

Abstract
The ability to make sensitive measurements of protein–protein interaction kinetics in single neurons is critical for understanding the molecular and cellular basis of neuronal function. We have developed a reporter technology based on the differential induction of Escherichia coli TEM-1 β-lactamase (Bla) enzymatic activity that can function as a sensor of the interaction state of two target proteins within single neurons in vivo. To modulate Bla enzymatic activity, we first split the enzyme into two separate, complementary protein fragments that we identified by using a functional screening approach based on circular permutation of the Bla enzyme. The split enzyme was then brought together by the phosphorylation-dependent association of the kinase inducible domain of the cAMP response element binding protein (CREB) and the KIX domain of the CREB binding protein. Using an intracellular substrate whose fluorescence spectrum changes after hydrolysis by Bla, we performed time-lapse ratiometric imaging measurements of Bla enzymatic induction after association of the CREB and CREB binding protein interaction domains. This approach permits direct imaging of protein–protein interactions in single cells with high signal discrimination.
New technologies that permit dynamic protein–protein interactions to be monitored in single neurons over time are likely to have a major impact on our ability to understand the molecular and cellular basis of nervous system development and function. Recently, several experimental approaches have been developed for detecting protein–protein interactions in single cells, including fluorescence resonance energy transfer (1–3), fluorescence correlation spectroscopy (4), bioluminescence resonance energy transfer (5), fluorescence lifetime imaging microscopy (6), and protein fragment complementation assays. Although each of these technologies has been used successfully to detect interactions between highly abundant proteins in vitro and in vivo, further improvements in the sensitivity of these approaches are required before dynamic interactions between low abundance proteins can be routinely studied inside single cells with standard light microscopy techniques.
Enzyme fragment complementation assays (ECAs) are a promising approach for addressing the technical challenge of monitoring protein–protein interactions with high sensitivity in single cells (7–9). In all ECAs, the biological function of a specific enzyme is disrupted by splitting the reporter protein into two distinct fragments. Functional activity is then reconstituted by bringing the two fragments back together in a controlled manner. This is accomplished by fusing the enzyme fragments to two heterologous protein domains to generate chimeric proteins that have the capacity to interact with one another. If the interaction of the two heterologous domains restores the activity of the enzyme by bringing the two enzyme fragments into close spatial proximity, then this restoration of enzymatic activity can be used to monitor the interaction of the two heterologous protein domains. A strength of the ECA approach is the capacity to amplify the signal associated with each protein–protein interaction event. Because the induction of enzymatic activity initiates the catalytic turnover of a substrate at a rate proportional to the enzymatic activity of the reporter enzyme, the ECA approach has the potential to provide significant signal amplification.
A limitation of existing ECAs for quantitative in vivo time-lapse imaging studies of protein–protein interactions has been the lack of suitable substrates that allow quantitative measurements to be performed under physiological conditions. Given this need for substrates, an exciting technological advance has been the development of an intracellular reporter substrate called CCF-2 whose fluorescence properties change after hydrolysis by the TEM-1 β-lactamase (Bla) enzyme from Escherichia coli (10). CCF-2 is available as an acetoxymethyl ester (CCF-2/AM) that is easily loaded into and retained by mammalian cells. The development of CCF-2/AM has made Bla an attractive target for use as an ECA that would permit real-time kinetics to be measured for protein–protein interactions in single mammalian cells. Two reports of ECAs based on TEM-1 Bla have been published recently that demonstrate the utility of Bla complementation for studying constitutive protein–protein interactions as well as the rapamycin-induced interaction of FKBP12 (FK506 binding protein 12) and FRP (FKBP-rapamycin binding domain of FKBP-rapamycin-associated protein) in vitro and in transfected cells (11, 12). In the present article, we report the use of an alternative Bla-based ECA that has allowed us to extend the Bla-based ECA approach to perform time-lapse imaging measurements of dynamic phosphorylation-dependent protein–protein interactions in single neurons. In addition, we have derived an analytic expression to quantitatively characterize changes in a protein–protein association state by measuring relative changes in Bla enzymatic activity as a function of time. These results suggest that the described Bla-based ECA is a useful tool for measuring dynamic phosphorylation-dependent protein–protein interactions at the single cell level.
Materials and Methods
DNA Constructs.
PCR amplification strategies, primer sequences, plasmid sequences, and plasmid maps are available on request for the following plasmid constructs: pBla C-T7 (WT Bla), pCP-7, pCP-8, pCP-9, pCP-11, pCP-12, pCP-13, pCP-14, pKIX-(Bla-N), p(Bla-C)-kinase inducible domain (KID), p(Bla-C)-KID-M1, pGWK-(Bla-C)-KID, pGWK-(Bla-C)-KID-M1, and pRSV CHO protein kinase A (PKA) C-alpha 1 (13). All expression constructs have C-terminal T7 epitope tags except for pKIX-(Bla-N) that has a N-terminal hemagglutinin epitope tag. The KIX and KID sequences encompass amino acids 574–686 from murine cAMP response element binding protein (CREB) binding protein (CBP) and 100–170 from rat α-CREB, respectively.
