nM pre-tRNAAsp.
fVarying the concentration of KCl using C domain in lieu of PRNA.
gK4n3 = K2K3n3.
hValues were determined from a global fit of the data as described in Figures 3 ▶–5 ▶ using the following equation: n3log K3 = n3log K4 - log K2.
iValue obtained from the linearization of the data for 1–800 mM LiCl.
Previously, we described the specificity of the His-tagged P protein for different PRNAs at moderate ionic strength (Fig. 2 ▶, inset). To test for nonspecific binding at either low or high ionic strength, we measured the KD between the His-tagged P protein and either a small DNA oligomer (TA ATACGACTCACTATAG) or the S. cerevisiae RPR1 RNA. At 15 mM KCl, 10 mM MgCl2, 10 mM imidazole, 50 mM Tris (pH 8), the observed KD for the P protein–PRNA complex is 65 nM. Under the same conditions, varying the concentration of P protein (1 nM–5 μM), a maximal amount of <30% of the DNA oligomer and ~35% of RPR1 RNA bound to P protein at 5 μM. Because the affinity is so weak, the KD for each nucleic acid can only be estimated at ~9 μM by assuming an endpoint of 90% bound (data not shown). When the concentration of KCl is increased to 800 mM, the observed PRNA–P protein KD is 440 nM. Under these conditions, <25% of the DNA oligomer and ~35% of RPR1 RNA bind to P protein at a concentration of 5 μM, indicative of a significantly larger KD (estimated at 10 μM). Taken together, these data demonstrate that the observed affinity between P protein and PRNA at the extreme ionic conditions is significantly higher than a nonspecific interaction between the P protein and other nucleic acids and reflects a specific binding site.
The “activation” is primarily due to stabilization of the PRNA by environmental cations
In many cases, RNA–protein complexes are proposed to form using an induced fit rather than a “lock-and-key” mechanism (Leulliot and Varani 2001), which brings about three possibilities. One scenario is that on formation of the RNA–protein complex, the RNA undergoes a conformational change or structural stabilization, which may be a large macroscopic rearrangement or a localized movement. An example of this can be seen in the S15–rRNA complex, where a helical rearrangement occurs in the RNA in the presence of Mg2+ and the S15 protein compared with the free form of the RNA (Williamson 2000). Another possibility is that the protein undergoes a conformational change when it binds to the RNA, as observed for the N antiter-mination protein of bacteriophage λ when it binds to its cognate boxB hairpin RNA (Van Gilst et al. 1997; Mogridge et al. 1998). The final option is a mutually induced fit where the structures of both components alter, as seen in the U1A–UTR complex (Williamson 2000; Leulliot and Varani 2001).
The enhancement of the PRNA•P protein binding affinity by ions at low ionic strength could be due to stabilization of the protein alone, the RNA alone, both components, and/or the protein•RNA complex. Previous studies have shown that the B. subtilis P protein is intrinsically unstructured in the absence of PRNA or other anions, but it folds into a specific structure on addition of anions (Henkels et al. 2001). The concentration of anion required to fold the P protein follows the anion binding series, indicating that specific ionic interactions are important for structure stabilization (Henkels et al. 2001), as observed for acid-denatured Sac7d (McCrary et al. 1998). To test whether the activation observed in the holoenzyme complex formation is due mainly to stabilization of the native fold of the P protein, we determined the dependence of the intraholoenzyme dissociation constant as a function of the ionic strength in the presence of 1 mM ATP. At 1 mM ATP, the P protein is completely folded in solution (Henkels et al. 2001). If the enhancement of the stability of the holoenzyme complex is solely due to stabilization of the protein structure by anions, then the activation by anions should disappear or shift to lower concentrations in the presence of 1 mM ATP. This did not occur (Fig. 4 ▶; Table 1 ▶). Although the ionic strength at submillimolar conditions is difficult to maintain, these data clearly demonstrate qualitatively that addition of ATP has little effect on the values of the apparent KD at low ionic strength. The value of K1 is unchanged (0.07 ± 0.01 M) although n1 increases (>5), perhaps because of partial coordination of the Mg2+ ions by ATP at low ionic strength. Furthermore, the presence of ATP has virtually no effect on K2 (12 ± 3 nM in 1 mM ATP and 13 ± 2 nM in the absence of ATP). At high concentrations of salt, the presence of ATP modestly enhances the value of K4 (0.5 ± 0.1 M), but the value of n3 is unaffected (Table 1 ▶). These data indicate that activation of the holoenzyme complex formation at low ionic strength is not mainly due to stabilization of the P protein structure. Most likely, the presence of the poly-anionic PRNA is sufficient to stabilize the native fold of the P protein on complex formation.
FIGURE 4.

Measurement of the intraholoenzyme KD in the presence of ATP or LiCl. The observed KD was determined in 50 mM Tris-HCl (pH 8), 10 mM MgCl2, 10 mM imidazole, and 0.1–800 mM KCl at room temperature in the absence (•) and presence of 1 mM ATP (○) or with 0.1–800 mM LiCl (□) used in lieu of KCl. The ionic strength was determined as described in Equation 3, and Equation 1 was fit to the data. The results of these fits are listed in Table 1 ▶.
To determine if the ionic stabilization of the PRNA•P protein complex is due to stabilization of RNA structure, we monitored the dependence of the intraholoenzyme KD as a function of ionic strength by varying the concentration of LiCl (0.1–800 mM) in lieu of KCl. Because of its higher charge density, Li+ ions previously have been shown to be more effective at stabilizing RNA structure in general than the K+ cation (Urbanke et al. 1975; Labuda and Augustyniak 1977; Torrent et al. 1994; Gluick et al. 1997). Figure 4 ▶ shows a plot of the log KD, app as a function of the log ionic strength. The substitution of lithium for potassium significantly decreases the dependence of the observed KD on the ionic strength, although the apparent KD at intermediate salt concentration (0.08–0.25 M) is essentially unaffected by the substitution of Li+ for K+. Most important, the “activation” portion of the curve is shifted to significantly lower ionic strength and almost disappears completely. The K1 is decreased significantly by the addition of Li+, as is expected if the activation portion of the curve reflects stabilization of the native RNA fold. The other major effect of Li+ is that the competition with cations at high ionic strength is less co-operative because n3 = 1. Lithium also modestly decreases K2 (8 nM) and K4 (0.2 M; Table 1 ▶). In summary, these data suggest that the stabilization of the PRNA•P protein complex at low ionic strength is mainly due to stabilization of the folded structure of PRNA by added cations.
The catalytic domain displays a similar ionic interaction with the P protein
PRNA contains two domains that fold independently of one another (Pan 1995), with each having specific functions. The catalytic domain of B. subtilis PRNA contains all of the essential components necessary for enzymatic catalysis (Loria and Pan 1996, 2001), although the pre-tRNA affinity is decreased because the S domain contacts the T-stem/loop region of tRNA (Loria and Pan 1996; Qin et al. 2001). The C domain, unlike the full-length wild-type PRNA, folds without any kinetic traps into the catalytically active, native structure at Mg2+ concentrations (0.4–1.2 mM MgCl2) significantly lower than required for folding of wild-type PRNA (Pan 1995; Fang et al. 1999). Furthermore, the C domain has been demonstrated previously to bind P protein (Loria and Pan 2001). We determined the observed KD of the C domain–P protein complex over the same range of concentrations of KCl (0.1–800 mM; Fig. 3 ▶). The C domain holoenzyme exhibits three phases similar to those observed for wild-type holoenzyme. The main difference is in the activation phase where the K1 (0.06 ± 0.01 M) is decreased slightly compared with that of wild type and the value of n1 (≫5; Table 1 ▶) is increased. These data provide additional evidence that the “activation” limb is dependent on the stability of PRNA because the C domain remains folded at lower concentrations of Mg2+ than wild-type PRNA. The K2 value is essentially identical to that of the wild-type PRNA (12.0 ± 0.8 nM and 13 ± 2 nM, respectively), whereas the K4 and log K3n3 values are slightly higher (0.44 ± 0.02 M and 6.6 ± 0.1, respectively; Table 1 ▶). These data show that the affinities of the C domain and the wild-type PRNA for P protein show similar behavior at moderate and high ionic strength. At low ionic strength, the C domain displays a higher cooperativity in ion binding, which could be explained by an increase in RNA structural stability as compared with that of the wild-type PRNA.
