

Abstract
The transcription factor Pax5 (BSAP) is required for the expression of a B-cell-specific genetic program and for B-cell differentiation, and also to suppress genes of alternative lineages. The molecular mechanism by which repression of myeloid genes occurs during early B-lineage restriction is unknown and in this study we addressed this question. One of the genes repressed by Pax5 in B cells is the colony-stimulating factor receptor 1 gene (csf1r or c-fms). We examined the changes in chromatin caused by Pax5 activity, and we show that Pax5 is directly recruited to c-fms resulting in the rapid loss of RNA polymerase II binding, followed by loss of transcription factor binding and DNaseI hypersensitivity at all cis-regulatory elements. We also show that Pax5 targets the basal transcription machinery of c-fms by interacting with a binding site within the major transcription start sites. Our results support a model by which Pax5 does not lead to major alterations in chromatin modification, but inhibits transcription by interfering with the action of myeloid transcription factors.
Keywords: B-lineage restriction, c-fms, chromatin, gene silencing, Pax5
Introduction
All blood cells originate from hematopoietic stem cells (HSCs), which in the adult mammalian organism reside in the bone marrow and have the capacity to self-renew as well as differentiate (Weissman et al, 2001). Differentiating HSCs undergo a gradual loss of developmental potential in favor of functional specialization. This generates defined precursor cell types, which give rise to functionally specialized blood cells, and this differentiation process is concurrent with the execution of different genetic programs. A conceptual breakthrough in how cell fate decisions are made came from experiments demonstrating that HSCs express a lineage promiscuous gene expression program and that the chromatin of genes not yet expressed in precursor cells is already partially reorganized (Jimenez et al, 1992; Enver and Greaves, 1998; Kontaraki et al, 2000; Tagoh et al, 2004a). Once cells start to differentiate into specific blood cell types, genes for inappropriate lineages are silenced, whereas lineage-appropriate genes are further activated. Lineage-specific transcription factors play an essential role in this process. It is well known that shifting the balance of specific transcription factors in hematopoietic precursor cells dictates the outcome of cell differentiation (reviewed in Orkin, 2000; Graf, 2002) and this principle holds true for all differentiation decisions in all multicellular organisms that have been studied so far. Overexpression of the transcription factor PU.1 in multipotent progenitor cells shifts differentiation toward myeloid cells (Nerlov and Graf, 1998; DeKoter and Singh, 2000; Yamada et al, 2001; McIvor et al, 2003). PU.1 functions in opposition to GATA-1, which regulates erythropoieses and inhibits GATA-1 activity (Kulessa et al, 1995; Nerlov et al, 2000; Zhang et al, 2000; Heyworth et al, 2002). It has even been shown that the overexpression of myeloid-specific transcription factors in mature B cells can reprogram these cells into macrophages (Xie et al, 2004).
Recent experiments have identified the B-cell-specific transcription factor Pax5 as an important transcription factor regulating commitment to the B lymphoid lineage and have given crucial insights into how transcription factors regulate cell fate decisions. Pax5 null cells in the bone marrow of knockout mice are blocked in B-cell differentiation (Urbánek et al, 1994; Nutt et al, 1997a; Hayashi et al, 2003), demonstrating that Pax5 is required for the activation of B-cell-specific genes (Nutt et al, 1998). Subsequent experiments have shown that the conditional elimination of Pax5 in immature and mature B cells leads to the re-expression of myeloid-specific genes. This includes the colony-stimulating factor 1 receptor gene (csf1r or c-fms) or the myeloperoxidase gene (mpo), confirming that not only activators but also repressors are required for the establishment of a specific genetic program (Nutt et al, 1999; Mikkola et al, 2002; Tagoh et al, 2004b). These experiments also showed that Pax5 is able to act as activator and repressor in one cell type. How Pax5 represses a subset of lymphoid-specific genes has been studied in some detail. It interacts with the Groucho co-repressor family member Grg4 to repress the activity of the immunoglobulin heavy chain enhancer (Eberhard et al, 2000; Linderson et al, 2004). However, ectopic expression of Pax5 in the myeloid lineage has no effect on myelopoiesis (Souabni et al, 2002; Cotta et al, 2003) and the molecular mechanism by which Pax5 represses myeloid-specific genes in B cells is not known.
Transcription factors do not act alone to establish genetic programs. They interact with a specific chromatin architecture and cooperate with chromatin-modifying complexes. To gain insight into how lineage-inappropriate genes are silenced at the chromatin level, we studied silencing of c-fms during B lymphopoiesis (Tagoh et al, 2004b). Expression of this growth factor receptor is crucial for macrophage differentiation and is developmentally regulated (Dai et al, 2002; Tagoh et al, 2002). c-fms is a target of the transcription factor PU.1 and is already expressed at a low level in HSCs. It is upregulated during macrophage differentiation and is silenced in B cells, despite the presence of PU.1 in both macrophages and B cells. We recently showed that c-fms can be reactivated in mature B lymphoid cells by conditional inactivation of Pax5, and this ability correlates with a partially active chromatin structure of this gene (Tagoh et al, 2004b). Although no transcription factors were bound to c-fms cis-regulatory elements in mature B cells, its chromatin was still DNaseI accessible, nucleosomes were positioned in the active conformation and the DNA of the cores of cis-elements was unmethylated. We concluded from these results (i) that Pax5 has to be present throughout B lymphopoiesis to keep c-fms in a silent state and (ii) that it is difficult to epigenetically silence a gene in dividing cells while transcriptional activators for lineage-specific genes (such as PU.1) are still present.
In the study presented here, we wanted to gain insight into the molecular mechanism by which Pax5 silences the c-fms gene. We show that expression of Pax5 in c-fms expressing multipotent Pax5−/− precursor cells leads to a repression of c-fms, which mimics what is happening during B-cell differentiation. After Pax5 induction, RNA polymerase II is lost immediately, thereafter transcription factor binding becomes unstable and factors are slowly lost from all c-fms cis-elements with a concomitant loss of DNaseI hypersensitivity. We identified the c-fms promoter as the major Pax5 response element and show that Pax5 is recruited to sequences binding the basal transcription machinery in vivo. We demonstrate by electrophoretic mobility shift assays (EMSAs) and transfection assays that Pax5 binding to this site is specific and is required for repression. Pax5 is also recruited to the promoter of the mpo gene, indicating that the mechanism of c-fms inactivation in B-cell precursors is shared by at least one other myeloid-specific gene.
