

Abstract
The vaccinia-related kinases (VRKs) comprise a branch of the casein kinase family whose members are characterized by homology to the vaccinia virus B1 kinase. The VRK orthologues encoded by Caenorhabditis elegans and Drosophila melanogaster play an essential role in cell division; however, substrates that mediate this role have yet to be elucidated. VRK1 can complement the temperature sensitivity of a vaccinia B1 mutant, implying that VRK1 and B1 have overlapping substrate specificity. Herein, we demonstrate that B1, VRK1, and VRK2 efficiently phosphorylate the extreme N′ terminus of the BAF protein (Barrier to Autointegration Factor). BAF binds to both DNA and LEM domain-containing proteins of the inner nuclear membrane; in lower eukaryotes, BAF has been shown to play an important role during the reassembly of the nuclear envelope at the end of mitosis. We demonstrate that phosphorylation of ser4 and/or thr2/thr3 abrogates the interaction of BAF with DNA and reduces its interaction with the LEM domain. Coexpression of VRK1 and GFP-BAF greatly diminishes the association of BAF with the nuclear chromatin/matrix and leads to its dispersal throughout the cell. Cumulatively, our data suggest that the VRKs may modulate the association of BAF with nuclear components and hence play a role in maintaining appropriate nuclear architecture.
INTRODUCTION
Dynamic protein phosphorylation is mediated by the actions of numerous protein kinases and protein phosphatases. The centrality of this modification as a modulator of cellular function is underscored by the observation that the human genome is thought to encode ∼600 kinases and perhaps one-third that many phosphatases (Manning et al., 2002; MacKeigan et al., 2005). Phosphorylation can modulate such diverse properties as protein localization, protein turnover, enzymatic activity, and protein-protein and protein-nucleic acid interaction. Our laboratory has recently become interested in a family of three proteins known as VRKs, or vaccinia-related kinases, which exhibit a high degree of homology to the B1 protein kinase encoded by vaccinia virus (Nezu et al., 1997; Zelko et al., 1998; Nichols and Traktman, 2004). B1 plays a poorly understood, but essential, role in enabling viral DNA replication to occur; temperature-sensitive mutants affected in B1 fail to accomplish DNA replication at the nonpermissive temperature (Rempel and Traktman, 1992). Interestingly, we have shown that expression of enzymatically active VRK1 from the tsB1 vaccinia virus genome restores viral DNA replication, whereas a catalytically inactive mutant of VRK1 cannot rescue the virus (Boyle and Traktman, 2004). Thus, B1 and VRK1 must share common substrate(s) whose phosphorylation is required for viral replication to occur.
We have therefore initiated further studies of the VRKs as a means to understand the role of B1 and vice versa. Mammalian genomes encode three VRK proteins: VRK1, which is nuclear and enzymatically active; VRK2, which associates with the endoplasmic reticulum (ER) and nuclear envelope and is enzymatically active; and VRK3, which is nuclear and enzymatically inactive due to key amino acid substitutions in catalytic domains (Nichols and Traktman, 2004). Although the participation of VRK1 in the phosphorylation of stress-related transcription factors has been documented (Lopez-Borges and Lazo, 2000; Barcia et al., 2002; Vega et al., 2004; Sevilla et al., 2004a, 2004b), a full appreciation of the biological roles of the VRK proteins awaits genetic dissection. In contrast, recent genetic and pseudogenetic studies of Caenorhabditis elegans and Drosophila melanogaster, each of which encodes a single VRK ortholog, have provided significant clues as to the cellular function of these proteins. The D. melanogaster VRK gene, named ballchen/NHK1, has been shown to encode a protein that possesses histone phosphorylation activity (Aihara et al., 2004). Furthermore, a strain with a hypomorphic mutation in NHK1 shows a recessive phenotype characterized by sterility in both male and female flies due to defects in meiotic spindle formation (Ivanovska et al., 2005). Other independently isolated NHK1 mutations result in flies having oocytes with multiple spindles and increased chromosome condensation during prophase (Cullen et al., 2005). NHK1 hypomorphs also display mitotic defects such as increased mitotic index, overcondensation of chromosomes, and aberrant spindle formation (Ivanovska et al., 2005; Cullen et al., 2005). The function of the C. elegans VRK homolog has been ablated through the use of siRNA-mediated depletion; loss of the VRK protein results in early embryonic lethality. These embryos arrest at the earliest cell division, unable to complete nuclear division and cytokinesis (www.wormbase.org).
One of the most important but also most daunting challenges posed by the study of protein kinases is the identification of bona fide substrates. The studies described in this article emerged from our attempt to screen for cellular substrates that could be phosphorylated by both B1 and VRK1. Using this approach, we have identified the barrier to autointegration factor (BAF) as a substrate for VRK1, VRK2, and the B1 kinase. BAF is a 10-kDa protein that was originally identified in mammalian cells by virtue of its association with the preintegration complexes (PICs) of retroviruses (Lee and Craigie, 1998). In the cytoplasm, it appears to bind to the proviral DNA within the PICs and prevent autointegration (Lee and Craigie, 1994, 1998); once within the nucleus, it appears to facilitate the integration of the proviral DNA into the host chromosome (Suzuki and Craigie, 2002). The functional form of BAF is a homodimer that binds to double-stranded DNA in a sequence-independent manner, making contacts with the phosphate backbone at the minor groove (Zheng et al., 2000; Bradley et al., 2005). BAF dimers are capable of associating with DNA in higher order structures, which can lead to cross-bridging and condensation of the DNA (Lee and Craigie, 1998; Zheng et al., 2000; Suzuki and Craigie, 2002). The solution structure of BAF has been solved by NMR spectroscopy (Cai et al., 1998) and the crystal structure of BAF bound to DNA has very recently been reported (Bradley et al., 2005).
In addition to binding to DNA, BAF can bind to protein components of the inner nuclear membrane (INM) via their LEM domains (Furukawa, 1999; Lee et al., 2001; Shumaker et al., 2001; Holaska et al., 2003; Mansharamani and Wilson, 2005). The latter domain was originally identified in the Lap2, emerin, and MAN1 proteins (Lin et al., 2000). The simultaneous interaction with both DNA and these INM proteins underlies the proposed functions of BAF in interphase and mitosis (Shumaker et al., 2001; Shimi et al., 2004). During interphase, BAF is found predominantly at the INM (Shimi et al., 2004), where it is thought to assist in the anchoring of chromatin to the nuclear periphery. As cells enter mitosis, BAF assumes a diffuse localization, until it begins to colocalize with the core region of chromosome telomeres at late anaphase (Haraguchi et al., 2001; Shimi et al., 2004). At this stage, BAF has been shown to be essential for reassembly of the nuclear envelope, perhaps by recruiting the INM proteins to the membranes that coalesce around the segregating chromosomes (Haraguchi et al., 2001).
BAF is also conserved in lower eukaryotes, and has been studied in both C. elegans and D. melanogaster. Genetic and pseudogenetic studies of BAF in these organisms also provide experimental support for a role for BAF during mitosis (Furukawa et al., 2003). When the D. melanogaster BAF gene was mutated by imprecise P element excision, the resulting null mutants had an embryonic lethal phenotype characterized by mitotic dysfunction (Furukawa et al., 2003). In addition, aberrations in the appearance of interphase chromatin were observed (Furukawa et al., 2003). RNAi-mediated depletion of the BAF protein in C. elegans also engendered an embryonic lethal phenotype before the 100-cell stage (Margalit et al., 2005). The two most striking features of these embryos were the presence of anaphase chromatin bridges and the aberrant condensation of interphase chromatin (Margalit et al., 2005). In sum, these data provide strong support for a role for BAF in the intranuclear organization of chromatin and the enclosure of chromosomes within the reassembling nuclear envelope. BAF has also been shown to be essential for the localization of the LEM domain-containing proteins to the inner nuclear envelope; in presence of a dominant negative BAF (G25E), these proteins are retained in the ER during nuclear reassembly (Haraguchi et al., 2001). These data underscore the importance of BAF in the maintenance of a correct nuclear architecture.
The model described above implies that the association of BAF with chromosomes and with the proteins of the INM must change as the cell cycle progresses. We speculated that protein phosphorylation might modulate these interactions. The studies described herein demonstrate that, in vitro, the VRK enzymes can phosphorylate the N′ terminus of BAF in a manner that has a modest effect on the interaction of BAF with LEM domain-containing proteins, and a dramatic effect on its interaction with DNA. Strikingly, VRK1 can modulate the phosphorylation status and the intracellular localization of BAF in vivo.
MATERIALS AND METHODS
Cell Culture and Reagents
Human thymidine kinase-negative 143B osteosarcoma cells (TK-) and African green monkey BSC40 kidney epithelial cells were maintained as monolayers in DMEM, supplemented with 5% fetal bovine serum (FBS; GIBCO/Invitrogen, Carlsbad, CA) in the presence of 5% CO2. Human osteosarcoma U2OS cells (kindly provided by J. L. Taylor, Medical College of Wisconsin) were maintained in DMEM with 10% FBS. All chemicals were of ultrapure quality from Sigma-Aldrich (St. Louis, MO). Enzymes for nucleic acid manipulation were from Roche (Indianapolis, IN) or Invitrogen. Purified GST-p53 was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). [γ-32P]ATP and 32PPi were obtained from Perkin Elmer-Cetus (Boston, MA). The following plasmids were used in this study: p3XFLAG-7.1 (Sigma), pAcGFP1-C3 (Clontech, Palo Alto, CA), pET15b (Novagen, Madison, WI), pGEX6P-1 (Amersham, Piscataway, NJ) and pTM1-3XFLAG (Nichols and Traktman, 2004; Punjabi and Traktman, 2005). The following commercial antibodies were used in this study: rabbit α-GST (Zymed; San Francisco, CA), affinity-purified M2 α-FLAG monoclonal antibody (mAb; Sigma), and 4A6 α-Myc (Upstate, Lake Placid, NY). Oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA). α-BAF sera were generous gifts from Katherine Wilson (Johns Hopkins Medical School, Baltimore, MD) and Kazuhiro Furukawa (Niigata University, Japan).
