

Abstract
The IL-18 binding protein (IL-18BP) is a circulating inhibitor of the proinflammatory cytokine IL-18. It is constitutively expressed in mononuclear cells, and elevated expression is induced by IFN-γ. In this study, we characterized the IL-18BP promoter. We first showed that induction is at the transcriptional level and requires de novo protein synthesis. The IL-18BP promoter resides within 1.6 kb DNA upstream of the first exon and includes at least six regulatory elements. We identified in the basal promoter a gamma-activated sequence (GAS) proximal to the transcription start site (base 1), followed by an IFN regulatory factor 1 response element (IRF-E) and two CCAAT/enhancer binding protein β (C/EBPβ) sites, all of which are essential for basal promoter activity. Furthermore, GAS and IRF-E were essential for IFN-γ-induced transcription. Indeed, sera of IRF-1-deficient mice lacked basal and IFN-γ-induced IL-18BP. We found that after induction of IRF-1 by IFN-γ, it formed a complex with C/EBPβ, which bound to the IRF-E and GAS-containing proximal DNA. In contrast, the IFN-γ-induced signal transducer and activator of transcription 1 dimer did not associate with this GAS. In addition, we identified a silencer element and a distal enhancer at bases −1081 to −1272, which was also physically associated with IRF-1. The IRF-1–C/EBPβ complex described here probably plays a fundamental role in regulating additional IFN-γ-responsive genes.
Interleukin18 (IL-18) is a cytokine that initiates and promotes host defense and inflammation after infection or injury. IL-18 is a member of the IL-1 superfamily, having a distinct receptor, shared only by the recently identified IL-1 homologue IL1H4 (1). The hallmark of IL-18 activity is the activation and proliferation of T helper 1 and natural killer cells, manifested by induction of T helper 1 cytokines, e.g., IFN-γ. However, it was recently shown that IL-18 activity is mediated not only through IFN-γ, as IL-18 was involved in protection of IFN-γ-deficient mice from listeria infection (2).
IL-18 binding protein (IL-18BP) (3) belongs to a growing family of circulating binding proteins, including osteoprotegerin (4) and cytokine-like factor 1 (5), which are not variants of their cell-bound receptors. Rather, IL-18BP is a secreted protein consisting of a single Ig-like domain that bears little homology to either chain of the IL-18 receptor complex. IL-18BP binds IL-18 with high affinity (0.4 nM) (6) and neutralizes its biological activities in vitro and in vivo (3). Viral homologues of IL-18BP are encoded by most members of the Poxvirus family and were shown to bind and to neutralize human IL-18 (3, 7).
In humans, IL-18BP is constitutively expressed in the spleen and circulates at plasma concentrations of 2.5 ng/ml (8). Serum IL-18BP is significantly elevated during sepsis, indicating its role in regulating immune responses in vivo (8). Indeed, IL-18BP is induced by IFN-γ in various cells, suggesting that it serves as a negative feedback inhibitor of the IL-18-mediated immune response (9, 10). Here, we characterized the IL-18BP promoter and its activation by two IFN-γ-induced transcription factors: IFN regulatory factor 1 (IRF-1) and CCAAT/enhancer binding protein β (C/EBPβ). We show a physical association between these two transcription factors.
Materials and Methods
Cells and Reagents.
Human epithelial WISH cells, HaCaT keratinocytes, U-937 histiocytic lymphoma cells, and Hep G2 hepatocellular carcinoma cells were from the American Type Culture Collection. Cells were grown in DMEM supplemented with 10% FBS in humidified 5% CO2 at 37°C. Peripheral blood mononuclear cells were obtained from healthy subjects after their informed consent. Rabbit polyclonal antibodies to the various transcription factors were from Santa Cruz Biotechnology. Recombinant human cytokines were purchased from Cytolab (Rehovot, Israel). FuGene 6 transfection reagent was from Roche Molecular Biochemicals. All other reagents were purchased from Sigma or as indicated.
ELISA for IL-18BP.
Human IL-18BP was assayed by a double antibody ELISA as described (8). Mouse IL-18BP was measured by a double antibody ELISA using antigen-affinity-purified rabbit polyclonal antibody to murine IL-18BP and biotinylated antibody obtained from Cytolab.
RNA Isolation and RT-PCR.
After treatment in serum-free medium, Hep G2 and WISH cells (106) were harvested at the indicated times, and total RNA was extracted by using TRI reagent. cDNA was prepared by using random hexamers and SuperscriptII (Invitrogen) according to the manufacturer's instructions. PCR was performed with the following primers: human IL-18BP, 5′-CACGTCGTCACTCTCCTGG and 5′-CGACGTGACGCTGGACAAC; human IRF-1, 5′-GACCCTGGCTAGAGATGCAG and 5′-GAGCTGCTGAGTCCATCAG; and human β-actin, 5′-GTGGGGCGCCCCAGGCACCA and 5′-CTCCTTAATGTCACGCACGATTTC.
Amplifications were done by initial denaturation (92°C, 2 min), 28 cycles of denaturation (92°C, 45 s), annealing (62°C, 1 min) and extension (72°C, 1.5 min), and final extension (72°C, 10 min). The resulting PCR products were resolved by agarose (1%) gel electrophoresis.
5′ RACE.
