

Abstract
To provide a global analysis of gene expression in the aging heart, we monitored the expression of 9,977 genes simultaneously in 5- and 30-month-old male B6C3F1 mice by using high-density oligonucleotide microarrays and several statistical techniques. Aging was associated with transcriptional alterations consistent with a metabolic shift from fatty acid to carbohydrate metabolism, increased expression of extracellular matrix genes, and reduced protein synthesis. Caloric restriction (CR) started at 14 months of age resulted in a 19% global inhibition of age-related changes in gene expression. Interestingly, CR also resulted in alterations in gene expression consistent with preserved fatty acid metabolism, reduced endogenous DNA damage, decreased innate immune activity, apoptosis modulation, and a marked cytoskeletal reorganization. These observations provide evidence that aging of the heart is associated with specific transcriptional alterations, and that CR initiated in middle age may retard heart aging by inducing a profound transcriptional reprogramming.
When started either early in life or at middle age, caloric restriction (CR) increases average and maximum lifespan, and reduces the incidence and delays the onset of spontaneous cancers and several other age-related diseases (1). Additionally, CR reduces the age-associated increase in reactive oxygen species (ROS)-induced molecular damage (2–5). Previously, we have used high-density oligonucleotide arrays to define aging and CR-related transcriptional alterations in mouse skeletal muscle (6) and brain (7). These reports provided evidence for an age-associated stress response characterized by the induction of heat-shock factors and other oxidative stress-induced transcripts. CR prevented these age-related alterations completely or partially. Both studies provided further support for the concept that oxidative stress may be an important, and perhaps underlying cause of the aging process of postmitotic tissues.
The cardiac myocyte is the most energy demanding cell in the body, contracting constantly, 3 billion times or more in the average human lifespan (8), requiring large supplies of high-energy phosphates (9). Age-related changes in human and rodent hearts include a reduction in the number of myocytes (10, 11), myocyte hypertrophy (11, 12), cardiac fibrosis (13), lipofuscin pigment accumulation (14), a reduction in calcium transport across sarcoplasmic reticulum membrane (15), and alterations in the response to β-adrenergic stimulation (16). Collectively, these alterations likely contribute to age-related heart diseases being the leading cause of mortality in the U.S. (17). CR reduces the severity of spontaneous cardiomyopathy in male Sprague–Dawley rats (18) and prevents age-associated alterations in late diastolic function in B6D2F1 mice (19). At the molecular level, CR reduces the concentration of both 8-hydroxydeoxyguanosine in DNA (20) and dityrosine cross-linking of proteins (21) in the heart of aging mice, and prevents somatic mitochondrial genomic rearrangements associated with aging (22).
To infer mechanisms of aging in the heart and their possible modification by middle age-onset CR, we obtained global gene expression profiles by monitoring mRNA levels for 9,977 genes, using high-density oligonucleotide arrays. To understand the effects of aging on gene expression, the transcriptional activities of hearts were measured in 5- and 30-month-old mice. To determine the effects of CR on aging, transcriptional alterations were measured for 30-month-old mice on CR since 14 months of age.
Materials and Methods
Animals and Dietary Manipulations.
Male B6C3F1 mice (6–7 weeks of age) were purchased from Harlan Sprague–Dawley. Mice were housed singly and provided acidified water ad libitum. The control group was fed 98 kcal/week of AIN-M semipurified diet (Bioserv, Frenchtown, NJ), which is ≈15% less than the average ad libitum intake. The CR group was gradually restricted from 14 months of age, being fed 78 kcal/week for the first month of CR and 58 kcal/week (a 41% CR) thereafter. The restricted diet was nearly isocaloric to the normal diet, but enriched in protein, vitamins, and minerals to avoid malnutrition (23). Animals were fed three times per week, a strategy supported by data showing that the frequency and timing of meals for mice on CR does not influence longevity (24). Mice were euthanized by cervical dislocation. Tissues were frozen in liquid nitrogen and stored at −80°C until use.
Target RNA Preparation, Hybridization, and Preliminary Data Analysis by Affymetrix Algorithms.
Total RNA was extracted from frozen heart tissues and polyadenylate [poly(A)+] RNAs were isolated as described (6). Detailed procedures for target preparation and array image data analysis are available in Supporting Text, which is published as supporting information on the PNAS web site, www.pnas.org. We used five animals per group, and hybridized each sample to independent DNA chips, because previous work from our laboratory suggests that variability between individuals is higher than variability observed in replicate hybridizations of the same samples (25).
Statistical Analysis.
To statistically evaluate the effects of age and CR on gene expression levels, we conducted a multistep evaluation procedure. In the first step, we used nonparametric bootstrap hypothesis testing (26) to obtain a frequentist P value for each gene. Part of our reason for having five animals per group is that n = 5 in a two-group comparison offers a sufficient number (>15,000) of unique bootstrap resamples. We used 10,000 resamples to compute P values to the level of 10−4. Gene expression change was called significant when the P value was <0.05.
In the second step, we used a mixture modeling approach (27) that begins by using frequentist P values, but then incorporates them into a mixture model and ultimately produces posterior probabilities (pp) for each gene. The pp is the Bayesian probability that a gene with a frequentist P value less than or equal to that observed is a gene for which there is a real difference between groups in expression level. This procedure provides an omnibus test of whether there is any overall effect of treatment on gene expression levels, an estimate of the total number of genes that have their expression levels altered, and a model describing the distribution of effects on gene expression levels (27).