Plasmid Construction
Construction of Plasmids for Expression of WT and Mutant BAF Proteins. 1) BAF in vitro expression plasmid: The BAF open reading frame (ORF) was PCR amplified from oligo dT-primed cDNA generated from hTK-143 RNA as described (Nichols and Traktman, 2004), using BAFMTTSQUP and BAF DN. The product was digested with NdeI/BamHI and ligated with pTM1-3XFLAG digested similarly and treated with calf intestinal alkaline phosphatase (CIP). 2) BAF bacterial expression plasmids: pET15b-BAF was generously provided by Dr. Robert Craigie (NIH). Mutations at the amino terminus of BAF were introduced by PCR amplification with BAF-DN in combination with the appropriate upstream mutagenic primer BAFmxxxq-UP (Table 1). PCR products were digested with NdeI/BamHI and ligated with pET15b plasmid that had been digested similarly and treated with CIP. Transformants of the BL21(DE3)pLysS strain of E. coli were generated and used for BAF expression. 3) BAF eukaryotic expression constructs: For expression in mammalian cells, wild-type (WT) BAF was amplified with BAF-DN and the appropriate upstream mutagenic primer BAFmxxxq-UP (Table 1); these PCR products were digested with HindIII/BamHI and ligated to p3XFLAG or pAcGFP1-C3 plasmid DNA that been similarly digested and treated with CIP.
Table 1.
Table of primers
| Primera,b | Sequence |
|---|---|
| BAFMTTSQ UP | TAGGATCCCATATGACAACCTCCCAAAAGC |
| BAFMTTAQ UP | TAGAATTCaagcttCATATGACAACCGCCCAAAAGCACCGAGAC |
| BAFMAASQ UP | TAGAATTCaagcttCATATGGCAGCCTCCCAAAAGCACCGAGAC |
| BAFMTTDQ UP | TAGAATTCaagcttCATATGACAACCGACCAAAAGCACCGAGAC |
| BAFMAAAQ UP | TAGAATTCaagcttCATATGGCAGCCGCCCAAAAGCACCGAGAC |
| BAF DN | GCTGAATTCGGATCCTCACAAGAAGGCG |
| hVRK1Asp UP | TCAGACggtaccACATATGCCTCGTGTAAAGC |
| D177A UP | GCATGGACTATCAAGGCCTCAAATCTTC |
| D177A DN | CCTTGATAGCTCCATGCACATACTCATGC |
| hVRK1 DN | CAGGATCCTTACTTCTGGACTC |
| Emerin UP1 | CGATGGAAGCTTCATATGGACAACTACGCAGATC |
| Emerin DN | TAGGATCCCTAGAAGGGGTTGCCTTCTTC |
| Emerin UP2 | CGTTGGATCCCATATGGACAACTACGCAGATC |
| Emerin LEM DN | AGGATCCCTAGAAGCTATAAGAGGAGG |
Primer-introduced mutations are indicated by underlined and boldface type
Relevant restriction sites used in this study are indicated as follows: BamHI, boldface type; NdeI, italicized; HindIII, lowercase; Asp718/KpnI lowercase boldface type
Construction of Plasmids for Expression of Emerin. GST-LEM: The emerin ORF was amplified by PCR using oligo dT-primed cDNA generated from hTK-143 RNA with the Emerin-UP1 and Emerin-DN primers. This product was digested with HindIII/BamHI and cloned into a similarly digested p3XFLAG vector. This plasmid served as the template for PCR amplification with Emerin-UP2 and Emerin LEM-DN primers; the 183-base pair product, which encodes 61 amino acids that encompass the LEM domain, was digested with BamHI. This product was ligated with pGEX6P-1 vector DNA that had been previously digested with BamHI and treated with CIP, generating the plasmid pGST-LEM. E. coli BL21 transformants containing pGST-LEM were generated and used for protein expression.
Construction of a Catalytically Inactive hVRK1 cDNA Allele. Using overlap PCR, the aspartic acid residue at amino acid 177 was altered to alanine. To produce this mutation, two sets of primer pairs were used to amplify the target region; 1) D177A DN introduced the sense strand mutation and was used with hVRK1Asp-UP, and 2) D177A-UP introduced the complement of the mutation and was used with hVRK1-DN. The pcDNA3-hVRK1MYC (Nichols and Traktman, 2004) plasmid was used as the template for these PCRs. A mixture of the PCR products from the two reactions served as the template in a second round of amplification performed with hVRK1Asp-UP and hVRK1-DN. The final product was digested with Asp718/BamHI and ligated with p3XFLAG that had been similarly digested and treated with CIP. A plasmid expressing a 3XFLAG-tagged version of WT VRK1 was amplified using the same outside primers and cloned into the same vector.
Purification of Recombinant Proteins
The purification of recombinant vvB1, hVRK1, and hVRK2 was performed as previously described (Nichols and Traktman, 2004). Recombinant WT BAF and mutant derivatives were prepared from the insoluble fraction of induced cultures essentially as described (Lee and Craigie, 1998; Umland et al., 2000), except that, after denaturing Ni2+-agarose purification and removal of the amino terminal 6Xhis-tag with thrombin, BAF dimers were isolated by gel filtration on Sephacryl S100 (Amersham) media and concentrated with Centriprep-10 filtration units (Millipore, Bedford, MA). To induce the expression of GST and GST-LEM, BL21 transformants were grown to an OD600 of 0.5 at 37°C and induced at 30°C by the addition of IPTG to a final concentration of 1 mM. Cells were lysed by sonication in bead binding buffer (50 mM sodium phosphate [pH 7.5], 150 mM KCl, 1 mM MgCl2, 1% Triton X-100, 10% glycerol, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mM phenylmethylsulfonyl fluoride [PMSF]). The soluble fraction was retrieved after centrifugation at 15000 × g for 20 min. The concentration of GST and GST-LEM within these lysates was determined by comparison with known amounts of GST in quantitative immunoblot analysis.
Affinity Purification of GST Proteins and GST Coprecipitations
A GST or GST-LEM matrix was generated by incubating lysates containing 3 μg GST or GST-LEM to 25 μl glutathione Sepharose 4B beads (GS-4B; Amersham) for 2 h at 4°C on an end-over-end rotator. Beads were washed twice with 20 bed volumes binding buffer and used in BAF-binding reactions. Binding reactions contained bead binding buffer, 25 μl of GS-4B-GST or -GST-LEM matrix, and unphosphorylated or phosphorylated BAF, in a total volume of ∼335 μl. BAF protein samples were prepared in kinase reactions as described in detail below, except that BAF was used at a concentration of 3 μM and hVRK1 at 30 nM when present. Kinase reactions were terminated by being placed on ice and brought to 10 mM EDTA. Unphosphorylated or phosphorylated BAF, 1.2 μg, was added to the immobilized GST or GST-LEM, and incubations were allowed to continue at 4°C for 2 h with end-over-end mixing. The beads were washed twice with bead-binding buffer; bound proteins were eluted with SDS-PAGE sample buffer, resolved electrophoretically, and subjected to immunoblot analysis with α-GST and α-BAF (K. Wilson) sera.
In Vitro Kinase Assays
Phosphorylation of Cell Lysates. BSC40 cells were disrupted by dounce homogenization in ice-cold homogenization buffer (0.25 M sucrose, 10 mM triethanolamine, 1 mM EDTA, and 10 mM KCl adjusted to pH 7.8 with 100 mM acetic acid), followed by the addition of Triton X-100, 1% final concentration. Lysate from ∼5 × 106 cells was heat-inactivated at 65°C for 20 min before the addition of kinase buffer (final concentrations: 50 mM Tris [pH 7.5], 1 mM dithiothreitol, 1 mM ATP [unless otherwise noted], 10 μCi [γ-32P]ATP, 3000 Ci/mmol) and 100 ng of B1 or hVRK1 kinase. Reactions were performed at room temperature for 30 min before being stopped by the addition of SDS-PAGE sample buffer. Reactions were then fractionated by SDS-PAGE; in some cases, radiolabeled proteins were detected directly by autoradiography. In other cases, the proteins were transferred electrophoretically to nitrocellulose, and the filter was analyzed both by autoradiography and immunoblot analysis with α-BAF sera (K. Furukawa).
Phosphorylation of Purified BAF or GST-p53. Kinase assays utilizing purified BAF or GST-p53 protein as a substrate were performed at room temperature in 25-μl reactions containing kinase buffer and the indicated concentrations of enzyme and WT or mutant protein. After the indicated times, reactions were stopped by the addition of SDS-PAGE sample buffer unless otherwise noted, resolved by SDS-PAGE, and visualized by silver-staining, autoradiography and PhosphorImager analysis (Molecular Dynamics, Sunnyvale, CA; Amersham).
Phosphoamino Acid Analysis. In vitro kinase reactions containing BAF and B1 or VRK1 kinase were carried out for various times in the presence of 20 μCi [γ32P]ATP and 125 μM rATP. Reactions were resolved on a 17% SDS-polyacrylamide gel, transferred to Immobilon P membrane, and visualized by autoradiography. Portions of the membrane corresponding to phosphorylated BAF were excised and hydrolyzed in situ in 100 μl of constant boiling HCl at 110°C for 1 h. Hydrolysates were resolved by thin layer electrophoresis on a HTLE-7000 system (C.B.S. Scientific, Del Mar, CA) at 1800 V for 35 min, as described previously (Traktman et al., 2000). Phosphoserine, phosphothreonine, and phosphotyrosine standards were included and visualized by ninhydrin staining; radiolabeled amino acids were visualized by autoradiography.
DNA-Binding Assays. Binding of BAF to DNA cellulose was assayed as described before (Segura-Totten et al., 2002). Briefly, kinase reactions containing BAF and either B1 or VRK1 kinase were stopped at the indicated times by the addition of 500 μl ice-cold binding buffer (20 mM Tris [pH 7.4], 1 mM EDTA, 100 mM NaCl [with the exception of Figure 5C, in which the NaCl concentration was varied] and 0.1% NP-40) and 50 μl native dsDNA cellulose beads (Amersham). Incubations were allowed to continue overnight at 4°C with end-over-end rotation. Beads were washed three times with binding buffer, and bound proteins were eluted by the addition of SDS sample buffer. Proteins were visualized by immunoblot analysis with α-BAF sera (K. Furukawa). For Figure 7A, 35S-labeled 3XFLAG-BAF was synthesized in vitro using a coupled transcription/translation system (TnT, Promega, Madison WI) programmed with pTM1-3XFLAG-BAF. Lysates were reacted with DNA cellulose as described above and analyzed by SDS-PAGE and autoradiography.
Figure 5.

Modification or mutation of ser4 and/or thr2,3 inhibits the BAF:DNA interaction. (A and B) Purified preparations of WT or mutant BAF dimers were incubated in the absence or presence of hVRK1 (A) or vvB1 (B), with or without ATP, for the indicated times. Ten percent of each reaction was removed and saved for analysis as the load, and the remainder was incubated with DNA cellulose beads in a buffer containing 100 mM NaCl. Both the load (L) and bound (B) fractions were resolved electrophoretically and subjected to immunoblot analysis with α-BAF serum. (C) Untreated preparations of WT BAF or the MTTAQ or MTTDQ variants were incubated with DNA cellulose beads in buffers containing differing concentrations of NaCl, as shown. The input (load) and bound proteins were analyzed as described above.