5′ RACE was performed with a 5′ RACE System (Invitrogen) according to the manufacturer's instructions. Briefly, total RNA from IFN-γ-treated WISH cells was reverse-transcribed with a primer complementary to nucleotides 89–70 of IL-18BPa mRNA (GenBank accession no. AF110799) followed by tailing of the newly synthesized ends with an anchor DNA. PCR was then performed with a forward primer complementary to the anchor DNA and a nested reverse primer complementary to nucleotides 31–11 of IL-18BPa mRNA. The PCR products were then subcloned and subjected to DNA sequence analysis.
Plasmids and Cloning.
The entire putative IL-18BPa promoter region of 1,601 bp was obtained by PCR of genomic DNA with a sense primer (S4753.pgl) containing a KpnI site (5′-CTATATGGTACCCACCCTTCCTTTTACTTTTTCC) and reverse primer (R1exA) containing a NheI site (5′-TATCGCTAGCCAGTCACACAGGGAGGCAGT). The PCR product was cloned into pGEM-T Easy vector (Promega) and verified by DNA sequence analysis. A KpnI–NheI fragment was isolated from the pGEM-T Easy clone and ligated into pGL3-Basic vector (Promega) with a Rapid DNA Ligation Kit (Roche Molecular Biochemicals) to give pGL3(1601). A series of 5′ truncated reporters [pGL3(1272), pGL3(1194), pGL3(1107), pGL3(656), pGL3(280), and pGL3(123)] was similarly prepared with the R1exA reverse primer and the following sense primers, respectively: S334.pgl, 5′-CTATATGGTACCCATGAACTAGACACCTAGAG; S415.pgl, 5′-CTATATGGTACCCTACAAGAAGTTTGAGATCA; S501.pgl, 5′-CTATATGGTACCCAGCCGTTGCACCCTCCCAATCAC; 1exD pgl, 5′-CTATATGGTACCGTCTTGGAGCTCCTAGAGG; S504.pgl, 5′-CTATATGGTACCCACCAAAGTCCTGACACTTG; and S139.pgl, 5′-TTGGTACCCACTGAACTTTGGCTAAAGC.
All PCR products were also cloned into pGL3-basic vector in the opposite orientation to serve as controls.
Site-Directed Mutagenesis by Overlap Extension.
Mutations were introduced by a PCR-based mutagenesis method (11). Primers S4753.pgl and R1exA and their mutagenic reverse-complementing primers were used for amplification of the promoter in two sections. PCRs were done with Taq (3.5 units) with the following program: 95°C for 5 min, followed by 24 cycles, each consisting of 60°C for 1 min, 72°C for 1 min, and 95°C for 1 min. The resulting PCR products, which overlap by ≈50 bp, were purified, mixed, denatured, (100°C, 10 min) and cooled on ice for 5 min. A PCR mix without primers was added, and the full-length mutated promoter region was obtained by the following PCR protocol: 72°C for 3 min (once), followed by six cycles of 95°C for 1 min, 60°C for 1 min, and 72°C for 1 min. The flanking primers (30 pmol of S4753.pgl and R1exA) were then added and five cycles of PCR were performed. Thereafter, the mutated promoter region was cloned into pGEM-T Easy vector, its DNA sequence was verified, and it was subcloned into a pGL3-basic vector. The following specific primers and their reverse complementing primers were used: for gamma-activated sequence (GAS) mutation, 5′-GAAATAGAGGGTCGGGGCAGTGCTGATATCGAAGGATTGCTCGGCATCC; for IRF-1 response element (IRF-E) mutation, 5′-GCTCAAGATTCCCTGGTTGATATCGAATTCTGAAATAGAGGGTCGGGG- CAG; for the first C/EBPβ mutation, 5′-CAGATGCCTGTTCATAGATATCCAAAGACCTGGCTG; and for the second C/EBPβ mutation, 5′-CTAGAGGGAAGCTTCGATATCGGAAGGCTCTTC. Mutated regions are underlined. Primers S4753.pgl and R1exA were used as a common flanking primers for mutated pGL3(1601) vectors; primers S334.pgl and R1exA were used as a common flanking primers for mutated pGL3(1272) vectors.
Transient Transfection Assays.
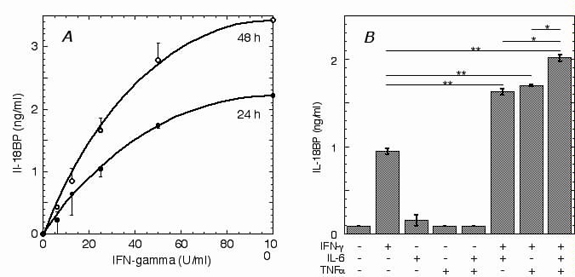
Hep G2 or WISH cells in six-well plates (5 × 105 per well) were transfected by using FuGene 6 and the indicated luciferase reporter vector (0.5 μg per well) and pSV40 β-galactosidase (0.2 μg per well, Promega) according to the manufacturer's instructions. In some cases, cotransfection was done with the following expression vectors: pCDNA3-IRF-1 (0.07–1.5 μg per well, kindly provided by B. Levi, Technion, Haifa, Israel); pCDNA3-C/EBPβ (0.5–2.5 μg per well, kindly provided by D. Zipori, Weizmann Institute of Science). After 16 h, cells were treated with either IFN-γ (100 units/ml), IL-6 (150 units/ml), tumor necrosis factor α (10 ng/ml), or their indicated combinations in serum-free medium for 24 h. Cells were then collected and lysed, and luciferase activity was measured. All results were normalized against β-galactosidase activity.