Finally, we estimated the effect of CR on aging by using a statistical procedure. First, we constructed an index that used all genes to create a function on which old and young animals were highly separated. We converted all genes to unit variance, and then assigned them a coefficient in the function that was proportional to their mean difference across the young and old groups. Once the index was so constructed, we were able to test whether the young and old animals differed significantly on the index. We conducted 100 simulations under the global null hypothesis to determine the P value. Once it was determined that the young and old groups differed significantly, we were next able to determine whether, on this same function, the CR group was significantly different from the old controls and, if so, what percent of the way on this function they had moved toward being like the young controls. This provided a quantitative estimate of the extent to which CR inhibits the age-induced changes in gene expression levels. These estimates, as well as the significance levels, were again determined by simulation. This percent reduction was calculated as:
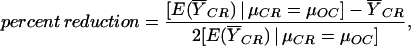 |
where E(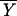 CR)|μCR = μOC is the expected value of the mean index score for the CR group under the null hypothesis of there being no difference between the CR group and the old control group, and
CR)|μCR = μOC is the expected value of the mean index score for the CR group under the null hypothesis of there being no difference between the CR group and the old control group, and 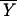 CR is the observed mean index value for the CR group.
CR is the observed mean index value for the CR group.
Results
Overview of Aging- and CR-Induced Gene Expression Patterns.
Of the 9,977 genes studied, 5,701 (57%) genes were expressed in the 5-month-old control group, 5,669 (57%) genes in the 30-month-old control group, and 5,253 (53%) genes in the 30-month-old CR group based on the Affymetrix algorithm. According to the bootstrap test, 996 (10%) transcripts were significantly changed by aging, whereas 2,075 (21%) transcripts were significantly changed by CR. The number of genes was further reduced to 312 and 831 in the aging and CR comparisons, respectively, after filtering by detection decision and average FC (1.5 or higher). The proportions of genes changed by aging and CR were 27% and 47%, respectively, as estimated by the mixture model analysis, which takes into account the P value distribution (Fig. 1 Upper). As long as the P value for a given gene was <0.05, there was a 63% chance in the comparison between the 30- and 5-month-old control group, and an 87% chance in the comparison between the 30-month-old CR and 30-month-old controls, that the gene is truly differentially expressed (the posterior probability; Fig. 1 Lower). The estimate for the global CR inhibition effect on aging was ≈19% when using the percent reduction equation described in Statistical Analysis (Fig. 2). Table 1 contains selected genes that are significantly changed in expression by aging (P < 0.05), and display a fold change equal or greater than 1.5. Tables 2–6 contain all genes meeting statistical criteria and gene identifier numbers, and are published as supporting information on the PNAS web site.
Fig 1.

Distribution of P values and pp for 9,977 genes in the (a) aging comparison (30- and 5-month-old control fed animals) and (b) CR comparison (30-month-old CR and 30-month-old control diet-fed animals). (Upper) The distribution of the P values for all genes representing in the oligonucleotide array. The fitted model was a mixture of one uniform (…) and one beta (—) distribution. The null (uniform) proportions were 0.7285 (a) and 0.5304 (b), and the alternative (beta) proportions were 0.2715 (a) and 0.4696 (b). (Lower) The pp of the 9,977 genes along with their corresponding P values.
Fig 2.

Global estimation of CR effect on aging. Both aging and CR significantly affected global gene expression patterns. Old and young means would fall with 99% confidence in the region between the green lines if there were no global effects of aging on gene expression. The region between blue lines denotes the 99% confidence interval for a global effect of CR on gene expression. The upper orange line denotes the expected value of the CR mean if there was no aging retardation by CR. The lower orange line denotes the expected value of the CR mean if CR completely eliminated all effects of aging (i.e., if the population CR mean equaled the population young mean). The estimate of the percent CR inhibition in the transcriptional changes associated with the aging process was ≈19%.
Table 1.