As previously mentioned, the wild-type B. subtilis PRNA–P protein can form dimers under stoichiometric, micromolar concentrations of PRNA and P protein in the absence of substrate and at low salt. Under the conditions of the magnetocapture assay (excess P protein, nanomolar P RNA), dimerization is not predicted to occur, except perhaps at very low ionic strength. Hydroxyl-radical footprinting data indicate that the dimer interface is in the S domain adjacent to the binding site of the T-stem/loop of tRNA (Barrera et al. 2002) and therefore the C domain should not dimerize. The similarity between the ionic strength dependence of the affinity of the P protein for the C domain and for wild-type PRNA at moderate and high salt concentrations confirms that dimerization is not occurring under these conditions. The differential affinity at low ionic strength could potentially reflect dimerization of the PRNA–P protein complex, although dimer formation is predicted to increase the observed affinity whereas the affinity of P protein for wild-type PRNA is lower than for the C domain under all conditions (Fig. 3 ▶).
Addition of substrate stabilizes the P protein•PRNA complex
The affinity of the B. subtilis holoenzyme for pre-tRNAAsp, but not tRNAAsp, is enhanced 104-fold compared with PRNA alone (Kurz et al. 1998). Photocrosslinking and hydroxyl radical cleavage studies have shown that the 5′ leader of the pre-tRNA interacts with the central cleft of the P protein (Niranjanakumari et al. 1998b; Tsai et al. 2003; S. Niranjanakumari, J.J. Day-Storms, R.A. Venters, M. Ahmed, and C.A. Fierke, in prep.). However, previous measurements of the E. coli holoenzyme show no change in the affinity of the C5 protein for the M1 RNA in the presence of either pre-tRNATyr or tRNATyr (Talbot and Altman 1994a). To determine the effect of substrate on the affinity of the holoenzyme complex of B. subtilis RNase P, we measured the intraholoenzyme KD as a function of ionic strength in the presence of pre-tRNAAsp. For these experiments, calcium was substituted for magnesium to maintain the E•pre-tRNA complex throughout the experiment; this substitution decreases the pre-tRNA cleavage rate constant by ~04-fold (Smith and Pace 1993) without decreasing the affinity of PRNA for pre-tRNA (Smith et al. 1992; Hardt et al. 1993). Furthermore, the structure of the ribozyme–substrate complex is unchanged in the presence of Ca2+, as indicated by intramolecular crosslinking patterns (Harris et al. 1997). The KD of RNase P holoenzyme for pre-tRNAAsp in 10 mM Ca2+ and 100 mM NH4Cl is 0.4 nM (Kurz et al. 1998). To avoid altering the ionic strength in our binding experiments, we added a subsaturating concentration of substrate (0.1 nM). Under these conditions, the P protein•PRNA KD was determined using the magnetocapture assay at different concentrations of KCl (25–800 mM). The shape of the curve is similar to that of the holoenzyme in the absence of substrate (Fig. 5 ▶). The most significant change is that the PRNA•P protein affinity at moderate and high ionic strength is enhanced about 10-fold; K2 is decreased to 1.3 nM as compared with 13 nM (Table 1 ▶). In contrast, addition of tRNAAsp has little effect on the intraholoenzyme KD (Fig. 5 ▶). At 100 mM KCl, the observed intraholoenzyme KD is 18 nM in the presence or absence of mature tRNA and 1.8 nM in the presence of pre-tRNAAsp. In 200 mM KCl, the observed KDs are 22 nM and 2.5 nM for addition of mature tRNA and substrate, respectively. These data are consistent with the enhanced affinity of the pre-tRNA to the holoenzyme complex compared with PRNA (Kurz et al. 1998), although the magnitude is smaller, presumably because of both the subsaturating pre-tRNA concentration and the altered ionic conditions.
FIGURE 5.

The effect of pre-tRNA substrate and mature tRNA product on P protein•PRNA affinity. The observed KD was determined in 50 mM Tris-HCl (pH 8), 10 mM imidazole, and 0.1–800 mM KCl at room temperature in 10 mM MgCl2 (•) or 0.1 nM pre-tRNAAsp with 10 mM CaCl2 (○) to inhibit catalysis. For the experiments using mature tRNA (□), the conditions were identical to those of the pre-tRNAAsp experiments except that the KCl was varied from 100 to 200 mM. The pre-tRNA or mature tRNA was added after the PRNA and P protein had been in solution for a minimum of 15 min. The ionic strength was determined as described in Equation 3, and Equation 1 was fit to the data. The results of these fits are listed in Table 1 ▶.
The addition of pre-tRNA also increases the value of K1 modestly (approximately twofold), suggesting that pre-tRNA destabilizes the folded structure of PRNA. This could be caused by a myriad of possibilities, including that the pre-tRNA is competing for the low concentration of cations to effectively decrease the ionic strength at low salt. Taken together, these data indicate that the presence of substrate increases the apparent affinity between the P protein and the PRNA without greatly affecting the affinity of the holoenzyme for cations (K4 = 0.4 M; Table 1 ▶) or the number of cations displaced in the formation of the holoenzyme complex.
Number of ionic interactions is independent of cation
As previously stated, the decrease in the affinity of PRNA for P protein at high ionic strength can be described by the association constant K3 in Scheme 1 ▶, where cations dissociate from PRNA when the P protein binds. Under high ionic strength solution conditions, the approximation of the observed KD,app, [Z]n3 / (K3)n3 can be linearized by taking the logarithm of both sides: log KD,app = n3 log[Z]-n3 log K3. This type of relationship has been frequently observed for proteins binding to nucleic acids, and these earlier studies have manipulated this relationship to further investigate the effects of monovalent and/or divalent cations on complex assembly (deHaseth et al. 1977, 1978; Record et al. 1977, 1978). In this equation, the slope of the line, n3, approximates m′ψ. The symbol m′ signifies the number of ion pairs in the RNA–protein binding site, most likely formed between positively charged protein side chains and the negatively charged phosphodiester backbone of RNA, which stabilize complex formation. Furthermore, ψ is the thermodynamic ion association parameter per nucleic acid phosphate group that indicates the fraction of counterion bound thermodynamically to each phosphate (Record et al. 1976). Figure 6 ▶ depicts the dependence of the log KD, app (reflecting K3) on the log of the ionic strength for the destabilization phase. The n3 calculated from the slope of this plot is 3.1 ± 0.4 when [KCl] is varied, which is smaller than but within error of the n3 value obtained from a global fit of the data (4.0 ± 0.5). By using the ψ value determined for poly(A)•poly(U) as an approximation for RNA (0.89; Record et al. 1976, 1978), the m′ value is 3.5. The y-intercept of this linear fit reflects the value of -n3 log K3 and is similar to that obtained from a global fit of the data (6.3 ± 0.1 and 6.2 ± 0.1, respectively; Tables 1 ▶, 2 ▶). The presence of substrate has little to no effect on n3 and m′ (3.2 ± 0.9 and 3.6, respectively). However, the value of log (K3)n3 obtained either from the linear fit (data not shown) or from the global fit of the data is increased by a factor of 10 (7.3 ± 0.3 in the presence of substrate versus 6.3 ± 0.3 in the absence of substrate), demonstrating that the substrate enhances the affinity of P protein. The removal of the S domain increases the log (K3)n3 slightly as compared with wild-type PRNA (6.66 ± 0.05 linear fit; Fig. 6 ▶, Table 2 ▶). By looking at only the high ionic strength data, the n3 value decreases to 2.8 ± 0.2; consequently, the m′ is 3.1 ± 0.2. This decrease could signify two possibilities; the first is a decrease in the cooperativity of dissociation of ions at high ionic strength as compared with wild-type PRNA, and the other is that there is a decrease in the number of ionic interactions under these conditions on the deletion of the S domain. In summary, these data indicate that three to four ion pairs stabilize the PRNA–P protein interface, demonstrating that the P protein/PRNA interaction contains important ionic components.
