

Abstract
We used gain-of-function and null synaptic signaling network mutants to investigate the relationship of the Gαq and Gαs pathways to synaptic vesicle priming and to each other. Genetic epistasis studies using Gαq gain-of-function and null mutations, along with a mutation that blocks synaptic vesicle priming and the synaptic vesicle priming stimulator phorbol ester, suggest that the Gαq pathway generates the core, obligatory signals for synaptic vesicle priming. In contrast, the Gαs pathway is not required for the core priming function, because steady-state levels of neurotransmitter release are not significantly altered in animals lacking a neuronal Gαs pathway, even though these animals are strongly paralyzed as a result of functional (nondevelopmental) defects. However, our genetic analysis indicates that these two functionally distinct pathways converge and that they do so downstream of DAG production. Further linking the two pathways, our epistasis analysis of a ric-8 null mutant suggests that RIC-8 (a receptor-independent Gα guanine nucleotide exchange factor) is required to maintain both the Gαq vesicle priming pathway and the neuronal Gαs pathway in a functional state. We propose that the neuronal Gαs pathway transduces critical positional information onto the core Gαq pathway to stabilize the priming of selected synapses that are optimal for locomotion.
ONE proposed mechanism for establishing and modifying behaviors or memories involves presynaptic changes in synaptic strength, and regulating the amount of synaptic-vesicle-mediated neurotransmitter release at synapses is thought to be critical for changing presynaptic strength (Kandel and Pittenger 1999; Lin and Scheller 2000; Sudhof 2000). Electrophysiological studies have defined a “readily releasable,” or “primed,” pool of synaptic vesicles, the size of which determines release probability and, therefore, presynaptic strength (Sudhof 2000). The term “primed” can be applied either to an individual synaptic vesicle or to a synapse (i.e., a primed synapse is one containing a relatively large pool of primed synaptic vesicles). A major challenge is to understand the layout and logic of the network of signaling pathways that controls synaptic vesicle and synapse priming.
The Gαq and the Gαs pathways are two major signaling pathways that have been implicated in positively regulating synaptic vesicle and/or synapse priming; however, the relationship of the Gαq and the Gαs pathways to each other is poorly understood. Both pathways have been reported to increase the pool of readily releasable, primed synaptic vesicles (Trudeau et al. 1996; Chen and Regehr 1997; Stevens and Sullivan 1998; Kuromi and Kidokoro 2000; Waters and Smith 2000). These electrophysiological studies thus show that both pathways can positively affect priming, but they do not reveal how the two pathways interact and why they both exist. Highlighting this point, two recent studies showed that the Gαs pathway is not even required for synaptic vesicle priming, because evoked release remains normal, or even increases, when Gαs null synapses are subjected to low frequency stimulation (Hou et al. 2003; Renden and Broadie 2003).
Providing important insights and yet further adding to the puzzlement about the relative roles of the Gαq and Gαs pathways, recent research suggests that both pathways, directly or indirectly, affect the synaptic abundance of UNC-13, a large, conserved protein that binds diacylglycerol (DAG) (Maruyama and Brenner 1991), interacts with the synaptic vesicle fusion machinery (Betz et al. 1997; Sassa et al. 1999), and is required for synaptic vesicle priming (Aravamudan et al. 1999; Augustin et al. 1999; Richmond et al. 1999, 2001). Intriguing studies by Aravamudan and Broadie (2003) and Speese et al. (2003) suggest that both DAG (produced by the Gαq pathway) and cAMP (produced by the Gαs pathway) increase the abundance of Drosophila UNC-13 at synapses via a proteasome-dependent mechanism, and earlier studies had already implicated the Gαq and Gαo pathways in the regulation of UNC-13 abundance at synapses (Lackner et al. 1999; Nurrish et al. 1999).
The above physiological and molecular studies show that altering the intracellular concentration of components of the Gαq and the Gαs pathways can alter synaptic vesicle and/or synapse priming and can also alter the synaptic abundance of the UNC-13 priming protein; however, questions of fundamental importance remain unanswered. Most importantly, studies focusing on each pathway as a single entity have not revealed the relationship of these two major pathways to each other. A major obstacle has been the lack of experiments that examine the consequences of knocking out the Gαq pathway, with respect to both synaptic vesicle priming and how such a manipulation affects Gαs pathway function.
Investigating the relationship of the Gαq and Gαs pathways to each other in intact animals requires epistasis analysis, a powerful method in which, by carefully analyzing the phenotype of strategically constructed double mutants, one can often infer the upstream/downstream order of proteins in a signaling pathway or network (Avery and Wasserman 1992; Huang and Sternberg 1995). However, signaling pathway epistasis analysis has strict requirements, including the availability of null mutants, the availability of mutations that turn on as well as completely block the pathway, and the ability to demonstrate that the single and double mutants used for the analysis do not have secondary effects that interfere with the measured output.
Genetic studies of the Caenorhabditis elegans synaptic signaling network have produced a remarkable set of genetic mutations and tools, unique among model organisms in their scope, that are appropriate for use in epistasis studies of the Gαo, Gαq, and Gαs synaptic signaling pathways. These include mutations that completely knock out each of these three major pathways, mutations that knock out the UNC-13 synaptic vesicle priming protein, transgenic strains and native gain-of-function mutants in which each pathway is strongly hyperactivated, a variety of mutations in negative regulatory components that indirectly lead to hyperactivation of each pathway, and transgenic and pharmacological manipulations that allow each pathway to be acutely activated in whole animals (see Table 1 and references therein). Adding to these tools, and facilitating their use in epistasis analysis is the amazing ability of C. elegans mutants lacking these pathways in neurons to remain alive for long periods of time and in some cases even to reproduce as homozygotes under standard or specially controlled laboratory conditions.
TABLE 1.