Preparation of Nuclear and Cytoplasmic Extracts.
Cells were washed three times with ice-cold PBS and immediately frozen in liquid nitrogen. Cell pellets were resuspended in four packed cell volumes of cytoplasmic buffer (10 mM Hepes, pH 7.9/10 mM NaCl/0.1 mM EDTA/5% glycerol/1.5 mM MgCl2/1 mM DTT/0.5 mM PMSF/50 mM NaF/0.1 mM Na3VO4/2 mM EGTA/10 mM EDTA/10 mM Na2MoO4/2 μg/ml each of leupeptin, pepstatin, and aprotinin). The lysate was centrifuged (3,000 × g, 10 min), and the supernatant containing the cytoplasmic proteins was collected. The pellet was resuspended in 2.5 packed cell volumes of nuclear buffer (identical to cytoplasmic buffer except that NaCl was increased to 0.42 M). Nuclear debris was removed by centrifugation (15,000 × g, 20 min, 4°C), and aliquots of the supernatant were frozen in liquid nitrogen and stored at −80°C. Protein concentration was determined by a BCA Protein assay reagent kit (Pierce) using BSA as a standard.
Electrophoretic Mobility-Shift Assays (EMSAs).
Double-stranded oligonucleotides corresponding to selected response elements (10 pmol) were labeled with 3′ dATP[α-32P] by polynucleotide kinase (New England Biolabs). Nuclear extracts (5 μg of protein) were preincubated (15 min, 0°C) together with poly(dI-dC) (2 mg/ml, final concentration, Amersham Pharmacia) in 20 μl EMSA buffer (20 mM Hepes, pH 7.5/5 mM MgCl2/2 mM EDTA/5 mM DTT/5% glycerol). A labeled probe (3 × 104 cpm) was then added and incubation continued for an additional 30 min at room temperature. For supershift assays, samples were incubated with the indicated antibodies (4 μg, 1 h, 0°C) before addition of the probe. A 200-fold excess of WT and mutated competitors were added together with the relevant probe. Reaction mixtures were then electrophoresed in 5% nondenaturing polyacrylamide gels. Gels were vacuum-dried and autoradiographed overnight at −80°C.
Immunoprecipitation (IP) and Immunoblot (IB) Analysis.
Nuclear or cytoplasmic protein extracts (80 μg) were incubated with the indicated polyclonal antibodies (6 μg, overnight, 4°C). Protein G Sepharose beads (Amersham Pharmacia) were then added for 1 h at room temperature. The beads were then isolated, washed, and boiled in SDS/PAGE sample buffer containing 25 mM DTT, and the supernatant was resolved by SDS/PAGE (10% acrylamide). The gel was then blotted onto a nitrocellulose membrane, and proteins were detected with the indicated antibodies. Immune complexes were identified with a Super Signal (Pierce) detection kit.
Results
The IL-18BP Gene Is Transcriptionally Regulated by IFN-γ, Requiring de Novo Protein Synthesis.
We first characterized the induction of IL-18BP by IFN-γ and several other cytokines in various human cell lines and peripheral blood mononuclear cells. As reported (9, 10), IFN-γ induced both IL-18BP mRNA and protein in these cells. IFN-γ induced IL-18BP in a dose-dependent manner, exhibiting an EC50 at 50 units/ml in WISH and Hep G2 cells. IL-18BP apparently accumulated in the culture supernatants, as its concentration was higher at 48 h compared with that at 24 h. IFN-α2, IFN-β, IL-1, IL-6, IL-12, IL-18, and tumor necrosis factor α did not induce IL-18BP directly; however, in Hep G2 cells, IL-6 and tumor necrosis factor α acted synergistically with IFN-γ, providing a statistically significant increase of IL-18BP (see Fig. 6, which is published as supporting information on the PNAS web site, www.pnas.org). IFN-γ did not induce IL-18BP in undifferentiated U-937 cells; however, after differentiation with phorbol ester (10 ng/ml, 24 h) into macrophage-like cells, a basal level of IL-18BP (0.07 ± 0.01 ng/ml) was detected, and increased by 4.6 ± 0.05-fold on induction with IFN-γ (100 units/ml, 24 h), further increasing at 96 h. Human peripheral blood mononuclear cells constitutively produced IL-18BP (0.7–1.5 ng/ml), and treatment with IFN-γ (100 units/ml) increased the level of IL-18BP by 1.7 ± 0.1-fold and 2.1 ± 0.3-fold at 24 and 48 h, respectively (P < 0.05, n = 4). No effect on IL-18BP production was seen on pretreatment of the peripheral blood mononuclear cells with phorbol ester.