Figure 7.

Expression of WT 3XFLAG-VRK1 results in a release of WT BAF from the insoluble nuclear matrix fraction of U2OS cells. (A) 35S-labeled 3XFLAG-BAF, synthesized using an vitro transcription/translation protocol, was incubated with DNA cellulose beads. Ten percent of the input (load, L) and all of the bound (B) protein were resolved electrophoretically and visualized by autoradiography. (B) U2OS cells transfected with a plasmid directing the expression of 3XFLAG-BAF were metabolically labeled with 32PPi from 20 to 24 h posttransfection. Cell lysates were subjected to immunoprecipitation with α-BAF antibody; immunoprecipitates were resolved electrophoretically and transferred to nitrocellulose. Total BAF protein was visualized by immunoblot analysis using α-BAF serum (left box); radiolabeled BAF was visualized by autoradiographic analysis of the same filter (right box). The open and filled arrowheads indicate the unphosphorylated and phosphorylated forms of BAF, respectively. (C) 3XFLAG-BAF, 3XFLAG-MAASQ, 3XFLAG-MTTAQ, and 3XFLAG-MAAAQ were expressed in U2OS cells by transient transfection and immunoprecipitated using rabbit α-FLAG antibody. Immunoprecipitates were then treated with λ-phosphatase (P'ase) or mock-treated and subjected to immunoblot analysis using mouse α-FLAG antibody. (D) U2OS cells were transfected with plasmids directing the expression of 3XFLAG-BAF or 3XFLAG-BAF-MAAAQ with or without plasmids directing the expression of hVRK1 or hVRK1-D177A. At 24 h posttransfection, cells were lysed in buffer containing 0.5% Triton X-100 and separated into soluble and insoluble fractions by low-speed centrifugation. Equal volumes of both fractions were subjected to immunoblot analysis with α-FLAG antibody. The open and filled arrowheads indicate the unphosphorylated and phosphorylated forms of BAF, respectively.
Immunofluorescence
U2OS cells, 3 × 105, were seeded in a one-well chamber slide (Lab-Tek, Naperville, IL) 1 d before transfection. Transfection complexes contained 10 μl of Lipofectamine 2000 (Invitrogen) with 2 μg either p3XFLAG-hVRK1 or p3XFLAG-hVRK1 D177A and 2 μg of either pAcGFP-BAF or pAcGFP-BAF MAAAQ. In some cases, the BAF-expressing plasmid was administered individually along with empty vector. At 24 h posttransfection, cells were fixed in 4% PFA, 4% sucrose in phosphate-buffered saline (PBS; 140 mM NaCl, 2 mM KCL, 10 mM Na2HPO4, 1 mM KH2PO4, [pH 7.4]) at room temperature for 15 min, permeabilized with 0.2% Triton X-100 in PBS for 5 min at room temperature and then blocked in 10% BSA in PBS for 30 min. Cells were then reacted with α-FLAG antibody at a dilution of 1:200 in 3% BSA in PBS, followed by goat anti-mouse-594 (Molecular Probes) at 1:300 in PBS. DNA was stained with DAPI; GFP-BAF was detected by direct fluorescence on Nikon Eclipse TE2000-U microscope (Melville, NY). Images were pseudocolored and labeled using Canvas 8 (Deneba Systems, Miami, FL).
Subcellular Fractionation and Immunoprecipitation
Cell Fractionation. Approximately 3 × 106 U2OS cells were transfected with 4 μg of p3XFLAG-BAF or the indicated mutant plasmid and 4 μg of p3XFLAG-hVRK1, p3XFLAG-hVRK1 D177A, or empty vector using Lipofectamine, 2000. At 24 h posttransfection the cells were resuspended in ice-cold fractionation buffer (20 mM Tris pH 7.4, 5 mM MgCl2, 5 mM DTT, 150 mM KCl, and 0.5% Triton X-100) containing 1 mM NaF, 1 mM sodium orthovanadate, 40 mM β-glycerol phosphate, 1 μg/ml leupeptin, 1 μg/ml pepstatin, and 1 mM PMSF. After 10 min of incubation on ice, sucrose was added to 10% and the insoluble fraction was retrieved by centrifugation at 1000 × g for 10 min. The supernatant, which contained the cytoplasmic/nucleosolic fraction, and the pellet were resolved by SDS-PAGE and subjected to immunoblot analysis with α-FLAG M2 antibody (Sigma). When indicated, cells were radiolabeled by replacing the media with phosphate-free DMEM (MP Biomedical, Irvine, CA) supplemented with 100 μCi/ml 32PPi from 20 to 24 h posttransfection, and 3XFLAG-BAF was then retrieved from the soluble fraction of radio-labeled extracts using M2 affinity agarose, washed, and eluted in SDS sample buffer before analysis by immunoblot and autoradiography.
Immunoprecipitation and Phosphatase Treatment. Transfected cells were harvested by scraping in PBS and lysed by incubation in 1 ml immunoprecipitation buffer (50 mM Tris [pH 7.4], 500 mM NaCl, 1% Triton-X 100, 0.1% SDS, 1 mM EDTA with 1 mM NaF, 40 mM β-glycerol phosphate and protease inhibitors) for 15 min. Particulate matter was removed by centrifugation at 1000 × g for 10 min. 3XFLAG BAF and mutants were immunoprecipitated from the soluble fraction with rabbit α-FLAG and protein A-Sepharose. Immune complexes were washed twice with immunoprecipitation buffer and twice with phosphatase pretreatment buffer (25 mM Tris [pH 7.4], 150 mM NaCl, and 0.01% Brij-35; Pierce, Rockford, IL). On the final wash in this buffer, complexes were divided and treated with or without λ protein phosphatase (New England Biolabs; Ipswich, MA) in the provided buffer. Immunoprecipitates treated thus were analyzed by immunoblot analysis with mouse α-FLAG.
Computer Analysis
Autoradiography films were scanned using an Epson 1680 scanner (Epson, Long Beach, CA). All sequence analyses were performed with Lasergene Software (DNASTAR, Madison, WI). Figures were labeled using Canvas software. Where immunoblot signal intensity comparisons are presented (i.e., Figures 4 and 5), chemiluminescent signals were gathered with a FlourChem 8800 and densitometry determined with FluorChem 8900 software (Alpha Innotech, San Leandro, CA.). The BAF structural model in Figure 8 was created using Cn3D 4.1 software and BAF structure PDB 1CI4.
Figure 4.
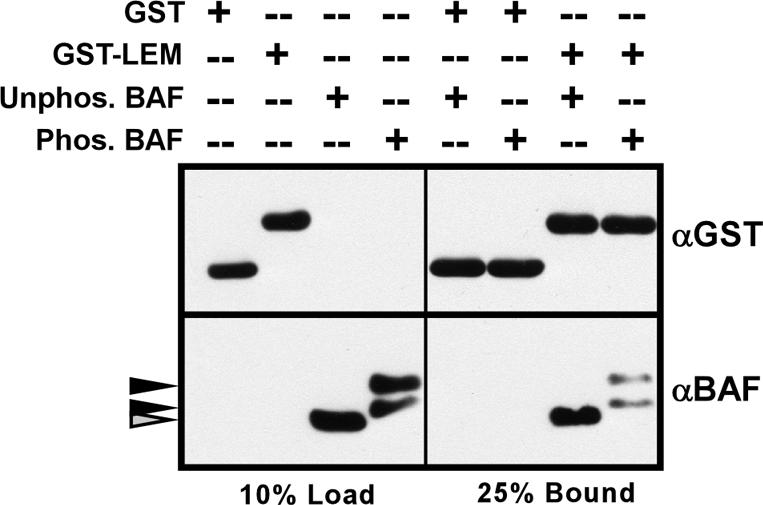
Phosphorylation of BAF decreases its association with the LEM domain. GST or GST-LEM were bound to glutathione agarose beads to generate affinity matrices, which were then incubated with untreated BAF (unphos. BAF) or BAF that had been phosphorylated to completion with hVRK1 (phos. BAF). Ten percent of the input proteins (load) and 25% of the bound proteins (bound) were resolved by SDS-PAGE and analyzed by immunoblot with α-GST and α-BAF.
Figure 8.

Model of BAF regulation. The interaction of BAF with LEM domain-containing proteins within the inner nuclear membrane, and with DNA, can be negatively regulated through the phosphorylation of its N terminus by VRK1.
RESULTS
VRKs represent a distinct branch of the casein kinase family of serine/threonine kinases; they were so named because of their high homology to the vaccinia B1 serine/threonine kinase. The homology between these viral and eukaryotic kinases led to the hypothesis that they might have overlapping substrate specificities and hence overlapping functions. Indeed, human, mouse and Drosophila VRK1 can complement the DNA replication defect that characterizes nonpermissive infections performed with a vaccinia virus mutant encoding a temperature-sensitive B1 protein (Boyle and Traktman, 2004 and K. Boyle and P. Trakman, unpublished results). Although there are no data implicating the VRKs in cellular DNA replication per se, siRNA depletion of C. elegans VRK leads to an early embryonic lethality characterized by impaired nuclear division (www.wormbase.org). Likewise, mutations in the D. melanogaster homolog of VRK (NHK1) result in defects in chromosomal segregation during mitosis and meiosis (Cullen et al., 2005; Ivanovska et al., 2005). The work described herein, therefore, was stimulated by our hypothesis that the identification of substrates that could be readily phosphorylated by both VRK1 and B1 would provide important clues as to the biological roles performed by these kinases.
Phosphorylation of Whole Cell Lysates by the VRK1 and B1 Kinases
As a first attempt to identify unknown cellular substrates for VRK1 and B1, we performed in vitro kinase assays using extracts of BSC40 cells as a source of substrates. The extracts were heat-inactivated before being assayed so that only the exogenously added kinases would direct the incorporation of 32P-ATP into target substrates (Figure 1A). Few if any phosphorylated species were observed when no exogenous kinase was added; purified B1 kinase, in contrast, phosphorylated many proteins within these extracts. When purified VRK1 kinase was utilized in the same assay, only two phosphorylated proteins were observed. A 50-kDa species corresponding to autophosphorylated VRK1 was seen, as was a radiolabeled band migrating at ∼10 kDa. Interestingly, B1 also appeared to target a protein(s) of the same electrophoretic mobility (arrow).
Figure 1.