Results
Chromatin of all cis-regulatory elements of c-fms is reorganized in Pax5−/− cells
c-fms mRNA can be found in HSCs and restricted common lymphoid progenitor cells, but not in pro-B cells (Tagoh et al, 2004b). c-fms is expressed at a low level in Pax5−/− cells (Figure 1A) and it was previously shown that c-fms expression is reactivated by the conditional inactivation of Pax5 in pro-B cells (Mikkola et al, 2002). However, it was unknown to date whether all or only a subset of cis-regulatory elements respond to the removal of Pax5, form DNaseI hypersensitive sites (DHSs) in chromatin and bind transcription factors. We therefore needed to define the full complement of DHSs on the c-fms locus. We have previously mapped three hypersensitive regions in the promoter and the first intron, which coincided with sequences highly conserved between human and mouse (Figure 1B) (Himes et al, 2001). The intronic elements, the c-fms intronic regulatory element (FIRE) and an upstream DHS (FIRE-1) are sufficient for the correct expression of c-fms in transgenic mice and stably transfected cells (Himes et al, 2001; Sasmono et al, 2003). The most important element is FIRE, which has enhancer activity and which is crucial for correct c-fms regulation. Here we extended our analysis up to the next downstream located gene. We found one additional macrophage-specific DHS in intron 10, but this site was only partly conserved and had no cis-regulatory function on its own, in the context of the other cis-elements or when linked up to a heterologous promoter, and no DHS was seen in Pax5−/− cells (data not shown). Next, we mapped the DHS across the c-fms locus in wild-type (wt) or Pax5-deficient pro-B cells (Figure 1B and Supplementary Figure 1). Our results clearly show that there were no DHSs in Pax5+/+ cells and the same regions as in myeloid cells became DNaseI hypersensitive in Pax5−/− cells. We then investigated transcription factor occupancy of c-fms cis-regulatory elements by DMS in vivo footprinting in purified pro-B cells from the bone marrow of Pax5 mutant as well as RAG2−/− mice (Pax5+/+). In RAG2−/− mice, B-cell differentiation is blocked at the same pro-B-cell stage as in Pax5−/− mice, but these pro-B cells do not possess alternative differentiation potential. As shown in Supplementary Figure 2 and before (Tagoh et al, 2004b), Pax5+/+ pro-B cells showed no transcription factor binding at the promoter and FIRE. In contrast, we saw consistent alterations in DMS reactivity at the PU.1 binding sites of the c-fms promoter and FIRE in Pax5−/− progenitor cells. Interestingly, transcription factor occupancy at the Runx1/Egr-2/ets element at FIRE was only partial, similar to what we saw in committed lymphoid progenitor cells (Tagoh et al, 2004b). Taken together, we conclude that all cis-regulatory elements of the c-fms locus respond to Pax5 and bind transcription factor once this factor is removed.
Figure 1.
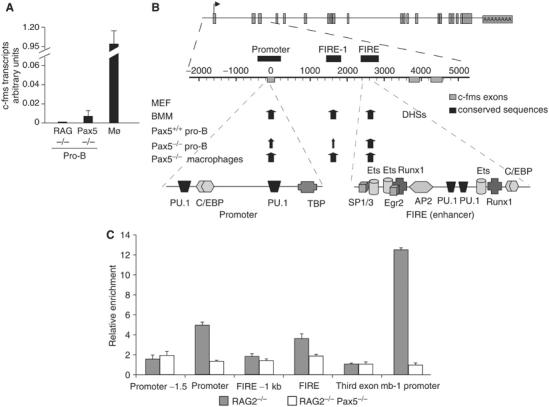
(A) Expression of c-fms mRNA in macrophages (Mø), wt (RAG2−/−) pro-B cells and Pax5−/− pro-B cells as measured by real-time PCR. The level of expression in Mø was set as one. (B) Map of the c-fms locus. Top panel: position of introns, exons and the transcription start. Lower panel: expanded view of the c-fms regulatory region indicating the position of confirmed transcription factor binding sites and DHSs. The intensity of DHSs is indicated by differently sized arrows. MEF: mouse embryo fibroblasts; BMM: bone marrow-derived macrophages. (C) Endogenous Pax5 protein is recruited to c-fms regulatory regions in pro-B cells. ChIP assay with an antibody against the Pax5 paired domain with pro-B cells derived from either RAG2−/− or RAG2−/−Pax5−/− mice. The value of enrichment was corrected for input and then normalized to the control gene. The data represent the means of two independent chromatin preparations analyzed in triplicate.
Pax5 is recruited to the c-fms locus in pro-B cells and the induction of Pax5 in Pax5−/− cells leads to loss of transcription factor and RNA polymerase II binding
Pax5 exerts its activating and repressing effect on B-cell-specific genes by directly binding to specific cis-regulatory elements of these genes. To confirm that this was also true for myeloid-specific genes, we performed chromatin immunoprecipitation (ChIP) experiments with pro-B cells isolated from RAG2−/− mice and from RAG2−/−Pax5−/− mice (Figure 1C). Enrichment of DNA fragments after immunoprecipitation was quantified by real-time PCR using primers specific for different c-fms cis-regulatory elements and for the B-cell-specific mb-1 promoter as a positive control. The data show clearly that the Pax5 protein is recruited to two sites in the c-fms locus, the promoter and FIRE, whereby the signal at the promoter was the strongest. To determine the order of events occurring in chromatin during early phases of the silencing of c-fms by Pax5, we employed a Pax5−/− cell line that expressed an inducible Pax5-estrogen receptor fusion protein (Pax5-ER) (Nutt et al, 1998). ChIP assays showed that Pax5-ER was rapidly recruited to c-fms regulatory elements, again with a preference for the promoter (Figure 2A). Induction of Pax5 with 4-hydroxy-tamoxifen (OHT) resulted in a reduction of c-fms mRNA within 24 h, whereas the B-cell-specific Pax5 target gene Cd19 was induced (Figure 2B). A Pax5-ER fusion protein (ΔPax5-ER) lacking the DNA binding domain (DBD), which has previously been shown to be unable to rescue Pax5 activity (Nutt et al, 1998), could not bind to c-fms promoter and alter its expression (Supplementary Figure 3). Figure 2C depicts a ChIP assay showing that Pax5 induction results in a rapid reduction of RNA polymerase II binding to the c-fms promoter to the background levels observed in 3T3 cells, which we have shown not to express c-fms mRNA and not to bind RNA polymerase II and TATA-binding protein (Follows et al, 2003). This indicates that the process of transcription is disrupted immediately, in contrast to the decay of the c-fms mRNA steady-state level, which is also controlled by post-transcriptional mechanisms. Our results also prove that the low level of c-fms mRNA expression in multipotent precursor cells compared to macrophages is the result of reduced RNA-polymerase II occupancy and not post-transcriptional events. We next wanted to know whether Pax5 also binds to other myeloid-specific genes, such as the myeloperoxidase (mpo) gene. We therefore measured mpo expression as well as RNA polymerase II and Pax5-ER recruitment to its promoter after Pax5 induction. Similar to the c-fms promoter, we also saw rapid binding of Pax5-ER to the mpo promoter followed by a gradual loss of RNA polymerase II binding (Supplementary Figure 4). This indicates that the mechanism of repression by Pax5 is shared by at least one other myeloid-specific gene.
Figure 2.