Summary of mutations used for epistasis analysis of the Gαq and Gαs pathways
| Desired pathway manipulation |
Mutation(s) or drug used for pathway manipulation |
Mutation type | Affected protein | Effect of mutation or drug | Reference for mutation description |
|---|---|---|---|---|---|
| Block Gαq pathway | egl-30(ad810) | Probable null | EGL-30 (Gαq) | Knocks out EGL-30 (Gαq): paralysis | Brundage et al. (1996) |
| Block Gαq pathway at the downstream synaptic vesicle priming mechanism |
unc-13(s69) | Strong loss of function (near null) |
UNC-13 | Strongly reduces UNC-13: paralysis |
Richmond et al. (1999); Kohn et al. (2000) |
| Activate Gαq pathway | egl-30(tg26) | Strong gain of function | EGL-30 (Gαq) | Activates EGL-30 (Gαq): hyperactive locomotion |
Doi and Iwasaki (2002) |
| eat-16(ce71) | Probable null | EAT-16 (RGS protein for EGL-30) |
Increases activity of EGL-30 (Gαq) by knocking out the EAT-16 RGS protein: hyperactive locomotion |
This study | |
| goa-1(sa734) | Null | GOA-1 (Gαo) | Increases activity of EGL-30 pathway by an unknown mechanism (point of intersection unknown): hyperactive locomotion |
Robatzek and Thomas (2000) |
|
| Activate Gαq pathway at the downstream synaptic vesicle priming mechanism |
Phorbol myristate acetate (phorbol ester) |
NA | Synaptic vesicle priming proteins: effects require UNC-13 |
Activates synaptic vesicle priming; bypasses the requirement for Gαq: hyperactive locomotion |
Lackner et al. (1999); this study; see also Introduction for references |
| Block Gαs pathway | acy-1(pk1279) | Null | ACY-1 (adenylyl cyclase) |
Knocks out ACY-1: near paralysis | Moorman and Plasterk (2002) |
| Activate one or more components of the Gαs pathway |
gsa-1(ce81) | Strong gain of function | GSA-1 (Gαs) | Hyperactivates Gαs by inhibiting GTP hydrolysis: hyperactive locomotion |
Schade et al. (2005) |
| gsa-1(ce94) | Strongest GSA-1 gain of function |
GSA-1 (Gαs) | Strongly hyperactivates Gαs: hyperactive locomotion |
Schade et al. (2005) | |
| acy-1(md1756) | Strong gain of function | ACY-1 (adenylyl cyclase) |
Activates ACY-1; probably increases cAMP: hyperactive locomotion |
Schade et al. (2005) | |
| acy-1(ce2) | Strong gain of function | ACY-1 (adenylyl cyclase) |
Activates ACY-1; probably increases cAMP: hyperactive locomotion |
Schade et al. (2005) | |
| kin-2(ce179) | Strong reduction of function |
KIN-2 (regulatory subunit protein kinase A) |
Makes protein kinase A holoenzyme extremely hypersensitive to cAMP: hyperactive locomotion |
Schade et al. (2005) | |
| Strongly reduce receptor-independent activation of the Gαq pathway |
ric-8(md303) | Strong reduction of function |
RIC-8 (synembryn) | Strongly reduces activation of the Gαq pathway; may also affect the Gαs pathway: near paralysis |
Miller et al. (2000); Schade et al. (2005) |
| Knock out receptor-independent activation of both the Gαq and the Gαs pathways |
ric-8(ok98) | Null | RIC-8 (synembryn) | Blocks activation of both the Gαq and the Gαs pathways: paralysis |
This study |
Previous epistasis studies revealed that the Gαo pathway is strongly dependent on the Gαq pathway to exert its strong inhibitory effects on locomotion and neurotransmitter release (Hajdu-Cronin et al. 1999; Miller et al. 1999). In a prelude to the current study, a pair of high-resolution forward genetic screens revealed an apparent link between the Gαs pathway and the Gαo/Gαq signaling network (Schade et al. 2005, accompanying article in this issue); however, that study did not directly address the relationship of the Gαq and Gαs pathways to each other.
In this study, we use gain-of-function and null synaptic signaling network mutants to investigate the relationship of Gαq and Gαs pathways to synaptic vesicle priming and to each other. Genetic epistasis studies using Gαq gain-of-function and null mutations, along with a mutation that blocks synaptic vesicle priming and the synaptic vesicle priming stimulator phorbol ester, suggest that the Gαq pathway generates the core, obligatory signals for synaptic vesicle priming. In contrast, the Gαs pathway is not required for the core priming function, because steady-state levels of neurotransmitter release are not significantly altered in animals lacking a neuronal Gαs pathway, even though these animals are strongly paralyzed as a result of functional (nondevelopmental) defects. However, our genetic analysis indicates that these two functionally distinct pathways converge and that they do so downstream of DAG production. Further linking the two pathways, our epistasis analysis of a ric-8 null mutant suggests that RIC-8 (a receptor-independent Gα guanine nucleotide exchange factor, GEF) is required to maintain both the Gαq vesicle priming pathway and the neuronal Gαs pathway in a functional state. We propose that the neuronal Gαs pathway transduces critical positional information onto the Gαq priming pathway to stabilize the priming of selected synapses that are optimal for locomotion.
MATERIALS AND METHODS
Worm culture and observation:
Unless otherwise specified, wild-type worms were C. elegans variety Bristol, strain N2. All culture media were made with Sigma A-7002 agar. All other worm cultures were based on previously described methods (Brenner 1974). Worms were observed and manipulated using Olympus SZX-12 stereomicroscopes equipped with ×1.2, 0.13 numerical aperture plan apochromatic objectives.
Strains:
Alleles used in genetic analysis and for double-mutant strain construction are described and referenced in Table 1, except for egl-30(ad805) (Brundage et al. 1996) and eat-16(ce71), which we describe here for the first time. eat-16(ce71), which we isolated in a screen for hyperactive mutants (Schade et al. 2005), contains a Q333stop mutation and is our best candidate for an eat-16 null mutant, because it is predicted to block production of the C-terminal 30% of the protein, including ∼75% of the RGS domain that is required for EAT-16's function (Hajdu-Cronin et al. 1999). All mutants used in this study have been outcrossed at least three times to reduce secondary mutations.
The mutations goa-1(sa734) and egl-30(tg26) were maintained at 20° or room temperature, because they were found to not grow at the standard long-term storage temperature of 14°. The larval lethal mutations egl-30(ad810) and acy-1(pk1279) were maintained as heterozygotes. egl-30(ad810) was outcrossed from DA1096 egl-30(ad810)/szT1 [lon-2(e678) I; +/szT1 X] and maintained as egl-30(ad810)/+ with no special balancing mutations. Since the mutation is semidominant (as a result of haplo-insufficiency), we identified ad810/+ heterozygotes on the basis of their larger than normal size and their significant Egl phenotype. Stock cultures were produced by picking ∼10 large Egl ad810/+ animals to each of two culture plates and allowing the plates to starve at 14° for long-term storage. Chunks from this plate were transferred to fresh culture plates as needed and grown for 2 days at room temperature; ad810/+ animals were chosen on the basis of their phenotype and used to generate homozygotes. acy-1(pk1279) was maintained over the closely linked mutation dpy-17(e164). These animals were maintained semiweekly in continuous culture for injections and double-mutant strain construction by cloning 6–10 wild larvae to individual culture plates, growing them 3 days at room temperature or 4 days at 20°, and saving plates that segregated paralyzed larvae and Dpy-17 animals. The ric-8(ok98) null mutation was initially maintained in a heterozygous state by selecting animals that segregated straight paralyzed larvae and subsequently balanced under the flanking genetic markers lin-1(e1275) unc-33(e204) and outcrossed three times.
Long PCR products and plasmids:
All long PCR products were produced via Expand 20 Kb+ (Roche) amplification of purified N2 genomic DNA, according to the manufacturer's instructions. The boundaries of the 10.0-kb ric-8 gene rescuing PCR product KG372/373 and the 9.88-kb fusion PCR product KG430/431 are shown in Figure 6A. The fusion PCR product was produced using the method of Hobert (2002) and excludes the small upstream gene that is in an operon with ric-8. To construct KG#80 [myo-3::acy-1(+) cDNA], we applied reverse transcriptase to purified C. elegans mRNA and synthesized the 1155-bp 5′ part of the acy-1 cDNA. This fragment was then fused to the partial cDNA clone yk35d9, using an internal SphI site and a 5′ site that had been engineered into the 5′ primer. The 3.8-kb acy-1 coding region was then amplified using Pfu Ultra polymerase and primers engineered with restriction sites and cloned into AgeI/XhoI-cut pPD96.52, a C. elegans muscle expression vector. The acy-1 coding region of this construct was sequenced and a clone was chosen that contained no mutations. To make KG#62 [rab-3::acy-1(+)], the acy-1 coding region was cloned into the KG#59 rab-3 expression vector (Schade et al. 2005), again using AgeI/XhoI. To make KG#82 [hsp-16-2::acy-1(gf)], the acy-1 (P260S) gain-of-function cDNA (Schade et al. 2005) was cloned, using AgeI/XhoI, into the heat-shock-inducible expression vector KG#45, which is the same as pPD96.52, except that the myo-3 promoter is replaced with the hsp-16-2 promoter (taken from pPD49.78). KG#81 [myo-3::acy-1(gf) cDNA] and KG#83 [rab-3::acy-1(gf) cDNA] have been described (Schade et al. 2005). To make KG#87 [myo-3::unc-18(+) cDNA] and KG#88 [rab-3::unc-18(+) cDNA], we applied reverse transcriptase to purified C. elegans mRNA and synthesized the full-length unc-18 coding region cDNA and then amplified this fragment using Accuprime Pfx (Invitrogen, San Diego) and primers engineered with AgeI/XhoI restriction sites. This fragment was then cloned into like-digested pPD96.52 (a body-wall muscle expression vector) to make KG#87 [myo-3::unc-18(+) cDNA]. After sequencing the unc-18 coding region in KG#87 to confirm the absence of PCR-introduced mutations in the chosen clone, we used AgeI/XhoI to transfer the unc-18 cDNA into the rab-3 expression vector KG#59 to make KG#88 [rab-3::unc-18(+) cDNA].