BAF is a substrate for phosphorylation by recombinant B1 and VRK1 kinases. (A) Phosphorylation of heat-killed cell lysates by B1 and VRK1 kinases. Heat-killed lysates of BSC40 cells were incubated with [γ-32P]ATP in the absence (no kinase) or presence of purified vvB1 or hVRK1 kinases. Reaction products were resolved electrophoretically and visualized by autoradiography; the numbers at the left indicate the molecular weights of 14C-labeled protein standards. Autophosphorylated hVRK1, and a protein postulated to be BAF, are also indicated. (B) The BAF protein appears to become phosphorylated in lysates treated with B1 or hVRK1. The reactions shown in A were transferred to nitrocellulose and subjected to immunoblot analysis with an α-BAF serum (left panel) and autoradiography. Unmodified BAF is indicated with a black arrow; the two immunoreactive BAF species that comigrate with the 32P-labeled signal are indicated with gray arrows. (C) Recombinant GST-p53 or dimeric BAF (1 μM) were tested for their ability to serve as hVRK1 substrates by performing in vitro kinase assays in the presence of [γ-32P]ATP for 0-60 min. A graphic representation of the incorporated radiolabel is shown. The corresponding silver-stained gel and autoradiograph of the reactions performed with GST-p53 is shown in the inset.
Data generated in a yeast two-hybrid screen of the entire Drosophila proteome had previously identified three proteins as able to interact with the Drosophila VRK protein (Alvarez et al., 1995; Giot et al., 2003). One was a protein of unknown function, predicted to contain a bipartite nuclear localization sequence and to have an unusually high (17%) proline content. A second protein identified was norpA, which has been previously shown to have phospholipase activity (Alvarez et al., 1995). The third protein, which showed the strongest interaction with dVRK, was the D. melanogaster ortholog of the DNA-binding protein Barrier to Autointegration Factor (BAF). Mammalian BAF was originally identified as a 10-kDa cellular protein that interacted with retrovirus preinteraction complexes and prevented intramolecular recombination by the proviral DNA, thereby promoting integration of the proviral DNA into the cellular genome (Lee and Craigie, 1994, 1998). Because BAF has a molecular weight similar to the protein we observed being phosphorylated by VRK1 and B1 in whole cell lysates (Figure 1A), we hypothesized that the latter protein might indeed be BAF. We therefore obtained an α-BAF antibody from K. Furukawa, which we used to perform immunoblot analyses of the same lysates shown in Figure 1A. Immunoreactive species were visualized by chemiluminescent detection, and phosphorylated species were visualized by autoradiographic analysis of the same filter. We observed three immunoreactive bands in the samples that had been phosphorylated by the exogenous kinases: one band comigrated with the band seen in untreated samples and the other two comigrated with the 32P-labeled bands.
VRK1 Phosphorylates Purified BAF In Vitro
The data described above strongly suggest that BAF present within cellular extracts becomes phosphorylated when incubated with the VRK1 or B1 kinases. To confirm this directly, we purified recombinant BAF in a dimeric form and used it as a substrate for in vitro kinase assays. These assays contained 40 nM VRK1, 1 μM BAF, and [γ-32P]ATP and were performed for 0-60 min. For comparison, we performed parallel assays using a GST-p53 fusion protein as a substrate. Previous studies have described VRK1-mediated phosphorylation of GST fusion proteins containing fragments of the tumor suppressor protein p53 (Lopez-Borges and Lazo, 2000; Barcia et al., 2002; Vega et al., 2004); in particular, VRK1 has been shown to phosphorylate p53 on thr18 (Lopez-Borges and Lazo, 2000). The reaction products were electrophoretically resolved, silver-stained to ensure that equal levels of protein were present, and then excised from the gel and subjected to Çerenkov counting. As shown in Figure 1C, VRK1-mediated phosphorylation of BAF was rapid and robust. In contrast, the autoradiographic signal for GST-p53 was nearly undetectable (inset), and at 60 min, ∼180-fold more radiolabel had been incorporated into BAF than into p53. Thus, VRK1 phosphorylates BAF with an efficiency that greatly exceeds what is seen for GST-p53.
BAF Is Phosphorylated on ser4 and Less Efficiently on thr2 and/or 3
The extreme N′ terminus of BAF is highly conserved in different species and contains two thr and one ser residues that represent potential targets for phosphorylation (thr2,3 and ser4). As an initial test of whether this region of the molecule might contain the site(s) of phosphorylation, we used a coupled in vitro transcription/translation system to express WT BAF and a variant in which residues 2-4 had been replaced by an ala residue, and determined that VRK1 could phosphorylate the former but not the latter protein (our unpublished data). To precisely map the sites of N′-terminal phosphorylation and determine the biochemical impact of this modification, we generated alleles of BAF that contained the following amino acid substitutions: MTTSQ (WT) → MTTA4Q, MA2A3SQ, MTTD4Q, and MA2A3A4Q. Using previously established protocols (Lee and Craigie, 1998; Umland et al., 2000), we induced the expression of these proteins in E. coli and prepared purified preparations of the dimeric form of the WT and mutant BAF proteins. Each of the proteins analyzed displayed comparable chromatographic profiles.
VRK1-mediated Phosphorylation of BAF. We analyzed the ability of VRK1 to phosphorylate the WT and mutant forms of BAF by performing in vitro kinase assays for 0-120 min. Reactions containing 3 nM VRK1 and 3 μM BAF were resolved by SDS-PAGE and the BAF protein was visualized by silver-staining (top panels) and autoradiography (bottom panels), as shown in Figure 2A. For WT BAF, as the time course of phosphorylation progressed, we observed the original BAF protein (gray arrowhead) being converted to two more slowly migrating species (black and white arrowheads). The middle species (black arrowhead) was observed after only 1 min of phosphorylation, and 50% of the BAF protein was present in this form after 2 min of phosphorylation. By 10 min, all of the protein had been shifted to this middle species (corresponding to 1 phosphorylation event/∼0.5 s). At this time, the most slowly migrating form (white arrowhead) had also begun to appear; at 120 min, perhaps 20% of the total protein was present in this uppermost form. The autoradiograph shows that both the middle and upper forms did indeed represent phosphorylated species. A more quantitative profile of phosphorylation is shown in Figure 2C, which shows a graphic representation of the level of phosphorylation determined by phosphorimage quantitation of each lane. The plot for VRK1-mediated phosphorylation of WT BAF is shown with black circles: a biphasic profile is seen. The first phase, which is of ∼10 min in duration, represents rapid and robust phosphorylation, which corresponds to the generation of the middle species shown in Figure 2A. A second slower phase then ensues, with a gradual rise in 32P incorporation that continues until 120 min; this phase corresponds to the conversion of some of the BAF protein to the uppermost form seen in Figure 2A.
Figure 2.

In vitro phosphorylation of recombinant BAF by human VRK1, VRK2, and vvB1. Preparations of WT (MTTSQ) and mutant (MAAAQ, MAASQ, MTTAQ, MTTDQ) BAF dimers (3 μM) were used as substrates in in vitro kinase reactions performed with hVRK1 (A and C), B1 (B and D), or VRK2 (E) in the presence of [γ-32P]ATP. In all cases, the substrate was present at 3 μM and the enzyme at 3 nM; reactions were performed for 0-120 min, as indicated. Reaction products were analyzed by SDS-PAGE and visualized by silver staining and autoradiography (A and B); phosphorylation was also quantitated by phosphorimage analysis, as shown graphically in C-E. For A and B, the gray arrowheads indicate the input protein, and the black and white arrowheads indicate electrophoretically distinct, phosphorylated forms of BAF.
We then performed parallel analyses of the mutant forms of BAF in which the N′-terminal ser and/or thr residues had been changed to ala. When thr2, thr3, and ser4 were simultaneously changed to ala, the ability of VRK1 to phosphorylate BAF was lost (Figure 2A, and lines with filled squares in Figure 2C). These data confirm that all of the sites phosphorylated by VRK1 lie within this extreme N′ terminus of BAF. Next, we examined the MAASQ allele, in which the two thr residues had been altered. As shown in Figure 2A, the middle species (black arrowhead) appeared with the same kinetics seen with WT BAF, and all of the BAF protein was converted to this species. As shown in Figure 2C (compare lines with filled triangles and filled circles), the initial, rapid phase of phosphorylation of the MAASQ protein was indistinguishable from what we observed for WT BAF. However, in this case, there was no second, slow phase of phosphorylation—the levels of 32P incorporated had reached a plateau after 10 min of phosphorylation, and no upper species (white arrowhead) was seen by silver staining or autoradiography. Thus, we conclude that the initial, rapid phase of phosphorylation of WT BAF (and the appearance of the middle form seen in Figure 2A) represents phosphorylation of ser4; the second phase of phosphorylation represents slow and incomplete phosphorylation of thr2 and/or thr3. This conclusion was supported by the analysis of the MTTAQ protein, in which the target ser residue had been altered. Here, the rate of phosphorylation was significantly slower (6-fold reduction in slope; Figure 2C, open circles) and the ultimate level of 32P incorporation was approximately one-half of that seen with WT BAF. Examination of the silver-stained gel and the autoradiograph confirms that a single shifted species accumulated slowly. These data confirm that ser4 is the preferred site of phosphorylation and that phosphorylation of thr2 and/or thr3 does occur, albeit at a significantly slower rate. Finally, we examined the phosphorylation rate of the MTTDQ form of BAF, in which ser4 had been replaced with an asp in an attempt to observe the rate of thr2 and/or thr3 phosphorylation when a negative charge mimicking “prephosphorylation” was present at residue 4. Under these circumstances, the rate of phosphorylation of thr2 and thr3 is slowed even further, suggesting that the initial rapid phosphorylation of ser4 by VRK1 may interfere with subsequent phosphorylation of thr2 and/or thr3. These data may explain why only ∼20% of WT BAF undergoes the thr phosphorylation events that generate the uppermost species, whereas all of the MTTAQ protein can be shifted to a phosphorylated form.