Cell Culture and Transfection.
293T cells, 293 cells, and cortical neurons were cultured and transfected by standard methods as described (14). For more details, see Supporting Materials and Methods, which is published as supporting information on the PNAS web site, www.pnas.org.
Western Blotting.
Immunoblots were performed by using standard protocols. The anti-T7 antibody is available from Novagen, and the anti-hemagglutinin antibody is available from Roche Molecular Biochemicals.
Nitrocefin Assay.
Bla and circularly permuted Bla constructs were expressed in 293T cells. After 24 h, the cells were washed twice by using 4°C PBS, and then lysed at 4°C by using 200 μl luciferase lysis buffer (Promega). Lysates were cleared by using centrifugation at 14,000 rpm for 5 min at 4°C. A 100 μM nitrocefin working solution was prepared at 4°C by diluting a 0.01 M nitrocefin solution (in 100% DMSO) 1:100 in PBS. Bla enzymatic activity assays were performed by combining 50 μl of cell lysate with 450 μl of 100 μM nitrocefin working solution and monitoring the 500-nm absorbance at 25°C every 2 s for a total of 400 s with a UV/vis spectrometer (Beckman DU-600).
Imaging Bla Enzymatic Activity with CCF-2.
Cells were loaded with CCF-2/AM (Vertex, Cambridge, MA) prepared in Tyrodes solution (140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 25 mM Hepes, and 10 mM glucose) according to the manufacturer's instructions. When stimulating 293 cells with forskolin (Sigma) and 8-(4-chlorophenylthio)cAMP (CPT-cAMP) (Sigma), these drugs were added to the CCF-2/AM loading solution at final concentrations of 10 μM and 300 μM, respectively. Cortical neurons were stimulated by perfusing with Tyrodes solution containing 10 μM forskolin and 300 μM CPT-cAMP ≈350–400 s after starting time-lapse imaging. Imaging was performed at 25°C on an inverted epifluorescence microscope (Nikon Eclipse) equipped with a xenon arc lamp, an emission filter wheel, and a cooled charge-coupled device camera. open lab software (Improvision, Lexington, MA) was used to collect either single or time-lapse images at 460 nm and 530 nm as noted in the text. Images were background-subtracted, and the ratio of the 460-nm/530-nm images was calculated pixel by pixel. open lab software or the National Institutes of Health image program (http://rsb.info.nih.gov/nih-image/) was used to quantify the average fluorescence emission ratio of single cells. CCF-2 ratio time courses in neurons were converted into a measure of effective Bla enzymatic activity, kBla(t), by using igor pro software. The measured values for FS460, FP460, FS530, and FP530 were 227, 3,296, 1,695, and 692, respectively. These represent relative CCF-2 molecular fluorescence values that scale equally according to the concentration of CCF-2 in a cell. A rolling two-point average of the CCF-2 R460/530 was used to reduce point-to-point variation in each kBla(t) plot.
Results
Bla Circular Permutation Screen for Candidate Complementation Domains.
To create an ECA based on TEM-1 Bla, the first step was to identify sites where we could disrupt the primary amino acid sequence of Bla to separate the enzyme into two complementary protein fragments (15). We engineered circularly permuted forms of Bla where the native N- and C-terminal ends of Bla were joined by an 8-aa linker (16, 17), and new N- and C-terminal ends were created by splitting the Bla protein at an internal site (18, 19) (Fig. 1A). Each circularly permuted Bla enzyme contained a pair of complementary Bla fragments joined at the native N- and C-terminal ends by a linker. We reasoned that if a given circularly permuted Bla protein was enzymatically active, then the two Bla fragments contained within that circularly permuted protein must be properly folded. Moreover, these two Bla protein fragments would be likely to fold correctly when expressed as independent proteins and might reconstitute Bla enzymatic activity when brought into close spatial proximity by the interaction of two heterologous proteins that were fused to the Bla fragments.
Fig 1.

Circular permutation strategy used to develop the KID/KIX-BEAM assay. (A) Circularly permuted Bla proteins were created by joining the native Bla N- and C-terminal ends (N and C) with an 8-aa linker (AAGSKNAA), and introducing new N- and C-terminal ends (N′ and C′) at sites within primary amino acid sequence. Removal of the linker yielded a pair of complementary N- and C-terminal Bla protein fragments Bla-N and Bla-C. The Bla-N fragment was fused to the C-terminal end of the KIX domain of CBP and the Bla-C fragment was fused to the N-terminal end of the KID of CREB by using a triple alanine linker (AAA). The KID and the KIX domain can interact after phosphorylation of the Ser-133 site (P) within the KID. The KIX-(Bla-N) and the (Bla-C)-KID proteins together comprised the KID/KIX-BEAM proteins. (B) The enzymatic activity of WT Bla as well as the CP-7, CP-8, CP-9, CP-11, CP-12, CP-13, and CP-14 circularly permuted enzymes was measured by monitoring the increase in nitrocefin absorbance at 500 nm as a function of time. The slope of the curve near time 0 for the nitrocefin absorbance at 500 nm versus time was used to estimate the relative Bla enzymatic activity as a percentage of WT Bla (Table 1).