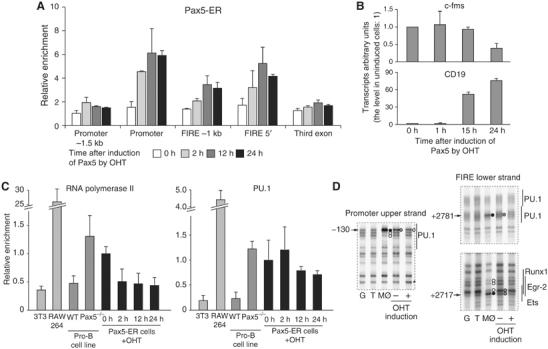
(A) Pax5-ER fusion protein is rapidly recruited to c-fms cis-regulatory elements. ChIP assay with a human ERα antibody. The value of enrichment was corrected for input and normalized to the control gene. The data represent the means of three independent chromatin preparations analyzed in triplicate. (B) c-fms mRNA expression is repressed after Pax5 induction with OHT (upper panel), whereas CD19 mRNA is induced (lower panel). The scale was set with the levels in non-induced cells as one. (C) RNA polymerase II binding to the c-fms promoter is reduced to background levels as seen in NIH3T3 cells (3T3) or wt pro-B cells (wt) after Pax5 induction (left panel). PU.1 is only starting to be lost after 12 h of OHT induction (right panel). The recruitment of these proteins was measured by a ChIP assay in the indicated cell types. The experiment also demonstrates that PU.1 and RNA polymerase II binding is only partial (compare with the signals obtained with RAW264 cells). The value of enrichment was corrected for input and then normalized to the control gene. Relative enrichment in non-induced Pax5-ER cells was presented as one. The data represent the means of three independent chromatin preparations analyzed in triplicate. (D) DMS in vivo footprinting experiment examining transcription factor binding in the indicated cell types. G: DMS-piperidine cleavage reaction performed on naked DNA; T: CD3+ thymocytes; Mø: bone marrow-derived macrophages; −/+: Pax5-ER cells without OHT (−) or after 24 h of OHT treatment (+). Black circles indicate DMS hyper-reactivity, gray circles indicate different degrees of weak hyper-reactivity and white circles indicate DMS hypo-reactivity. The numbers indicate the nucleotide position relative to the ATG. Positions of transcription factor binding sites are indicated.
To investigate the effect of Pax5 recruitment on transcription factor occupancy, we performed a ChIP assay examining the binding of PU.1 to the c-fms promoter (Figure 2C, right panel). PU.1 is an essential factor regulating c-fms expression, and elimination of this factor abolishes c-fms expression and blocks macrophage differentiation (Scott et al, 1994; Simon et al, 1996). PU.1 occupancy at the c-fms promoter in Pax5−/− cells was only partial as compared to macrophages and was further reduced after 24 h of Pax5 induction, but not with the same kinetics as RNA polymerase II. The same partial binding and slow reduction in binding was seen with DMS footprinting (Figure 2D). The DMS hyper-reactive band at −130 bp that is indicative of PU.1 binding was strongly reduced after 24 h of OHT treatment, but was not completely absent (compare with the signal observed with T cells). We also noted increased DMS reactivity of a G at −113 bp after OHT induction (marked by an asterisk). By contrast, transcription factors binding to the FIRE enhancer were completely lost (Figure 2D, right panel). This held true for PU.1 binding at +2781 bp, and for other transcription factors such as those binding to the Runx1/Egr-2/ets cluster at +2717 bp.
In order to gain insights into the mechanism of repression activity by Pax5 at the level of chromatin, we studied the effect of Pax5 induction on different features of c-fms chromatin fine structure (Figures 3 and 4 and Supplementary Figures 5 and 6). We first examined the effect of Pax5 induction on DNaseI hypersensitivity using a novel high-resolution ligation-mediated PCR (LM-PCR) assay. To this end, we treated uninduced, induced and control cells with DNaseI, followed by direct linker ligation and amplification. This method only amplifies double-strand cuts that are indicative of highly distorted regions in chromatin, which allows the enzyme to simultaneously access opposite DNA strands. Figure 3A shows that the region over the main transcription start sites at the promoter was strongly DNaseI hypersensitive, and this hypersensitivity was progressively but not immediately lost after Pax5 induction. The same held true for the FIRE enhancer. This loss of DNaseI hypersensitivity was not observed with a ribosomal RNA gene analyzed as a control (Supplementary Figure 5A). General DNaseI accessibility at these elements, which is an indication of the extent of chromatin condensation as measured by single-strand specific LM-PCR, was not altered (Supplementary Figure 6). The c-fms promoter in c-fms non-expressing cells such as 3T3 cells is organized in a positioned nucleosome, which is relocated once the gene is expressed, thus exposing the different transcription start sites to RNA polymerase II binding (Tagoh et al, 2004b). Pax5−/− cells show a partially shifted nucleosome, and nucleosome positioning was not altered by induction of Pax5 as measured by LM-PCR on micrococcal nuclease (MNase)-digested chromatin (Figure 4). This is in keeping with our earlier data showing that c-fms chromatin is still in a partially active and accessible conformation in B cells (Tagoh et al, 2004b). Repression of gene expression often involves the acquisition of inactive histone marks and the loss of active histone marks such as histone H3 lysine 4 methylation or lysine 9 acetylation, respectively (Jenuwein and Allis, 2001). We have previously shown that in contrast to macrophages, histone H3 at c-fms chromatin in immature B lymphoid and myeloid cells is not hyperacetylated at lysine 9 (Tagoh et al, 2004b). However, we had not measured histone modifications in multipotent cells that express a low level of c-fms, such as Pax5−/− cells. The ChIP experiment shown in Figure 3B clearly shows that also in multipotent precursor cells c-fms chromatin was neither hyperacetylated nor hypermethylated at H3 lysine 9 and this modification state did not change after Pax5 induction.
Figure 3.
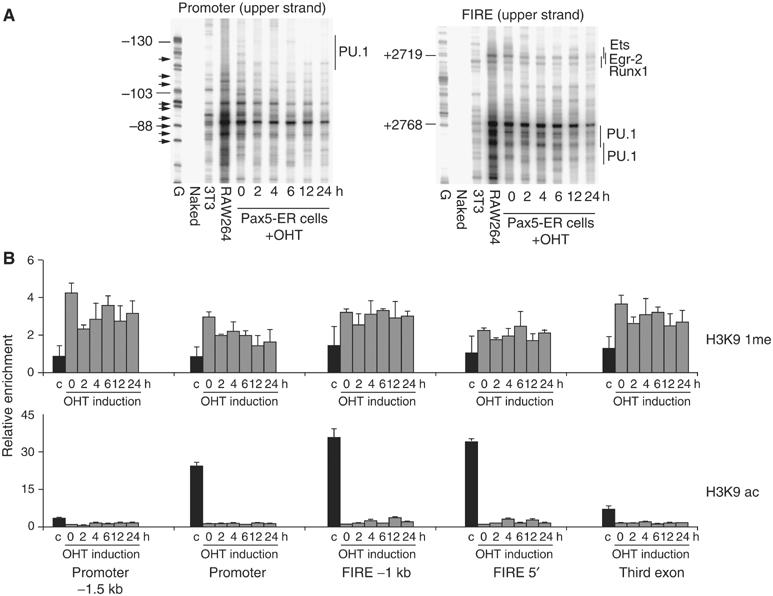
(A) DNaseI hypersensitivity at the c-fms promoter and FIRE is reduced after Pax5 induction. Double-strand-specific LM-PCR was performed on DNaseI-treated NIH3T3 cells, RAW264 macrophage cells and Pax5-ER cells without or after a time course of OHT induction. From the left to the right: DMS-treated naked DNA (G), DNaseI-treated naked DNA (naked), DNA prepared from DNaseI-treated 3T3 cells, RAW 264 cells and from Pax5-ER cells induced with OHT for the indicated time points. Arrows in promoter panel represent multiple transcription start sites. The internal control confirming equal DNaseI digestion levels can be found in Supplementary Figure 4A. (B) Histone H3 lysine 9 modification is not altered by the induction of Pax5 activity. ChIP assay measuring histone H3 lysine 9 monomethylation and H3 lysine 9 acetylation in macrophages (black bars) and in Pax5-ER cells during a time course of OHT induction (gray bars) across the c-fms locus (for relative locations, see map in Figure 1A). Relative enrichment was corrected for input and then normalized to control gene. The data represent the means of two independent chromatin preparations analyzed in triplicate.