FIGURE 6.

Characterization of the ionic interaction of the holoenzyme complex at high ionic strength. This is a plot of the intraholoenzyme KD measured at high ionic strength (“destabilization”). The experiments varying the concentration of KCl using wild-type PRNA (•) or the catalytic domain (▴) are described in the legend of Figure 3 ▶ The experiments where the ionic strength was altered by varying concentrations of MgCl2 (○) also included 200 mM KCl. The Co(NH3)6Cl3 experiments (▪) included 100 mM KCl and varying Co(NH3)6Cl3. The resulting slope of the line is m′ψ (= n3). The m′ values are 3.5 for wild-type PRNA (•), 3.6 for Mg2+ (○), 3.8 for Co3+ (▪), and 3.1 for catalytic domain (▴). The y-intercept is equal to log K3n3, and the calculated values for n3 and log K3n3 are listed in Table 2 ▶.
TABLE 2.
Effects of different cations on ionic interactionsa
| KClb | KCl (+pre-tRNA)c | C domaind | MgCl2e | Co(NH4)6Cl3f | |
| n3 | 3.1 ± 0.4 | 3.2 ± 0.9 | 2.8 ± 0.2 | 3.2 ± 0.5 | 3.4 ± 0.2 |
| n3logK3 | 6.3 ± 0.1 | 7.3 ± 0.3 | 6.66 ± 0.05 | 6.2 ± 0.1 | 5.8 ± 0.2 |
aThe apparent intraholoenzyme KDs were measured at varying ionic conditions as described in the legend of Figure 6 ▶.
bVarying the concentration of KCl.
cVarying the concentration of KCl in the presence of 0.1 nM pre-tRNAAsp.
dVarying the concentration of KCl using C domain in lieu of PRNA.
eVarying the concentration of MgCl2.
fVarying the concentration of Co(NH4)6Cl3.
To determine the effect of di- and trivalent cations on the stability of the holoenzyme complex, we quantitated the apparent intraholoenzyme KD as a function of either the con-centration of MgCl2 or Co(NH3)6Cl3. At 200 mM KCl, log KD is linearly dependent on the log ionic strength when the concentration of Mg2+ is varied (Fig. 6 ▶) with a slope, n3, equal to 3.2 ± 0.5 (Table 2 ▶). This value of n3 is virtually identical to that determined from varying KCl in the absence of substrate. This similarity suggests that the effect of cations on KD is due to nonspecific electrostatic interactions. The m′ value for MgCl2 is calculated as 3.6 ± 0.6. MgCl2 also has no effect on log (K3)n3 as compared with KCl (6.2 ± 0.1; Fig. 6 ▶). For the cobalt hexammine experiments, the KCl concentration was decreased to 100 mM. Figure 6 ▶ illustrates the linear dependence of log KD on ionic strength at varying concentrations of cobalt hexammine. The slope (n3 = 3.4 ± 0.2) is virtually identical to that measured under the other conditions, and m′ is 3.8 ± 0.2. Under these experimental conditions, log (K3)n3 decreases slightly (to 5.8 ± 0.2), indicating that the P protein competes less well with cobalt hexammine than potassium for interacting with PRNA.
To further substantiate that binding affinity is not dependent on the identity of the cation, we measured the intraholoenzyme KD at a constant ionic strength with the ratio of [KCl]/[MgCl2] varied from 920:1 to 0.08:1. The buffer contained 50 mM Tris-HCl (pH 8), 10 mM imidazole, 0.25–65 mM MgCl2, and 5–230 mM KCl. Over this entire range, the values of KD,app vary no more than twofold (15 ± 5 nM to 30 ± 13 nM) without a consistent trend. These data demonstrate that the value of the KD,app is dependent on the ionic strength but not dependent on the concentration of either [K+] or [Mg2+] (data not shown). In total, these data indicate that the interaction between the P protein and PRNA contains a major electrostatic component. Varying the cation, moreover, has little effect on either the number of ionic interactions between the protein and RNA at the binding site with the formation of three or four important ion pairs or the value of K3. These properties are consistent with the P protein displacing cations that interact nonspecifically with PRNA.
DISCUSSION
Comparison of gel-shift assay and magnetocapture assay
Talbot and Altman (1994a) previously monitored the formation of E. coli holoenzyme using a gel-shift (or gel retardation) assay. The values they observed were 0.4 nM for both the E. coli holoenzyme and the B. subtilis holoenzyme complex and 1.4 nM for the E. coli–B. subtilis heterologous complex, independent of ionic conditions. A number of differences exist between this method and the magnetocapture-based assay described here. First, the assay conditions were considerably different. The holoenzyme in the gel-shift assay was reconstituted in 400 mM NH4OAc and 10 mM Mg(OAc)2 at 37°C and then loaded onto a 5% polyacryl-amide gel with a low salt running buffer (1 mM Mg(OAc)2, 50 mM K-Hepes at pH 8.0) and electrophoresed for 3–4 h at 4°C. This method assumes that the amount of holoenzyme complex does not change on dilution into the loading buffer in the well prior to entering the gel (Garner and Revzin 1981) and that the complex formation in the gel is not altered because of either the lower salt in the gel buffer or a “caging” effect where dissociated components are not allowed to diffuse away once in the gel matrix (Fried and Crothers 1981). These conditions may also enhance dimerization or aggregation of the holoenzyme complex (Fang et al. 2001).
The magnetocapture assay described here has several advantages. First, the salt concentration is maintained throughout the affinity measurement and it is relatively straightforward to alter the solution conditions. However, some limitations on the solution conditions are imposed by the nature of the His-tag/Ni2+ interaction, such as variation in pH is limited; divalent metal ion chelators, such as EDTA, should be excluded from the binding buffer; and nickel reducing agents, such as sulfhydryl-containing reagents, and buffers containing secondary or tertiary amines should be limited. Under the experimental conditions used in this report, the concentration of Tris (50 mM) is not significant enough to chelate the nickel ions away from the NTA resin. Low concentrations of imidazole or histidine can be used to help reduce nonspecific interactions with the resin. Furthermore, ionic interactions between the protein or RNA and the bead should be considered. In our experiment, RNA does not bind to the beads in the absence of P protein, and the measured affinity is dependent on the nature of the specific ions in solution, that is, lithium versus potassium, not just the ionic strength. Consequently, the ionic interaction between the protein and the bead is not a significant factor for the range of ionic strengths we used. Second, lengthy separation times are not needed, although the dissociation rate constant must be less than the amount of time needed to separate the beads from the bulk solution (<1 min). Third, this assay is easily adaptable for measurement of a wide range of protein–nucleic acid affinities requiring only the addition of a His-tag to the protein. One caveat is that interaction with the NTA resin could potentially alter the affinity of the P protein/PRNA complex. However, the identical affinity measured with the His-tag located at the N and C terminus of P protein suggests that specific interactions with the resin do not occur. Furthermore, this limitation could be minimized by addition of a linker peptide between the protein and the His-tag. In setting up this assay, the optimal incubation time to equilibrate the ribonucleoprotein complex and to bind the protein to the Ni2+ bead should be determined. Taken together, the magnetocapture assay is a potentially powerful tool for determining the affinity of protein–nucleic acid complexes.