The functional activity of each circularly permuted enzyme was assessed by transfecting an expression construct encoding a particular circularly permuted Bla enzyme into 293T cells. We then determined the Bla enzymatic activity level in lysates derived from these transfected cells by using the Bla substrate, nitrocefin (11, 20, 21). Nitrocefin undergoes a color change from yellow to red after hydrolysis by Bla, permitting Bla enzymatic activity to be measured by monitoring the nitrocefin absorbance at 500 nm as a function of time. Because to a first approximation nitrocefin hydrolysis by Bla follows first-order Michaelis–Menten kinetics, the initial slope of the 500-nm absorbance near time 0 provides an estimate of the maximal enzymatic rate for each circularly permuted enzyme when the active site is saturated with nitrocefin (22).
The enzymatic activities of the different circularly permuted Bla enzymes varied over a wide range (Fig. 1B). Lysates derived from transfected 293T cells expressing the three most active enzymes (CP-8, CP-13, and CP-14) displayed 15%, 4%, and 9%, respectively, of the enzymatic activity of WT Bla (Table 1). CP-7, CP-9, CP-11, and CP-12, however, hydrolyzed the nitrocefin substrate biphasically near time 0, suggesting a second-order, non-Michaelis–Menten reaction. For these cases, an upper bound for the relative rates was derived by taking the maximal slope of the 500-nm absorbance versus time near 400 s, yielding values of <3.5%, <1.2%, <0.1%, and <2.0%, respectively, of the enzymatic activity of WT Bla. We found by Western blotting that the 30-kDa WT Bla enzyme and the different circularly permuted Bla enzymes all were expressed at approximately equal levels, suggesting that the relative enzymatic activities observed in the nitrocefin assay were intrinsic enzymatic properties and not a consequence of different enzyme concentrations (data not shown).
Table 1.
Summary of relative enzymatic activities for circularly permuted Bla proteins
| Enzyme name | New terminal ends, C′/N′ | Relative rate, % of WT |
|---|---|---|
| WT Bla | — | 100 |
| CP-7 | 254/255 | <3.5 |
| CP-8 | 198/199 | 16 |
| CP-9 | 187/188 | <1.2 |
| CP-11 | 215/216 | <0.1 |
| CP-12 | 217/218 | <2.0 |
| CP-13 | 268/269 | 4.4 |
| CP-14 | 269/270 | 9.0 |
N′ and C′ denote the new N-terminal and C-terminal ends, respectively, of each circularly permuted enzyme and are numbered according to the accepted convention for class A Bla proteins (15).
Development of an Inducible ECA Based on TEM-1 Bla.
We next determined whether removal of the linker joining the native N- and C-terminal ends in each of the different circularly permuted enzymes would yield a pair of Bla fragments that could function as an ECA. For this purpose, each candidate protein fragment pair was fused to the KID of the transcription factor CREB (23, 24) and the KIX domain of the transcriptional coactivator CBP (25). We used the KID and KIX domain to test our Bla-based ECA because these domains have been both well studied in neurons and shown to interact with one another specifically in a phosphorylation-dependent manner (26–28). Previous studies have shown that the exposure of neurons to extracellular stimuli that elevate the level of intracellular cAMP induce CREB phosphorylation at Ser-133 within the KID (29, 30). The phosphorylation of CREB at Ser-133 leads to the interaction of the KID of CREB with the transcriptional cofactor CBP via the KIX domain. The binding of CBP to Ser-133-phosphorylated CREB serves to activate transcription of target genes that are important for neuronal survival and synaptic plasticity.
Although we tested several of the candidate Bla fragment pairs for their ability to function in the context of an ECA, in this study we focus specifically on a Bla fragment pair derived from the CP-13 circularly permuted enzyme. CP-13 was selected because the two complementary protein fragments derived from this protein did not interact constitutively, and because this enzyme exhibited relatively low enzymatic activity levels that allowed us to monitor Bla activity for prolonged periods of time in single cells without completely hydrolyzing the CCF-2 substrate. Removal of the linker from CP-13 yields two Bla fragments. The N-terminal Bla fragment comprising the amino acids His-26 to Ser-268 (Bla-N) was fused to the C-terminal end of the KIX domain and was termed the KIX-(Bla-N) protein. The C-terminal Bla fragment comprising the amino acids Gln-269 to Trp-290 (Bla-C) was fused to the N-terminal end of the KID and was termed the (Bla-C)-KID protein. As we demonstrate in the experiments described below, the (Bla-C)-KID and KIX-(Bla-N) proteins, when coexpressed in cortical neurons under conditions where the KID is phosphorylated at Ser-133, were able to interact in a manner that led to the reconstitution of Bla enzymatic activity. We have termed the heterologous Bla reporter ECA composed of the (Bla-C)-KID and KIX-(Bla-N) proteins the KID/KIX Bla enzymatic activity modulator, or KID/KIX-BEAM.