List of selected genes altered in the expression by aging
| Gene | FC, age | %CR effect | P value | pp |
|---|---|---|---|---|
| Glucose/carbohydrate/energy metabolism | ||||
| Creatine kinase, brain | 1.7 | 86 | 0.0016 | 0.81 |
| Homologous to human glucan (1,4-alpha-), branching enzyme 1 | 1.6 | 8 | 0.0427 | 0.64 |
| Phosphofructokinase, liver, B-type | 1.5 | 105 | 0.0005 | 0.85 |
| Pyruvate dehydrogenase kinase 4 | −8.5 | 139 | 0.0020 | 0.80 |
| Fructose 1,6-bisphosphatase 2 | −5.0 | 22 | 0.0013 | 0.82 |
| Uncoupling protein 3, mitochondrial | −2.6 | 146 | 0.0038 | 0.77 |
| Fatty acid metabolism | ||||
| Fatty acid transport into cytosol and mitochondrial matrix | ||||
| Solute carrier family 27 (fatty acid transporter), member 1 | −2.0 | 25 | 0.0055 | 0.76 |
| CD36 antigen | −1.8 | −14 | 0.0363 | 0.65 |
| Carnitine O-palmitoyl transferase I, mitochondrial liver isoform | −3.3 | 46 | 0.0004 | 0.86 |
| Carnitine acetyltransferase | −1.6 | −15 | 0.0211 | 0.68 |
| Mitochondrial carnitine/acylcarnitine translocase | −1.5 | 73 | 0.0011 | 0.82 |
| Mitochondrial β-oxidation | ||||
| Acyl-CoA thioesterase 1, cytosolic | −5.1 | 71 | 0.0007 | 0.84 |
| Lipase, hormone sensitive | −4.6 | 89 | 0.0073 | 0.74 |
| Acyl-Coenzyme A oxidase 1, palmitoyl | −1.8 | 38 | 0.0213 | 0.68 |
| Carboxylesterase 3 | −1.6 | 182 | 0.0154 | 0.70 |
| Peroxisomal delta3, delta2-enoyl-coenzyme A isomerase | −1.6 | 174 | 0.0054 | 0.76 |
| Enoyl coenzyme A hydratase 1, peroxisomal | −1.5 | 13 | 0.0486 | 0.63 |
| Dodecenoyl-coenzyme A delta isomerase | −1.5 | 26 | 0.0164 | 0.70 |
| Alpha-methylacyl-CoA racemase | 4.5 | 32 | 0.0103 | 0.72 |
| Neurodegeneration | ||||
| Cyclin-dependent kinase 5 | 6.3 | 74 | 0.0051 | 0.76 |
| Synuclein, alpha | 6.0 | −80 | 0.0046 | 0.76 |
| Protein synthesis, modification, and turnover | ||||
| Eukaryotic translation initiation factor 2, subunit 3, structural gene Y-linked | −2.2 | −10 | 0.0030 | 0.78 |
| Eukaryotic translation initiation factor 4E binding protein 1 | −1.7 | 19 | 0.0002 | 0.88 |
| Homologous to human eIF4B | −1.6 | 10 | 0.0122 | 0.71 |
| Proteasome (prosome, macropain) 28, subunit 3 | 2.7 | 129 | 0.0262 | 0.67 |
| Peptidylprolyl isomerase C | 2.4 | 7 | 0.0062 | 0.75 |
| Ubiquitin specific protease 23 | 1.6 | 210 | 0.0085 | 0.73 |
| Ubiquitin-like I (sentrin) activating enzyme EIA | 1.5 | 115 | 0.0334 | 0.65 |
| Extracellular matrix proteins | ||||
| Procollagen, type XV | 3.4 | 134 | 0.0285 | 0.66 |
| Procollagen C-proteinase enhancer protein | 2.1 | 60 | 0.0315 | 0.66 |
| Type VI collagen alpha 3 subunit | 2.1 | 189 | 0.0134 | 0.71 |
| Collagen alpha 1 (IV) | 1.9 | 59 | 0.0007 | 0.84 |
| Hyaluronan synthase 1 | 1.7 | 0 | 0.0388 | 0.64 |
| Serine (or cysteine) proteinase inhibitor, clade H, member 1 | 1.5 | 231 | 0.0048 | 0.76 |
| Structure, structural modulation, cell–cell interaction, and muscle contraction | ||||
| Smoothelin | 15.2 | 81 | 0.0275 | 0.67 |
| Claudin-5 | 8.1 | 71 | 0.0069 | 0.74 |
| Sodium channel, voltage-gated, type 1, beta polypeptide | 5.9 | 77 | 0.0064 | 0.75 |
| SWI/SNF related, matrix-associated, actin-dependent regulator of chromatin, subfamily, member 1 | 4.2 | 23 | 0.0405 | 0.64 |
| Actin, alpha 2, smooth muscle, aorta | 2.2 | 28 | 0.0230 | 0.68 |
| Integrin alpha 6 | 1.9 | 93 | 0.0033 | 0.78 |
| SWI/SNF related, matrix-associated, actin-F171 dependent, regulator of chromatin, subfamily e, member 1 | 1.7 | 38 | 0.0209 | 0.68 |
| Gap junction membrane channel protein alpha 1 | 1.6 | 159 | 0.0073 | 0.74 |
| Intercellular adhesion molecule 2 | 1.6 | 162 | 0.0273 | 0.67 |
| Troponin T1, skeletal, slow | 1.6 | 21 | 0.0005 | 0.85 |
| Calponin 2 | 1.6 | 67 | 0.0223 | 0.68 |
| Ankyrin 1, erythroid | 1.5 | 63 | 0.0338 | 0.65 |
| Type 2 desmoglein | −3.3 | 14 | 0.0086 | 0.73 |
| NAD(P)+ arginine ADP-ribosyltransferase | −1.6 | 167 | 0.0209 | 0.68 |
| Gap junction membrane channel protein beta 5 | −1.6 | 12 | 0.0131 | 0.71 |
| Neuroepithelial cell transforming gene 1 | −1.6 | 28 | 0.0018 | 0.81 |
| Dynein, cytoplasmic, heavy chain 1 | −1.5 | 39 | 0.0069 | 0.74 |
This table lists the genes of selected classes that were significantly (P value < 0.05, FC ≥ 1.5) altered in expression with aging. The complete list of genes that were significantly changed by aging, including gene identifier numbers, can be found in the supporting information. %CR effect was computed as ((O − CR)/(O − Y)) × 100, where O, CR, and Y are the average intensities of the 30-month-old control, 30-month-old CR, and 5-month-old control groups, respectively.
Significantly different between CR and O groups.
Aging Results in Expression of Genes Encoding Structural Proteins.