Figure 6.—
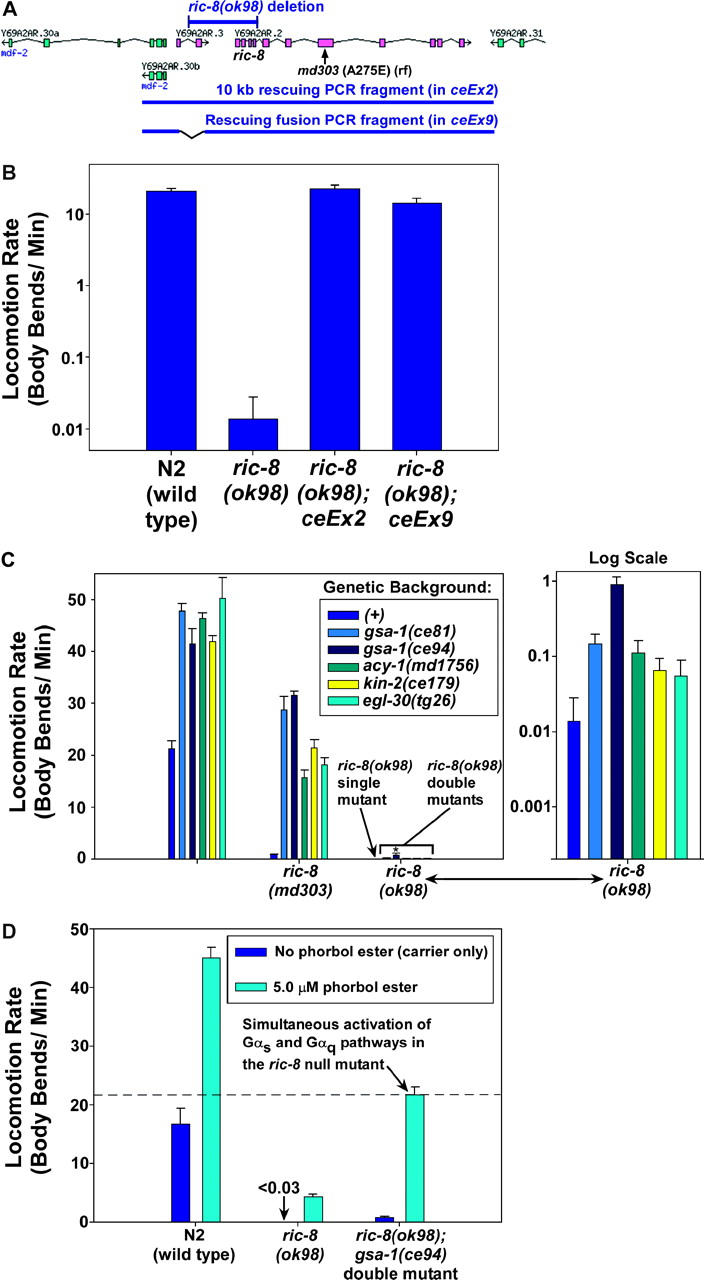
Epistasis analysis using the ric-8 null mutant suggests an unexpected role for RIC-8 in neuronal Gαs pathway activation. (A) Scale drawing of the region corresponding to the two-gene ric-8-containing operon (magenta) and flanking genes (cyan). The ric-8(ok98) deletion destroys the ric-8 gene and a small upstream gene that is in an operon with ric-8. Regions corresponding to the ok98 deletion, rescuing PCR fragments, and the ric-8(md303) missense mutation are indicated. The 1945-bp ok98 deletion begins 1346 bp upstream of ric-8 initiator Met and extends to just past the fourth exon. Note that the fusion PCR fragment used in the ceEx9 rescuing transgene does not contain the upstream gene. Also, note that the ok98 deletion may not affect the promoter for the operon and that transcription could start with the first exon of Y69A2AR.3 and continue with the fifth exon of ric-8. However, this would result in a frameshift. The deletion, therefore, is a ric-8 null mutation. (B) The paralysis of ric-8(ok98) mutants can be rescued with a PCR fragment that includes both genes and with a fusion PCR fragment that only restores the ric-8 gene. A log-scale graph of the mean locomotion rates is shown, expressed as body bends per minute, of strains homozygous for the ric-8(ok98) deletion, with or without rescuing transgenes. A wild-type control is shown for comparison. Error bars represent the standard error of the mean for 8–10 animals. (C) Mutations that singly activate the Gαs or Gαq pathways do not strongly suppress the ric-8 null mutant. The mean locomotion rates of various strains, expressed as body bends per minute, are shown. Strains homozygous for ric-8(md303) or ric-8(ok98) are grouped together as indicated. Dark-blue bars within each set represent strains carrying no additional mutations (genetic background (+)). Bars of other colors represent double mutants in which the second mutation activates a component of the Gαs or Gαq pathways. The first group of bars (unlabeled) represents wild-type and single-mutant control strains. Data for the ric-8(ok98)-containing strains are redisplayed on a log scale in the graph to the right. Only the gsa-1(ce81) and gsa-1(ce94) gain-of-function mutations caused significant suppression of the ric-8(ok98) null mutant (P-values of 0.040 and 0.013, respectively, when compared to the ric-8(ok98) single mutant). Note that the gsa-1(ce94) mutation most strongly suppresses the paralysis of ric-8(ok98); however, even this double mutant has a locomotion rate that is only ∼2% of the gsa-1(ce94) single mutant. All of the other mutations, including gsa-1(ce81), are significantly weaker suppressors of the paralysis of ric-8(ok98). Error bars represent the standard error of the mean for 8–10 animals. Statistical comparisons use the unpaired t-test with Welch correction. (D) Simultaneous activation of both the Gαq and the Gαs pathways strongly suppresses the paralysis of the ric-8 null mutant. The mean locomotion rates are shown, expressed as body bends per minute of various strains on plates containing 5 μm phorbol myristate acetate (cyan bars) or vehicle only (0.06% ethanol; dark-blue bars). The N2 wild-type strain is shown for comparison. Error bars represent the standard error of the mean for 8–10 animals. See also supplemental QuickTime movies for Figure 6 at http://www.genetics.org/supplemental/.