We then tested whether the induction of IL-18BP mRNA occurred at the transcriptional level. IL-18BP mRNA was detectable in Hep G2 cells by semiquantitative RT-PCR after 3 h of treatment with IFN-γ and after 5 h in HaCaT and WISH cells. Pretreatment of Hep G2 and WISH cells with actinomycin D abolished the expression of IL-18BP mRNA at all time points, indicating that induction was at the transcriptional level (Fig. 1A). Because accumulation of IL-18BP after IFN-γ treatment was apparent mainly after 24 h and later, we studied the dependence of its mRNA expression on IFN-γ-induced proteins. Indeed, pretreatment of the cells with the protein synthesis inhibitor cycloheximide abolished the induction of IL-18BP mRNA (Fig. 1B). This result indicates that de novo translation is essential for IL-18BP gene expression.
Fig 1.

Induction of IL-18BP by IFN-γ is at the transcriptional level and depends on de novo protein synthesis. (A) Semiquantitative RT-PCR of IL-18BP mRNA from Hep G2 cells that were preincubated with actinomycin D (Act. D, 1 μg/ml, 30 min), washed, and incubated with IFN-γ (100 units/ml) for the indicated times. RT-PCR of β-actin mRNA is shown as a control. (B) Semiquantitative RT-PCR of IL-18BP mRNA from Hep G2 cells that were preincubated with cycloheximide (CHX, 20 μg/ml) and IFN-γ (100 units/ml) for the indicated times.
Defining the Transcription Start Site of IL-18BPa and Its Promoter Region.
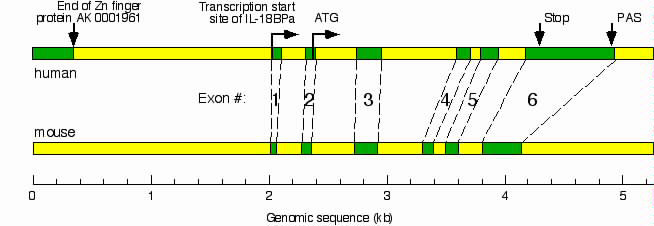
To map the IL-18BP promoter, we determined the transcription start site of human IL-18BPa mRNA by 5′ RACE. Only one PCR product, corresponding to IL-18BPa, the most abundant splice variant, was obtained. DNA sequence analysis of this product revealed an additional exon of 50 bp at the 5′ end of human IL-18BPa mRNA, corresponding to positions 785–835 of the genomic IL-18BP DNA sequence (GenBank accession no. AF110798). We generated an exon-intron map by comparing the genomic DNA with the mRNA to which the new 5′ exon was added. A similar map was generated for the mouse IL-18BP gene by comparing the mouse genomic DNA (Celera accession no. GA_x5J8B7W5P47, positions 1,190,001–1,205,000) with the mouse IL-18BPd mRNA (GenBank accession no. AF110803). We found that the exon-intron organization of the human and mouse IL-18BP genes are highly conserved (see Fig. 7, which is published as supporting information on the PNAS web site).
Having identified the transcription start site of IL-18BPa (base 1), we studied human genomic DNA upstream of base 1 (chromosome 11q clone:RP11–757C15, GenBank accession no. AP000719.4, upstream of base 152,178). Comparison of this DNA to the EST database at the National Center for Biotechnology Information by using the blast program revealed an upstream gene at the + strand, coding for a zinc-finger protein (GenBank accession no. AK001961). The deposited mRNA sequence of this Zn finger protein was further elongated by computer analysis using the instant race program (www.LabOnWeb.com), which scanned an extensive collection of human ESTs. The program placed the 3′ end mRNA of the Zn finger protein at nucleotide 150,517 of the genomic clone RP11–757C15, thereby limiting the potential upstream regulatory sequence of the IL-18BPa to 1,661 bases upstream of base 1 (see Fig. 7).
Proximal GAS, IRF-E, and Two C/EBPβ Sites Are Required for Basal IL-18BPa Gene Expression.
To further characterize the promoter region, we constructed a series of luciferase reporter vectors containing up to 1,601 bp corresponding to the DNA sequence upstream of base 1 and extending 50 bp into the first exon of IL-18BPa. In addition, we generated luciferase reporter vectors bearing truncated or mutated forms of this DNA (Fig. 2A). Luciferase activity in human Hep G2 cells transfected with a vector containing the entire 1,601-bp upstream DNA [pGL3(1601)] was 10.3 ± 0.9-fold higher than that obtained with the empty pGL3 vector. Such activity was not observed when the same DNA was inserted in the opposite orientation [pGL3(−1601)]. This result demonstrates that the 1,601-bp DNA upstream of base 1 has basal promoter activity. This promoter lacked a TATA box, but had several GC–rich domains near the transcription start site at bases −3 to −9, −39 to −48, and −123 to −132. We then constructed a series of luciferase reporter vectors with progressive truncations at the 5′ end of the inserted promoter. All constructs, including pGL3(123), were at least as effective as pGL3(1601) in supporting basal promoter activity.
Fig 2.

Basal and IFN-γ-induced activity of luciferase reporter vectors carrying the human IL-18BP promoter. Insert size, extending upstream of the transcription start site (+1), is shown in parentheses. Circled numbers represent the various response elements: 1, GAS; 2, IRF-E; 3, C/EBP-E (two sites); 4, silencer; 5, distal enhancer. Filled squares depict mutation in a specific response element. Hep G2 cells were cotransfected with the indicated reporter vector and pSV40 β-galactosidase. All luciferase values were normalized to β-galactosidase activity. (A) Luciferase activity in extracts of uninduced cells relative to that of cells transfected with pGL3-basic vector. (B) Luciferase activity in cells transfected with the indicated vectors and induced with IFN-γ. Fold induction is over basal activity as given in A.