Figure 4.
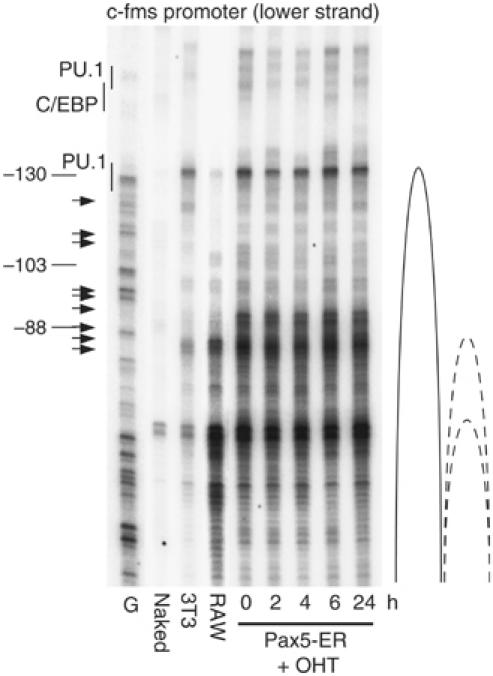
Nucleosome positioning at the c-fms promoter is in a partially active conformation in Pax5−/− cells and is unchanged after Pax5 induction. Naked DNA and chromatin prepared from the indicated cell populations was treated with MNase and double-strand breaks were detected by LM-PCR using primers specific for the c-fms promoter. Horizontal arrows indicate the positions of transcription start sites and transcription factor binding sites are shown on the left. Alternate nucleosome positions in the different cell types are depicted on the right. The internal control confirming equal MNase digestion levels can be found in Supplementary Figure 4B.
The c-fms promoter is the main cis-regulatory element mediating Pax5 repression
The ChIP assay in Figure 1C indicates that the c-fms promoter and FIRE were directly targeted by Pax5. However, it is known that interacting elements could co-precipitate after formaldehyde crosslinking (Murrell et al, 2004). To determine the main element mediating the repression, we performed transient transfection experiments in RAW264 macrophage cells, where c-fms promoter activity was high and where repression could be easily measured. We assayed luciferase constructs carrying combinations of c-fms cis-regulatory elements coupled to the c-fms promoter or the SV40 promoter. We co-transfected the full-length Pax5 cDNA, and also a construct expressing the Pax5 DBD alone, which is sufficient to repress c-fms in Pax5−/− cells (D Eberhard and M Busslinger, unpublished observations). Figure 5A shows that the SV40 promoter was activated by Pax5 overexpression, whereas the c-fms promoter was repressed. Repression of the c-fms promoter was also observed with the Pax5 DBD; however, activation of the SV40 promoter was abolished owing to the lack of an activation domain. Addition of the intronic elements did not abrogate SV40 promoter activation, and did not change the extent of c-fms promoter repression. From these experiments, we conclude that the promoter is the main cis-regulatory element targeted by Pax5. We next performed a deletion analysis to define the Pax5 response element more precisely and localized it to a 35 bp region between −135 and −100 bp relative to the ATG (Figure 5B). Pax5 repressed transcription in all constructs carrying trans-activator (mostly PU.1) binding sites, but repression was lost once we deleted the most proximal PU.1 site.
Figure 5.
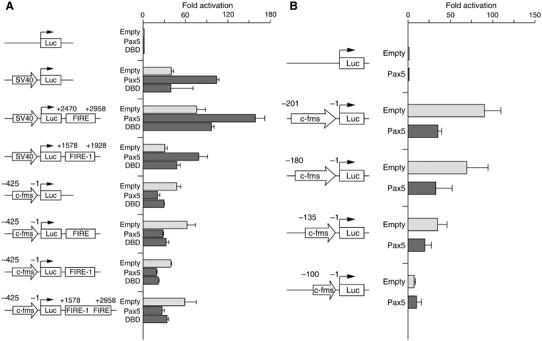
(A) The promoter is the main target of Pax5 repression and the DBD is sufficient for repression. Transient transfection studies with luciferase constructs carrying different combinations of the indicated cis-regulatory elements were carried out in RAW264 cells by co-transfecting with either an empty expression vector or vectors encoding full-length Pax5 or the Pax5 DBD. (B) A 35 bp region in the promoter mediates Pax5 repression. Similar expression levels of transfected Pax5 were controlled by Western blot (Supplementary Figure 8). The fold activation was normalized to the promoter-less reporter gene. The data represent the mean value of three independent experiments.
Pax5 binds to region harboring the main c-fms transcription start sites and represses transcription without abrogating PU.1 binding
To define the Pax5 binding site and to identify the mechanism by which Pax5 represses c-fms, we performed EMSAs with a 51 bp oligonucleotide harboring the Pax5 responsive region and recombinant Pax5 protein (DBD) (Figure 6A). Figure 6B demonstrates that the Pax5 DBD bound to this oligonucleotide. Pax5 DBD bound with approximately the same affinity to the binding site for the B-cell-specific gene Pax5 target gene mb-1, where Pax5 acts as an activator. However, compared to the Pax5 binding site in the B-cell-specific CD19 gene, the binding affinity was significantly lower (Supplementary Figure 7). In order to test whether binding was specific and to obtain information about binding site specificity, we performed EMSAs with oligonucleotides in which specific bases were mutated (Figure 6B), and tested the effect of these mutations on the transcriptional activity of the c-fms promoter in the presence and absence of Pax5 (Figure 6C). Mutation 1 abrogated binding of PU.1 (data not shown) but not of Pax5. A construct harboring this mutation had the same low basal activity as the −100 bp promoter in a transfection assay and was not further repressed by Pax5. This indicates that the elimination of PU.1 binding and the repression by Pax5 reduce transcription to a similarly low level. Constructs that harbored mutations 4 and 10 that did not eliminate Pax5 binding, or mutation 7 that showed weaker binding activity were still repressed by Pax5. Mutations 11, 5, 9 and 8 that eliminated Pax5 binding compromised repression by the full-length Pax5 protein or the DBD alone (Figure 6C and data not shown). The guanine base at −113 bp that was altered by mutation 5 is most likely in direct contact with Pax5, because it became reproducibly hyper-reactive to DMS after Pax5 induction (see Figure 2D, marked with an asterisk).
Figure 6.
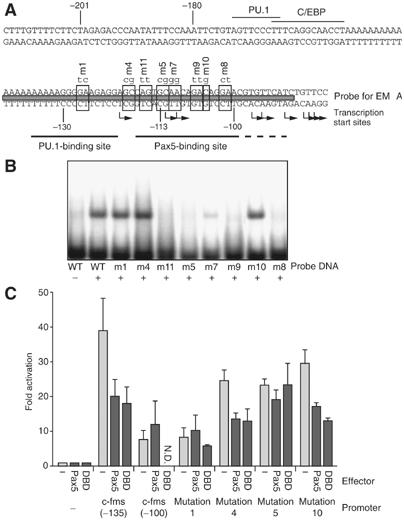
(A) DNA sequence of the c-fms promoter. Indicated are the transcription factor binding sites, the position of the probe used for EMSA, the position of mutations and the different transcription start sites. (B) Pax5 binds to c-fms promoter in a sequence-specific manner. EMSA was performed with an oligonucleotide spanning from −140 to −90 bp and with different mutations as indicated in (A) and recombinant Pax5 DBD. (C) The transcriptional repression by Pax5 is abolished by the mutation of the Pax5 binding site. Point mutations were introduced at the indicated bases within the −135 bp c-fms promoter construct (also see Supplementary Table 1) and these constructs were transfected together with a Pax5 expression plasmid or the empty vector into RAW264 cells. Similar expression levels of transfected Pax5 were controlled by Western blot (Supplementary Figure 8). The data represent the mean value of three independent experiments.