Production of transgenes:
Transgenic strains bearing extrachromosomal arrays were produced by the method of Mello et al. (1991). pBluescript carrier DNA was used, if necessary, to bring the final concentration of DNA in the injection mixture to 175 ng/μl. ceEx2 and ceEx9 were produced by injecting lin-1(e1275) unc-33(e204)/ric-8(ok98) animals with the KG372/373 and KG430/431 PCR products, respectively, at 10 and 60 ng/μl, respectively, and using pPD118.20 [myo-3::GFP] as a co-injection marker. Putative rescued ric-8(ok98) homozgyotes were cloned as green adult F2 progeny of green F1's that segregated Lin-1 and Unc-33. Lines whose only nongreen progeny were paralyzed larvae were saved and tested by PCR for ric-8(ok98) homozygosity. All remaining experimental DNAs in this study, with the exception of the unc-18 cDNAs, were injected into the acy-1 null heterozygous strain NL1999 acy-1(pk1279)/dpy-17(e164) (Moorman and Plasterk 2002), which was maintained in a continuous growing culture as described above. All experimental DNAs were co-injected with a GFP-bearing plasmid driven by the same promoter as that driving the experimental DNA. For injections involving rab-3-promoted coding regions, KG#68 [rab-3::GFP] and KG#67 ttx-3::GFP (gift of Oliver Hobert) were mixed at 50 and 25 ng/μl, respectively, to reduce nonspecific intestinal expression. F1 progeny of injected animals were screened, and GFP-positive adult animals were selected from plates segregating Dpy-17, pk1279 homozygotes, and wild type, and these animals were cloned to separate plates. In the next generation, lines segregating green F2's, no Dpy-17, and whose only nongreen progeny were paralyzed larvae (that had lost the transgene) were kept as candidate-rescued pk1279 homozygotes (in all cases the larval lethality was rescued to some extent). A total of 12 GFP-positive adults from each of these lines were cloned, and a line with good expression and transmittance was saved to establish a stock. ceEx67 [myo-3::acy-1(+) high copy #] and ceEx108 [myo-3::acy-1(+) low copy #] were produced by injecting NL1999's with KG#80 at 15 and 5 ng/μl, respectively. To make ceEx87 [myo-3::acy-1(gf)], we injected NL1999 with KG#81 at 15 ng/μl. To make ceEX76 [myo-3::acy-1(+) HS::acy-1(gf)], we injected NL1999 with a mixture of KG#80 (15 ng/μl) and KG#82 (10 ng/μl). To make ceEx100, we injected unc-18(e81) mutants with KG#87 (15 ng/μl) and selected GFP-positive F2's. All of these injections included pPD118.20 [myo-3::GFP] (15 ng/μl) as the cotransformation marker. To make ceEx97 [rab-3::acy-1(+)], we injected NL1999 with KG#62 (50 ng/μl). To make ceEx98 [rab-3::acy-1(gf)], we injected NL1999 with KG#83 (25 ng/μl). To make ceEx102, we injected unc-18(e81) mutants with KG#88 (25 ng/μl) and selected GFP-positive F2's. The latter three transgenes all included KG#68 [rab-3::GFP] and KG#67 ttx-3::GFP (gift of Oliver Hobert), mixed at 50 and 25 ng/μl, respectively, as the cotransformation markers.
Double-mutant strain construction and verification:
Unless otherwise specified, double mutants were constructed using standard genetic methods without additional marker mutations, and homozygosity of mutations, where necessary, was confirmed by PCR and/or sequencing. We constructed egl-30(ad810); acy-1(md1756) double mutants by crossing md1756/+ males to ad810/+ hermaphrodites. L4 progeny of this cross were then cloned and, from plates segregating both mutant phenotypes, 30 putative ad810/+; md1756 animals were cloned. From this group, plates that segregated ad810 larval homozygotes were tested for md1756 homozygosity by PCR and sequencing of the region containing the md1756 sequence change, testing lysates derived from multiple animals from each plate. Cultures found to be homozygous for md1756 and segregating ad810; md1756 double-mutant larvae were then expanded and maintained by cloning more putative ad810/+; md1756 animals and by checking for proper segregation. After collecting double mutants for documentation and assays (see below), a portion of the population was used to confirm homozygosity of both mutations by double amplification PCR using nested primers, followed by sequence analysis. We constructed egl-30(ad810); kin-2(ce179) and all of the acy-1(pk1279)-containing double mutants using similar methods. To make the egl-30(tg26); acy-1(pk1279); ceEx108 [myo-3::acy-1(+) myo-3::GFP] strain, tg26/+ males were crossed to pk1279; ceEx108 hermaphrodites, and 20 GFP-positive, hyperactive L4 hermaphrodite cross progeny were cloned and grown 4 days at room temperature. From the progeny of these animals, 42 GFP-positive putative tg26 homozygotes were cloned to 24-well culture plates and grown 4–5 days at room temperature. Wells containing no wild-type animals and segregating nongreen pk1279 larval lethals (from loss of the transgenic array) were identified and, from these wells, 25 GFP-positive putative tg26; ceEx108; pk1279/+ animals were cloned to individual culture plates. Properly segregating plates were used to maintain and expand the population for documentation and assays. The homozygous presence of the pk1279 deletion in all acy-1(pk1279)-containing strains was verified (using a portion of each population) by duplicate reactions of double-amplification PCR using nested primers, with both sets of primers completely internal to the deletion and completely derived from introns that would not be present in the cDNA rescued strains. These amplifications were done in parallel with control strains without the deletion, and all reactions contained the same number and same stage of animals in each tube. ric-8(ok98) double mutants were constructed by crossing mut-x/+ males (where mut-x is a generic mutation) to the heterozygous balanced strain lin-1(e1275) unc-33(e204)/ ric-8(ok98) and then cloning putative mut-x/mut-x; ric-8(ok98)/+ animals from the grand-progeny of this cross. From plates homozygous for mut-x, we chose staged mut-x; ric-8(ok98) double mutants (which were always paralyzed or near-paralyzed and sterile) for confirmation by PCR and sequencing and for the assays described herein.
Locomotion assays:
Standard locomotion assays were performed as previously described, using standardized plates and a standardized definition of a body bend (Miller et al. 1999). Exaggerated movements in which the animal doubles back on itself during reversal such that the tail touches the anterior of the body in a figure-eight pattern were scored as three body bends [this became relevant in some strains containing the egl-30 gain-of-function mutation, double mutants containing the acy-1(pk1279) mutation in combination with mutations that activate the egl-30 pathway (i.e., Figure 5B), and some strains treated with phorbol esters]. For coiling movements, a body bend was counted every 90° around the circle. To assay egl-30(ad810) and acy-1(pk1279)-containing strains, synchronized, larval-arrested, homozygous animals were collected by plating 14–20 heterozygous parental gravid adult animals on each of 7–10 culture plates seeded with a 1-cm-diameter lawn of OP-50 bacteria. These animals were plated in late afternoon and allowed to lay eggs for 24 hr before killing the parents. The eggs were then allowed to hatch overnight, producing a population of 6- to 30-hr-old larvae. Animals homozygous for egl-30(ad810) or acy-1(pk1279) were chosen from this population on the basis of their paralyzed phenotype and transferred to a standard locomotion assay plate (Miller et al. 1999) using a worm pick laden with a moist dab of bacteria. Approximately 60–100 animals were collected in this way for each strain to have enough animals for video and photo documentation, locomotion assays, genotype confirmation by PCR and sequencing, and population half-life studies (Table 2; see below). For locomotion assays, 10 animals were transferred in moist bacteria to a fresh, room-temperature equilibrated locomotion assay plate. After a 30-min stabilization period, body bends were counted for 6 min for each of the 10 animals. Identically staged N2 (wild type) and single-mutant control animals were produced and assayed in the same manner. egl-30(ad810) single mutants and egl-30(ad810); kin-2(ce179) double mutants were also assayed by a track locomotion assay, in which 30–70 animals were plated on a locomotion plate and allowed to sit at room temperature for 160 min, after which the body bends produced by each animal during this time were counted by following the short track that each animal made during that period.