Analysis of the 1,601-base DNA sequence by the program tfsearch identified a GAS at bases −24 to −32 (Fig. 2A). Further analysis revealed an IRF-E spanning bases −57 to −69 and two C/EBPβ response elements (C/EBP-E) at bases −309 to −322 and −621 to −634. All of these elements were essential for basal transcription, as mutation of any one of these sites [pGL3(1601mGAS), pGL3(1272mIRF), pGL3(1272mC/EBP-E1), or pGL3(1272mC/EBP-E2)] totally abolished the basal activity of the full-length promoter. In contrast, elimination of C/EBP-E1 and C/EBP-E2 by progressive truncation of the promoter did not reduce basal promoter activity. It should be noted, however, that luciferase reporter vectors containing the full-length promoter are more faithful models of the actual promoter as compared with truncated constructs. These data revealed four response elements within the IL-18BP promoter that are essential for basal promoter activity.
Proximal GAS, IRF-E, and C/EBP-E Regulate the Induction of IL-18BPa by IFN-γ.
We then studied the mechanism of IL-18BP gene induction by IFN-γ with the aid of the luciferase reporter vectors (Fig. 2B). After transfection of Hep G2 cells with pGL3(123), IFN-γ increased the luciferase activity at 24 h by 33-fold over the basal expression level of this vector, demonstrating that the IRF-E–GAS pair mediates the IL-18BP gene induction. Inclusion of C/EBP-E1 and C/EBP-E2 [pGL3(656)] significantly increased the induction of luciferase activity by IFN-γ to 88-fold over the basal activity, demonstrating the importance of these elements in the response to IFN-γ. In contrast, inclusion of additional upstream DNA [pGL3(1107)] abolished the induction of luciferase activity above its basal level. This result suggests that a silencer element resides within bases −656 to −1107. Further extension of the promoter by 88 bases [pGL3(1272)] restored the response to IFN-γ, suggesting that bases −1107 to −1272 contain an enhancer element, and its activation by IFN-γ suppresses the effect of the neighboring silencer. Further extension of the sequence did not affect basal or IFN-γ-induced activity, suggesting that all upstream regulatory sequences are located between bases −1 to −1272.
We then measured serum IL-18BP in IRF-1-deficient C57BL/6 mice (The Jackson Laboratory) before and after administration of murine IFN-γ. Basal serum IL-18BP in WT C57BL/6 mice was 9.1 ± 1.9 ng/ml and was significantly increased to 22.4 ± 2.2 ng/ml after treatment with IFN-γ. In contrast, serum IL-18BP in IRF-1-deficient mice was below the limit of detection and increased to only 0.7 ± 1.15 ng/ml on treatment with IFN-γ (Fig. 3). This result confirmed the importance of IRF-1 as a mediator of basal as well as IFN-γ-induced expression of IL-18BP.
Fig 3.

IRF-1 is essential for IL-18BP expression in mice. Serum IL-18BP was measured in control C57BL/6 and C57BL/6 IRF-1−/− mice that were injected i.p. with murine IFN-γ (53,000 units per mouse). Mice were bled before injection and 24 h postinjection, and serum IL-18BP was determined by ELISA. Data represent mean ± SD (n = 6 for each group). The differences between serum IL-18BP in control and IRF-1-deficient mice, as well as those between control and IFN-γ-induced C57BL/6 mice, were statistically significant (P < 0.05).
An IFN-γ-Induced Complex of C/EBPβ and IRF-1 Binds to the Promoter.
We then used EMSA to identify protein–DNA interactions among the various response elements within the IL-18BP promoter. Labeled double-stranded DNA probes corresponding to bases −33 to −75 (containing the IRF-E) and −8 to −55 (containing the GAS) were allowed to bind with nuclear extracts of control and IFN-γ-treated cells. A complex of the IRF-E-containing probe and nuclear protein(s) was apparent after incubation of cells for 1 h with IFN-γ, and maximal response was seen at 3 h (Fig. 4A, lanes 2–5). No such complex was obtained with extracts of untreated cells (Fig. 4A, lane 1). As expected, addition of antibodies to IRF-E caused a “supershift,” whereas control anti-signal transducer and activator of transcription 1 (STAT1) antibodies had no effect (data not shown). In contrast to IRF-E, the GAS-containing probe was constitutively associated with a protein (Fig. 4A, lane 6) and this complex was enhanced on induction of cells with IFN-γ for 3–6 h (Fig. 4A, lanes 7 and 8). GAS was expected to bind the IFN-γ-induced STAT1 dimer (12). Nevertheless, this complex was not affected by antibodies to STAT1 (Fig. 4A, lane 10), suggesting that it consisted of other transcription factors. STAT1 binding was also not observed with nuclear extracts of cells treated with IFN-γ for only 15 or 30 min (data not shown). Surprisingly, this complex was abolished by antibodies to C/EBPβ (Fig. 4A, lane 9) and was supershifted with antibodies to IRF-1, suggesting a possible interaction between the GAS-containing probe and these two transcription factors (Fig. 4A, lane 11). Hence the GAS-containing DNA probe appears to bind C/EBPβ despite the lack of a consensus C/EBP-E. The partial supershifting with anti-IRF-1 could stem from epitope masking by the interaction between IRF-1 and C/EBPβ.