B1-mediated Phosphorylation of BAF. In light of our observation that the B1 kinase could also phosphorylate BAF in whole cell lysates, we sought to compare its target sites with those found for VRK1. Purified B1 was therefore incubated with WT and mutants forms of BAF in kinase assays similar to those described for VRK1. Again, phosphorylation was monitored both by visualizing electrophoretic shifts (Figure 2B) and by quantitating 32P incorporation (Figure 2D). As we had seen with VRK1, B1 induced the formation of two phosphorylated forms of BAF that migrated more slowly during SDS-PAGE. The rate of B1-mediated phosphorylation of WT BAF was also similar: 3 nM kinase could phosphorylate 3 μM WT BAF in ∼10 min, as measured by a complete shift of the unphosphorylated BAF to the middle protein species (black arrowhead; Figure 2B, lane 5). B1 could not phosphorylate the BAF-MAAAQ protein, indicating that the site(s) for B1-mediated phosphorylation were also contained within the extreme N′ terminus of BAF (Figure 2D, filled squares). Phosphorylation of the MAASQ variant by B1 closely approximated what had been seen with VRK1; the initial rate of phosphorylation was the same as for WT BAF (Figure 2D, filled triangles vs. filled circles), but the second, slow phase of phosphorylation was absent and hence a lower plateau was reached. Again, phosphorylation of the MTTAQ variant proceeded significantly more slowly, with a 10-fold decrease in the initial reaction rate. These results for B1 closely mirrored those found for VRK1 and indicate that B1 targets ser4 preferentially, but is able to modify thr2 and/or 3 given additional time. Finally, we assayed the rate at which B1 phosphorylates the BAF-MTTDQ variant of BAF, in order to determine whether substituting a negatively charged residue at the ser4 position would impair the phosphorylation on thr2 and/or thr3, as we had observed for VRK1. However, two shifted forms were observed after phosphorylation (black and white arrowhead, Figure 2B), and the rate of phosphorylation was indistinguishable from that observed for the MTTAQ variant (Figure 2D, compare open triangles with open circles). Thus, the presence of the asp residue at position 4 adversely affected the phosphorylation of thr2 and/or thr3 by VRK1, but not by B1.
BAF Is Also Phosphorylated by VRK2. We previously described the relationship of the VRK paralogs to each other and to the B1 kinase (Nichols and Traktman, 2004). The VRK1 and VRK2 enzymes share a significant amount of identity within their catalytic domains, and both are indeed catalytically active. Although VRK1 is predominantly nuclear, VRK2 is found associated with the ER and the nuclear envelope. Because the catalytic domains of VRK1 and VRK2 are closely related, we also assayed purified VRK2 kinase for its ability to phosphorylate WT and variant preparations of BAF in vitro. These data are shown graphically in Figure 2E. VRK2 does indeed phosphorylate WT BAF with similar kinetics to VRK1, and moreover, the pattern of phosphorylation obtained with the various BAF mutants is indistinguishable from what was observed with VRK1 (compare Figure 2E with 2C).
Phosphoamino Acid Analysis. Further confirmation that ser4 is the preferred site of phosphorylation by VRK1 and B1 was provided by performing phosphoamino acid analysis of phosphorylated WT BAF or BAF-MTTAQ. In these reactions, the total concentration of ATP was reduced eightfold to allow us to increase the specific activity and hence augment the radioactive signal. Under these more limiting conditions, only a single electrophoretically shifted species was observed after VRK1-mediated phosphorylation of WT BAF (unpublished data). The phosphoamino acid analysis indicates that VRK1 only phosphorylates ser under these conditions (Figure 3, lanes 1-3). Phosphoserine could be detected by 1 min after the initiation of the kinase assay and increased in abundance throughout the 60-min time course; in contrast, even after 60 min, <1% of the radiolabel was found at the position of phosphothreonine. When the same analysis was performed on BAF-MTTAQ (Figure 3, lanes 4-6), phosphoserine was no longer observed, as would have been expected from the results shown in Figure 2. In contrast, the appearance of phosphothreonine was accelerated. Thus, phosphorylation of ser4 appears to diminish the efficiency with which thr 2 and/or thr3 are phosphorylated.
Figure 3.
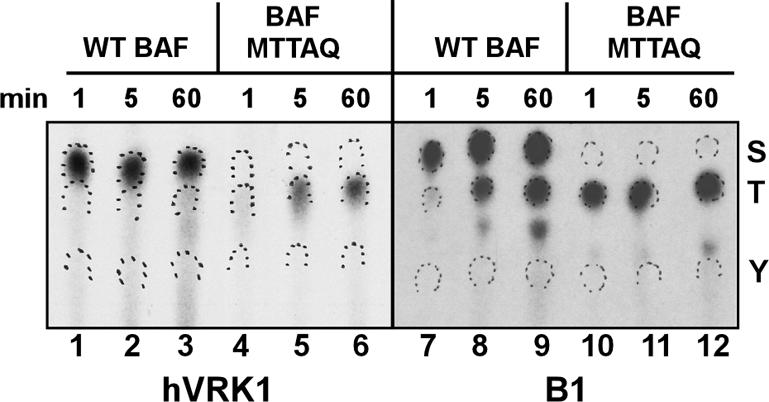
Phosphoamino acid analysis of B1- and VRK1-phosphorylated BAF proteins. WT BAF and the MTTAQ variant were phosphorylated in vitro by VRK1 or vvB1 for 1, 5, and 60 min. Reaction products were resolved electrophoretically, transferred to PVDF, excised, and processed for hydrolysis in situ. The hydrolysates were subjected to high-voltage thin-layer electrophoresis in the presence of phosphoamino acid markers (S, phosphoserine; T, phosphothreonine; Y, phosphotyrosine). Radiolabeled phosphoamino acids were visualized by autoradiography; the markers were visualized by color development and are demarcated by ink wickets.
When WT BAF was incubated with B1 (Figure 3, lanes 7-9), phosphoserine was detected in significant levels after 1 min of incubation and increased in intensity during the rest of the time course; phosphothreonine was observed in significant levels after 5 min of incubation and also increased substantially during the rest of the time course. As expected, we observed no phosphoserine when kinase reactions containing BAF-MTTAQ and B1 were performed. Instead, phosphothreonine was observed in significant levels after 1 min of incubation (Figure 3, lane 10). Thus, the B1 kinase also phosphorylates thr2 and/or thr3 more rapidly in the absence of ser4 phosphorylation, but its overall preference for ser4 versus thr2/3 is less absolute than is VRK1s under these reaction conditions.
Phosphorylation of BAF Has a Modest Effect on Its Interaction with the LEM Domain
BAF has been shown to form specific interactions with three components of the inner nuclear envelope, Lap2, emerin, and MAN1, through a common motif known as the LEM domain (Acharya et al., 1996; Lee et al., 2001; Shumaker et al., 2001; Holaska et al., 2003; Mansharamani and Wilson, 2005). We were interested in determining whether phosphorylation of BAF might affect its interaction with the LEM domain. Glutathione sepharose beads containing immobilized GST or GST-LEM were incubated with BAF or phosphorylated BAF, and the efficiency with which the BAF species bound to the matrix was determined by immunoblot assay (Figure 4). We consistently observed that 2-3-fold more untreated BAF bound to the immobilized GST-LEM than did phosphorylated BAF. Thus, phosphorylation of BAF has a modest but reproducible impact on the BAF-LEM interaction.
Phosphorylation of ser4 and/or thr2/3 Disrupts BAF's Ability to Bind DNA
A second and important property of BAF is its ability to bind dsDNA tightly and without sequence preference. Previous structure/function studies have implicated lys6 as participating in DNA binding (Harris and Engelman, 2000; Umland et al., 2000; Segura-Totten et al., 2002); the proximity of this residue to the MTTSQ motif was provocative. We therefore probed the ability of untreated and phosphorylated BAF to bind to a DNA cellulose affinity resin at near physiological concentrations of NaCl (100 mM). BAF, 3 μM, was preincubated with or without ATP, in the presence or absence of VRK1, for 7.5 or 90 min. These time points, selected based on the time courses shown in Figure 2, were chosen to represent either partially (primarily ser4) or fully (ser4 and thr2/3) phosphorylated BAF. Parallel experiments were performed using the B1 kinase, except that only a 60-min time point, representing full phosphorylation of BAF, was used. Each of these samples was then incubated with DNA-cellulose, and the load and bound fractions were subjected to immunoblot analysis (Figure 5). We observed that ∼50% of the untreated BAF bound to the resin under these reaction conditions; prior incubation with ATP alone or kinase alone did not alter this profile. However, if BAF had been phosphorylated by either B1 or VRK1, its ability to bind to DNA was severely inhibited; <0.5% of the input BAF bound to the resin (Figure 5, A and B, top panels). This effect was seen after 7.5 or 90 min of incubation with VRK1, suggesting that phosphorylation of ser4 was sufficient to abrogate DNA binding.
To gain further insight into the relationship between phosphorylation and the loss of DNA binding, similar studies were performed with the BAF variants described above in Figure 2. Untreated BAF-MTTAQ and BAF-MAASQ bound to the DNA cellulose with similar efficiency to WT BAF, and both proteins lost this ability after phosphorylation. These data indicate that phosphorylation of either ser4 or thr2/3 by VRK1 or B1 is capable of inhibiting BAF's ability to bind DNA. The DNA binding ability of BAF-MAAAQ, in contrast, was unaffected by incubation with kinase + ATP. Somewhat surprisingly, untreated BAF-MTTDQ protein bound to DNA cellulose with the same efficiency as WT BAF, despite the presence of an asp residue at the position of ser4. Apparently, the presence of a negative charge at this position does not have the same effect as acquisition of a phosphate group. However, when the MTTDQ protein was phosphorylated at thr2/3 with VRK1 or B1, it did lose its ability to bind to the DNA cellulose.
Because of the unexpected observation that BAF-MTTDQ was still able to bind to DNA cellulose under our experimental conditions, we performed additional binding studies in which WT and mutant BAF proteins were applied to the DNA cellulose resin in the presence of increasing concentrations of salt (Figure 5C). For WT BAF, raising the concentration of NaCl to 700 mM salt only led to a 20% decrement in the amount of protein that bound to the DNA cellulose resin. These data agree with the previous observation that BAF remains bound to retroviral PICs until >750 mM NaCl is applied (Suzuki and Craigie, 2002). The binding of the BAF-MTTAQ protein was somewhat more salt-sensitive; increasing the salt concentration to 300, 500, or 700 mM NaCl led to 38, 73, and 81% decreases in the amount of protein that bound to the resin, respectively. The BAF-MTTDQ protein was significantly more salt-sensitive; in the presence of 300 mM salt, we observed an 80% decrease in the amount of BAF bound to the DNA cellulose, and there was a >97% reduction when the salt was raised to 500 or 700 mM NaCl. These data confirm that ser4 has an important role in mediating the BAF-DNA interaction, and that modifications to this residue have an inhibitory impact that follows the hierarchy ser→pser > ser→asp > ser→ala.
The Phosphorylation Status and Subcellular Distribution of BAF Changes upon Overexpression of VRK1
The data described above provide strong evidence that BAF is an excellent substrate for VRK1-mediated phosphorylation and that this phosphorylation has a modest effect on the interaction of BAF with LEM-containing proteins and a dramatic effect on its interaction with DNA. To obtain preliminary corroboration that VRK1 would affect BAF in a similar manner in vivo, we generated reagents that allowed the expression of epitope-tagged VRK1 and/or BAF in cultured cells. We prepared vectors that directed the expression of 3XFLAG-BAF, 3XFLAG-BAF-MAAAQ, GFP-BAF, and GFP-BAF-MAAAQ. We also generated constructs expressing 3XFLAG-VRK1 and 3XFLAG-VRK1D177A; the amino acid substitution in the latter allele affects a critical residue involved in coordinating ATP via a Mg2+ salt bridge and hence ablates catalytic activity.