Several genes involved in structural roles, such as extracellular matrix (ECM) components, collagen deposition, cell adhesion, and cell growth, were induced as a result of the aging process. These include Smoothelin B, a cytoskeletal marker of smooth muscle cell differentiation (28), Calponin 2, a smooth muscle contraction modulator, and Troponin T1, which couples troponin C to actin-myosin contractile machinery (29). Genes encoding junctional proteins were also up-regulated, including Claudin-5, a component of tight junctions, and Connexin 43, a component of gap junctions. Some collagen family members were also up-regulated, including Type XV collagen alpha 1, Type VI collagen alpha 3, Type IV collagen alpha 1, and Type VI collagen alpha 2. These observations are consistent with previous findings that the aging heart undergoes ECM protein deposition (30), fibrosis (31), and cardiomyocyte hypertrophy (11).
Aging Results in Expression of Genes Involved in Neurodegeneration.
Aging also resulted in a striking up-regulation of Cyclin-dependent kinase 5 (Cdk5). Aberrant Cdk5 expression has been observed in skeletal muscle following nerve injury (32), and also in a number of neurodegenerative conditions (33). Another surprising finding was a 6-fold increase in the expression of Alpha synuclein, a major component of Lewy bodies (LBs), a marker of neurodegeneration found in Parkinson's disease. LBs, as well as ubiquitin-positive degenerating neurites, are strongly immunoreactive with antibodies against Alpha synuclein (34, 35). The heart has dual sympathetic and parasympathetic innervation, and recent findings have linked Alpha synuclein expression in the heart with sympathetic cardiac dysfunction in Parkinson's disease patients (36).
Aging Results in Down-Regulation of Genes Involved in Fatty Acid Metabolism.
Several genes involved in lipid metabolism and fatty acid oxidation (FAO), the major energy source in the adult heart, were altered in expression as a result of aging (Table 1 and Fig. 3). There was a concerted down-regulation of genes involved in fatty acid transport to mitochondria, including Carnitine O-palmitoyl transferase I (Cpt1), Carnitine acetyltransferase (Crat), Mitochondrial carnitine/acylcarnitine translocase, and Carnitine palmitoyltransferase 2 (Cpt2). Cpt1 esterifies the long-chain fatty acids to carnitine (37), whereas Crat is specific for short-chain fatty acids. Carnitine/acylcarnitine translocases transport fatty acids across the mitochondrial membranes (38), and Cpt2 releases the fatty acids from carnitine at the inner mitochondrial membrane. Also down-regulated were Solute carrier family 27, which catalyzes the transfer of long-chain fatty acids across the plasma membrane (39), and CD36 antigen, which encodes a fatty acid translocase. Another group of genes showing a decrease in expression in the old heart is involved in lipolysis; this group includes Triacylglycerol hydrolase, which mobilizes triacylglycerol from storage pools (40), Lipase, hormone sensitive, which catalyzes the rate-limiting step in adipose tissue lipolysis (41), and Acyl-CoA thioesterase 1, cytosolic, which modulates the cellular levels of fatty acid-CoA ligands. Aging also down-regulated several genes involved in fatty acid β-oxidation in mitochondria (Table 1). Taken as a whole, the striking down-regulation of genes involved in fatty acid metabolism is consistent with previous observations of impaired mitochondrial function and decreased palmitoylcarnitine-supported respiration in heart mitochondria from aged rats (42, 43).
Fig 3.

Genes involved in β-oxidation are reduced in expression as a result of aging. The transcriptional patterns also suggest that glucose utilization is increased as a result of aging. Genes encoding enzymes involved in each metabolic step are italicized. All genes shown were altered in expression as a result of aging, and the alteration in expression was either completely or partially prevented by CR. For fold-change data for each gene, and the specific percentage inhibition of the age-related change by CR, see Table 1. Fold change for phosphoglycerate kinase and enolase was 1.1-fold (P < 0.05) and is not listed in Table 1.
Aging Results in a Transcriptional Shift Toward Carbohydrate Metabolism.
A key enzyme involved in fuel selection is Pyruvate dehydrogenase kinase isoenzyme 4 (PDK4). PDK4 inhibits pyruvate dehydrogenase and thus minimizes carbohydrate oxidation by preventing the flow of glycolytic products into the tricarboxylic acid cycle (44). We observed a dramatic down-regulation (8.5 fold) of PDK4, suggesting a metabolic shift toward glycolysis in the aging heart. This is also supported by the finding that the gene encoding Fructose 1,6-bisphosphatase 2, a key enzyme in gluconeogenesis, is markedly depressed (5-fold) in the aging heart. Phosphofructokinase, an allosteric enzyme that controls the pace of glycolysis by converting fructose 6-phosphate to fructose 1,6-bisphosphate is also induced in the aging heart (Fig. 3).
CR Prevents Many Age-Related Alterations in Gene Expression in the Heart.
To determine the influences of CR effects on gene expression, 30-month-old mice on CR from 14 months of age were compared with 30-month-old control fed mice. CR prevented most aging-related gene expression changes in the main functional classes either completely or partially (Table 1). Specifically, CR prevented either completely or partially 134 of the 177 (75%) genes observed to be altered as a result of aging. For example, CR completely prevented the age-related increases in expression of several collagen family members (Table 1). CR also completely prevented the age-related down-regulation of PDK4 and the up-regulation of Phosphofructokinase, whereas the 5-fold up-regulation of Fructose 1,6-bisphosphatase 2 was only marginally prevented (22%).
CR Markedly Suppresses the Expression of Genes Encoding Structural Proteins.