Fig 4.

Physical association and role of IRF-1 and C/EBPβ in IL-18BP gene induction. (A) EMSA of double-stranded DNA probes corresponding to bases −33 to −75 (IRF-E, lanes 1–5) and −8 to −55 (GAS, lanes 6–11). Hep G2 cells were treated with IFN-γ for the indicated times, and nuclear extracts were allowed to react with the IRF-E or GAS probes. Shifted bands are indicated by filled arrowheads. The GAS complex was also subjected to supershift with the indicated antibodies. The supershifted band is indicated by an open arrowhead. (B) Semiquantitative RT-PCR of IL-18BP mRNA from Hep G2 cells that were transfected with the indicated combinations of IRF-1 or C/EBPβ expression vectors. Where indicated, IFN-γ was added and cells were harvested 5 h later. Values were normalized to β-actin mRNA. (C) Luciferase activity in cells transfected with the luciferase reporter vector pGL3(1272), containing the complete IL-18BP promoter, together with the indicated concentrations of pCDNA3-IRF-1 (circles) and a fixed concentration of pCDNA3-C/EBPβ concentration of pCDNA3-IRF-1 (1 μg/106 cells). Alternatively, cells were transfected with the indicated concentrations ofpCDNA3-C/EBPβ (squares) and a fixed pCDNA3-IRF-1 (0.1 μg/106 cells). Luciferase activity was normalized by the β-galactosidase activity. (D) Immunoblots of nuclear and cytoplasmic extracts of cells treated with IFN-γ (100 units/ml, 2 h). Extracts were immunoprecipitated and immunoblotted with the indicated antibodies.
To further study the role of IRF-1 and C/EBPβ in IL-18BP gene induction, we measured IL-18BPa mRNA by semiquantitative RT-PCR after transfection with IRF-1 and C/EBPβ expression vectors (Fig. 4B). Neither overexpression of the transcription factors singly or in combination in Hep G2 cells induced basal levels of IL-18BP mRNA. This result suggests that additional IFN-γ-induced factors are required for activation of the IL-18BP gene. Transfection of the cells with either one of the expression vectors followed by their induction with IFN-γ also had no effect (IRF-1) or even reduced (C/EBPβ) IL-18BP mRNA compared with IFN-γ alone. In contrast, coexpression of the two transcription factors increased the induction of IL-18BP mRNA by IFN-γ. This result suggests that both IRF-1 and C/EBPβ are involved in activation of the IL-18BP gene by IFN-γ. To further study the possible interaction between IRF-1 and C/EBPβ, we performed a titration of luciferase activity by cotransfecting cells with pGL3(1272), a fixed amount of the IRF-1 expression vector, and varying amounts of the C/EBPβ expression vector. Similarly, we measured luciferase activity when the C/EBPβ vector was kept constant and with varying amounts of the IRF-1 vector. In both cases, a bell-shaped dose–response curve was obtained, suggesting that optimal IL-18BP induction requires a fixed molar ratio between these two transcription factors (Fig. 4C).
These findings prompted us to test for a possible physical association between IRF-1 and C/EBPβ (Fig. 4D). IP followed by IB of nuclear and cytoplasmic proteins from IFN-γ-treated cells with antibodies to C/EBPβ revealed that this protein is constitutively expressed in Hep G2 cells and spontaneously translocates to the nucleus (Fig. 4D Top). In contrast, IP and IB of cell extracts with antibodies to IRF-1 revealed that IFN-γ induced the expression of IRF-1 and its translocation to the nucleus (Fig. 4D Middle). After IFN-γ treatment, IP with antibodies to C/EBPβ followed by IB with antibodies to IRF-1 revealed the presence of a stable IRF-1-C/EBPβ complex in the nuclear fraction (Fig. 4D Lower).
The Two Other C/EBP-Es Constitutively Interact with C/EBPβ.
Our EMSA studies showed that the proximal GAS-containing sequence and its adjacent IRF-E bind a complex of C/EBPβ and IRF-1. The two C/EBPβ sites at positions −309 to −322 and −621 to −634 do not have an adjacent IRF-E. Indeed, EMSA of a probe corresponding to the C/EBPβ sites at positions −309 to −322 revealed a retarded band (Fig. 5A, filled arrowhead) that was supershifted by antibodies to C/EBPβ (Fig. 5A, open arrowhead) but not by antibodies to IRF-1. Hence, these experiments indicate that this site binds C/EBPβ alone, and not its complex with IRF-1. Furthermore, this band was generated with nuclear extract of uninduced Hep G2 cells that constitutively express C/EBPβ but lack IRF-1. In fact, IFN-γ did not increase the expression of C/EBPβ in these cells (Fig. 4D), and consequently it did not increase the intensity of the retarded band (Fig. 5A). These experiments indicate that that C/EBPβ is constitutively bound to this C/EBP-E. Similar results were obtained with the more distal C/EBP-E (data not shown).
Fig 5.