Figure 5.—
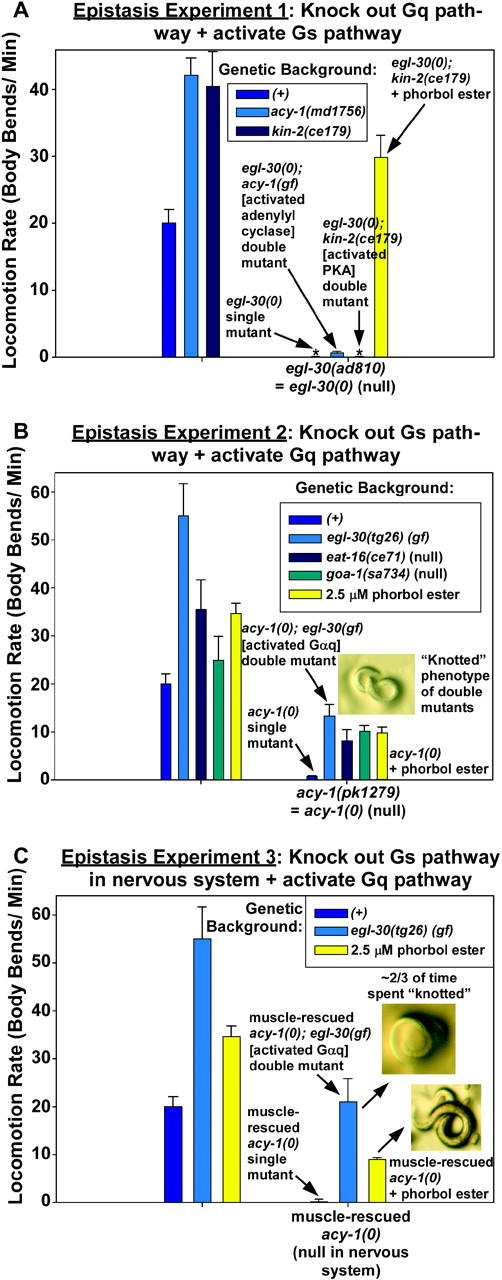
The Gαs pathway converges with the Gαq pathway downstream of DAG production, and hyperactivation of the Gαq pathway in the absence of a neuronal Gαs pathway severely disrupts coordinated locomotion. (A) The effects on locomotion rate caused by activating the Gαs pathway can be largely blocked by knocking out the Gαq synaptic vesicle priming pathway. Comparison of the mean locomotion rates of strains is shown in an egl-30(+) background (first set of bars) and an egl-30(ad810) null mutant background (second set of bars). Arrows point out the egl-30 null single mutant and double mutants containing the egl-30 null mutation in combination with mutations that activate acy-1 or PKA. Note that the acy-1 gain-of-function mutation causes a small, but significant, improvement to the locomotion rate of the egl-30 null; however, this amounts to only ∼2% of the acy-1 gain-of-function single mutant (shown in the left set of bars). In comparison, the locomotion rate of the egl-30 null is completely unaffected by the kin-2 mutation (which hyperactivates protein kinase A), yet the paralysis of this double mutant can still be completely rescued with phorbol esters (last bar; yellow). The values of the two bars with asterisks are 0.024 body bends/minute for egl-30(ad810) and 0.025 body bends/minute for egl-30(ad810); kin-2(ce179). Error bars represent the standard error of the mean for populations of 10 early larval stage animals. (B) Mutations or conditions that activate the Gαq pathway only partially suppress the paralysis caused by a block of the Gαs pathway. Comparison of the mean locomotion rates of strains in an acy-1(+) background (first set of bars) and an acy-1(pk1279) null mutant background (second set of bars) is shown. Arrows point out the acy-1 null single mutant and double mutants containing the acy-1 null mutation in combination with various mutations that activate the Gαq pathway or in combination with phorbol esters, which is the last bar. Note that all of the mutations that activate the Gαq pathway and even phorbol esters, which strongly suppress the more paralyzed egl-30 null, really only partially suppress the acy-1 null. The photograph shows the tightly knotted posture in which all of these double mutants (but the not the acy-1 null single mutant) spend about two-thirds of their time. Error bars represent the standard error of the mean for populations of 10 early larval stage animals. (C) The neuronal Gαs pathway is required for the Gαq pathway (and the DAG that it produces) to properly exert its effects. Comparison of the mean locomotion rates of strains in an acy-1(+) background (first set of bars) and a muscle-rescued acy-1(pk1279) null mutant background (second set of bars) is shown. These animals are null for acy-1 in their nervous system, but they contain the ceEx108 [myo-3::acy-1(+) cDNA] transgene that rescues acy-1 in muscle cells and also partially rescues the larval lethality caused by the acy-1 null mutation. Arrows point out the muscle-rescued acy-1 null single mutant and double mutants containing the muscle-rescued acy-1 null mutation in combination with a strong egl-30(gf) mutation that activates the Gαq pathway or in combination with phorbol esters. Note that both of these conditions only partially suppress paralysis of the acy-1 null. The photographs show the tightly knotted posture that dominates as a consequence of activating the Gαq pathway in the absence of a neuronal Gαs pathway. Error bars represent the standard error of the mean for populations of 10 early larval stage animals. See also supplemental QuickTime movies for Figure 5 at http://www.genetics.org/supplemental/.
TABLE 2.
Population half-lives and percentage reaching adulthood of larval arrested single and double mutants used in this study
| Genotype | Population half-life (days)a |
Fraction reaching adulthoodb |
% reaching adulthood |
No. of days monitored |
|---|---|---|---|---|
| egl-30(ad810) | 7 | 0/33 | 0 | 20 |
| egl-30(ad810); acy-1(md1756) | 18 | 2/53 | 3.8 | 20 |
| egl-30(ad810); kin-2(ce179) | 11 | 0/45 | 0 | 21 |
| acy-1(pk1279) | 11 | 0/24 | 0 | 25 |
| egl-30(tg26); acy-1(pk1279) | 30 | 1/57 | 1.8 | 30 |
| eat-16(ce71); acy-1(pk1279) | 18 | 0/50 | 0 | 21 |
| goa-1(sa734); acy-1(pk1279) | 13 | 0/6 | 0 | 16 |
Defined as the number of days needed for half of the original population to die. Animals were plated as 6- to 30-hr-old larvae.
Denominator is the sample size.
The heat-shock locomotion assays in this study were performed as described (Schade et al. 2005) and the heat shock and recovery times are specified in Figure 3. For phorbol ester locomotion assays, culture plates containing 5 μm phorbol myristate acetate (PMA; CalBiochem 52440; a 5 mg/ml stock solution was prepared in ethanol and stored at −35°) were prepared by adding the phorbol ester stock solution or ethanol carrier for control plates directly to 60° equilibrated molten media in a disposable flask with stirring. Plates were poured by hand in a chemical fume hood, allowed to solidify for 4 hr, and then spread with OP-50 culture. Plates were stored upright for ∼40 hr in covered plastic containers (separate containers for control and phorbol ester plates) before using for the assays. At this point, if plates were not immediately used for assays, they were inverted and sealed in plastic wrap for up to 3 days, with no detectable change in the response of wild-type animals. To assay, five animals were loaded on each of eight plates at 7-min intervals. After the elapsed time (usually 2.5–3 hr for adults), one of the five animals was chosen to count body bends for 6 min, and the remaining plates were assayed at staggered 7-min intervals. Control plates were loaded and assayed in parallel with the phorbol ester plates. All assays performed on larval-stage animals were done using 2.5 μm phorbol myristate acetate (instead of the 5.0-μm concentration used for adults), and larval animals were assayed 2–2.5 hr after plating (instead of 2.5–3 hr for adult assays). Synchronized larval animals for this assay were collected as described above.