Stabilization of RNA structure by cations
Unlike DNA, RNA has a much less rigid secondary and tertiary structure granted by the presence of the 2′-hydroxyl. For the RNA molecule to fold into a biologically relevant structure, it has to overcome both energetic constraints where its polyanionic characteristics are counterproductive to favorable base-stacking and van der Waal interactions and topological constraints due to local secondary structures competing against a global fold (Thirumalai et al. 2001). Cations are necessary to overcome these competing forces so that an ion core, homologous to the hydrophobic core in protein folding, exists as a framework around which RNA can fold (Tinoco and Kieft 1997).
Two different ion–RNA interactions are possible. The first is diffuse binding or association where the cation is electrostatically attracted to the high anionic charge of the RNA molecule and cation–cation repulsion is minimal. Additionally, such ions typically remain fully hydrated (Misra and Draper 2001; Draper 2004), decreasing the energetic cost of dehydration. A diffusely bound ion can become kinetically trapped in a specific location, but it still maintains an outer-sphere contact, potentially forming hydrogen bonds with the RNA surface. The second class is a chelated ion or a site-specific ion. These ions often form inner-sphere contacts with RNA functional groups and are partially desolvated. Therefore, the energetic cost can be high. Consequently, diffuse ions dominate the overall thermodynamic stabilization of RNA structure (Misra and Draper 2001; Draper 2004).
At low ionic strength, the affinity of PRNA for P protein decreases. The disappearance of this effect when Li+ is substituted for K+ suggests that this decreased affinity is caused by destabilization of the native structure of PRNA at low ionic strength. Cations with a higher charge/ionic radius ratio have previously been demonstrated to stabilize RNA structure better than cations with a lower charge/radius ratio. This is not only true for RNA secondary structure, as seen in the mRNA pseudoknot from Moloney murine leukemia virus responsible for the repression of the gag gene stop codon (Gluick et al. 1997), but it is also evident in stabilizing the tertiary structure of tRNAPhe (Urbanke et al. 1975; Labuda and Augustyniak 1977) and the quaternary structure of VL30 RNA dimers from murine leukemia virus (Torrent et al. 1994). Pan and colleagues have demonstrated that there are a number of intermediates and kinetic traps in the folding of PRNA, and nonnative structures can predominate at low Mg2+ concentrations (Pan and Sosnick 1997). In our experiments, PRNA may similarly form a nonnative structure at low ionic strength that has low affinity for P protein.
The PRNA•P protein binding site
At high ionic strength, the observed intraholoenzyme KD increases as the ionic strength increases, indicating an ionic interaction. The slope of the line in Figure 6 ▶ indicates that three to four important ionic interactions stabilize the formation of the PRNA–P protein complex. Furthermore, the small dependence of these interactions on the cation type suggests that the protein displaces diffusely bound, rather than specifically bound, cations.
Only when lithium is substituted for potassium is the n3 value different (1.1 ± 0.6; Table 1 ▶). These data suggest either that only one to two important ion pairs (m′) stabilize the holoenzyme complex under these conditions or that the plateau region extends from 10−1.6 to 10−0.4 M ionic strength because of an increase in K3 (see Equation 1), suggesting that a larger value for n3 would be observed at higher concentrations of LiCl. Unfortunately, it is not possible to access high enough concentrations of LiCl to test this hypothesis without precipitating either the PRNA and/or the P protein.
Molecular dynamics simulations with the RNA binding domain of the U1A spliceosomal protein–hairpin RNA complex (Hermann and Westhof 1999) have shown that the cations cluster around the RNA and compete with the protein at the binding site, and we propose a similar mechanism for complex formation in RNase P. Ribonucleoprotein complexes can form hydrogen bonds and van der Waal contacts between protein side chains and either the phosphate backbone or the nucleotide bases. Unlike DNA–protein complexes, which have a preference for contacts between the nucleic acid backbone and the protein, RNA–protein complexes make base-protein and backbone-protein contacts with equal frequency. Ionic interactions between proteins and RNA most often include interactions by positively charged lysine and arginine residues and the negatively charge phosphate backbone (Jones et al. 2001).
The crystal structure of the B. subtilis P protein (Stams et al. 1998) illuminates possible RNA binding regions. One RNA binding region is the central cleft formed from an α helix and a β sheet lined with aromatic residues. Crosslinking studies (Niranjanakumari et al. 1998b) and affinity measurements (Crary et al. 1998) demonstrate that this region of the protein interacts with the 5′ leader sequence of pre-tRNA. Consistent with this, we demonstrate here that addition of the substrate enhances the apparent P protein–PRNA affinity, likely a result of direct interaction of the pre-tRNA with both PRNA and P protein because product tRNA has no effect on complex affinity (Fig. 5 ▶). Taken together, these results indicate that the central cleft interacts with the leader of pre-tRNA and therefore cannot be a site for the PRNA/P protein interaction.
A second potential RNA binding region on P protein is the highly conserved, left-handed βαβ crossover and helix called the RNR motif because of the conserved arginine, asparagine, arginine sequence motif, consisting of the sequence Lys-Xaa4–5-Ala-Xaa2-Arg-Asn-Xaa2-(Lys or Arg)-Arg-Xaa2-(Arg or Lys) (Stams et al. 1998). Engineering site-specific Cys mutations to anchor hydroxyl radical reducing agents such as EDTA-Fe in E. coli (Tsai et al. 2003) and Cu-phenanthroline in B. subtilis (S. Niranjanakumari, J.J. Day-Storms, R.A. Venters, M. Ahmed, and C.A. Fierke, in prep.) has demonstrated that the RNR motif is near helices P3 and P4, the putative enzyme active site, in both the E. coli and B. subtilis RNase P holoenzymes (Tsai et al. 2003; S. Niranjanakumari, J.J. Day-Storms, R.A. Venters, M. Ahmed, and C.A. Fierke, in prep.). In particular, labels attached to the K64C mutant (Cys 66 in E. coli) in the RNR motif cleaved at either C370 or A371 (B. subtilis numbering) in J19/4 (J2/4 in E. coli), where C370 is universally conserved in all cellular PRNAs and A371 is invariant in bacterial PRNAs (Frank and Pace 1998). Additionally, radicals anchored to the N terminus of the P protein cleave helix P3 of both RNAs. Consistent with this interaction, helix P3 in S. cerevisiae RNase P interacts with Pop1p, one of the nine essential protein subunits of the yeast holoenzyme (Ziehler et al. 2001).
The ionic strength dependence of the intraholoenzyme KD (Fig. 6 ▶; Tables 1 ▶, 2 ▶) shows that three to four ionic interactions stabilize the PRNA/P protein complex formation. Based on multiple sequence alignments of the left-handed βαβ crossover into the helix of the RNR motif in gram-positive and gram-negative bacteria (Gopalan et al. 1997), two amino acids, R60 and R65 (B. subtilis numbering) are completely conserved, suggesting that these two positively charged residues could play a role in the ionic interaction at the PRNA–P protein binding site. Additionally, both K53 and R68 (B. subtilis numbering) are conserved as either Arg or Lys in bacteria (Gopalan et al. 1997; Tsai et al. 2003), and the charged side chain at K64 (B. subtilis numbering) is conserved in bacteria except in the case of Mycobacterium leprae P protein, where it is an Ala (Gopalan et al. 1997). These residues could also be involved in charge–charge interactions in RNase P holoenzyme. A site-directed spin label incorporated at position E. coli K54 (K53, B. subtilis) has decreased mobility, as indicated by EPR spectroscopy, on addition of the E. coli M1 RNA, further implicating its potential involvement in a protein–RNA interaction (Gopalan et al. 1999). In summary, the RNR motif and, possibly, the N terminus are the area(s) to probe to localize the ionic interactions between the PRNA and P protein.