In addition to preventing aging-related changes in gene expression, CR shifted transcriptional activity of genes that were not changed in expression in response to aging in the hearts of the control group. The largest transcriptional class affected by CR encodes genes involved in structural roles, including ECM proteins, and genes related to actin and tubulin structural modulation (see Tables 3 and 4). CR suppressed the expression of type I, V, and VI collagens. In addition, CR down-regulated genes related to posttranslational modification of collagen, such as Prolyl 4-hydroxylase alpha-1 subunit and Procollagen-proline 2-oxoglutarate 4-dioxygenase alpha II polypeptide. CR also down-regulated Laminin beta 2, a major component of the basement membrane, and two actin components, Actin alpha 1 and Actin gamma. In addition, CR down-regulated the expression of several actin-binding and actin cytoskeleton-modulating genes, including Tropomyosin 2, Plastin 2, Fascin homolog 1, Cofilin 1, and Gelsolin. Tropomyosin stabilizes the actin cytoskeleton, whereas plastins and fascins organize filamentous actins into bundles (45–47). Cofilin enhances actin filament turnover (48), and gelsolin regulates actin assembly (49, 50). These findings suggest major structural remodeling in response to CR.
CR Markedly Suppresses the Expression of Genes Involved in Immunity.
CR suppressed the expression of genes encoding several major histocompatibility complex (MHC) components (see Table 3); MHC class I H-2B1, H-2Q2, H-2Q4alpha, H-2Kf, H-2K2, and H-2D(b)alpha. Complement-related genes, which are involved in innate immunity, were also down-regulated in a concerted fashion, including the three genes encoding the complement component 1, q subunits C1qa, C1qb, and C1qc. C1qbp (Complement component 1 binding protein), an inhibitor of C1q, was up-regulated in expression by adult-onset CR. Previously, McGeer and colleagues (51) demonstrated that myocardial tissue locally expresses complement, and that this expression significantly increases in response to ischemia and reperfusion. Our results suggest that CR lowers the expression of immune-related genes, which in turn may be associated with a lower proinflammatory status in the CR heart.
CR Modulates the Expression of Genes Involved in DNA Repair and Apoptosis in the Heart.
CR down-regulated the expression of several DNA damage-inducible transcripts, including Chop-10, Gadd45a, and a DNA polymerase beta homolog, suggesting lowered endogenous DNA damage in the heart (see Table 3). In contrast, CR induced the expression of some constitutive DNA repair genes, including genes encoding Rad50, Rad21, and Xpc. CR reduced the expression of several apoptotic genes, including down-regulation of the proapoptotic factors Bax, Bad, DNA fragmentation factor, and Caspase 9 and 11. CR up-regulated the expression of the Mouse inhibitor of apoptosis protein-2, the apoptosis inhibitor Bcl-x, and two poorly characterized apoptosis-related genes, a Fas homolog and Programmed cell death 1. These observations suggest that CR modulates apoptosis in the heart, perhaps by enhancing DNA repair and reducing endogenous DNA damage.
Discussion
The cardiac myocyte is the most energetically demanding cell in the body, contracting repeatedly throughout the life span and requiring large amounts of ATP and rapid ATP turnover (9, 52). In adult hearts, fatty acids are the major energy source, whereas fetal hearts primarily use glucose (53). Age-associated reductions in lipid oxidation by heart mitochondria have been previously reported in rats (54, 55). Our results at the transcriptional level are in agreement with these observations, because we detected a marked down-regulation in transcripts involved in β-oxidation (Fig. 3). These observations suggest a shift with aging toward the use of glucose and pyruvate as energy sources, as reflected by transcriptional up-regulation of genes related to glycolysis in aged hearts (Fig. 3). Aging also increased the transcription of genes related to glucose uptake, such as Glut4 (1.4-fold; data not shown), and down-regulated the transcription of the gene encoding a glycolysis inhibitory enzyme, PDK4 that phosphorylates mitochondrial pyruvate dehydrogenase and inhibits its activity (56). Interestingly, these observations are in contrast with our previous observations in C57Bl6 mouse skeletal muscle, where aging down-regulated the expression of genes associated with glycolysis, such as Alpha enolase, Glucose 6-phosphate isomerase, and Glycerophosphate dehydrogenase (6). Clearly, aging in different tissues leads to unique transcriptional and metabolic alterations.
The postnatal activation of the mitochondrial energy production pathway involves the expression of nuclear genes encoding enzymes involved in fatty acid oxidation (FAO), and other proteins important in mitochondrial energy pathways (57). During the development of cardiac hypertrophy, the myocardial energy substrate preference shifts back toward the fetal pattern, with a corresponding reduction in the expression of FAO enzyme genes. Therefore, the transcriptional patterns associated with aging are consistent with an adult to fetal transcriptional shift typical of cardiac hypertrophy. In fact, when measuring heart weights before tissue processing for the microarray experiment, we observed an increase in average weights in the young and old groups from 214 mg to 278 mg, respectively. Although not statistically significant (P = 0.19), this age-related trend was not observed in the CR group (average weight 211 mg). Other transcriptional alterations observed in aged B6C3F1 mice consistent with a state of cardiac hypertrophy as determined by previous DNA microarray profiling studies (58, 59) include alterations in the expression of alpha actin isoforms, myosin heavy chain, and multiple collagen-encoding genes (Table 1, and see supporting information).