Figure 3.—

Functional defects, rather than permanent developmental defects, cause paralysis in mutants lacking a neuronal Gαs pathway. Mean locomotion rates are shown, expressed as body bends per minute of wild-type and two transgenic strains containing the acy-1(pk1279) null mutation. Dark blue and cyan bars indicate locomotion rates without or with heat-shock treatment, respectively. Numbers above asterisks state the mean locomotion rates for nearly paralyzed strains. Note that heat-shock induction of the acy-1(gf) cDNA in muscle-rescued acy-1 null adults (genotype acy-1(pk1279); ceEx76) improves their locomotion rate by ∼275-fold, but the same treatment does not affect the locomotion rates of the wild-type and muscle-rescued acy-1 null control strains, which do not contain the heat-shock-promoter-driven acy-1 (gf) cDNA. Error bars represent the standard error of the mean for populations of eight animals. See also supplemental QuickTime movies for Figure 3 at http://www.genetics.org/supplemental/.
Video production and imaging:
Videos of worms on agar plates containing OP-50 bacterial lawns were captured and converted as described (Schade et al. 2005). Still images of mutants were collected using an Olympus C3040 digital camera mounted on an Olympus SZX-12 stereomicroscope. High-magnification larval images were obtained using a high resolution ×2, 0.275 NA plan apochromatic objective.
Drug sensitivity assays:
Aldicarb sensitivity assays using the population growth rate method were performed as described (Miller et al. 1999). Only larvae that were uniformly GFP positive in body-wall muscle cells were chosen for the acy-1(pk1279); ceEx76 and acy-1(pk1279); ceEx108 aldicarb sensitivity assays (Figure 4). Aldicarb acute paralysis assays on solid media were performed as previously described (Lackner et al. 1999; Nurrish et al. 1999). Aldicarb was added to a final concentration of 1 mm from a 10-mm stock solution in ddH2O (allowing ∼2–3 hr for dissolving before adding to the 55° cooled molten media), and media was made with 10% less water than normal to compensate for the large drug volume. Aldicarb-containing plates were seeded with OP-50 on the day that they were poured and stored at room temperature for 2 days, lid side up, before using. Animals designated for plates containing PMA + aldicarb were preincubated with 2.5 μm PMA to allow the PMA time course to maximize before beginning the aldicarb + PMA time course. These phorbol ester plates were produced as described (Schade et al. 2005). Preexposure time for N2 (wild type) was 1 hr and for egl-30(ad810) was 2.5 hr (on the basis of past experience of maximal response). Three such preincubation plates were produced at staggered times, so they would be available for each of the three independent trials of the aldicarb paralysis assay. Twenty animals were used for each strain/condition per trial. Six- to 30-hr-old N2 (wild type) and egl-30(ad810) homozygous larvae were collected for this assay as described in Locomotion assays.
Figure 4.—
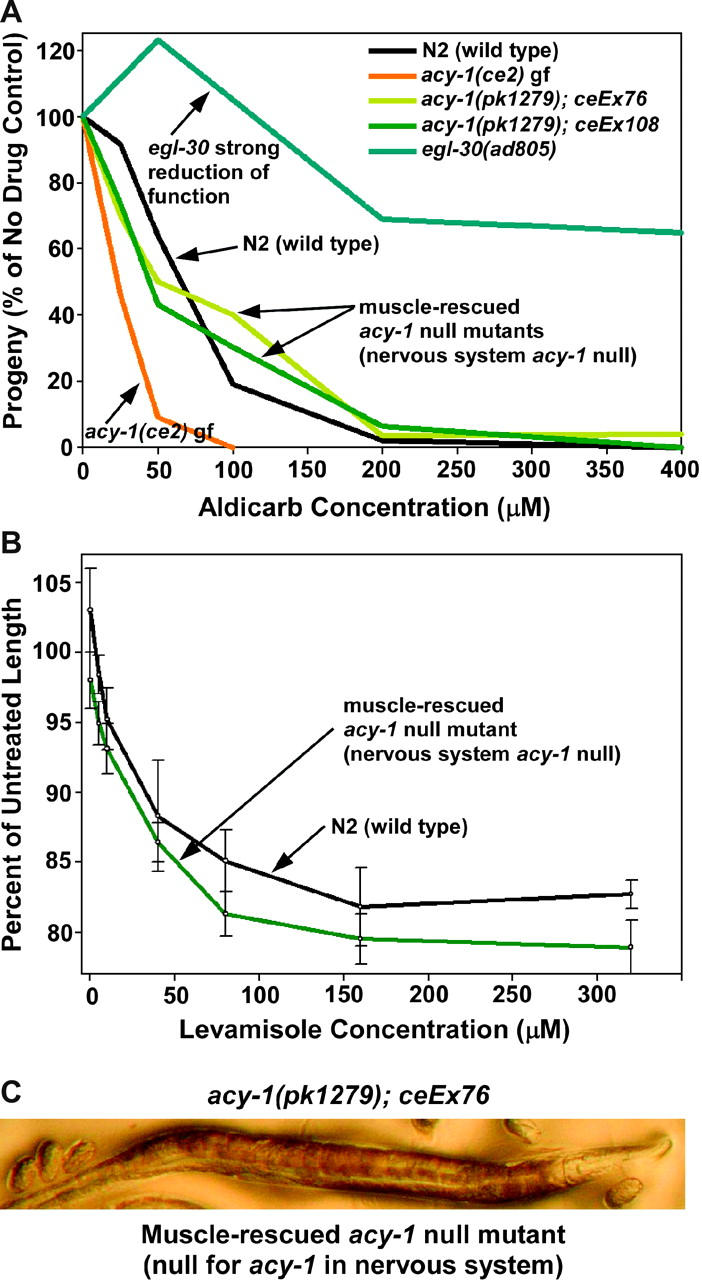
Knocking out the Gαs pathway in neurons does not significantly affect steady-state neurotransmitter release. (A) Comparison of the population growth rates of strains on various concentrations of aldicarb. One hundred percent represents the number of progeny produced from a starting population of L1 larvae over a 96-hr period in the absence of aldicarb (carrier only). Note that activating the Gαs pathway via a gain-of-function mutation in acy-1 causes hypersensitivity to aldicarb, which indicates increased neurotransmitter release; however, the opposite is not true: strains lacking the Gαs pathway in neurons exhibit levels of steady-state release that are superficially similar to the wild-type strain. For comparison, note that strongly reducing the function of the EGL-30 Gαq vesicle priming pathway causes strong resistance to aldicarb, which indicates strongly decreased neurotransmitter release. Curves are representative of duplicate experiments. (B) Animals lacking a neuronal Gαs pathway exhibit a near-normal postsynaptic response to the acetylcholine receptor agonist levamisole. The graph compares the contraction of individual N2 (wild type) or acy-1(pk1279); ceEx108 animals after a 20-min exposure to various concentrations of levamisole. Each data point represents the mean contraction of four to six individual animals and error bars represent standard errors. Animals lacking a neuronal Gαs pathway appear slightly hypersensitive to levamisole, although the difference is not statistically significant at any one data point. (C) The straight, paralyzed phenotype exhibited by strains lacking the Gαs pathway in neurons. Why are these animals nearly paralyzed if they are capable of releasing near-normal amounts of neurotransmitter?