Analysis of endogenous VRK1, or transiently expressed variants containing N′ or C′ epitope tags, has revealed that the protein is found to be localized throughout the nucleus. (Lopez-Borges and Lazo, 2000; Sevilla et al., 2004a, 2004b; Nichols and Traktman, 2004). This nuclear localization was seen once again when we transiently expressed 3XFLAG-VRK1 on its own (unpublished data). Similar, transiently expressed GFP-BAF or GFP-BAF MAAAQ localized to the nucleus when expressed alone (Figure 6), as expected from previous studies of BAF localization (Segura-Totten et al., 2002; Shimi et al., 2004).
Figure 6.

Expression of WT VRK1 results in the relocalization of WT GFP-BAF to the cytoplasm of U2OS cells. U2OS cells were transfected with plasmids directing the synthesis of GFP-BAF or GFP-BAF-MAAAQ, with or without plasmids directing the synthesis of 3XFLAG-hVRK1 or 3XFLAG-hVRK1D177A. At 24 h posttransfection, cells were fixed and stained with α-FLAG mAb and a fluorescent-tagged secondary antibody, and DAPI. Representative fields were photographed using filters appropriate to visualize GFP-BAF, VRK, and DNA. Bar, 50 μm.
When 3XFLAG-VRK1 and GFP-BAF were introduced together, there was an increased pool of cytoplasmic BAF; indeed BAF appeared to be found throughout the cell (arrows, Figure 6). Surprisingly, in these cells, we also observed that VRK1 was now dispersed throughout the cell. This change in BAF localization was not seen when GFP-BAF was coexpressed with the catalytically null VRK1D77A, indicating that VRK1-mediated phosphorylation events were driving the relocalization of BAF. Support for the conclusion that it was the phosphorylation of BAF itself that was causing its relocalization came from our observation that the GFP-BAF MAAAQ protein retained its nuclear localization when coexpressed with active VRK1 (or the catalytically inactive variant; unpublished data). Because BAF-MAAAQ cannot be phosphorylated by VRK1, these cumulative data imply that the VRK1-mediated phosphorylation of BAF alters its cellular localization.
We next sought to gain more direct evidence that the intracellular localization of BAF was correlated with, or regulated by, its phosphorylation status. To this end, we used constructs encoding 3XFLAG BAF and first confirmed that this epitope-tagged version retained the DNA-binding properties of untagged BAF. 3XFLAG-BAF was synthesized in vitro using a coupled transcription/translation protocol and then incubated with DNA cellulose. Figure 7A shows that 35S-labeled 3XFLAG BAF does indeed bind to DNA cellulose. We then transiently transfected cells with the plasmid directing the expression of 3XFLAG-BAF, and metabolically labeled the cells with 32PPi in order to monitor the phosphorylation of BAF in vivo. Cell lysates were subjected to immunoprecipitation with FLAG-agarose resin, and the bound proteins were resolved electrophoretically, transferred to nitrocellulose, and visualized with both immunoblot analysis and autoradiography (Figure 7B). Two electrophoretically distinct forms of BAF were seen on the immunoblot (black and white arrowheads), and a single radiolabeled species that comigrated with the more slowly migrating species (black arrowhead) was observed on the autoradiograph. These data confirmed that transiently expressed BAF was indeed being phosphorylated in vivo by endogenous kinases. To examine whether this phosphorylation was affecting thr2,3 and/or ser4, we performed parallel transfections with plasmids expressing WT BAF (MTTSQ) or variants in which thr2,3 and/or ser4 were mutated to ala (MTTAQ, MAASQ, MAAAQ). The epitope-tagged proteins were retrieved by immunoprecipitation, left untreated or treated with λ phosphatase, and then resolved electrophoretically and visualized by immunoblot analysis. As shown in Figure 7C, the WT protein appeared as two distinct species of nearly equivalent abundance; upon phosphatase treatment, the abundance of the more slowly migrating species was dramatically reduced (compare lanes 1 and 5). A similar profile was seen for the MAASQ protein (Figure 7C, lanes 2 and 6), although in this case the upper band was somewhat less abundant in the untreated sample and absent after phosphatase treatment. For the MTTAQ protein, a faint but distinct upper band was observed, and this species was also eliminated by phosphatase treatment (Figure 7C, lanes 3 and 7). These data provided confirmation that BAF was indeed being phosphorylated in vivo, and supported the conclusion that thr 2,3 and ser4 are minor and major determinants, respectively, of the efficiency with which this modification occurs. For the MAAAQ treatment, only one species was observed, and its mobility was not affected by phosphatase treatment (Figure 7C, lanes 4 and 8). This variant, which was not a substrate for VRK-mediated phosphorylation in vitro (Figure 2), also does not appear to undergo detectable phosphorylation in vivo.
These data also confirmed that we could use immunoblot analysis to monitor the electrophoretically distinct unphosphorylated and phosphorylated BAF proteins. We therefore used this assay to monitor the subcellular fractionation of WT BAF and the MAAAQ variant. Cells were therefore transfected with plasmids directing the expression of 3XFLAG-BAF or 3XFLAG-BAF MAAAQ, alone or with plasmids directing the expression of VRK1 or VRK1D177A, and then subjected to a limited Triton X-100 extraction followed by centrifugation. The supernatant fraction has been shown to contain soluble cytoplasmic and nucleoplasmic proteins, whereas the pellet contains proteins associated with the nuclear matrix, intermediate filaments, and chromatin (Okuno et al., 2001; Zaidi et al., 2001; Dreuillet et al., 2002; Yoshizawa-Sugata et al., 2005). These fractions were then subjected to immunoblot analysis with α-FLAG serum (Figure 7D). When 3XFLAG-BAF was expressed alone, it was found in both the soluble and pellet fractions. However, whereas the soluble fraction contained equivalent amounts of the unphosphorylated and phosphorylated forms of BAF, the pellet fraction was highly enriched for unphosphorylated BAF (lanes 1 and 2). The profile was quite different when VRK1 was coexpressed with 3XFLAG-BAF. First, the soluble fraction was now significantly enriched in phosphorylated BAF (compare lanes 3 and 1), and there was a sharp decrease in the total amount of BAF present in the pellet fraction (compare lanes 4 and 2). These data indicate that, in the presence of VRK1, significantly more BAF is phosphorylated and that this protein is no longer fixed within an insoluble nuclear compartment, but is in fact soluble. As would be expected from this conclusion, expression of VRK1D177A did not alter either the phosphorylation status or fractionation profile of BAF (compare lanes 5,6 with 1,2). When we examined the profile of 3XFLAG-BAF MAAAQ, we found a single electrophoretic species corresponding to unphosphorylated BAF; this protein, too, partitioned to both the soluble and pellet fractions. Because this form of BAF cannot be phosphorylated by VRK1, it was not surprising that we observed no impact on its electrophoretic mobility or subcellular fractionation when either VRK1 or VRK1D177A was coexpressed (lanes 7-12). Together, these data indicate that the extreme N′ terminus of BAF undergoes phosphorylation in vivo and that this phosphorylation can be readily modulated by the VRK1 kinase. They further indicate that both soluble and insoluble pools of BAF are present within cells. It is predominantly the unphosphorylated BAF that is present in the insoluble pool, which probably corresponds to an association with the nuclear matrix/chromatin. The phosphorylated BAF is not found associated with these nuclear compartments, which is consistent with our findings that, in vitro, phosphorylation of BAF diminishes its interaction with LEM-containing proteins and virtually abolishes its interaction with DNA (Figure 8).
DISCUSSION
Elucidation of the interactions between kinases and their substrates is an essential step in gaining an understanding of the intricate intracellular signal transduction cascades that regulate cell biology. The cellular VRKs, and the related vaccinia virus B1 kinase, define a distinct branch of the casein kinase family whose biological roles remain to be fully resolved. While screening cell lysates for common substrates that could be phosphorylated by these enzymes, we identified a 10-kDa protein that we were able to identify as BAF. By performing an extensive series of in vitro kinase reactions, we were able to determine that VRK1, VRK2, and B1 can all phosphorylate the extreme N′ terminus of BAF. Interestingly, the motif surrounding the phosphorylation sites, MTTSQ, bears no resemblance to target sequences within proteins that have been previously shown to be phosphorylated by VRK1 (p53, ATF2, cJUN) or B1 (vaccinia H5 protein; Brown et al., 2000; Lopez-Borges and Lazo, 2000; Sevilla et al., 2004a, 2004b). Within BAF, ser4 is the preferred target for these kinases, but thr2 and/or thr3 can also be phosphorylated, albeit at a slower rate. Phosphorylation of BAF occurs very rapidly and to completion, indicating that BAF is modified very efficiently by these kinases. Coincident with the submission of this article, Bengtsson and Wilson (2005) published a report documenting the phosphorylation of endogenous BAF in HeLa cells and Xenopus oocytes. When recombinant BAF was added to Xenopus extracts, the predominant site of phosphorylation was shown to be ser4, although 2D gel analysis suggested that posttranslational modification was also occurring at other sites. Our data are consistent with and extend these findings.
Analysis of the structure of BAF by both NMR and x-ray crystallography has provided a significant amount of insight into the organization of the BAF dimer (Cai et al., 1998; Umland et al., 2000). The LEM-binding domain has a saddle-like conformation that is generated from both subunits of the BAF dimer (Segura-Totten et al., 2002). Although this domain is quite removed from the N′-termini that undergo phosphorylation, mutations that affect several regions of the protein have been shown to have an impact on the BAF-LEM interaction (Segura-Totten et al., 2002). We were therefore not too surprised to find that phosphorylation of BAF diminished the efficiency with which it interacted with a GST-LEM fusion protein to a modest, but reproducible, extent. We assume that phosphorylation induces a subtle conformational change that in turn affects the LEM-binding region. Bengtsson and Wilson (2005) have recently reported that, in vitro, nonphosphorylated BAF interacts with lamin A and enhances the interaction of emerin with lamin A, whereas both of these activities are greatly reduced for phosphorylated BAF (or a ser4→glu variant designed to mimic constitutive phosphorylation). Cumulatively, the data from the Wilson laboratory and our laboratory suggest that phosphorylation can modulate the interaction of BAF with its known protein partners.