KID/KIX-BEAM ECA Characterization.
We tested the interaction of the KIX/KID-BEAM proteins in 293T cells where there is no endogenous Bla expression. To measure intracellular Bla enzymatic activity levels, we loaded transfected 293T cells with CCF-2/AM and captured digital images of the fluorescence emission at 460 nm and 530 nm. In the absence of intracellular Bla activity, strong CCF-2 emission at 530 nm is observed after excitation of CCF-2 with 409-nm light. In contrast, intracellular Bla enzymatic activity causes CCF-2 to become hydrolyzed, resulting in a shift of the CCF-2 fluorescence emission maximum from 530 nm to 447 nm. The change in the CCF-2 emission spectrum after hydrolysis by Bla permits relative Bla enzymatic activity within each cell to be quantitatively measured by calculating the ratio of the cellular fluorescence at 460 nm (Bla activity present) divided by cellular fluorescence at 530 nm (Bla activity absent).
We initially tested the interaction of the KIX/KID-BEAM proteins in 293T cells where the KID is constitutively phosphorylated because of the presence of constitutively active CREB kinases. We expected that the KID/KIX-BEAM proteins would interact constitutively in 293T cells because the Ser-133 phosphorylated form of the KID interacts constitutively with the KIX domain. Therefore, if the two Bla protein fragments could effectively complement each other, coexpression of the (Bla-C)-KID and KIX-(Bla-N) proteins in 293T cells should result in a significant increase in Bla enzymatic activity compared with the activity detected in cells expressing either of these two proteins alone.
293T cells expressing the KIX-(Bla-N) protein alone exhibited low levels of Bla enzymatic activity relative to cells expressing WT Bla (Fig. 2 A and B). This finding implied that the Bla-N fragment retained some enzymatic function, albeit significantly less than WT Bla. 293T cells expressing the (Bla-C)-KID protein alone were completely inactive, consistent with the Bla-C protein fragment lacking the Bla enzyme active site. Cells expressing the KID/KIX-BEAM proteins exhibited a dramatic increase in intracellular Bla activity relative to both the untransfected and cells expressing the KIX-(Bla-N) protein alone. This increase in intracellular Bla enzymatic activity observed in cells expressing the KID/KIX-BEAM proteins was a consequence of the interaction of the KIX domain with the Ser-133 phosphorylated form of the KID because the mutation of Ser-133 to an alanine within the (Bla-C)-KID protein (KID/KIX-BEAM-M1) abolished the increase in Bla enzymatic activity while not affecting the level of (Bla-C)-KID expression (data not shown).
Fig 2.

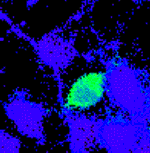
293T and 293 cells expressing the KID/KIX-BEAM proteins show increased Bla enzymatic activity under conditions where the KID Ser-133 site is phosphorylated. (A) Ratiometric images of the CCF-2 fluorescence emission at 460 nm divided by the CCF-2 fluorescence emission at 530 nm (R460/530) were collected for 293T cells expressing no exogenous protein (untransfected), the KIX-(Bla-N) protein alone, the KID/KIX-BEAM proteins, the M1-KID/KIX-BEAM proteins where the KID Ser-133 site is replaced by alanine, or WT Bla. The pseudocolor images were collected after 1 h of CCF-2 loading and are shaded such that the degree of CCF-2 hydrolysis increases from blue to red (NT = untransfected; BEAM = KID/KIX-BEAM; M1-BEAM = M1-KID/KIX-BEAM; WT = WT Bla). (B) The CCF-2 R460/530 was measured for single cells in four fields (n > 1,000) for each of the conditions in A. Each bar of a histogram shows the relative percentage of cells that have a particular range of CCF-2 R460/530 values. (C) Histograms showing the percentage of 293 cells within a range of CCF-2 R460/530 values. Cells expressing either the KID/KIX-BEAM or the M1-KID/KIX-BEAM proteins were untreated (KID/KIX-BEAM; n = 123), cotransfected with constitutively active PKA plasmid (KID/KIX-BEAM + PKA; n = 332), or treated with 10 μM forskolin plus 300 μM CPT-cAMP (F/C) for 1 h (KID/KIX-BEAM + F/C, n = 254; and M1-KID/KIX-BEAM + F/C, n = 299). For cells expressing the KID/KIX-BEAM proteins, either coexpression of constitutively active PKA or treatment with 10 μM forskolin plus 300 μM CPT-cAMP caused a marked increase in the number of cells exhibiting higher levels of Bla enzymatic activity. Cells expressing the M1-KID/KIX-BEAM proteins where the KID Ser-133 is replaced by alanine were indistinguishable after stimulation with 10 μM forskolin and 300 μM CPT-cAMP (M1-KID/KIX-BEAM + F/C) from untreated cells expressing the KID/KIX-BEAM proteins (KID/KIX-BEAM). (D) The percentage of cells exhibiting Bla enzymatic activity for each of the conditions presented in C. Cells were judged to be enzymatically active if the average cellular R460/530 was >0.4, which corresponded to the average R460/530 observed in untransfected cells.