To measure body-wall muscle contraction in response to levamisole, 24-well culture plates were prepared containing standard media in the first and third rows and standard media containing various concentrations of levamisole in the remaining rows (six identical wells containing the same drug concentration in each row). Levamisole (dispensed from a 100-mm aqueous stock) was added to a beaker of 55° molten media and dispensed into the wells using a 25-ml pipette rinsed with hot water between sets of wells. Wells were completely filled with media. Within 2 hr of solidifying, the media in the wells was spotted with 10 μl of an OP-50 bacterial culture. Plates were dried ∼1 hr in a 37° room with their lids off and then returned to room temperature and used for the assay ∼40 hr later. To assay contraction, mature adults from growing cultures (not previously starved) were singly placed in the rows containing no drug and, after a 10-min equilibration, were photographed at ×60 using an Olymplus C3040 digital camera attached to the stereomicroscope. Animals were then transferred at 20-sec intervals to their intended wells containing levamisole, which were in the rows immediately below the no-drug wells, such that each drug concentration had an “n” of six. Twenty minutes after loading the first animal on its levamisole well, each animal was photographed at 20-sec intervals. Images were cropped with Paintshop Pro to reduce size and then imported into Canvas 9.0. The curve tool was used to trace the center line of each animal along its entire length, and the length of the line was determined using the object specifications “tool.”
Measurements of population half-lives and percentage reaching adulthood:
To measure population half-lives of various larval lethal strains, ∼25–60 homozygous mutant or double-mutant animals were collected as described in Locomotion assays and transferred to a locomotion assay plate in a dab of moist bacteria. The starting age of the animals in each population ranged from 6 to 30 hr. Populations were monitored daily or every other day for the indicated number of days (Table 2), and dead animals were counted and removed. Living animals and the number of adults were also counted. “Dead” was considered to be a total lack of any observable movement or response to the touch of a pick. In most cases, death by this definition was also accompanied by an obvious change in the appearance of the animal. To assay the percentage reaching adulthood of the acy-1(pk1279) transgenic strains in Table 3, the indicated number of uniformly GFP positive 6- to 30-hr-old larvae from each strain was transferred to a locomotion assay plate. The plate was checked twice daily for the indicated number of days, and L3 and later-stage animals were transferred to a fresh plate for further observation. The number of adults on each plate was counted, and the counted animals were removed and killed.
TABLE 3.
Expression of theacy-1 cDNA in either the muscle or the nervous system partially rescues the larval arrest ofacy-1 null mutants
| Genotype | Tissue specificity of transgene |
Dosagea | Fraction reaching adulthoodb |
% reaching adulthood |
No. of days monitored |
|---|---|---|---|---|---|
| acy-1(pk1279) | No transgene | NA | 0/24 | 0 | 25 |
| acy-1(pk1279); ceEx108 [myo-3::acy-1(+)] | Muscle | Low | 20/37 | 54 | 5 |
| acy-1(pk1279); ceEx67 [myo-3::acy-1(+)] | Muscle | High | 47/101 | 47 | 5 |
| acy-1(pk1279); ceEx87 [myo-3::acy-1(gf)] | Muscle | High | 55/82 | 67 | 5 |
| acy-1(pk1279); ceEx97 [rab-3::acy-1(+)] | Nervous system | High | 11/33 | 33 | 5 |
| acy-1(pk1279); ceEx98 [rab-3::acy-1(gf)] | Nervous system | High | 6/64 | 9.4 | 7 |
Low dosage represents an injection concentration of 5 ng/μl; high dosage represents 15 ng/μl for myo-3-driven expression, 50 ng/μl for rab-3-driven expression of the wild-type acy-1 cDNA, and 15 ng/μl for rab-3-driven expression of the gain-of-function acy-1 cDNA.
This is the fraction of animals with uniform expression of the cotransformation GFP marker (either myo-3::GFP or rab-3::GFP, depending on the experiment) that reach adulthood. Denominator equals sample size.
RESULTS
The presynaptic Gαq pathway is the core synaptic vesicle priming pathway:
Hyperactivating the Gαq pathway through the egl-30(tg26) gain-of-function mutation causes strongly hyperactive locomotion (Figure 1B). A model of the C. elegans EGL-30 (Gαq) pathway predicts that this is caused by increased synaptic vesicle priming, in part via DAG-mediated activation of the UNC-13 synaptic vesicle priming protein (Figure 1A); however, a prior study suggests that UNC-13-mediated synaptic vesicle priming is not dependent on DAG binding to UNC-13, because eliminating the ability of UNC-13 to bind DAG does not significantly affect neurotransmitter release (Rhee et al. 2002). Nevertheless, if the neuronal Gαq pathway ultimately exerts its effects through UNC-13 or through an UNC-13-dependent mechanism, then a strong egl-30 (Gαq) gain-of-function mutation should have little or no effect on the phenotype conferred by a strong reduction of mutation in UNC-13, and this indeed is the case. egl-30(tg26) unc-13(s69) double mutants are just as paralyzed as unc-13(s69) single mutants, which move at ∼1 body bend per 3 min (∼1.7% of the wild-type rate; Figure 1B).