The N′ terminus of BAF is known to be intimately involved in the interaction of BAF with DNA. In fact, analysis of a BAF-DNA cocrystal has revealed that ser4 makes contact with the phosphate backbone of the DNA via a water molecule (Bradley et al., 2005). Phosphorylation of ser4, or indeed thr2/3, virtually ablated the ability of BAF to bind to DNA. By varying the concentrations of NaCl present in our DNA-binding assays, we were able to show that the BAF-DNA interaction was moderately weakened by mutation of ser4 to ala and further weakened by mutation of ser4 to asp. The latter mutation, which might have been predicted to mimic phosphorylation, was not as deleterious to the BAF-DNA interaction as was phosphorylation. Because lys6, gly25, and gly27, in concert with other amino acids, form hydrogen bonds with phosphate moieties on the DNA backbone (Bradley et al., 2005), it may be that a phosphate moiety on ser4 competes directly for these interactions.
In sum, our in vitro data indicated that B1 and both VRK1 and 2 could stoichiometrically convert BAF to a phosphorylated form and that this modification blocked the DNA-BAF interaction and diminished the LEM-BAF interaction. To test the biological relevance of these findings, we monitored the impact of 3XFLAG-VRK1 expression on the behavior of 3XFLAG-BAF or GFP-BAF in tissue culture cells. Although GFP-BAF was predominantly nuclear in its localization, coexpression of catalytically active VRK1 led to a striking redistribution of the protein throughout the cell. This effect was not observed when a catalytically inactive VRK1 mutant was coexpressed with GFP-BAF. Likewise, the nuclear localization of the GFP-MAAAQ variant, which cannot be phosphorylated by the VRK or B1 kinases, was unaffected by coexpression of catalytically active VRK1. We speculated that VRK-mediated phosphorylation of BAF released it from nuclear DNA, causing it to assume a diffuse localization throughout the cell.
We therefore performed more direct analyses of the impact of VRK1 expression on the phosphorylation status and nuclear anchoring of 3XFLAG-BAF. When transiently expressed in cells, 3XFLAG-BAF was found in two electrophoretically distinct forms that represent unphosphorylated and phosphorylated species. When these cells were fractionated using a method known to separate soluble cytoplasmic and nucleoplasmic proteins from proteins rendered insoluble because of an association with chromatin or the nuclear matrix, the soluble fraction contained an equal amount of both forms of BAF. In contrast, the insoluble fraction was highly enriched in unphosphorylated BAF, which is what would be expected if phosphorylation prevented the association of BAF with proteins of the inner nuclear membrane and with DNA. When VRK1 was coexpressed with BAF, two changes were observed: first, the amount of BAF recovered in the insoluble fraction decreased dramatically and second, the BAF found in the soluble fraction was largely in the phosphorylated form. Neither of these changes occurred when a catalytically null variant of VRK1 was introduced along with BAF. These data provide strong evidence that VRK1 can phosphorylate BAF in vivo. The exclusion of phosphorylated BAF from the insoluble fraction is consistent with our demonstration that, in vitro, phosphorylated BAF does not bind to DNA and binds to LEM domain-containing proteins with reduced efficiency.
The demonstration that phosphorylation affects the interactions of BAF with protein and DNA is quite provocative, because BAF plays distinct roles in regulating nuclear architecture during different phases of the cell cycle. During interphase, BAF is implicated in anchoring chromatin to the nuclear periphery and has been shown to interact with chromatin-bound transcription factors (Wang et al., 2002; Holaska et al., 2003). This nuclear pool of BAF would be predicted to be unphosphorylated. We hypothesize that this same pool of BAF becomes phosphorylated during prophase, so that it is released from chromatin and from the proteins of the inner nuclear membrane, permitting chromatin condensation and nuclear envelope breakdown to begin. This model is consistent with previous reports demonstrating that BAF is largely nuclear during interphase and assumes a diffuse localization early in mitosis as the nuclear membrane disassembles (Haraguchi et al., 2001; Dechat et al., 2004). In contrast, BAF localizes to the core regions of chromosomes at telophase, where it facilitates the recruitment of LAP2 and/or emerin from the ER during the initial stages of nuclear membrane reassembly (Haraguchi et al., 2001). We would presume, therefore, that BAF must become dephosphorylated at the onset of telophase. This model for the cell cycle regulation of BAF closely mirrors other dynamic processes that occur at the nuclear envelope. At the onset of mitosis, disassembly of the nuclear lamina is triggered by sequential phosphorylation events mediated by protein kinase C (PKC) followed by the CDK1/cyclinB complex (Heald and McKeon, 1990; Peter et al., 1990; Foisner and Gerace, 1993; Goss et al., 1994; Thompson and Fields, 1996; Collas et al., 1997). The LEM domain-containing proteins Lap2α and emerin have also been reported to be differentially phosphorylated in a cell cycle-dependent manner; these modifications are capable of inducing dissociation from DNA and BAF, respectively (Gajewski et al., 2004; Hirano et al., 2005). These phosphorylation events are mediated by multiple kinases and are part of a wave of mitotic action that occurs during nuclear envelope breakdown, which is later reversed at the onset of nuclear reassembly by protein phosphatases such as protein phosphatase-1 (Thompson et al., 1997).
Our in vitro and in vivo data strongly implicate the VRK kinases in this cycle of BAF phosphorylation. It remains to be determined whether the localization and/or activity of VRK1 or VRK2 are themselves regulated in a cell cycle-dependent manner. It has been shown that the Drosophila VRK ortholog, NHK1, phosphorylates the H2A histone exclusively during mitosis (Aihara et al., 2004), suggesting that its activity may peak at this stage of the cell cycle. Our data also suggest that overexpression of VRK1, or mis-regulation of its activity, could have highly deleterious effects on various aspects of the cell cycle, including nuclear disassembly, chromosome partitioning, and nuclear reassembly. The studies presented herein set the stage for further dissection of the temporal and spatial interactions between the VRKs and BAF, and for consideration of the full implication of these interactions for regulation of nuclear structure and function.
Acknowledgments
We thank T. Haraguchi, M. Segura-Totten, and members of the Traktman laboratory for stimulating discussions. M.S.W. is supported by a postdoctoral fellowship from the Great Lakes Regional Center for Excellence in Biodefense and Emerging Infectious Diseases (GLRCE).
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05-12-1179) on February 22, 2006.
References
- Acharya, S., Wilson, T., Gradia, S., Kane, M. F., Guerrette, S., Marsischky, G. T., Kolodner, R., and Fishel, R. (1996). hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc. Natl. Acad. Sci. USA 93, 13629-13634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aihara, H. et al. (2004). Nucleosomal histone kinase-1 phosphorylates H2A Thr 119 during mitosis in the early Drosophila embryo. Genes Dev. 18, 877-888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez, R. A., Ghalayini, A. J., Xu, P., Hardcastle, A., Bhattacharya, S., Rao, P. N., Pettenati, M. J., Anderson, R. E., and Baehr, W. (1995). cDNA sequence and gene locus of the human retinal phosphoinositide-specific phospholipase-C beta 4 (PLCB4). Genomics 29, 53-61. [DOI] [PubMed] [Google Scholar]
- Barcia, R., Lopez-Borges, S., Vega, F. M., and Lazo, P. A. (2002). Kinetic properties of p53 phosphorylation by the human vaccinia-related kinase 1. Arch. Biochem. Biophys. 399, 1-5. [DOI] [PubMed] [Google Scholar]
- Bengtsson, L., and Wilson, K. L. (2005). BAF Phosphorylation on Ser-4 regulates emerin binding to lamin A in vitro and emerin localization in vivo. Mol. Biol. Cell (Dec 21 Epub ahead of print). [DOI] [PMC free article] [PubMed]
- Boyle, K., and Traktman, P. (2004). Members of a novel family of mammalian protein kinases complement the DNA- phenotype of a vaccinia virus ts mutant defective in the B1 kinase. J. Virol. 78, 1992-2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley, C. M., Ronning, D. R., Ghirlando, R., Craigie, R., and Dyda, F. (2005). Structural basis for DNA bridging by barrier-to-autointegration factor. Nat. Struct. Mol. Biol. 12, 935-936. (Epub 2005 Sep 11). [DOI] [PubMed] [Google Scholar]
- Brown, N. G., Nick, M. D., Beaud, G., Hardie, G., and Leader, D. P. (2000). Identification of sites phosphorylated by the vaccinia virus B1R kinase in viral protein H5R. BMC Biochem. 1, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, M., Huang, Y., Zheng, R., Wei, S. Q., Ghirlando, R., Lee, M. S., Craigie, R., Gronenborn, A. M., and Clore, G. M. (1998). Solution structure of the cellular factor BAF responsible for protecting retroviral DNA from autointegration. Nat. Struct. Biol. 5, 903-909. [DOI] [PubMed] [Google Scholar]
- Collas, P., Thompson, L., Fields, A. P., Poccia, D. L., and Courvalin, J. C. (1997). PKC-mediated interphase lamin B phosphorylation and solubilization. J. Biol. Chem. 272, 21274-21280. [DOI] [PubMed] [Google Scholar]
- Cullen, C. F., Brittle, A. L., Ito, T., and Ohkura, H. (2005). The conserved kinase NHK-1 is essential for mitotic progression and unifying acentrosomal meiotic spindles in Drosophila melanogaster. J. Cell Biol. 171, 593-602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dechat, T., Gajewski, A., Korbei, B., Gerlich, D., Daigle, N., Haraguchi, T., Furukawa, K., Ellenberg, J., and Foisner, R. (2004). LAP2alpha and BAF transiently localize to telomeres and specific regions on chromatin during nuclear assembly. J. Cell Sci. 117, 6117-6128. [DOI] [PubMed] [Google Scholar]
- Dreuillet, C., Tillit, J., Kress, M., and Ernoult-Lange, M. (2002). In vivo and in vitro interaction between human transcription factor MOK2 and nuclear lamin A/C. Nucleic Acids Res. 30, 4634-4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foisner, R., and Gerace, L. (1993). Integral membrane proteins of the nuclear envelope interact with lamins and chromosomes, and binding is modulated by mitotic phosphorylation. Cell 73, 1267-1279. [DOI] [PubMed] [Google Scholar]