MATERIALS AND METHODS
Nucleic acid and protein preparations
Unmodified pre-tRNAAsp containing a 10-nucleotide 5′ leader sequence and mature tRNAAsp was prepared from a linearized plas-mid by in vitro transcription with T7 RNA polymerase (Milligan and Uhlenbeck 1989) as previously described (Beebe and Fierke 1994). B. subtilis PRNA, B. subtilis C domain, and S. cerevisiae RPR1 RNA were transcribed in the presence of excess guanosine to form RNA transcripts with a 5′ hydroxyl (Ziehler et al. 2000) and were subsequently labeled at the 5′ end with 32P-γ-ATP (MP Life Technologies) using T4 polynucleotide kinase (New England Bio-labs). The DNA oligomer containing the sequence TAATAC GACTCACTATAG was purchased from Invitrogen Life Technologies and was subsequently labeled at the 5′ end with 32P-γ-ATP as previously described. Purified His-tagged E. coli C5 protein (Rivera-Leon et al. 1995) was a generous gift from Dr. Jeffrey C. Kurz. The gene for B. subtilis P protein was cloned into pET28b such that the expressed protein contained either a C-terminal (pET28b-PP-hisC) or an N-terminal (pET28b-PP-hisN) hexa-histidine tag. Recombinant C-terminal and N-terminal His-tagged P proteins were purified from BL21(DE3)pLysSpET28b-PP cells induced with 1 mM isopropylthio-β-D-galactopyranoside followed by incubation at 37°C for 4 h. The cells were washed, resuspended with Buffer A (50 mM Tris-HCl at pH 8.0, 0.5M NaCl, 5 mM imidazole), and lysed by French press. The lysate was centrifuged at 10,000 rpm for 10 min to remove any insoluble cell debris. The clarified lysate was loaded onto a 5 mL Ni-NTA agarose column pre-equilibrated with Buffer A and washed with Buffer A containing 20 mM imidazole. The bound His-tagged P-protein was eluted with Buffer A containing 250 mM imidazole. The eluted protein was diluted threefold with Buffer B (50 mM potassium phosphate buffer at pH 8.0, 5 mM EDTA, 0.1 M NaCl), loaded onto a 5 mL P-11 phosphocellulose column pre-equilibrated with buffer B, and washed with Buffer B containing 0.8 M NaCl. The bound protein was eluted with Buffer B containing 2 M NaCl. Eluted P protein was dialyzed against 50 mM Tris-Cl (pH 8.0), 100 mM NaCl. The protein concentration was determined as described previously (Niranjanakumari et al. 1998a).
Magnetocapture assay
Refolded, 32P-5′ end-labeled PRNA, RPR1 RNA (for the heterologous studies and nonspecific interaction studies), or C domain at trace concentrations (<0.1 nM) in 50 mM Tris (pH 8), 10 mM MgCl2, 10 mM imidazole, and 100 mM KCl was mixed with various concentrations (0.1 nM–2 μM) of His-tagged P protein (Fig. 1 ▶, step A). The solution was incubated for 30 min at 25°C to allow the equilibrium formation of the holoenzyme complex (Fig. 1 ▶, step B). Then magnetic agarose beads containing nickel coordinated by NTA (Qiagen) were added (Fig. 1 ▶, step C). Typically, 10–20 μL of magnetic beads were added to a final solution volume of 300–1000 μL in a microfuge tube or 300 μL of solution with 10 μL of magnetic beads per well in a standard 96-well plate. The mixture was continuously agitated by either an end-over-end rotator (microfuge tubes) or by a platform shaker (96-well plates) for 1 h at room temperature. This agitation is necessary to ensure that the beads remain suspended in solution. One hour of incubation was sufficient to bind the His-tagged holoenzyme complex; no increase in bound radioactivity was observed for longer incubation times. The magnetic beads were separated from the bulk solution using an external magnet (Fig. 1 ▶, steps D, E). The radiolabeled PRNA (reflecting the holoenzyme complex) was eluted from the beads by incubation in 100 mM EDTA, 2% (v/v) SDS for 5 min at 95°C. Cerenkov scintillation counting was used to quantitate the radioactivity of the bound and free PRNA. The radioactivity bound to the beads went from a low of <5% (cpmbackground) in the absence of P protein to a high of 70%–90% at saturating P protein (cpmendpoint). The fraction of bound RNA was determined as previously described (Beebe and Fierke 1994; Kurz et al. 1998), where ([protein · RNA] / [RNAtotal]) = (cpmeluate − cpmbackground) /(cpmendpoint − cpmbackground). The Ni-NTA magnetic beads functioned much better than Ni-NTA spin columns (Qiagen) where the radiolabeled PRNA was retained in the column in the absence of protein, presumably by binding to the column filter. All data were fit with Kaleidagraph (Synergy Software) curve-fitting program. The dissociation constants were determined using Equation 2.
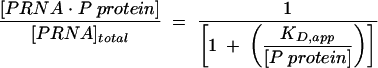 |
(2) |
Intraholoenzyme KD measured as a function of ionic strength
The individual assays were performed as previously described, but the ionic strength was altered by varying the concentration of KCl. The dependence of the apparent dissociation constant on ionic strength was fit globally as described in Equation 1 and Scheme 1 ▶ assuming complete cooperativity in ion binding. The KD,app is the dependent variable, and the ionic strength, Z, was the independent variable. Furthermore, at high ionic strength, the KD,app is approximated by log KD,app = n3log [Z] - n3log K3 to determine the value of n3log K3.
The ionic strength was calculated using Equation 3 (Ellis and Morrison 1982) where [Ii] is the concentration of ion Ii, and Zi is the ionic charge of ion i. It should be noted that both the cation and the anion must be treated separately. This equation was used to determine the ionic strength for all experiments including those where ATP, Co(NH3)6Cl3, and/or MgCl2 were included. The intraholoenzyme dissociation constant was determined by varying the
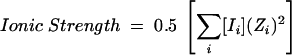 |
(3) |
concentration of KCl or LiCl from 0.1 to 800 mM in the presence of 50 mM Tris-HCl (pH 8), 10 mM imidazole, and/or 10 mM MgCl2. To test the possibility that the KD is enhanced by stabilization of the protein structure by anions, the experimental conditions were identical to those of the KCl titration except that 1 mM ATP was added. To monitor the effects of divalent and trivalent cations on intraholoenzyme affinity, we titrated either 0.1–100 mM MgCl2 or 0.1–20 mM Co(NH3)6Cl3 into the solution containing 50 mM Tris-HCl, 10 mM imidazole, and either 200 mM KCl (for MgCl2) or 100 mM KCl (for Co(NH3)6Cl3). To measure nonspecific binding of nucleic acids at low and high ionic strength, we determined the observed KD between either trace 5′ end-labeled RPR1 RNA or DNA oligomer and His-tagged P protein as previously described using buffers containing 50 mM Tris-HCl (pH 8), 10 mM imidazole, 10 mM MgCl2, and either 15 mM KCl (for low ionic strength) or 800 mM KCl (for high ionic strength).
Measurement of intraholoenzyme KD in the presence of pre-tRNAAsp or mature tRNAAsp
The intraholoenzyme KD was determined in the presence of either 0.1 nM pre-tRNAAsp or 0.1 nM mature tRNAAsp as a function of ionic strength by varying the concentration of KCl. As previously stated, 50 mM Tris-HCl and 10 mM imidazole were included in the solution buffer but 10 mM CaCl2 replaced the 10 mM MgCl2. The KD was determined as previously described.