With its paired domain Pax5 makes extended and variable contacts to the DNA bases and phosphate backbone (Czerny et al, 1993; Garvie et al, 2001). From our in vivo data, it was already clear that Pax5 recruitment parallels the loss of RNA polymerase II from the c-fms promoter. However, the same experiments also showed a loss of PU.1, albeit with slower kinetics (Figure 2C). We therefore wanted to investigate whether destabilization of PU.1 binding contributes to the repression of the c-fms promoter by Pax5. To this end, we performed EMSAs with a probe harboring the Pax5 responsive region, nuclear extracts from a wt pro-B-cell line and increasing amounts of recombinant Pax5 (Figure 7). B cells contain PU.1 as well as Pax5, and both bind to the c-fms promoter as demonstrated by EMSA (Figure 7A and B). Specificity of binding was confirmed by abrogation of binding with Pax5- and PU.1-specific antibodies (Figure 7A) and by competition with an excess of Pax5 or PU.1 consensus oligonucleotide (Figure 7B). No Pax5-specific band was observed with extracts from Pax5−/− cells, whereas the PU.1-specific band was still present (data not shown). The addition of increasing amounts of recombinant Pax5 led to the disappearance of both bands, indicating that recombinant and wt Pax5 compete for the same binding site. We also saw the appearance of a new higher molecular weight complex (asterisks in Figure 7B). This new complex contained Pax5 and PU.1, as shown by incubation of the reactions with antibodies specific for PU.1 and Pax5 (data not shown).
Figure 7.
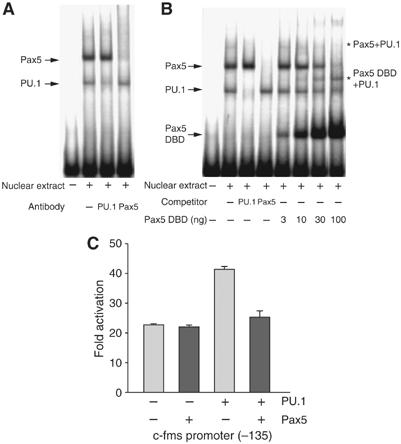
(A) Both PU.1 and Pax5 from pro-B-cell nuclei bind to the c-fms promoter. EMSA was performed with the 140–90 bp probe and 6 μg of nuclear extract from wt pro-B cells. DNA–protein complexes specific for Pax5 and PU.1 indicated by arrows were completely or partially disrupted by addition of antibodies against PU.1 or Pax5 (lanes 3 and 4). (B) Pax5 and PU.1 form a ternary complex at the c-fms promoter. EMSA was performed with the 140–90 bp oligonucleotide, pro-B cells nuclear extract, recombinant Pax5 DBD as well as PU.1- and Pax5-specific competitors. The positions of DNA–protein complexes specific for Pax5, Pax5-DBD and PU.1 are indicated by arrows and the position of higher molecular weight complexes is indicated by asterisks. (C) Pax5 expression abrogates PU.1-dependent transcriptional stimulation in A20 mouse mature B cells. A 35 bp region in the promoter was trans-activated by PU.1 and this activation was diminished by co-transfecting with Pax5.
The data outlined so far indicate that Pax5 binding did not immediately interfere with PU.1 binding in vivo and in vitro, but led to a rapid loss of RNA polymerase II and repression of transcription in vivo. However, removal of PU.1 also abrogated Pax5 repression and reduced c-fms promoter activity to the same background level. This indicated that Pax5 could block activated expression rather than repressing it altogether. To this end, we performed co-transfection experiments in a B-cell line using the −135 bp minimal c-fms promoter carrying the Pax5 response element. Owing to the presence of endogenous Pax5, the activity of the c-fms promoter was repressed to the same lowered activity as in RAW264 cells overexpressing Pax5 (Figure 5B); however, this repression could be overcome by overexpression of PU.1, resulting in an approximately two-fold stimulation of reporter gene activity (Figure 7C). This stimulation was dependent on the PU.1 binding site (data not shown). When Pax5 was coexpressed, this stimulation was abrogated and c-fms promoter activity was reduced to background levels and was not further repressed. These experiments clearly demonstrate that in B cells PU.1 is in direct competition with Pax5 and that Pax5 interferes with PU.1 activity.
A model of Pax5 interaction with the c-fms promoter
In contrast to a previously published report (Maitra and Atchison, 2000), our data show that binding of Pax5 to DNA is required to repress c-fms promoter activity. In order to understand how Pax5 binds to its recognition sequence at the c-fms promoter, we modelled the interaction based on the crystal structure of the Pax5 paired domain bound to the mb-1 promoter (Garvie et al, 2001, PDB code 1K78) (Figure 8A). Our model is consistent with the previously determined Pax5 consensus binding sequence (Czerny and Busslinger, 1995) and shows the N- and C-terminal subdomains of the Pax5 paired domain binding in the major groove and the linker binding in an extended conformation in the minor groove of the c-fms promoter. Our model predicts that many of the major groove base-specific contacts observed in the Pax5-mb-1 DNA structure (Garvie et al, 2001) will be preserved at the Pax5-c-fms DNA interface, and is consistent with the outcome of the mutational analysis.
Figure 8.
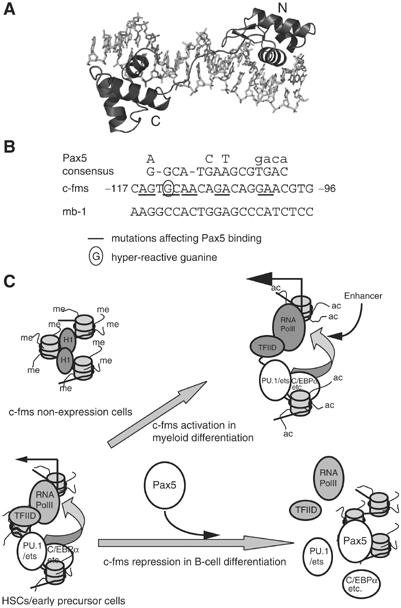
(A) Model for the PAX5/c-fms promoter interaction based on Garvie et al (2001) (PDB code 1K78). The N- and C-terminal subdomains of the PAX5 paired domain are shaded dark gray, and the c-fms promoter DNA is shaded light gray. The sequence of the PAX5 binding site of the c-fms promoter is shown on the right. The illustration was prepared using PYMOL (DeLano Scientific, pymol.sourceforge.net). (B) Alignments of the Pax5 consensus sequence as in Czerny and Busslinger (1995) with c-fms promoter sequences. (C) Model of the different transcriptional states of the c-fms promoter during hematopoiesis. Transcription factors are shown as spheres and nucleosomes are depicted as barrels with DNA wrapped around them and with protruding N-terminal tails. The central promoter nucleosome can be either absent (in myeloid cells) or shows alternate positions as indicated by altered DNA contacts.