- Furukawa, K. (1999). LAP2 binding protein 1 (L2BP1/BAF) is a candidate mediator of LAP2-chromatin interaction. J. Cell Sci. 112(Pt 15), 2485-2492. [DOI] [PubMed] [Google Scholar]
- Furukawa, K., Sugiyama, S., Osouda, S., Goto, H., Inagaki, M., Horigome, T., Omata, S., McConnell, M., Fisher, P. A., Nishida, Y. (2003). Barrier-to-autointegration factor plays crucial roles in cell cycle progression and nuclear organization in Drosophila. J. Cell Sci. 116, 3811-3823. [DOI] [PubMed] [Google Scholar]
- Gajewski, A., Csaszar, E., and Foisner, R. (2004). A phosphorylation cluster in the chromatin-binding region regulates chromosome association of LAP2alpha. J. Biol. Chem. 279, 35813-35821. [DOI] [PubMed] [Google Scholar]
- Giot, L. et al. (2003). A protein interaction map of Drosophila melanogaster. Science 302, 1727-1736. [DOI] [PubMed] [Google Scholar]
- Goss, V. L., Hocevar, B. A., Thompson, L. J., Stratton, C. A., Burns, D. J., and Fields, A. P. (1994). Identification of nuclear beta II PKC as a mitotic lamin kinase. J. Biol. Chem. 269, 19074-19080. [PubMed] [Google Scholar]
- Haraguchi, T., Koujin, T., Segura-Totten, M., Lee, K. K., Matsuoka, Y., Yoneda, Y., Wilson, K. L., and Hiraoka, Y. (2001). BAF is required for emerin assembly into the reforming nuclear envelope. J. Cell Sci. 114, 4575-4585. [DOI] [PubMed] [Google Scholar]
- Harris, D., and Engelman, A. (2000). Both the structure and DNA binding function of the barrier-to-autointegration factor contribute to reconstitution of HIV type 1 integration in vitro. J. Biol. Chem. 275, 39671-39677. [DOI] [PubMed] [Google Scholar]
- Heald, R., and McKeon, F. (1990). Mutations of phosphorylation sites in lamin A that prevent nuclear lamina disassembly in mitosis. Cell 61, 579-589. [DOI] [PubMed] [Google Scholar]
- Hirano, Y., Segawa, M., Ouchi, F. S., Yamakawa, Y., Furukawa, K., Takeyasu, K., Horigome, T. (2005). Dissociation of emerin from barrier-to-autointegration factor is regulated through mitotic phosphorylation of emerin in a Xenopus egg cell-free system. J. Biol. Chem. 280, 39925-39933. [DOI] [PubMed] [Google Scholar]
- Holaska, J. M., Lee, K. K., Kowalski, A. K., and Wilson, K. L. (2003). Transcriptional repressor germ cell-less (GCL) and barrier to autointegration factor (BAF) compete for binding to emerin in vitro. J. Biol. Chem. 278, 6969-6975. [DOI] [PubMed] [Google Scholar]
- Ivanovska, I., Khandan, T., Ito, T., and Orr-Weaver, T. L. (2005). A histone code in meiosis: the histone kinase, NHK-1, is required for proper chromosomal architecture in Drosophila oocytes. Genes Dev. 19, 2571-2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, K. K., Haraguchi, T., Lee, R. S., Koujin, T., Hiraoka, Y., and Wilson, K. L. (2001). Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J. Cell Sci. 114, 4567-4573. [DOI] [PubMed] [Google Scholar]
- Lee, M. S., and Craigie, R. (1994). Protection of retroviral DNA from autointegration: involvement of a cellular factor. Proc. Natl. Acad. Sci. USA 91, 9823-9827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, M. S., and Craigie, R. (1998). A previously unidentified host protein protects retroviral DNA from autointegration. Proc. Natl. Acad. Sci. USA 95, 1528-1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, F., Blake, D. L., Callebaut, I., Skerjanc, I. S., Holmer, L., McBurney, M. W., Paulin-Levasseur, M., and Worman, H. J. (2000). MAN1, an inner nuclear membrane protein that shares the LEM domain with lamina-associated polypeptide 2 and emerin. J. Biol. Chem. 275, 4840-4847. [DOI] [PubMed] [Google Scholar]
- Lopez-Borges, S., and Lazo, P. A. (2000). The human vaccinia-related kinase 1 (VRK1) phosphorylates threonine-18 within the mdm-2 binding site of the p53 tumour suppressor protein. Oncogene 19, 3656-3664. [DOI] [PubMed] [Google Scholar]
- MacKeigan, J. P., Murphy, L. O., and Blenis, J. (2005). Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat. Cell Biol. 7, 591-600. [DOI] [PubMed] [Google Scholar]
- Manning, G., Whyte, D. B., Martinez, R., Hunter, T., and Sudarsanam, S. (2002). The protein kinase complement of the human genome. Science 298, 1912-1934. [DOI] [PubMed] [Google Scholar]
- Mansharamani, M., and Wilson, K. L. (2005). Direct binding of nuclear membrane protein MAN1 to emerin in vitro and two modes of binding to barrier-to-autointegration factor. J. Biol. Chem. 280, 13863-13870. [DOI] [PubMed] [Google Scholar]
- Margalit, A., Segura-Totten, M., Gruenbaum, Y., and Wilson, K. L. (2005). Barrier-to-autointegration factor is required to segregate and enclose chromosomes within the nuclear envelope and assemble the nuclear lamina. Proc. Natl. Acad. Sci. USA 102, 3290-3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nezu, J., Oku, A., Jones, M. H., and Shimane, M. (1997). Identification of two novel human putative serine/threonine kinases, VRK1 and VRK2, with structural similarity to vaccinia virus B1R kinase. Genomics 45, 327-331. [DOI] [PubMed] [Google Scholar]
- Nichols, R. J., and Traktman, P. (2004). Characterization of three paralogous members of the Mammalian vaccinia related kinase family. J. Biol. Chem. 279, 7934-7946. [DOI] [PubMed] [Google Scholar]
- Okuno, Y., McNairn, A. J., den Elzen, N., Pines, J., and Gilbert, D. M. (2001). Stability, chromatin association and functional activity of mammalian pre-replication complex proteins during the cell cycle. EMBO J. 20, 4263-4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter, M., Nakagawa, J., Doree, M., Labbe, J. C., and Nigg, E. A. (1990). In vitro disassembly of the nuclear lamina and M phase-specific phosphorylation of lamins by cdc2 kinase. Cell 61, 591-602. [DOI] [PubMed] [Google Scholar]
- Punjabi, A., and Traktman, P. (2005). Cell biological and functional characterization of the vaccinia virus F10 kinase: implications for the mechanism of virion morphogenesis. J. Virol. 79, 2171-2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempel, R. E., and Traktman, P. (1992). Vaccinia virus B1 kinase: phenotypic analysis of temperature-sensitive mutants and enzymatic characterization of recombinant proteins. J. Virol. 66, 4413-4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segura-Totten, M., Kowalski, A. K., Craigie, R., and Wilson, K. L. (2002). Barrier-to-autointegration factor: major roles in chromatin decondensation and nuclear assembly. J. Cell Biol. 158, 475-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevilla, A., Santos, C. R., Barcia, R., Vega, F. M., and Lazo, P. A. (2004a). c-Jun phosphorylation by the human vaccinia-related kinase 1 (VRK1) and its cooperation with the N-terminal kinase of c-Jun (JNK). Oncogene 23, 8950-8958. [DOI] [PubMed] [Google Scholar]
- Sevilla, A., Santos, C. R., Vega, F. M., and Lazo, P. A. (2004b). Human vaccinia-related kinase 1 (VRK1) activates the ATF2 transcriptional activity by novel phosphorylation on Thr-73 and Ser-62 and cooperates with JNK. J. Biol. Chem. 279, 27458-27465. [DOI] [PubMed] [Google Scholar]
- Shimi, T., Koujin, T., Segura-Totten, M., Wilson, K. L., Haraguchi, T., and Hiraoka, Y. (2004). Dynamic interaction between BAF and emerin revealed by FRAP, FLIP, and FRET analyses in living HeLa cells. J. Struct. Biol. 147, 31-41. [DOI] [PubMed] [Google Scholar]
- Shumaker, D. K., Lee, K. K., Tanhehco, Y. C., Craigie, R., and Wilson, K. L. (2001). LAP2 binds to BAF. DNA complexes: requirement for the LEM domain and modulation by variable regions. EMBO J. 20, 1754-1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, Y., and Craigie, R. (2002). Regulatory mechanisms by which barrier-to-autointegration factor blocks autointegration and stimulates intermolecular integration of Moloney murine leukemia virus preintegration complexes. J. Virol. 76, 12376-12380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, L. J., Bollen, M., and Fields, A. P. (1997). Identification of protein phosphatase 1 as a mitotic lamin phosphatase. J. Biol. Chem. 272, 29693-29697. [DOI] [PubMed] [Google Scholar]
- Thompson, L. J., and Fields, A. P. (1996). betaII PKC is required for the G2/M phase transition of cell cycle. J. Biol. Chem. 271, 15045-15053. [DOI] [PubMed] [Google Scholar]
- Traktman, P., Liu, K., DeMasi, J., Rollins, R., Jesty, S., and Unger, B. (2000). Elucidating the essential role of the A14 phosphoprotein in vaccinia virus morphogenesis: construction and characterization of a tetracycline-inducible recombinant. J. Virol. 74, 3682-3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umland, T. C., Wei, S. Q., Craigie, R., and Davies, D. R. (2000). Structural basis of DNA bridging by barrier-to-autointegration factor. Biochemistry 39, 9130-9138. [DOI] [PubMed] [Google Scholar]
- Vega, F. M., Sevilla, A., and Lazo, P. A. (2004). p53 stabilization and accumulation induced by human vaccinia-related kinase 1. Mol. Cell Biol. 24, 10366-10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. et al. (2002). Barrier to autointegration factor interacts with the cone-rod homeobox and represses its transactivation function. J. Biol. Chem. 277, 43288-43300. [DOI] [PubMed] [Google Scholar]
- Yoshizawa-Sugata, N., Ishii, A., Taniyama, C., Matsui, E., Arai, K., and Masai, H. (2005). A second human Dbf4/ASK-related protein, Drf1/ASKL1, is required for efficient progression of S and M phases. J. Biol. Chem. 280, 13062-13070. [DOI] [PubMed] [Google Scholar]
- Zaidi, S. K., Javed, A., Choi, J. Y., van Wijnen, A. J., Stein, J. L., Lian, J. B., and Stein, G. S. (2001). A specific targeting signal directs Runx2/Cbfa1 to subnuclear domains and contributes to transactivation of the osteocalcin gene. J. Cell Sci. 114, 3093-3102. [DOI] [PubMed] [Google Scholar]
- Zelko, I., Kobayashi, R., Honkakoski, P., and Negishi, M. (1998). Molecular cloning and characterization of a novel nuclear protein kinase in mice. Arch. Biochem. Biophys. 352, 31-36. [DOI] [PubMed] [Google Scholar]
- Zheng, R., Ghirlando, R., Lee, M. S., Mizuuchi, K., Krause, M., and Craigie, R. (2000). Barrier-to-autointegration factor (BAF) bridges DNA in a discrete, higher-order nucleoprotein complex. Proc. Natl. Acad. Sci. USA 97, 8997-9002. [DOI] [PMC free article] [PubMed] [Google Scholar]