Age-related decreases in mRNA levels were also observed for several genes involved in protein synthesis. Translation initiation and elongation factors, including eIFs3y, eIF4ebp1, and Elongation factor 2 were reduced in expression. Aging was also associated with a concerted reduced mRNA levels for ribosomal components, including Ribosomal proteins L10a, L10, L19, L21, L44, S16, S11, S6, S5, S4x, S3a, and S3. These alterations displayed small fold changes between −1.3 and −1.1 (representing 10–30% decreases in mRNA levels), but were statistically significant (P < 0.05, and pp >60%). Possibly, these alterations reflect decreased protein synthesis as an adaptation to impaired ATP production. A decline in synthesis rate of protein in skeletal muscle has been observed from young to middle age in humans (60), and also in the hearts of aged rats (61). Our observations suggest that declining ribosomal activity may be a feature of aging in the heart.
Forty-four percent of the alterations in genes that were significantly decreased or increased in expression with aging by 50% or more were inhibited by at least 50% by adult-onset CR. In addition to preventing age-related changes in gene expression, CR also affected unique transcriptional categories that were not affected by aging. CR suppressed genes involved in antigen presentation, such as major histocompatibility complex (MHC) class I histocompatibility components. In addition, nonspecific lysosomal proteases, which produce peptide fragments for display by the MHC, were down-regulated by CR. Transcripts encoding complement C1q alpha, beta, and gamma chains were simultaneously down-regulated by CR, whereas the expression of C1qbp, which binds to the globular heads of C1q thus inhibiting C1 activation, was increased by CR. Based on these observations, it appears likely that CR suppresses the innate immune response. Because complement contributes to tissue damage induced by reperfusion (62), these findings suggest mechanisms involved in cardioprotection by CR.
CR also altered the expression of genes involved in apoptosis. Most of these alterations were consistent with a reduction in apoptosis-inducing signals in the heart, perhaps because of the fact that endogenous DNA damage, as determined by the concentration of 8-hydroxydeoxyguanosine, has been shown previously to be lower in multiple tissues of CR mice as compared with mice fed ad libitum diets (20). In both humans and animals, the aging process in the heart has been associated with a decrease in the total number of myocytes and reactive hypertrophy of the remaining cells (11, 63). Loss of myocytes in the aging heart may be due to apoptosis, which has been linked to cytochrome c release and oxidative stress (64). Based on the transcriptional profile of the hearts of CR mice, we suggest that CR protects myocytes from age-associated death, a conclusion that is consistent with the previous observation that CR prevents cardiomyopathy in rats (65, 66).
Surprisingly, we found no evidence that aging in the heart is associated with a transcriptional profile indicative of an oxidative stress response, as previously demonstrated for skeletal muscle (6) and brain (7) of C57Bl6 mice. As a whole, CR affects the expression of more genes in the heart than the aging process (21% vs. 10%). Possibly, alterations in the expression of genes involved in specific transcriptional classes, such as DNA repair, apoptosis, and immunity, mediate the effects of CR on the retardation of the aging process, whereas inhibition of most age-related alterations may simply reflect the fact that CR animals are physiologically younger. We also note that many of the alterations in gene expression observed with aging or CR may be associated with changes in cell populations in the heart. Importantly, when combined to our previous studies, our data suggest that each organ undergoes unique aging alterations at the transcriptional level. Therefore, to evaluate aging interventions by using DNA microarrays, it may be useful to identify transcriptional markers that represent various biochemical pathways altered with aging in specific tissues. The use of DNA microarrays representing the entire genome should allow for the rapid development of this resource.
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health Grants R01AG18922 and U24DK058776 and National Science Foundation Grant 0090286.
Abbreviations
CR, caloric restriction
pp, posterior probability
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Weindruch R. & Walford, R. L., (1988) The Retardation of Aging and Disease by Dietary Restriction (Charles C. Thomas, Springfield, IL).
- 2.Sohal R. S., Ku, H. H., Agarwal, S., Forster, M. J. & Lal, H. (1994) Mech. Ageing Dev. 74, 121-133. [DOI] [PubMed] [Google Scholar]
- 3.Koizumi A., Weindruch, R. & Walford, R. L. (1987) J. Nutr. 117, 361-367. [DOI] [PubMed] [Google Scholar]
- 4.Laganiere S. & Yu, B. P. (1987) Biochem. Biophys. Res. Commun. 145, 1185-1191. [DOI] [PubMed] [Google Scholar]
- 5.Lass A., Sohal, B. H., Weindruch, R., Forster, M. J. & Sohal, R. S. (1998) Free Radical Biol. Med. 25, 1089-1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee C. K., Klopp, R. G., Weindruch, R. & Prolla, T. A. (1999) Science 285, 1390-1393. [DOI] [PubMed] [Google Scholar]
- 7.Lee C. K., Weindruch, R. & Prolla, T. A. (2000) Nat. Genet. 25, 294-297. [DOI] [PubMed] [Google Scholar]
- 8.Severs N. J. (2000) BioEssays 22, 188-199. [DOI] [PubMed] [Google Scholar]
- 9.Jacobus W. E. (1985) Annu. Rev. Physiol. 47, 707-725. [DOI] [PubMed] [Google Scholar]
- 10.Anversa P., Palackal, T., Sonnenblick, E. H., Olivetti, G., Meggs, L. G. & Capasso, J. M. (1990) Circ. Res. 67, 871-885. [DOI] [PubMed] [Google Scholar]
- 11.Olivetti G., Melissari, M., Capasso, J. M. & Anversa, P. (1991) Circ. Res. 68, 1560-1568. [DOI] [PubMed] [Google Scholar]
- 12.Swynghedauw B., Besse, S., Assayag, P., Carre, F., Chevalier, B., Charlemagne, D., Delcayre, C., Hardouin, S., Heymes, C. & Moalic, J. M. (1995) Am. J. Cardiol. 76, 2D-7D. [DOI] [PubMed] [Google Scholar]
- 13.Cornwell G. G., Thomas, B. P. & Snyder, D. L. (1991) J. Gerontol. 46, B167-B170. [DOI] [PubMed] [Google Scholar]
- 14.Del Roso A., De Tata, V., Gori, Z. & Bergamini, E. (1991) Aging (Milano) 3, 19-23. [DOI] [PubMed] [Google Scholar]
- 15.Jiang M. T. & Narayanan, N. (1990) Mech. Ageing Dev. 54, 87-101. [DOI] [PubMed] [Google Scholar]
- 16.Xiao R. P., Spurgeon, H. A., O'Connor, F. & Lakatta, E. G. (1994) J. Clin. Invest. 94, 2051-2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Centers for Disease Control and Prevention (2002) Table LCWK1 (www.cdc.gov/nchs/data/nvsr/nvsr49/nvsr49_11.pdf).