Discussion
Pax5 acts on primed, but not yet fully active chromatin
We have previously studied expression, transcription factor occupancy and DNA-methylation status of the c-fms locus in HSCs, and early restricted myeloid as well as lymphoid progenitor cells, and we could show that c-fms mRNA is expressed and occupied by transcription factors in the entire early progenitor cell compartment (Tagoh et al, 2004b). However, our previous data also show that once differentiation along the lymphoid lineage has started, transcription factor binding becomes unstable and binding is lost at the pro-B-cell stage, coinciding with the onset of expression of Pax5. Chromatin in pro-B cells is still in a poised conformation, but lacks inactive histone marks and DNA is not methylated, indicating that c-fms in these cells is not epigenetically silenced. To date, we had not examined the histone modification status of c-fms in cells with multilineage differentiation potential. In multipotent Pax5−/− cells and similar to what is seen in early lymphoid progenitors, transcription factor occupancy was only partial, mRNA expression levels were low and chromatin showed a similar poised structure as in pro-B cells. We infer from these results that in multipotent progenitors and HSCs, c-fms is in a primed but not yet fully active state. We found that chromatin was accessible to transcription factor binding and that nucleosomes had been remodelled, but it had not yet been exposed to signals that activate transcription factors driving high-level expression and extensive chromatin modification.
Another important finding of this study is that the Pax5 DBD was sufficient for the repression of c-fms expression, in contrast to the requirements for the repression of B-cell-specific cis-regulatory elements such as the immunoglobulin heavy chain enhancer. We speculate that here Pax5 represses an active gene carrying stable enhancer complexes where chromatin is in the fully active conformation and which need to be rapidly silenced in response to differentiation stimuli. In this context, Pax5 cooperates with other transcription factors, such as PU.1, and recruits co-repressors that associate with histone deacetylases (Eberhard et al, 2000; Linderson et al, 2004). All these interacting proteins are bound by regions of Pax5 distinct from the paired domain, which are not required for the silencing of c-fms. In addition, the modification state of histones after Pax5 induction is not altered, and thus repression occurs by a different mechanism.
Pax5 destabilizes RNA polymerase II and interferes with PU.1 function
The c-fms promoter belongs to the class of TATA-less promoters and instead contains clusters of PU.1/ets recognition sites that were shown to be sufficient for macrophage-specific transcription initiation (Ross et al, 1998). Mutation of this binding site strongly reduces the activity of the minimal c-fms promoter (this study). It is likely that PU.1 directly contacts the basal transcription machinery. This is supported by biochemical experiments demonstrating that PU.1 can directly contact TFIID and this interaction requires the acidic activation domain of PU.1 (Hagemeier et al, 1993). From the data presented here, we suggest that the binding of Pax5 to DNA interferes with this interaction and renders the c-fms gene unresponsive to activators. As we show here, the consequence of this abrogation of activated transcription is a rapid loss of RNA-polymerase II binding, confirming published data demonstrating that the frequency of initiating a new transcription cycle is crucially dependent on the binding of upstream activators (Metivier et al, 2003). In addition, once bound, Pax5 occupies a region very close to the major transcription start sites and may further destabilize RNA polymerase II binding by steric interference. Albeit with slower kinetics, activator binding and DNaseI hypersensitivity are lost as well, suggesting that the interaction of PU.1 and polymerase II also stabilizes PU.1 binding. We suggest that in growing cells and after each transcription cycle, the repression by Pax5 occurs by a dynamic competitive mechanism rather than by a stable interaction. Figure 8 shows a model of how we envisage the different types of chromatin structure at the c-fms promoter in different hematopoietic cell types, based on the results presented here and previously (Tagoh et al, 2002, 2004b). In c-fms non-expressing cells such as T cells or 3T3 fibroblasts, the promoter is occupied by a specifically positioned nucleosome. DNA is methylated and chromatin carries inactive histone marks. Multipotent cells such as HSCs or Pax5−/− cells represent a primed state where some, but not all, transcription factor complexes are assembled. The nucleosome has moved, but chromatin is in a hypoacetylated modification state and mRNA expression levels are low. This primed state of the promoter is crucially dependent on the presence of PU.1. In parallel studies and using an inducible PU.1 expression system, we could show that c-fms is in an accessible but inactive state in the absence of PU.1. Once PU.1 is induced, the promoter is rapidly reorganized, but activation of the FIRE enhancer occurs with much slower kinetics and parallels cell differentiation (Krysinska et al, manuscript in preparation). This and the study presented here indicate that the crucial element driving low-level transcription in committed myeloid and lymphoid precursor cells is the promoter, while the enhancer is not yet fully assembled. In cells expressing c-fms at maximal levels, such as monocytes, the full complement of transcription factors has bound to all elements and chromatin is hyperacetylated. High-level expression is driven by FIRE, which only becomes fully occupied at the monoblast stage (Tagoh et al, 2002). In lymphoid precursors and pro-B cells, the expression levels of PU.1 and other myeloid factors are downregulated or already absent, but still can drive low-level c-fms expression in the absence of Pax5 (Mikkola et al, 2002; Tagoh et al, 2004b). Pax5 is continuously required to suppress this expression and it does so by blocking the promoter, and the same is most likely true for other genes such as mpo. We believe that a similar balanced interplay between antagonizing transcription factors regulating the activity of cis-regulatory elements of genes, which are primed in HSCs, but not yet fully active, lies at the heart of all cell fate decisions.
Materials and methods
Cell purification and tissue culture
Pro-B cells were purified from the bone marrow of 2-week-old Pax5−/− mice or 4-week-old RAG−/− mice as a c-kit+/B220+ fraction by FACS sorting following myeloid cell depletion using magnetic beads and CD11b, Gr1, ER-MP20 and TER-119 antibodies (Tagoh et al, 2004b). wt and Pax5−/− pro-B-cell lines were cultured on mitomycin C- (Kyowa Hakko) treated ST2 cells in Iscove's modified Dulbecco's medium (IMDM) (Invitrogen), supplemented with 100 U/ml penicillin, 100 mg/ml streptomycin (P/S), 50 μM 2-mercaptoethanol, 1 mM glutamine, 3% heat-inactivated fetal calf serum (FCS), 0.03% (w/v) primatone RL (Quest International) and 1% conditioned supernatant of rIL-7-secreting J558L cells (Rolink et al, 1993). Differentiation of Pax5−/− pro-B cells to macrophages was induced by removal of IL-7 from the culture and monocytic cells were transferred to fresh flasks at day 14 and cultured in IMDM, 10% FCS, 150 μM monothioglycerol, P/S and 10% M-CSF containing L cell conditioned medium. Pax5−/− pro-B cells harboring either Pax5-ER or ΔPax5-ER gene (Nutt et al, 1998) were cultured under the same conditions as Pax5−/− pro-B cells except that phenol red free IMDM was used. Functional Pax5 was induced by adding 1 μM OHT. 3T3 and RAW264 cells were cultured in IMDM with antibiotics and 10% FCS.