KID/KIX-BEAM Phosphorylation-Dependent Induction via PKA Activation.
We next asked whether the KID/KIX-BEAM proteins could be used to monitor the effects of an applied extracellular stimuli on the phosphorylation-dependent interaction of the KID and the KIX domain in single cells. Quiescent 293 cells, in contrast to 293T cells, exhibit a low level of constitutive CREB Ser-133 phosphorylation. However, the phosphorylation of CREB at Ser-133 is markedly increased on addition of extracellular agonists (forskolin and CPT-cAMP) that activate intracellular PKA. 293 cells were transfected with plasmids encoding the DsRed2 fluorescent protein as a transfection marker and either the KID/KIX-BEAM or M1-KID/KIX-BEAM proteins. Exposure of 293 cells expressing the KID/KIX-BEAM proteins to the PKA agonists forskolin and CPT-cAMP, or coexpression of the KID/KIX-BEAM proteins with constitutively active PKA, led to a significant increase in Bla enzymatic activity that was blocked by mutating Ser-133 to alanine in the (Bla-C)-KID protein (M1-KID/KIX-BEAM) (Fig. 2 C and D). This finding indicates that the KID/KIX- BEAM proteins can be used to monitor the activation of endogenous PKA at the single cell level. Moreover, these results suggest that the Bla-N and Bla-C proteins may be useful as a general in vivo reporter of the interaction between two proteins whose interaction state is regulated in a kinase-dependent manner.
Imaging KID/KIX-BEAM Interaction Dynamics in Primary Cortical Neuronal Cultures.
We next sought to determine whether the KID/KIX-BEAM proteins could be used as a intracellular sensor of the KID and the KIX domain interaction in cortical neurons. We cotransfected primary cortical neurons with the DsRed2 fluorescent protein along with either the KID/KIX-BEAM or M1-KID/KIX-BEAM proteins. We identified transfected neurons by DsRed2 fluorescence, collected time-lapse CCF-2 fluorescence images at 460 and 530 nm at ≈40-s intervals, and then converted these two emission wavelength images into CCF-2 460-nm/530-nm ratio images at each point in time. Neurons were treated with forskolin and CPT-cAMP 350–400 s after the start of the experiment to determine whether an increase in Bla enzymatic activity could be observed that was correlated with the induced association of the KID and KIX domain.
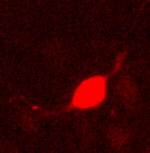
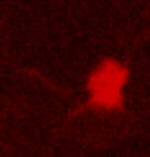
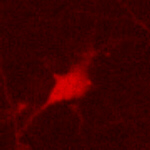
PKA-responsive cortical neurons expressing the KID/KIX-BEAM proteins (Fig. 3A) contained little or no hydrolyzed CCF-2 substrate before stimulation with forskolin and CPT-cAMP. However, the rate of CCF-2 hydrolysis increased dramatically in cortical neurons expressing the KID/KIX-BEAM proteins immediately after stimulation (Fig. 3B, see also Movies 1–4, which are published as supporting information on the PNAS web site). The enhanced rate of CCF-2 hydrolysis after PKA activation was sustained for the duration of the time period during which the PKA agonists were present. In contrast, none of the neurons expressing the M1-KID/KIX-BEAM proteins that contained the Ser-133–Ala mutant form of the KID-(Bla-C) protein exhibited any increase in Bla enzymatic activity after stimulation with PKA agonists (data not shown). This finding supports the conclusion that the increased enzymatic activity in neurons expressing the KID/KIX-BEAM proteins was caused by the PKA-mediated phosphorylation of Ser-133 within the KID.
Fig 3.

Time-lapse measurements of induced Bla enzymatic activity in single primary cortical neurons. (A) A neuron expressing the KID/KIX-BEAM proteins was identified by detecting fluorescence emission characteristic of the coexpressed DsRed2 red fluorescent protein. (B) Pseudocolor images of the CCF-2 R460/530 captured during the time-lapse imaging experiment for the KID/KIX-BEAM-expressing neuron identified in A. The neuron was treated with 10 μM forskolin (fsk) plus 300 μM CPT-cAMP 360 s after the start of the experiment. Bla enzymatic activity increased significantly after treatment with PKA agonists (t = 880 s) and continued to increase over the course of the experiment (t = 1,500 s). (C) CCF-2 R460/530 plots as a function of time for three neurons expressing the KID/KIX-BEAM proteins. A sustained increase in the slope, dR(t)460/530/dt, was observed in the presence of PKA agonists for the period of time indicated by the black bar above the plots. (D) Plots of relative Bla enzymatic activity, kBla(t), for the three neurons whose R460/530 plots as a function of time are shown in C. The magnitude of the relative increase in Bla enzymatic activity for these three neurons after stimulation with PKA agonists was 3.25 ± 0.16-, 3.45 ± 0.06-, and 4.49 ± 0.12-fold, respectively. (E) Average relative increase in Bla enzymatic activity for 49 neurons expressing the KID/KIX-BEAM proteins. An induced 2.71 ± 0.08-fold change was observed after treatment with forskolin and CPT-cAMP. (F) The average of 53 individual kbla(t) plots for neurons expressing the M1-KID/KIX-BEAM proteins showed no change in relative enzymatic rate after treatment with forskolin and CPT-cAMP.