Acknowledgments
We thank Dr. Jeffrey C. Kurz for giving us His-tagged E. coli C5 protein, Dr. David R. Engelke for giving us useful plasmids and helpful discussions, and Dr. Tao Pan for giving us the plasmid encoding the C domain. We also thank Dr. John Hsieh for invaluable insight and Nissa Westerberg for thoughtful reading of the manuscript. This research was supported by National Institutes of Health (NIH) Grant GM55387. Partial support was provided by NIH Training Grants GM08487 and GM008270 (J.J.D.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
C domain, catalytic domain of PRNA
C5, protein component of E. coli RNase P
E, RNase P holoenzyme
EDTA, (ethylenedinitrilo)-tetraacetic acid
M1 RNA, RNA component of E. coli RNase P
NTA, nitrilotriacetic acid
P, mature tRNA
P protein, protein component of B. subtilis RNase P
Pre-tRNA, precursor tRNA
PRNA, RNA component of B. subtilis RNase P
RNase P, ribonuclease P
RPR1 RNA, RNA component of S. cerevisiae RNase P
S or pre-tRNAAsp, precursor tRNAAsp
S domain, specificity domain of PRNA
Tris, Tris-(hydroxymethyl)-aminomethane
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.7550104.
REFERENCES
- Apirion, D. and Miczak, A. 1993. RNA processing in prokaryotic cells. Bioessays 15: 113–120. [DOI] [PubMed] [Google Scholar]
- Barrera, A., Fang, X., Jacob, J., Casey, E., Thiyagarajan, P., and Pan, T. 2002. Dimeric and monomeric Bacillus subtilis RNase P holoenzyme in the absence and presence of pre-tRNA substrates. Biochemistry 41: 12986–12994. [DOI] [PubMed] [Google Scholar]
- Beebe, J.A. and Fierke, C.A. 1994. A kinetic mechanism for cleavage of precursor tRNA(Asp) catalyzed by the RNA component of Bacillus subtilis ribonuclease P. Biochemistry 33: 10294–10304. [DOI] [PubMed] [Google Scholar]
- Beebe, J.A., Kurz, J.C., and Fierke, C.A. 1996. Magnesium ions are required by Bacillus subtilis ribonuclease P RNA for both binding and cleaving precursor tRNAAsp. Biochemistry 35: 10493–10505. [DOI] [PubMed] [Google Scholar]
- Carey, J. and Uhlenbeck, O.C. 1983. Kinetic and thermodynamic characterization of the R17 coat protein-ribonucleic acid interaction. Biochemistry 22: 2610–2615. [DOI] [PubMed] [Google Scholar]
- Chamberlain, J.R., Lee, Y., Lane, W.S., and Engelke, D.R. 1998. Purification and characterization of the nuclear RNase P holoenzyme complex reveals extensive subunit overlap with RNase MRP. Genes & Dev. 12: 1678–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y., Li, X., and Gegenheimer, P. 1997. Ribonuclease P catalysis requires Mg2+ coordinated to the pro-RP oxygen of the scissile bond. Biochemistry 36: 2425–2438. [DOI] [PubMed] [Google Scholar]
- Christian, E.L. and Harris, M.E. 1999. The track of the pre-tRNA 5′ leader in the ribonuclease P ribozyme-substrate complex. Biochemistry 38: 12629–12638. [DOI] [PubMed] [Google Scholar]
- Christian, E.L., McPheeters, D.S., and Harris, M.E. 1998. Identification of individual nucleotides in the bacterial ribonuclease P ribozyme adjacent to the pre-tRNA cleavage site by short-range photo-cross-linking. Biochemistry 37: 17618–17628. [DOI] [PubMed] [Google Scholar]
- Christian, E.L., Kaye, N.M., and Harris, M.E. 2000. Helix P4 is a divalent metal ion binding site in the conserved core of the ribo-nuclease P ribozyme. RNA 6: 511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crary, S.M., Niranjanakumari, S., and Fierke, C.A. 1998. The protein component of Bacillus subtilis ribonuclease P increases catalytic efficiency by enhancing interactions with the 5′ leader sequence of pre-tRNAAsp. Biochemistry 37: 9409–9416. [DOI] [PubMed] [Google Scholar]
- Crary, S.M., Kurz, J.C., and Fierke, C.A. 2002. Specific phosphoro-thioate substitutions probe the active site of Bacillus subtilis ribo-nuclease P. RNA 8: 933–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deHaseth, P.L., Lohman, T.M., and Record Jr., M.T. 1977. Nonspecific interaction of lac repressor with DNA: An association reaction driven by counterion release. Biochemistry 16: 4783–4790. [DOI] [PubMed] [Google Scholar]
- deHaseth, P.L., Lohman, T.M., Burgess, R.R., and Record Jr., M.T. 1978. Nonspecific interactions of Escherichia coli RNA polymerase with native and denatured DNA: Differences in the binding behavior of core and holoenzyme. Biochemistry 17: 1612–1622. [DOI] [PubMed] [Google Scholar]
- Draper, D.E. 2004. A guide to ions and RNA structure. RNA 10: 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis, K.J. and Morrison, J.F. 1982. Buffers of constant ionic strength for studying pH-dependent processes. Methods Enzymol. 87: 405–426. [DOI] [PubMed] [Google Scholar]
- Fang, X.W., Pan, T., and Sosnick, T.R. 1999. Mg2+-dependent folding of a large ribozyme without kinetic traps. Nat. Struct. Biol. 6: 1091–1095. [DOI] [PubMed] [Google Scholar]
- Fang, X.W., Yang, X.J., Littrell, K., Niranjanakumari, S., Thiyagarajan, P., Fierke, C.A., Sosnick, T.R., and Pan, T. 2001. The Bacillus subtilis RNase P holoenzyme contains two RNase P RNA and two RNase P protein subunits. RNA 7: 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank, D.N. and Pace, N.R. 1998. Ribonuclease P: Unity and diversity in a tRNA processing ribozyme. Annu. Rev. Biochem. 67: 153–180. [DOI] [PubMed] [Google Scholar]
- Fried, M. and Crothers, D.M. 1981. Equilibria and kinetics of lac repressor-operator interactions by polyacrylamide gel electrophoresis. Nucleic Acids Res. 9: 6505–6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner, M.M. and Revzin, A. 1981. A gel electrophoresis method for quantifying the binding of proteins to specific DNA regions: Application to components of the Escherichia coli lactose operon regulatory system. Nucleic Acids Res. 9: 3047–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegenheimer, P. 1995. Structure, mechanism and evolution of chloroplast transfer RNA processing systems. Mol. Biol. Rep. 22: 147–150. [DOI] [PubMed] [Google Scholar]
- Gerstner, R.B., Pak, Y., and Draper, D.E. 2001. Recognition of 16S rRNA by ribosomal protein S4 from Bacillus stearothermophilus. Biochemistry 40: 7165–7173. [DOI] [PubMed] [Google Scholar]
- Gluick, T.C., Wills, N.M., Gesteland, R.F., and Draper, D.E. 1997. Folding of an mRNA pseudoknot required for stop codon readthrough: Effects of mono- and divalent ions on stability. Biochemistry 36: 16173–16186. [DOI] [PubMed] [Google Scholar]
- Gopalan, V., Baxevanis, A.D., Landsman, D., and Altman, S. 1997. Analysis of the functional role of conserved residues in the protein subunit of ribonuclease P from Escherichia coli. J. Mol. Biol. 267: 818–829. [DOI] [PubMed] [Google Scholar]
- Gopalan, V., Kuhne, H., Biswas, R., Li, H., Brudvig, G.W., and Altman, S. 1999. Mapping RNA-protein interactions in ribonuclease P from Escherichia coli using electron paramagnetic resonance spectroscopy. Biochemistry 38: 1705–1714. [DOI] [PubMed] [Google Scholar]