In vivo footprinting
In vivo DMS footprinting and LM-PCR were performed exactly as previously described (Tagoh et al, 2004b, 2006). For DNaseI and MNase accessibility assay, cell nuclei were prepared as described by Cockerill (2000) after crosslinking with 1% formaldehyde for 5 min and exposed to increasing amounts of DNaseI or MNase (Worthington). To detect single-strand lesions, purified DNA from nuclease-treated nuclei was directly subjected to LM-PCR (Tagoh et al, 2006). Double-strand breaks created by MNase were phosphorylated by T4-polynucleotide kinase and those created by DNaseI treatment were blunted by using mung bean nuclease (New England Biolabs) at 1 U/μg for 5 min at 30°C. The blunted/phosphorylated double-strand breaks were ligated to the linker and amplified. Primers used for LM-PCR are described by Tagoh et al (2004b) except for the ribosomal DNA promoter (see Supplementary Table 1).
Chromatin immunoprecipitation
Chromatin from cell lines was prepared as described (Lefevre et al, 2003) after crosslinking with 1% formaldehyde. Each precipitation was performed using chromatin from 107 cells and 1 μg of antibody.
Pro-B cells from the bone marrow of RAG2−/− and RAG2−/−Pax5−/− mice (Fuxa et al, 2004) were grown for 5 days on ST2 cells in the presence of IL-7 (Nutt et al, 1997b) prior to crosslinking with 1% formaldehyde for 10 min at room temperature. The cells were lysed in 1% SDS, 10 mM EDTA and 50 mM Tris (pH 8.0), and the crosslinked chromatin was sheared by sonication to an average length of 200–500 bp. The chromatin solution was diluted in 0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris pH 8.0 and 167 mM NaCl, and incubated overnight at 4°C with an affinity-purified antibody (5 μg) directed against the Pax5 paired domain (Adams et al, 1992). After addition of protein A-Sepharose beads, the immune complexes were collected by centrifugation, washed in RIPA buffer and eluted in 1% SDS and 100 mM NaHCO3 followed by reversal of the crosslinks by heating at 65°C for 6 h. Genomic DNA was isolated by phenol extraction and ethanol precipitation before real-time PCR detection of c-fms gene sequences. Precipitated DNA was quantified by using real-time quantitative PCR with SYBR Green. Primers used in this assay are listed in Tagoh et al (2004b). Primers specific for mpo are listed in Supplementary Table 1.
Additional information on materials and methods can be found in Supplementary data.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
Supplementary Figure 8
Supplementary Table 1
Acknowledgments
This work was supported by the Leukaemia Research Fund, the Biotechnology and Biological Sciences Research Council and Yorkshire Cancer Research. H Tagoh is a Kay Kendall Leukaemia Fund fellow. M Busslinger's research is supported by Boehringer Ingelheim. We thank Elisabeth Straszinski (Leeds) and Gabriele Stengl (Vienna) for cell sorting. Work in AJ Warren's laboratory is supported by the Leukaemia Research Fund and the MRC.
References
- Adams B, Dörfler P, Aguzzi A, Kozmik Z, Urbánek P, Maurer-Fogy I, Busslinger M (1992) Pax-5 encodes the transcription factor BSAP and is expressed in B lymphocytes, the developing CNS, and adult testis. Genes Dev 6: 1589–1607 [DOI] [PubMed] [Google Scholar]
- Cockerill PN (2000) Identification of DNaseI hypersensitive sites within nuclei. Methods Mol Biol 130: 29–46 [DOI] [PubMed] [Google Scholar]
- Cotta CV, Zhang Z, Kim HG, Klug CA (2003) Pax5 determines B- versus T-cell fate and does not block early myeloid-lineage development. Blood 101: 4342–4346 [DOI] [PubMed] [Google Scholar]
- Czerny T, Busslinger M (1995) DNA-binding and transactivation properties of Pax-6: three amino acids in the paired domain are responsible for the different sequence recognition of Pax-6 and BSAP (Pax-5). Mol Cell Biol 15: 2858–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czerny T, Schaffner G, Busslinger M (1993) DNA sequence recognition by Pax proteins: bipartite structure of the paired domain and its binding site. Genes Dev 7: 2048–2061 [DOI] [PubMed] [Google Scholar]
- Dai XM, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S, Sylvestre V, Stanley ER (2002) Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood 99: 111–120 [DOI] [PubMed] [Google Scholar]
- DeKoter RP, Singh H (2000) Regulation of B lymphocyte and macrophage development by graded expression of PU.1. Science 288: 1439–1441 [DOI] [PubMed] [Google Scholar]
- Eberhard D, Jimenez G, Heavey B, Busslinger M (2000) Transcriptional repression by Pax5 (BSAP) through interaction with corepressors of the Groucho family. EMBO J 19: 2292–2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enver T, Greaves M (1998) Loops, lineage, and leukemia. Cell 94: 9–12 [DOI] [PubMed] [Google Scholar]
- Follows GA, Tagoh H, Lefevre P, Morgan GJ, Bonifer C (2003) Differential transcription factor occupancy but evolutionarily conserved chromatin features at the human and mouse M-CSF (CSF-1) receptor loci. Nucleic Acids Res 31: 5805–5816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuxa M, Skok J, Souabni A, Salvagiotto G, Roldán E, Busslinger M (2004) Pax5 induces V-to-DJ rearrangements and locus contraction of the immunoglobulin heavy-chain gene. Genes Dev 18: 411–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvie CW, Hagman J, Wolberger C (2001) Structural studies of Ets-1/Pax5 complex formation on DNA. Mol Cell 8: 1267–1276 [DOI] [PubMed] [Google Scholar]
- Graf T (2002) Differentiation plasticity of hematopoietic cells. Blood 99: 3089–3101 [DOI] [PubMed] [Google Scholar]
- Hagemeier C, Bannister AJ, Cook A, Kouzarides T (1993) The activation domain of transcription factor PU.1 binds the retinoblastoma (RB) protein and the transcription factor TFIID in vitro: RB shows sequence similarity to TFIID and TFIIB. Proc Natl Acad Sci USA 90: 1580–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Yamamoto M, Nojima T, Goitsuka R, Kitamura D (2003) Distinct signaling requirements for Dmu selection, IgH allelic exclusion, pre-B cell transition, and tumor suppression in B cell progenitors. Immunity 18: 825–836 [DOI] [PubMed] [Google Scholar]
- Heyworth C, Pearson S, May G, Enver T (2002) Transcription factor-mediated lineage switching reveals plasticity in primary committed progenitor cells. EMBO J 21: 3770–3781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himes SR, Tagoh H, Goonetilleke N, Sasmono T, Oceandy D, Clark R, Bonifer C, Hume DA (2001) A highly conserved c-fms gene intronic element controls macrophage-specific and regulated expression. J Leukoc Biol 70: 812–820 [PubMed] [Google Scholar]
- Jenuwein T, Allis CD (2001) Translating the histone code. Science 293: 1074–1080 [DOI] [PubMed] [Google Scholar]