Involvement of additional response elements in IL-18BP induction. (A) EMSA with the proximal C/EBP-E and whole-cell extracts after treatment with IFN-γ. Where indicated, the extracts were supershifted with the indicated antibodies. (B) EMSA with a probe corresponding to the distal enhancer and whole-cell extracts after treatment with IFN-γ. Where indicated, the extracts were supershifted with the indicated antibodies, with or without competition with double-stranded DNA corresponding to the proximal half of the probe. Shifted bands are indicated by filled arrowheads and supershifted bands are indicated by open arrowheads.
The Distal Enhancer Interacts with the Basal Promoter Through IRF-1.
We then studied the regulatory role of the distal enhancer by EMSA with a 192-bp DNA probe, corresponding to nucleotides −1081 to −1272. Nuclear extract from control Hep G2 cells formed a complex with this probe (Fig. 5B, filled arrowhead). On treatment of the cells with IFN-γ, the complex was more intense and somewhat more retarded. We then attempted to supershift this complex with antibodies directed against IRF-1, C/EBPβ, and c-Fos. Of these, only anti-IRF-1 elicited a supershift (Fig. 5B, empty arrowhead). An unlabeled double-stranded DNA fragment corresponding to nucleotides −1083 to −1174 did not compete with the radiolabeled probe, indicating that the nuclear proteins were bound to sites located between residues −1175 and −1272. Because the only IRF-E was identified in the proximal region, this result suggests that the distal enhancer associates with the proximal IRF-E.
Discussion
In this study we characterized the human IL-18BP promoter and studied its activation by IFN-γ. IL-18BP is a potent inhibitor of IL-18, an inducer of T helper 1 cytokines such as IFN-γ. Thus, induction of IL-18BP by IFN-γ provides a negative feedback mechanism that shuts off IL-18-elicited immune responses. To avoid premature termination of IL-18 activity, induction of IL-18BP must occur after some delay. Indeed, we show here that induction was not mediated by the rapid IFN-γ-induced Janus kinase-STAT signaling pathway. Rather, the induction required de novo synthesis of the transcription factor IRF-1, which together with C/EBPβ activates the IL-18BP promoter.
Previously, it was shown that IL-18BP mRNA and protein are elevated in cultures of IFN-γ-treated cells (9). Here we show that this increase is regulated by IFN-γ at the transcriptional level and requires additional IFN-γ-induced factors. Furthermore, we have demonstrated elevation of serum IL-18BP in vivo in IFN-γ-treated mice, indicating that this pathway is of physiological significance. We have recently shown that serum IL-18BP is elevated in sepsis patients compared with healthy individuals and that IL-18BP significantly reduces free serum IL-18 (8). The high levels of IFN-γ and other proinflammatory cytokines, such as tumor necrosis factor α and IL-6, in sepsis probably led to induction of IL-18BP in these patients. Yet, this induction is quite specific, as it was not triggered by many other proinflammatory cytokines.
As in the case of many other genes, several IL-18BP mRNA splice variants exist (3). At least three different transcription start sites have been identified (3), suggesting different mechanisms of gene regulation. Of these variants, IL-18BPa is the most abundant one; IL-18BPb and IL-18BPd are minor components that do not bind IL-18 and hence their physiological significance is unknown. IL-18BPc binds IL-18, but it is a minor component derived from a very large ≈8-kb mRNA, probably generated by incomplete mRNA splicing. It is possible however, that IL-18BPc may be induced under different physiological conditions compared with those required for IL-18BPa.
We have focused on the upstream regulatory 1.6-kb region of the IL-18BP gene and identified six regulatory sequences. This region appears to direct both the basal expression level seen in some tissues and cell types, as well as induction of the gene by IFN-γ. Successive truncation of this region suggests that a 123-bp DNA proximal to the transcription start site comprises the minimal promoter. However, other regulatory sequences within 1 kb upstream of this minimal promoter also contributed both to the extent of induction and its regulation, which are probably required for maintaining specificity and enabling more precise control of gene activity. It is noteworthy that the gene of IL-18BP, which seems to have a rather limited role as an inhibitor of IL-18, is regulated by such a complex mechanism.
The transcription factors IRF-1 and C/EBPβ are involved in regulation of inflammatory-associated genes (13, 14). Many cell types, e.g., Hep G2 cells, express a basal level of C/EBPβ but not IRF-1. However, these two transcription factors are induced at the transcriptional level by IFN-γ (13, 15). Binding of C/EBPβ to the proximal GAS-containing sequence was quite unexpected as no consensus C/EBP-E could be identified in this sequence. Yet, there is a partial homology between GAS and the C/EBP-E that might provide the basis for such binding: consensus C/EBP-E, TKNN-GMAAK (K = T or G; M = A, C, or T); the proximal GAS of IL-18BP gene, TCCCAG-AAG.
We have recently generated a point mutation of the G-26 to A, that disrupts the consensus sequence of C/EBP-E without affecting the consensus of GAS. On induction with IFN-γ, this mutant gave one-sixth of the luciferase activity as compared with WT pGL3(123). This result supports the hypothesis that the proximal GAS binds C/EBPβ.