- Guerrier-Takada, C. and Altman, S. 1984. Catalytic activity of an RNA molecule prepared by transcription in vitro. Science 223: 285–286. [DOI] [PubMed] [Google Scholar]
- Guerrier-Takada, C., Gardiner, K., Marsh, T., Pace, N., and Altman, S. 1983. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 35: 849–857. [DOI] [PubMed] [Google Scholar]
- Hardt, W.D., Schlegl, J., Erdmann, V.A., and Hartmann, R.K. 1993. Gel retardation analysis of E. coli M1 RNA-tRNA complexes. Nucleic Acids Res. 21: 3521–3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, M.E. and Pace, N.R. 1995. Identification of phosphates involved in catalysis by the ribozyme RNase P RNA. RNA 1: 210–218. [PMC free article] [PubMed] [Google Scholar]
- Harris, M.E., Kazantsev, A.V., Chen, J.L., and Pace, N.R. 1997. Analysis of the tertiary structure of the ribonuclease P ribozyme-substrate complex by site-specific photoaffinity crosslinking. RNA 3: 561–576. [PMC free article] [PubMed] [Google Scholar]
- Henkels, C.H., Kurz, J.C., Fierke, C.A., and Oas, T.G. 2001. Linked folding and anion binding of the Bacillus subtilis ribonuclease P protein. Biochemistry 40: 2777–2789. [DOI] [PubMed] [Google Scholar]
- Hermann, T. and Westhof, E. 1999. Simulations of the dynamics at an RNA-protein interface. Nat. Struct. Biol. 6: 540–544. [DOI] [PubMed] [Google Scholar]
- Hsieh, J., Andrews, A.J., and Fierke, C.A. 2004. Roles of protein subunits in RNA-protein complexes: Lessons from ribonuclease P. Biopolymers 73: 79–89. [DOI] [PubMed] [Google Scholar]
- Jones, S., Daley, D.T., Luscombe, N.M., Berman, H.M., and Thornton, J.M. 2001. Protein-RNA interactions: A structural analysis. Nucleic Acids Res. 29: 943–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye, N.M., Christian, E.L., and Harris, M.E. 2002. NAIM and site-specific functional group modification analysis of RNase P RNA: Magnesium dependent structure within the conserved P1-P4 multihelix junction contributes to catalysis. Biochemistry 41: 4533–4545. [DOI] [PubMed] [Google Scholar]
- Kazantsev, A.V., Krivenko, A.A., Harrington, D.J., Carter, R.J., Hol-brook, S.R., Adams, P.D., and Pace, N.R. 2003. High-resolution structure of RNase P protein from Thermotoga maritima. Proc. Natl. Acad. Sci. 100: 7497–7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasilnikov, A.S., Yang, X., Pan, T., and Mondragon, A. 2003. Crystal structure of the specificity domain of ribonuclease P. Nature 421: 760–764. [DOI] [PubMed] [Google Scholar]
- Krupp, G., Kahle, D., and Vogt, T. 1991. Sequence changes in both flanking sequences of a pre-tRNA influence the cleavage specificity of RNase P. J. Mol. Biol. 217: 637–648. [DOI] [PubMed] [Google Scholar]
- Kurz, J.C., Niranjanakumari, S., and Fierke, C.A. 1998. Protein component of Bacillus subtilis RNase P specifically enhances the affinity for precursor-tRNAAsp. Biochemistry 37: 2393–2400. [DOI] [PubMed] [Google Scholar]
- Labuda, D. and Augustyniak, J. 1977. Dependence of tRNA structure in solution upon ionic condition of the solvent. Fluorescence studies of monovalent cation binding to tRNAPhe from barley embryos. Eur. J. Biochem. 79: 303–307. [DOI] [PubMed] [Google Scholar]
- Lee, Y., Kindelberger, D.W., Lee, J.Y., McClennen, S., Chamberlain, J., and Engelke, D.R. 1997. Nuclear pre-tRNA terminal structure and RNase P recognition. RNA 3: 175–185. [PMC free article] [PubMed] [Google Scholar]
- Leulliot, N. and Varani, G. 2001. Current topics in RNA-protein recognition: Control of specificity and biological function through induced fit and conformational capture. Biochemistry 40: 7947–7956. [DOI] [PubMed] [Google Scholar]
- Loria, A. and Pan, T. 1996. Domain structure of the ribozyme from eubacterial ribonuclease P. RNA 2: 551–563. [PMC free article] [PubMed] [Google Scholar]
- ———. 1998. Recognition of the 5′ leader and the acceptor stem of a pre-tRNA substrate by the ribozyme from Bacillus subtilis RNase P. Biochemistry 37: 10126–10133. [DOI] [PubMed] [Google Scholar]
- ———. 2001. Modular construction for function of a ribonucleoprotein enzyme: The catalytic domain of Bacillus subtilis RNase P complexed with B. subtilis RNase P protein. Nucleic Acids Res. 29: 1892–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrary, B.S., Bedell, J., Edmondson, S.P., and Shriver, J.W. 1998. Linkage of protonation and anion binding to the folding of Sac7d. J. Mol. Biol. 276: 203–224. [DOI] [PubMed] [Google Scholar]
- Milligan, J.F. and Uhlenbeck, O.C. 1989. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol. 180: 51–62. [DOI] [PubMed] [Google Scholar]
- Misra, V.K. and Draper, D.E. 2001. A thermodynamic framework for Mg2+ binding to RNA. Proc. Natl. Acad. Sci. 98: 12456–12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogridge, J., Legault, P., Li, J., Van Oene, M.D., Kay, L.E., and Greenblatt, J. 1998. Independent ligand-induced folding of the RNA-binding domain and two functionally distinct antitermination regions in the phage λ N protein. Mol. Cell 1: 265–275. [DOI] [PubMed] [Google Scholar]
- Niranjanakumari, S., Kurz, J.C., and Fierke, C.A. 1998a. Expression, purification and characterization of the recombinant ribonuclease P protein component from Bacillus subtilis. Nucleic Acids Res. 26: 3090–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niranjanakumari, S., Stams, T., Crary, S.M., Christianson, D.W., and Fierke, C.A. 1998b. Protein component of the ribozyme ribonuclease P alters substrate recognition by directly contacting precursor tRNA. Proc. Natl. Acad. Sci. 95: 15212–15217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, T. 1995. Higher order folding and domain analysis of the ribozyme from Bacillus subtilis ribonuclease P. Biochemistry 34: 902–909. [DOI] [PubMed] [Google Scholar]
- Pan, T. and Jakacka, M. 1996. Multiple substrate binding sites in the ribozyme from Bacillus subtilis RNase P. EMBO J. 15: 2249–2255. [PMC free article] [PubMed] [Google Scholar]
- Pan, T. and Sosnick, T.R. 1997. Intermediates and kinetic traps in the folding of a large ribozyme revealed by circular dichroism and UV absorbance spectroscopies and catalytic activity. Nat. Struct. Biol. 4: 931–938. [DOI] [PubMed] [Google Scholar]
- Pannucci, J.A., Haas, E.S., Hall, T.A., Harris, J.K., and Brown, J.W. 1999. RNase P RNAs from some Archaea are catalytically active. Proc. Natl. Acad. Sci. 96: 7803–7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson, T., Cuzic, S., and Hartmann, R.K. 2003. Catalysis by RNase P RNA: Unique features and unprecedented active site plasticity. J. Biol. Chem. 278: 43394–43401. [DOI] [PubMed] [Google Scholar]
- Qin, H., Sosnick, T.R., and Pan, T. 2001. Modular construction of a tertiary RNA structure: The specificity domain of the Bacillus sub-tilis RNase P RNA. Biochemistry 40: 11202–11210. [DOI] [PubMed] [Google Scholar]