- 18.Kemi M., Keenan, K. P., McCoy, C., Hoe, C. M., Soper, K. A., Ballam, G. C. & van Zwieten, M. J. (2000) Toxicol. Pathol. 28, 285-296. [DOI] [PubMed] [Google Scholar]
- 19.Taffet G. E., Pham, T. T. & Hartley, C. J. (1997) J. Gerontol. A Biol. Sci. Med. Sci. 52, B285-B290. [DOI] [PubMed] [Google Scholar]
- 20.Sohal R. S., Agarwal, S., Candas, M., Forster, M. J. & Lal, H. (1994) Mech. Ageing Dev. 76, 215-224. [DOI] [PubMed] [Google Scholar]
- 21.Leeuwenburgh C., Wagner, P., Holloszy, J. O., Sohal, R. S. & Heinecke, J. W. (1997) Arch. Biochem. Biophys. 346, 74-80. [DOI] [PubMed] [Google Scholar]
- 22.Melov S., Hinerfeld, D., Esposito, L. & Wallace, D. C. (1997) Nucleic Acids Res. 25, 974-982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pugh T. D., Klopp, R. G. & Weindruch, R. (1999) Neurobiol. Aging 20, 157-165. [DOI] [PubMed] [Google Scholar]
- 24.Nelson W. & Halberg, F. (1986) J. Nutr. 116, 2244-2253. [DOI] [PubMed] [Google Scholar]
- 25.Weindruch R., Kayo, T., Lee, C. K. & Prolla, T. A. (2002) Mech. Ageing Dev. 123, 177-193. [DOI] [PubMed] [Google Scholar]
- 26.Hall P. & Wilson, S. R. (1991) Biometrics 47, 757-762. [Google Scholar]
- 27.Allison D. B., Gadbury, G. L., Heo, M., Fernandez, J. R., Lee, C. K., Prolla, T. A. & Weindruch, R. (2002) Comput. Statist. Data Anal. 39, 1-20. [Google Scholar]
- 28.Kramer J., Quensel, C., Meding, J., Cardoso, M. C. & Leonhardt, H. (2001) J. Vasc. Res. 38, 120-132. [DOI] [PubMed] [Google Scholar]
- 29.Honda H., Tamura, T., Hatori, K. & Matsuno, K. (1995) Biochim. Biophys. Acta 1251, 43-47. [DOI] [PubMed] [Google Scholar]
- 30.Burgess M. L., McCrea, J. C. & Hedrick, H. L. (2001) Mech. Ageing Dev. 122, 1739-1756. [DOI] [PubMed] [Google Scholar]
- 31.Wanagat J., Wolff, M. R. & Aiken, J. M. (2002) J. Mol. Cell. Cardiol. 34, 17-28. [DOI] [PubMed] [Google Scholar]
- 32.Fu W. Y., Fu, A. K., Lok, K. C., Ip, F. C. & Ip, N. Y. (2002) NeuroReport 13, 243-247. [DOI] [PubMed] [Google Scholar]
- 33.Dhavan R. & Tsai, L. H. (2001) Nat. Rev. Mol. Cell. Biol. 2, 749-759. [DOI] [PubMed] [Google Scholar]
- 34.Spillantini M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R. & Goedert, M. (1997) Nature 388, 839-840. [DOI] [PubMed] [Google Scholar]
- 35.Wakabayashi K., Hayashi, S., Kakita, A., Yamada, M., Toyoshima, Y., Yoshimoto, M. & Takahashi, H. (1998) Acta Neuropathol. 96, 445-452. [DOI] [PubMed] [Google Scholar]
- 36.Iwanaga K., Wakabayashi, K., Yoshimoto, M., Tomita, I., Satoh, H., Takashima, H., Satoh, A., Seto, M., Tsujihata, M. & Takahashi, H. (1999) Neurology 52, 1269-1271. [DOI] [PubMed] [Google Scholar]
- 37.Cox K. B., Johnson, K. R. & Wood, P. A. (1998) Mamm. Genome 9, 608-610. [DOI] [PubMed] [Google Scholar]
- 38.Ramsay R. R., Gandour, R. D. & van der Leij, F. R. (2001) Biochim. Biophys. Acta 1546, 21-43. [DOI] [PubMed] [Google Scholar]
- 39.Schaffer J. E. & Lodish, H. F. (1994) Cell 79, 427-436. [DOI] [PubMed] [Google Scholar]
- 40.Dolinsky V. W., Sipione, S., Lehner, R. & Vance, D. E. (2001) Biochim. Biophys. Acta 1532, 162-172. [DOI] [PubMed] [Google Scholar]
- 41.Li Z., Sumida, M., Birchbauer, A., Schotz, M. C. & Reue, K. (1994) Genomics 24, 259-265. [DOI] [PubMed] [Google Scholar]
- 42.Paradies G., Ruggiero, F. M., Petrosillo, G., Gadaleta, M. N. & Quagliariello, E. (1995) Mech. Ageing Dev. 84, 103-112. [DOI] [PubMed] [Google Scholar]