IRF-1 and other members of the IRF family were reported to form dimers among themselves and form complexes with other transcription factors, including NF-κB, HIF-1, p53, PCAF, GCN5, and CPB/p300 (13). Similarly, C/EBPβ was reported to associate with NF-κB, PU.1, and glucocorticoid receptors (16–18). Because these two proteins are induced by IFN-γ, our finding that IFN-γ also induces their association probably has much broader implications for IFN-γ-regulated genes. Interactions between other members of the IRF family and members of the C/EBP family are also likely, further extending the number of heterodimeric transcription factors that regulate the genome.
Supplementary Material
Acknowledgments
We thank Sara Barak for excellent technical assistance. This work was supported by the Serono Group of companies. M.R. is the Edna and Maurice Weiss Professor of Cytokine Research.
Abbreviations
IL-18BP, IL-18 binding protein
GAS, gamma-activated sequence
C/EBPβ, CCAAT/enhancer binding protein β
C/EBP-E, C/EBPβ response element
IRF-1, IFN regulatory factor 1
IRF-E, IRF-1 response element
STAT1, signal transducer and activator of transcription 1
EMSA, electrophoretic mobility-shift assay
IP, immunoprecipitation
IB, immunoblotting
References
- 1.Pan G., Risser, P., Mao, W., Baldwin, D. T., Zhong, A. W., Filvaroff, E., Yansura, D., Lewis, L., Eigenbrot, C., Henzel, W. J. & Vandlen, R. (2001) Cytokine 13, 1-7. [DOI] [PubMed] [Google Scholar]
- 2.Neighbors M., Xu, X., Barrat, F. J., Ruuls, S. R., Churakova, T., Debets, R., Bazan, J. F., Kastelein, R. A., Abrams, J. S. & O'Garra, A. (2001) J. Exp. Med. 194, 343-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Novick D., Kim, S.-H., Fantuzzi, G., Reznikov, L. L., Dinarello, C. A. & Rubinstein, M. (1999) Immunity 10, 127-136. [DOI] [PubMed] [Google Scholar]
- 4.Yasuda H., Shima, N., Nakagawa, N., Mochizuki, S. I., Yano, K., Fujise, N., Sato, Y., Goto, M., Yamaguchi, K., Kuriyama, M., et al. (1998) Endocrinology 139, 1329-1337. [DOI] [PubMed] [Google Scholar]
- 5.Elson G. C., Graber, P., Losberger, C., Herren, S., Gretener, D., Menoud, L. N., Wells, T. N., Kosco-Vilbois, M. H. & Gauchat, J. F. (1998) J. Immunol. 161, 1371-1379. [PubMed] [Google Scholar]
- 6.Kim S.-H., Eisenstein, M., Reznikov, L., Fantuzzi, G., Novick, D., Rubinstein, M. & Dinarello, C. A. (2000) Proc. Natl. Acad. Sci. USA 97, 1190-1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiang Y. & Moss, B. (1999) Virology 257, 297-302. [DOI] [PubMed] [Google Scholar]
- 8.Novick D., Schwartsburd, B., Pinkus, R., Suissa, D., Belzer, I., Sthoeger, Z., Keane, W. F., Chvatchko, Y., Kim, S. H., Fantuzzi, G., et al. (2001) Cytokine 14, 334-342. [DOI] [PubMed] [Google Scholar]
- 9.Muhl H., Kampfer, H., Bosmann, M., Frank, S., Radeke, H. & Pfeilschifter, J. (2000) Biochem. Biophys. Res. Commun. 267, 960-963. [DOI] [PubMed] [Google Scholar]
- 10.Paulukat J., Bosmann, M., Nold, M., Garkisch, S., Kampfer, H., Frank, S., Raedle, J., Zeuzem, S., Pfeilschifter, J. & Muhl, H. (2001) J. Immunol. 167, 7038-7043. [DOI] [PubMed] [Google Scholar]
- 11.Meza R., Nunez-Valdez, M. E., Sanchez, J. & Bravo, A. (1996) FEMS Microbiol. Lett. 145, 333-339. [DOI] [PubMed] [Google Scholar]
- 12.Shuai K., Horvath, C. M., Huang, L. H., Qureshi, S. A., Cowburn, D. & Darnell, J. E., Jr. (1994) Cell 76, 821-828. [DOI] [PubMed] [Google Scholar]
- 13.Kroger A., Koster, M., Schroeder, K., Hauser, H. & Mueller, P. P. (2002) J. Interferon Cytokine Res. 22, 5-14. [DOI] [PubMed] [Google Scholar]
- 14.Poli V. (1998) J. Biol. Chem. 273, 29279-29282. [DOI] [PubMed] [Google Scholar]
- 15.Niehof M., Streetz, K., Rakemann, T., Bischoff, S. C., Manns, M. P., Horn, F. & Trautwein, C. (2001) J. Biol. Chem. 276, 9016-9027. [DOI] [PubMed] [Google Scholar]
- 16.LeClair K. P., Blanar, M. A. & Sharp, P. A. (1992) Proc. Natl. Acad. Sci. USA 89, 8145-8149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagulapalli S., Pongubala, J. M. & Atchison, M. L. (1995) J. Immunol. 155, 4330-4338. [PubMed] [Google Scholar]
- 18.Nishio Y., Isshiki, H., Kishimoto, T. & Akira, S. (1993) Mol. Cell. Biol. 13, 1854-1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}