- Record Jr., M.T., Lohman, T.M., and De Haseth, P. 1976. Ion effects on ligand-nucleic acid interactions. J. Mol. Biol. 107: 145–158. [DOI] [PubMed] [Google Scholar]
- Record Jr., M.T., deHaseth, P.L., and Lohman, T.M. 1977. Interpretation of monovalent and divalent cation effects on the lac repressor-operator interaction. Biochemistry 16: 4791–4796. [DOI] [PubMed] [Google Scholar]
- Record Jr., M.T., Anderson, C.F., and Lohman, T.M. 1978. Thermodynamic analysis of ion effects on the binding and conformational equilibria of proteins and nucleic acids: The roles of ion association or release, screening, and ion effects on water activity. Q. Rev. Biophys. 11: 103–178. [DOI] [PubMed] [Google Scholar]
- Rivera-Leon, R., Green, C.J., and Vold, B.S. 1995. High-level expression of soluble recombinant RNase P protein from Escherichia coli. J. Bacteriol. 177: 2564–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossmanith, W. and Karwan, R.M. 1998. Characterization of human mitochondrial RNase P: Novel aspects in tRNA processing. Biochem. Biophys. Res. Commun. 247: 234–241. [DOI] [PubMed] [Google Scholar]
- Rossmanith, W. and Potuschak, T. 2001. Difference between mitochondrial RNase P and nuclear RNase P. Mol. Cell. Biol. 21: 8236–8236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rox, C., Feltens, R., Pfeiffer, T., and Hartmann, R.K. 2002. Potential contact sites between the protein and RNA subunit in the Bacillus subtilis RNase P holoenzyme. J. Mol. Biol. 315: 551–560. [DOI] [PubMed] [Google Scholar]
- Ryan, P.C. and Draper, D.E. 1989. Thermodynamics of protein-RNA recognition in a highly conserved region of the large-subunit ribosomal RNA. Biochemistry 28: 9949–9956. [DOI] [PubMed] [Google Scholar]
- Siew, D., Zahler, N.H., Cassano, A.G., Strobel, S.A., and Harris, M.E. 1999. Identification of adenosine functional groups involved in substrate binding by the ribonuclease P ribozyme. Biochemistry 38: 1873–1883. [DOI] [PubMed] [Google Scholar]
- Smith, D. and Pace, N.R. 1993. Multiple magnesium ions in the ribonuclease P reaction mechanism. Biochemistry 32: 5273–5281. [DOI] [PubMed] [Google Scholar]
- Smith, D., Burgin, A.B., Haas, E.S., and Pace, N.R. 1992. Influence of metal ions on the ribonuclease P reaction. Distinguishing substrate binding from catalysis. J. Biol. Chem. 267: 2429–2436. [PubMed] [Google Scholar]
- Spitzfaden, C., Nicholson, N., Jones, J.J., Guth, S., Lehr, R., Prescott, C.D., Hegg, L.A., and Eggleston, D.S. 2000. The structure of ribo-nuclease P protein from Staphylococcus aureus reveals a unique binding site for single-stranded RNA. J. Mol. Biol. 295: 105–115. [DOI] [PubMed] [Google Scholar]
- Stams, T., Niranjanakumari, S., Fierke, C.A., and Christianson, D.W. 1998. Ribonuclease P protein structure: Evolutionary origins in the translational apparatus. Science 280: 752–755. [DOI] [PubMed] [Google Scholar]
- Talbot, S.J. and Altman, S. 1994a. Gel retardation analysis of the interaction between C5 protein and M1 RNA in the formation of the ribonuclease P holoenzyme from Escherichia coli. Biochemistry 33: 1399–1405. [DOI] [PubMed] [Google Scholar]
- ———. 1994b. Kinetic and thermodynamic analysis of RNA-protein interactions in the RNase P holoenzyme from Escherichia coli. Biochemistry 33: 1406–1411. [DOI] [PubMed] [Google Scholar]
- Thirumalai, D., Lee, N., Woodson, S.A., and Klimov, D. 2001. Early events in RNA folding. Annu. Rev. Phys. Chem. 52: 751–762. [DOI] [PubMed] [Google Scholar]
- Thomas, B.C., Chamberlain, J., Engelke, D.R., and Gegenheimer, P. 2000. Evidence for an RNA-based catalytic mechanism in eukaryotic nuclear ribonuclease P. RNA 6: 554–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinoco Jr., I. and Kieft, J.S. 1997. The ion core in RNA folding. Nat. Struct. Biol. 4: 509–512. [DOI] [PubMed] [Google Scholar]
- Tobian, J.A., Drinkard, L., and Zosloff, M. 1985. tRNA nuclear transport: Defining the critical regions of human tRNAimet by point mutagenesis. Cell 43: 415–422. [DOI] [PubMed] [Google Scholar]
- Torrent, C., Bordet, T., and Darlix, J.L. 1994. Analytical study of rat retrotransposon VL30 RNA dimerization in vitro and packaging in murine leukemia virus. J. Mol. Biol. 240: 434–444. [DOI] [PubMed] [Google Scholar]
- Tsai, H.Y., Masquida, B., Biswas, R., Westhof, E., and Gopalan, V. 2003. Molecular modeling of the three-dimensional structure of the bacterial RNase P holoenzyme. J. Mol. Biol. 325: 661–675. [DOI] [PubMed] [Google Scholar]
- Ucci, J.W. and Cole, J.L. 2004. Global analysis of non-specific protein-nucleic interactions by sedimentation equilibrium. Biophys. Chem. 108: 127–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanke, C., Romer, R., and Maass, G. 1975. Tertiary structure of tRNAPhe (yeast): Kinetics and electrostatic repulsion. Eur. J. Biochem. 55: 439–444. [DOI] [PubMed] [Google Scholar]
- Van Gilst, M.R., Rees, W.A., Das, A., and von Hippel, P.H. 1997. Complexes of N antitermination protein of phage λ with specific and nonspecific RNA target sites on the nascent transcript. Biochemistry 36: 1514–1524. [DOI] [PubMed] [Google Scholar]
- Vartikar, J.V. and Draper, D.E. 1989. S4–16 S ribosomal RNA complex. Binding constant measurements and specific recognition of a 460-nucleotide region. J. Mol. Biol. 209: 221–234. [DOI] [PubMed] [Google Scholar]
- Warnecke, J.M., Held, R., Busch, S., and Hartmann, R.K. 1999. Role of metal ions in the hydrolysis reaction catalyzed by RNase P RNA from Bacillus subtilis. J. Mol. Biol. 290: 433–445. [DOI] [PubMed] [Google Scholar]
- Weeks, K.M. and Crothers, D.M. 1992. RNA binding assays for Tat-derived peptides: Implications for specificity. Biochemistry 31: 10281–10287. [DOI] [PubMed] [Google Scholar]
- Williamson, J.R. 2000. Induced fit in RNA-protein recognition. Nat. Struct. Biol. 7: 834–837. [DOI] [PubMed] [Google Scholar]
- Xiao, S., Houser-Scott, F., and Engelke, D.R. 2001. Eukaryotic ribo-nuclease P: Increased complexity to cope with the nuclear pre-tRNA pathway. J. Cell. Physiol. 187: 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, S., Scott, F., Fierke, C.A., and Engelke, D.R. 2002. Eukaryotic ribonuclease P: A plurality of ribonucleoprotein enzymes. Annu. Rev. Biochem. 71: 165–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahler, N.H., Christian, E.L., and Harris, M.E. 2003. Recognition of the 5′ leader of pre-tRNA substrates by the active site of ribonuclease P. RNA 9: 734–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziehler, W.A., Day, J.J., Fierke, C.A., and Engelke, D.R. 2000. Effects of 5′ leader and 3′ trailer structures on pre-tRNA processing by nuclear RNase P. Biochemistry 39: 9909–9916. [DOI] [PubMed] [Google Scholar]
- Ziehler, W.A., Morris, J., Scott, F.H., Millikin, C., and Engelke, D.R. 2001. An essential protein-binding domain of nuclear RNase P RNA. RNA 7: 565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]