Quantification of Real-Time Relative Bla Enzymatic Activity Induction Levels.
We next quantified the time course with which Bla enzymatic activity increased after PKA activation in individual neurons by analyzing the time-dependent change in the ratio of the CCF-2 fluorescence emission at 460 nm and 530 nm (R460/530). The R460/530 increases as Bla hydrolyzes CCF-2. Therefore, the rate of change of the R460/530 as a function of time is proportional to the Bla enzymatic activity in a neuron expressing the KID/KIX-BEAM proteins. In most neurons expressing the KID/KIX-BEAM proteins, the slope of the R460/530 versus time graph increased sharply immediately after the application of PKA agonists (Fig. 3C). In contrast, we observed no difference in the slope of the R460/530 versus time graph in neurons expressing the M1-KID/KIX-BEAM proteins (data not shown). Thus the level of Bla enzymatic activity in neurons expressing the KID/KIX-BEAM proteins increased significantly after phosphorylation of the (Bla-C)-KID protein by PKA in a manner that reflected the phosphorylation state of the KID Ser-133 site.
We next derived an equation that permitted us to quantify the effective increase in Bla enzymatic activity as a function of time. The rate of Bla enzymatic activity is proportional to the derivative of the R460/530 with respect to time (dR460/530/dt) multiplied by a scaling factor that accounts for the gradual depletion of CCF-2 during the course of an experiment. A scaling factor is necessary because the rate of CCF-2 hydrolysis depends not only on the enzymatic activity of Bla, but also on the concentration of unhydrolyzed CCF-2. The effective rate of CCF-2 hydrolysis, kBla(t), within a single neuron is given by the following equation (see the Appendix, which is published as supporting information on the PNAS web site, for the full derivation):
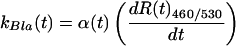 |
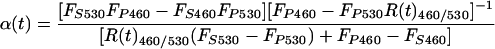 |
where FS460 and FP460 are the 460-nm fluorescence of the intact CCF-2 substrate (S) and hydrolysis products (P), respectively, and FS530 and FP530 are the 530-nm fluorescence of the intact and hydrolyzed forms of the CCF-2 substrate, respectively. Because FS460, FP460, FS530, and FP530 are intrinsic properties of CCF-2 and are not significantly altered by the relative concentration of CCF-2 in a cell, Eqs. 1 and 2 provide a measure of the relative Bla activity in a neuron that is independent of the intracellular CCF-2 concentration. Our analysis, however, did not compensate for other experimental variables such as the unique bleaching rates of the respective fluorochromes during the course of an experimental time course.
We calculated the relative change in the effective level of Bla enzymatic activity in neurons expressing either the KID/KIX-BEAM or M1-KID/KIX-BEAM proteins that were stimulated with PKA activators. Because the time-dependent scaling factor α(t) provided only a small correction to kBla(t) over the measured range of R460/530 values, the derived plots of kBla(t) exhibited much of the same functional character as plots of dR460/530/dt versus time. Neurons expressing the KID/KIX-BEAM proteins displayed up to a 4.5-fold increase in Bla enzymatic activity after stimulation with PKA agonists (Fig. 3D). The induction of Bla activity was apparent within 40 s after stimulation and peaked after approximately 6 min in accordance with previously reported time courses of endogenous CREB Ser-133 phosphorylation (31). The average response of 49 neurons expressing the KID/KIX-BEAM proteins was ≈3-fold (Fig. 3E). In contrast, none of the 53 neurons expressing the M1-KID/KIX-BEAM proteins showed any induction of Bla enzymatic activity (Fig. 3F). Taken together, these results indicate that the KID/KIX-BEAM assay can serve as an effective dynamic reporter of phosphorylation-dependent protein–protein interactions within intact cortical neurons.
Discussion
The KID/KIX-BEAM assay demonstrates that an ECA can be used to perform quantitative time-lapse imaging of the dynamic association of two proteins in single cells. By coupling the complementary Bla-C and Bla-N fragments to the KID of CREB and the KIX domain of CBP, respectively, we have shown that the KID/KIX-BEAM technique can serve as an intracellular sensor of the phosphorylation-dependent KID/KIX interaction. In addition, the signal amplification inherent to the KID/KIX-BEAM assay allows measurements to be performed by using a standard epifluorescence microscope equipped to capture digital images and does not require a significant investment in specialized equipment. Finally, we have derived an analytic expression that permitted relative changes in intracellular Bla enzymatic activity as a function of time to be directly correlated with either association or dissociation of the KID/KIX proteins after phosphorylation of the KID Ser-133 site.