- 43.McMillin J. B., Taffet, G. E., Taegtmeyer, H., Hudson, E. K. & Tate, C. A. (1993) Cardiovasc. Res. 27, 2222-2228. [DOI] [PubMed] [Google Scholar]
- 44.Buck M. J., Squire, T. L. & Andrews, M. T. (2002) Physiol. Genomics 8, 5-13. [DOI] [PubMed] [Google Scholar]
- 45.Otto J. J. (1994) Curr. Opin. Cell Biol. 6, 105-109. [DOI] [PubMed] [Google Scholar]
- 46.Edwards R. A., Herrera-Sosa, H., Otto, J. & Bryan, J. (1995) J. Biol. Chem. 270, 10764-10770. [DOI] [PubMed] [Google Scholar]
- 47.Arpin M., Friederich, E., Algrain, M., Vernel, F. & Louvard, D. (1994) J. Cell. Biol. 127, 1995-2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bamburg J. R. (1999) Annu. Rev. Cell. Dev. Biol. 15, 185-230. [DOI] [PubMed] [Google Scholar]
- 49.McGough A., Chiu, W. & Way, M. (1998) Biophys. J. 74, 764-772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kinosian H. J., Newman, J., Lincoln, B., Selden, L. A., Gershman, L. C. & Estes, J. E. (1998) Biophys. J. 75, 3101-3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yasojima K., Kilgore, K. S., Washington, R. A., Lucchesi, B. R. & McGeer, P. L. (1998) Circ. Res. 82, 1224-1230. [DOI] [PubMed] [Google Scholar]
- 52.Matthews P. M., Bland, J. L., Gadian, D. G. & Radda, G. K. (1981) Biochem. Biophys. Res. Commun. 103, 1052-1059. [DOI] [PubMed] [Google Scholar]
- 53.Neely J. R., Rovetto, M. J. & Oram, J. F. (1972) Prog. Cardiovasc. Dis. 15, 289-329. [DOI] [PubMed] [Google Scholar]
- 54.Hansford R. G. (1978) Biochem. J. 170, 285-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen J. C., Warshaw, J. B. & Sanadi, D. R. (1972) J. Cell. Physiol. 80, 141-148. [DOI] [PubMed] [Google Scholar]
- 56.Harris R. A., Hawes, J. W., Popov, K. M., Zhao, Y., Shimomura, Y., Sato, J., Jaskiewicz, J. & Hurley, T. D. (1997) Adv. Enzyme Regul. 37, 271-293. [DOI] [PubMed] [Google Scholar]
- 57.Lehman J. J. & Kelly, D. P. (2002) Clin. Exp. Pharmacol. Physiol. 29, 339-345. [DOI] [PubMed] [Google Scholar]
- 58.Shimkets R. A., Lowe, D. G., Tai, J. T., Sehl, P., Jin, H., Yang, R., Predki, P. F., Rothberg, B. E., Murtha, M. T., Roth, M. E., et al. (1999) Nat. Biotechnol. 17, 798-803. [DOI] [PubMed] [Google Scholar]
- 59.Friddle C. J., Koga, T., Rubin, E. M. & Bristow, J. (2000) Proc. Natl. Acad. Sci. USA 97, 6745-6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Balagopal P., Rooyackers, O. E., Adey, D. B., Ades, P. A. & Nair, K. S. (1997) Am. J. Physiol. 273, E790-E800. [DOI] [PubMed] [Google Scholar]
- 61.Goldspink D. F., Lewis, S. E. & Merry, B. J. (1986) Cardiovasc. Res. 20, 672-678. [DOI] [PubMed] [Google Scholar]
- 62.Elkayam U. (1994) Cardiol. Clin. 12, 73-85. [PubMed] [Google Scholar]
- 63.Olivetti G., Giordano, G., Corradi, D., Melissari, M., Lagrasta, C., Gambert, S. R. & Anversa, P. (1995) J. Am. Coll. Cardiol. 26, 1068-1079. [DOI] [PubMed] [Google Scholar]
- 64.Phaneuf S. & Leeuwenburgh, C. (2002) Am. J. Physiol. 282, R423-R430. [DOI] [PubMed] [Google Scholar]
- 65.Berg B. N. & Simms, H. S. (1960) J. Nutr. 71, 255. [PubMed] [Google Scholar]
- 66.Maeda H., Gleiser, C. A., Masoro, E. J., Murata, I., McMahan, C. A. & Yu, B. P. (1985) J. Gerontol. 40, 671-688. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.