- Jimenez G, Griffiths SD, Ford AM, Greaves MF, Enver T (1992) Activation of the beta-globin locus control region precedes commitment to the erythroid lineage. Proc Natl Acad Sci USA 89: 10618–10622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontaraki J, Chen HH, Riggs A, Bonifer C (2000) Chromatin fine structure profiles for a developmentally regulated gene: reorganization of the lysozyme locus before trans-activator binding and gene expression. Genes Dev 14: 2106–2122 [PMC free article] [PubMed] [Google Scholar]
- Kulessa H, Frampton J, Graf T (1995) GATA-1 reprograms avian myelomonocytic cell lines into eosinophils, thromboblasts, and erythroblasts. Genes Dev 9: 1250–1262 [DOI] [PubMed] [Google Scholar]
- Lefevre P, Melnik S, Wilson N, Riggs AD, Bonifer C (2003) Developmentally regulated recruitment of transcription factors and chromatin modification activities to chicken lysozyme cis-regulatory elements in vivo. Mol Cell Biol 23: 4386–4400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linderson Y, Eberhard D, Malin S, Johansson A, Busslinger M, Pettersson S (2004) Corecruitment of the Grg4 repressor by PU.1 is critical for Pax5-mediated repression of B-cell-specific genes. EMBO Rep 5: 291–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra S, Atchison M (2000) BSAP can repress enhancer activity by targeting PU.1 function. Mol Cell Biol 20: 1911–1922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIvor Z, Hein S, Fiegler H, Schroeder T, Stocking C, Just U, Cross M (2003) Transient expression of PU.1 commits multipotent progenitors to a myeloid fate whereas continued expression favors macrophage over granulocyte differentiation. Exp Hematol 31: 39–47 [DOI] [PubMed] [Google Scholar]
- Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, Gannon F (2003) Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 115: 751–763 [DOI] [PubMed] [Google Scholar]
- Mikkola I, Heavey B, Horcher M, Busslinger M (2002) Reversion of B cell commitment upon loss of Pax5 expression. Science 297: 110–113 [DOI] [PubMed] [Google Scholar]
- Murrell A, Heeson S, Reik W (2004) Interaction between differentially methylated regions partitions the imprinted genes Igf2 and H19 into parent-specific chromatin loops. Nat Genet 36: 889–893 [DOI] [PubMed] [Google Scholar]
- Nerlov C, Graf T (1998) PU.1 induces myeloid lineage commitment in multipotent hematopoietic progenitors. Genes Dev 12: 2403–2412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerlov C, Querfurth E, Kulessa H, Graf T (2000) GATA-1 interacts with the myeloid PU.1 transcription factor and represses PU.1-dependent transcription. Blood 95: 2543–2551 [PubMed] [Google Scholar]
- Nutt SL, Heavey B, Rolink AG, Busslinger M (1999) Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature 401: 556–562 [DOI] [PubMed] [Google Scholar]
- Nutt SL, Morrison AM, Dorfler P, Rolink A, Busslinger M (1998) Identification of BSAP (Pax-5) target genes in early B-cell development by loss- and gain-of-function experiments. EMBO J 17: 2319–2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt SL, Urbánek P, Rolink A, Busslinger M (1997a) Essential functions of Pax5 (BSAP) in pro-B cell development: difference between fetal and adult B lymphopoiesis and reduced V-to-DJ recombination at the IgH locus. Genes Dev 11: 476–491 [DOI] [PubMed] [Google Scholar]
- Nutt SL, Urbánek P, Rolink A, Busslinger M (1997b) Essential functions of Pax5 (BSAP) in pro-B cell development: difference between fetal and adult B lymphopoiesis and reduced V-to-DJ recombination at the IgH locus. Genes Dev 11: 476–491 [DOI] [PubMed] [Google Scholar]
- Orkin SH (2000) Diversification of haematopoietic stem cells to specific lineages. Nat Rev Genet 1: 57–64 [DOI] [PubMed] [Google Scholar]
- Rolink A, Haasner D, Nishikawa S, Melchers F (1993) Changes in frequencies of clonable pre B cells during life in different lymphoid organs of mice. Blood 81: 2290–2300 [PubMed] [Google Scholar]
- Ross IL, Yue X, Ostrowski MC, Hume DA (1998) Interaction between PU.1 and another Ets family transcription factor promotes macrophage-specific basal transcription initiation. J Biol Chem 273: 6662–6669 [DOI] [PubMed] [Google Scholar]
- Sasmono RT, Oceandy D, Pollard JW, Tong W, Pavli P, Wainwright BJ, Ostrowski MC, Himes SR, Hume DA (2003) A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood 101: 1155–1163 [DOI] [PubMed] [Google Scholar]
- Scott EW, Simon MC, Anastasi J, Singh H (1994) Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science 265: 1573–1577 [DOI] [PubMed] [Google Scholar]
- Simon MC, Olson M, Scott E, Hack A, Su G, Singh H (1996) Terminal myeloid gene expression and differentiation requires the transcription factor PU.1. Curr Top Microbiol Immunol 211: 113–119 [DOI] [PubMed] [Google Scholar]
- Souabni A, Cobaleda C, Schebesta M, Busslinger M (2002) Pax5 promotes B lymphopoiesis and blocks T cell development by repressing Notch1. Immunity 17: 781–793 [DOI] [PubMed] [Google Scholar]
- Tagoh H, Cockerill PN, Bonifer C (2006) In vivo genomic footprinting using LM-PCR methods. In Nuclear Reprogramming, Pells S (ed) pp 285–314. Totowa, NJ: Humana Press [DOI] [PubMed] [Google Scholar]
- Tagoh H, Himes R, Clarke D, Leenen PJ, Riggs AD, Hume D, Bonifer C (2002) Transcription factor complex formation and chromatin fine structure alterations at the murine c-fms (CSF-1 receptor) locus during maturation of myeloid precursor cells. Genes Dev 16: 1721–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagoh H, Melnik S, Lefevre P, Chong S, Riggs AD, Bonifer C (2004a) Dynamic reorganization of chromatin structure and selective DNA demethylation prior to stable enhancer complex formation during differentiation of primary hematopoietic cells in vitro. Blood 103: 2950–2955 [DOI] [PubMed] [Google Scholar]
- Tagoh H, Schebesta A, Lefevre P, Wilson N, Hume D, Busslinger M, Bonifer C (2004b) Epigenetic silencing of the c-fms locus during B-lymphopoiesis occurs in discrete steps and is reversible. EMBO J 23: 4275–4285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbánek P, Wang ZQ, Fetka I, Wagner EF, Busslinger M (1994) Complete block of early B cell differentiation and altered patterning of the posterior midbrain in mice lacking Pax5/BSAP. Cell 79: 901–912 [DOI] [PubMed] [Google Scholar]
- Weissman IL, Anderson DJ, Gage F (2001) Stem and progenitor cells: origins, phenotypes, lineage commitments, and transdifferentiations. Annu Rev Cell Dev Biol 17: 387–403 [DOI] [PubMed] [Google Scholar]
- Xie H, Ye M, Feng R, Graf T (2004) Stepwise reprogramming of B cells into macrophages. Cell 117: 663–676 [DOI] [PubMed] [Google Scholar]
- Yamada T, Abe M, Higashi T, Yamamoto H, Kihara-Negishi F, Sakurai T, Shirai T, Oikawa T (2001) Lineage switch induced by overexpression of Ets family transcription factor PU.1 in murine erythroleukemia cells. Blood 97: 2300–2307 [DOI] [PubMed] [Google Scholar]
- Zhang P, Zhang X, Iwama A, Yu C, Smith KA, Mueller BU, Narravula S, Torbett BE, Orkin SH, Tenen DG (2000) PU.1 inhibits GATA-1 function and erythroid differentiation by blocking GATA-1 DNA binding. Blood 96: 2641–2648 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
Supplementary Figure 8
Supplementary Table 1