The development of the KID/KIX-BEAM technology was assisted by using Bla circular permutation to identify specific sites within Bla where the protein could split to create two complementary protein fragments. The degree of enzymatic activity for a particular Bla circularly permuted enzyme correlated well with the enzyme's utility in the context of an ECA. As shown in this study, the CP-13 circularly permuted enzyme retained a significant fraction of the enzymatic activity exhibited by WT Bla and suggested that the protein fragments that arose from splitting the native Bla enzyme between amino acids Ser-268 and Gln-269 might function in an ECA. We have proceeded to demonstrate in this study that the Bla-N and Bla-C protein fragments from CP-13 when conjugated to the KIX domain and the KID, respectively can serve as a real-time phosphorylation-dependent reporter of the KID/KIX interaction in single neurons. Although the Bla protein fragments derived from the most enzymatically active circularly permuted enzyme, CP-8, have been reported to work well in a Bla-based ECA (11, 12), we judged that the diminished capacity for functional complementation exhibited in the CP-13 enzyme was better suited for developing an ECA for performing time-lapse imaging experiments over the longest possible period.
A unique combination of characteristics renders the KID/KIX-BEAM assay particularly well suited for in vivo imaging of protein–protein interactions in neurons. In developing an ECA for use in real-time imaging, a subtle balance between several, often contradictory, parameters needs to be achieved. Perhaps most critical is the requirement to balance enzyme efficiency versus the ability to monitor the protein–protein interaction under investigation for the longest period. In the case of the KID/KIX-BEAM ECA, although robust activation of the Bla reporter enzyme was desirable, the intracellular Bla enzymatic activity after association of the KID and KIX domain needed to be kept below a certain level to prevent the CCF-2 substrate from being completely hydrolyzed within a short period. Higher Bla enzymatic activity would improve the experimental signal-to-noise ratio of the assay, but would likely eliminate the capacity to measure dynamic changes in the KID/KIX association state for extended periods of time.
Although the KID/KIX-BEAM ECA has higher sensitivity than other methods for detecting protein–protein interaction dynamics in vivo, this technology is limited in its ability to detect the subcellular localization of the respective interacting proteins. The CCF-2 substrate and its hydrolysis products are freely diffusible, thereby limiting the capacity to monitor the accumulation of the CCF-2 hydrolysis products within a fixed region of a cell over time. However, lower-resolution questions such as those addressing the dynamic association/dissociation of two proteins localized to either the cytoplasmic or nuclear compartments should be possible. Moreover, by detecting and quantifying induced intracellular gradients of CCF-2 hydrolysis with time-lapse fluorescence confocal microscopy, it may be possible to localize a protein–protein interaction event to highly restricted regions of subcellular space. Even higher spatial resolution could be achieved by using either a Bla-based ECA or a variant of the CCF-2 substrate that could be targeted to specific subcellular regions.
In conclusion, the unprecedented signal amplification of the KID/KIX-BEAM technology represents a breakthrough in our ability to monitor protein–protein interaction dynamics at the single cell level with high temporal resolution. This general approach will be useful for addressing questions at the interface of molecular, cellular, and systems biology and represents a significant addition to existing methodologies for investigating protein–protein interactions in intact single cells.
Supplementary Material
Acknowledgments
We thank Jon M. Kornhauser and Adam J. Shaywitz for helpful discussions, Chia Haddad for technical assistance, and members of the Greenberg laboratory for critical reading of this manuscript. This work was supported by National Institutes of Health Mental Retardation Research Center Grant P30-HD18655, National Institutes of Health Grant NS28829 (to M.E.G.), and a McKnight Endowment for Neuroscience award (to R.E.D. and M.E.G.). M.E.G. acknowledges the generous contribution of the F. M. Kirby Foundation to the Division of Neuroscience at Children's Hospital. J.M.S. was supported by American Cancer Society Postdoctoral Fellowship Grant PF-01-009-01-DDC, and R.E.D was supported by a Burroughs Wellcome Fund Career Award in Biomedical Science.
Abbreviations
ECA, enzyme fragment complementation assay
Bla, β-lactamase
KID, kinase inducible domain
PKA, protein kinase A
CREB, cAMP response element binding protein
CBP, CREB binding protein
CPT-cAMP, 8-(4-chlorophenylthio)cAMP
BEAM, Bla enzymatic activity modulator
AM, acetoxymethyl ester
References
- 1.Mochizuki N., Yamashita, S., Kurokawa, K., Ohba, Y., Nagai, T., Miyawaki, A. & Matsuda, M. (2001) Nature 411, 1065-1068. [DOI] [PubMed] [Google Scholar]
- 2.Mayr B. M., Canettieri, G. & Montminy, M. R. (2001) Proc. Natl. Acad. Sci. USA 98, 10936-10941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel R. M., Chan, F. K., Zacharias, D. A., Swofford, R., Holmes, K. L., Tsien, R. Y. & Lenardo, M. J., (2000) Sci. STKE, http://stke.sciencemag.org/cgi/content/full/sigtrans;2000/38/pl1. [DOI] [PubMed]
- 4.Hess S. T., Huang, S., Heikal, A. A. & Webb, W. W. (2002) Biochemistry 41, 697-705. [DOI] [PubMed] [Google Scholar]
- 5.Xu Y., Piston, D. W. & Johnson, C. H. (1999) Proc. Natl. Acad. Sci. USA 96, 151-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harpur A. G., Wouters, F. S. & Bastiaens, P. I. (2001) Nat. Biotechnol. 19, 167-169. [DOI] [PubMed] [Google Scholar]
- 7.Michnick S. W., Remy, I., Campbell-Valois, F. X., Vallee-Belisle, A. & Pelletier, J. N. (2000) Methods Enzymol. 328, 208-230. [DOI] [PubMed] [Google Scholar]
- 8.Ozawa T. & Umezawa, Y. (2001) Curr. Opin. Chem. Biol. 5, 578-583. [DOI] [PubMed] [Google Scholar]
- 9.Rossi F. M., Blakely, B. T. & Blau, H. M. (2000) Trends Cell Biol. 10, 119-122. [DOI] [PubMed] [Google Scholar]
- 10.Zlokarnik G., Negulescu, P. A., Knapp, T. E., Mere, L., Burres, N., Feng, L., Whitney, M., Roemer, K. & Tsien, R. Y. (1998) Science 279, 84-88. [DOI] [PubMed] [Google Scholar]
- 11.Galarneau A., Primeau, M., Trudeau, L. E. & Michnick, S. W. (2002) Nat. Biotechnol. 20, 619-622. [DOI] [PubMed] [Google Scholar]
- 12.Wehrman T., Kleaveland, B., Her, J. H., Balint, R. F. & Blau, H. M. (2002) Proc. Natl. Acad. Sci. USA 99, 3469-3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maurer R. A. (1989) J. Biol. Chem. 264, 6870-6873. [PubMed] [Google Scholar]
- 14.Xia Z., Dudek, H., Miranti, C. K. & Greenberg, M. E. (1996) J. Neurosci. 16, 5425-5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ambler R. P., Coulson, A. F., Frere, J. M., Ghuysen, J. M., Joris, B., Forsman, M., Levesque, R. C., Tiraby, G. & Waley, S. G. (1991) Biochem. J. 276, 269-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jelsch C., Mourey, L., Masson, J. M. & Samama, J. P. (1993) Proteins 16, 364-383. [DOI] [PubMed] [Google Scholar]
- 17.Pieper U., Hayakawa, K., Li, Z. & Herzberg, O. (1997) Biochemistry 36, 8767-8774. [DOI] [PubMed] [Google Scholar]
- 18.Hennecke J., Sebbel, P. & Glockshuber, R. (1999) J. Mol. Biol. 286, 1197-1215. [DOI] [PubMed] [Google Scholar]
- 19.Topell S., Hennecke, J. & Glockshuber, R. (1999) FEBS Lett. 457, 283-289. [DOI] [PubMed] [Google Scholar]
- 20.O'Callaghan C. H., Morris, A., Kirby, S. M. & Shingler, A. H. (1972) Antimicrob. Agents Chemother. 1, 283-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shannon K. & Phillips, I. (1980) J. Antimicrob. Chemother. 6, 617-621. [DOI] [PubMed] [Google Scholar]
- 22.Stryer L., (1995) Biochemistry (Freeman, New York).
- 23.Brindle P., Linke, S. & Montminy, M. (1993) Nature 364, 821-824. [DOI] [PubMed] [Google Scholar]
- 24.Shaywitz A. J. & Greenberg, M. E. (1999) Annu. Rev. Biochem. 68, 821-861. [DOI] [PubMed] [Google Scholar]
- 25.Parker D., Ferreri, K., Nakajima, T., LaMorte, V. J., Evans, R., Koerber, S. C., Hoeger, C. & Montminy, M. R. (1996) Mol. Cell. Biol. 16, 694-703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwok R. P., Lundblad, J. R., Chrivia, J. C., Richards, J. P., Bachinger, H. P., Brennan, R. G., Roberts, S. G., Green, M. R. & Goodman, R. H. (1994) Nature 370, 223-226. [DOI] [PubMed] [Google Scholar]
- 27.Chrivia J. C., Kwok, R. P., Lamb, N., Hagiwara, M., Montminy, M. R. & Goodman, R. H. (1993) Nature 365, 855-859. [DOI] [PubMed] [Google Scholar]
- 28.Radhakrishnan I., Perez-Alvarado, G. C., Parker, D., Dyson, H. J., Montminy, M. R. & Wright, P. E. (1997) Cell 91, 741-752. [DOI] [PubMed] [Google Scholar]
- 29.Sheng M., Thompson, M. A. & Greenberg, M. E. (1991) Science 252, 1427-1430. [DOI] [PubMed] [Google Scholar]
- 30.Ginty D. D., Bonni, A. & Greenberg, M. E. (1994) Cell 77, 713-725. [DOI] [PubMed] [Google Scholar]
- 31.Bonni A., Ginty, D. D., Dudek, H. & Greenberg, M. E. (1995) Mol. Cell. Neurosci. 6, 168-183. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}