Figure 1.—
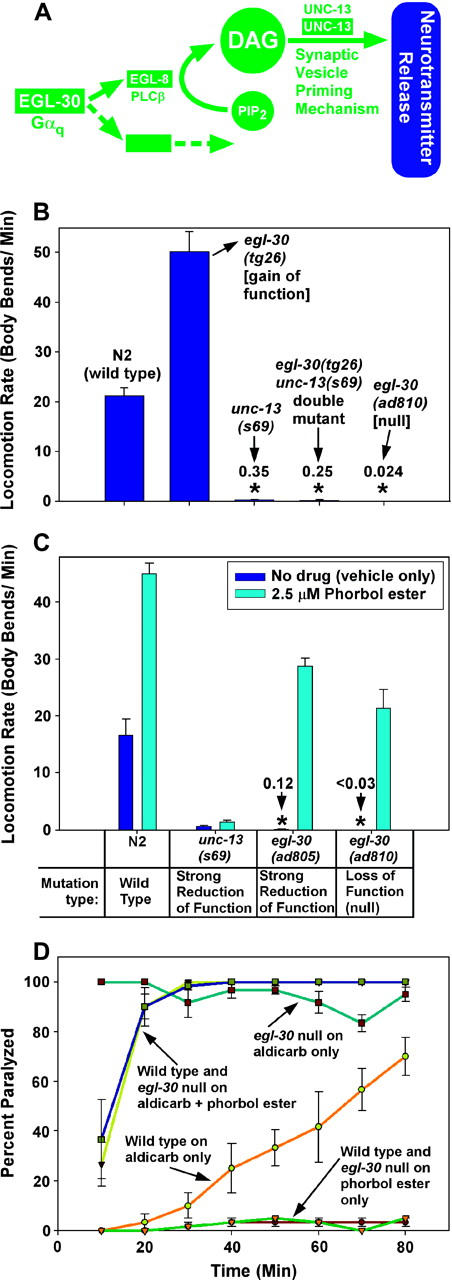
The presynaptic Gαq pathway is the core synaptic vesicle priming pathway. (A) A circuit model of the core EGL-30 (Gαq) pathway, in which EGL-30's action is mediated by EGL-8 (PLCβ) and one or more unidentified effectors. EGL-8 makes the small molecule DAG, which interacts with the synaptic vesicle priming mechanism by binding to, among other possible targets, the C1 domain of UNC-13. Not shown are other components of the synaptic signaling network that are known to regulate this pathway. This model is based on the following studies: Maruyama and Brenner (1991), Brundage et al. (1996), Hajdu-Cronin et al. (1999), Lackner et al. (1999), Miller et al. (1999), Nurrish et al. (1999), and Richmond et al. (1999)(2001). (B) Knocking out the EGL-30 (Gαq) pathway reduces locomotion rate to levels comparable to the synaptic vesicle priming loss-of-function mutant unc-13(s69), and the hyperactivated EGL-30 pathway is completely blocked, with respect to locomotion rate, by the unc-13(s69) mutation. Mean locomotion rates are shown, expressed as body bends per minute, of the indicated strains. Bars are not clearly visible for the three strains with very low locomotion rates, which are marked with asterisks. Numbers above the asterisks indicate actual body bends per minute. Error bars represent the standard error of the mean for populations of 8–10 animals. Data for egl-30(ad810) were derived from a track locomotion assay (see materials and methods) and are based on the movement of ∼80 animals over a 2-hr period. The means of the unc-13(s69) single mutant and the egl-30(tg26) unc-13(s69) double mutant are not significantly different. (C) Phorbol esters rescue the paralysis of egl-30 null mutants by a mechanism that requires synaptic vesicle priming. Mean locomotion rates are shown, expressed as body bends per minute of various strains on plates containing phorbol myristate acetate (cyan bars) or vehicle only (0.06% ethanol; dark blue bars). The N2 wild-type strain is shown for comparison. Bars are not clearly visible for the two egl-30 strains in the absence of phorbol ester treatment (asterisks). Numbers above the asterisks indicate actual body bends per minute. Error bars represent the standard error of the mean for populations of 8–10 animals. See also supplemental QuickTime movies for Figure 1 at http://www.genetics.org/supplemental/. (D) Phorbol esters induce similar levels of steady-state neurotransmitter release in wild-type and egl-30 null mutants. The percentage of animals that are paralyzed, over a time course, on plates containing aldicarb and/or phorbol esters. Strains and conditions are indicated with arrows. Note that both wild type and egl-30 null mutants that have been treated with phorbol esters are equally hypersensitive to the paralytic effects of aldicarb (their lines overlap over most of the time course), which suggests that they both release similar amounts of acetylcholine. Error bars represent the standard error of the mean for three independent populations of 20 animals each.
The above experiment shows that the neuronal Gαq pathway is completely dependent on the UNC-13 priming protein to exert its effects on locomotion, but it does not address whether or not the Gαq pathway is required for synaptic vesicle priming. If the main function of the presynaptic EGL-30 (Gαq) pathway is to activate synaptic vesicle priming via UNC-13 and other proteins, then knocking out the EGL-30 (Gαq) pathway should cause paralysis at least as severe as that seen in unc-13(s69) mutants, and that indeed is the case. The egl-30(ad810) mutation, a putative null nonsense mutation that results in strong paralysis and larval arrest (Brundage et al. 1996), reduces locomotion rate to ∼1 body bend per 42 min, a level that is ∼900-fold less than that of wild type (Figure 1B).
We next considered the possibility that the paralysis of egl-30 null mutants is largely or entirely caused by an inability to prime synaptic vesicles, which would block neurotransmitter release and thus block locomotion. Indeed, in unc-13(s69) mutants, which are significantly less paralyzed than egl-30 null mutants, evoked neurotransmitter release is essentially not detectable and spontaneous release is reduced ∼300-fold (Richmond et al. 1999). To test whether the paralysis of egl-30 null mutants is caused by a lack of primed synaptic vesicles, we incubated egl-30 null mutants on plates containing phorbol esters, which are DAG analogs known to prime synaptic vesicles (Stevens and Sullivan 1998; Waters and Smith 2000). Remarkably, we found that incubating egl-30 null mutants for only ∼2 hr on plates containing phorbol esters increased their locomotion rates ∼800-fold, to levels slightly greater than those seen with wild-type animals on the drug-free control plates (Figure 1C and Figure 1 supplemental movies at http://www.genetics.org/supplemental/). These results demonstrate that neither muscle nor permanent developmental defects contribute substantially to the paralysis of egl-30 null mutants. Remarkably, we also found that continuous culture on phorbol ester plates can rescue the larval arrest phenotype of egl-30 null mutants, because we were able to maintain a culture of egl-30 nulls for at least three generations on a plate containing phorbol esters. In addition, incubation on phorbol ester plates induced significantly hyperactive locomotion in adult animals carrying the strong reduction-of-function mutation egl-30(ad805) (Figure 1C and Figure 1 supplemental movies at http://www.genetics.org/supplemental/). The effects of phorbol ester on locomotion rate were strongly dependent on the synaptic vesicle priming protein UNC-13, because phorbol ester had only a small effect on the paralysis caused by the near-null unc-13(s69) mutation (Figure 1C), mutants of which contain appropriately docked but unprimed vesicles (Richmond et al. 1999, 2001).
To explain these results, we hypothesized that phorbol esters, by mimicking the signaling molecule DAG that is produced by the Gαq pathway, bypass the requirement for EGL-30 by restoring neurotransmitter release and thus rescuing the paralysis. Since acetylcholine (ACh) is the major excitatory neurotransmitter controlling locomotion rate in C. elegans, we tested this idea by placing phorbol-ester-treated egl-30 nulls on plates containing the acetylcholinesterase inhibitor aldicarb. Since the secreted ACh that accumulates in the presence of aldicarb is toxic, mutations that decrease or increase the steady-state rate of ACh release confer resistance or hypersensitivity, respectively, to aldicarb (Rand and Nonet 1997). The results of plating phorbol-ester-treated egl-30 nulls on plates containing aldicarb are shown in Figure 1D, which plots the percentage of animals that are paralyzed, over a time course, on plates containing various combinations of phorbol esters and/or aldicarb. On plates with phorbol esters only, neither wild-type animals nor egl-30 null mutants are paralyzed at any time point. On plates containing aldicarb only, wild-type worms gradually become paralyzed as the drug is adsorbed and takes effect. The egl-30 null mutant is paralyzed even in the absence of aldicarb, and aldicarb causes only a slight rescue of the paralysis, which is consistent with these animals releasing only a small amount of neurotransmitter. In contrast, both phorbol-ester-treated wild-type and phorbol-ester-treated egl-30 null mutants are strongly and equally hypersensitive to the paralytic effects of aldicarb. This suggests that phorbol esters completely bypass the steady-state neurotransmitter release defect of egl-30 nulls. From the results in this section we conclude that the presynaptic Gαq pathway and the core synaptic vesicle priming pathway are one and the same.
Both muscle and nervous system defects contribute to the larval lethality of mutants with a blocked Gαs pathway:
To prepare for investigating the relationship of the Gαq pathway to the Gαs pathway, we next analyzed mutants in which the Gαs pathway is blocked. A previous study produced the acy-1(pk1279) mutation and showed that it deletes the acy-1 gene and causes larval arrest and paralysis that can be rescued with the wild-type acy-1 gene (Moorman and Plasterk 2002). Interestingly, the acy-1(pk1279) null mutation actually increases life span; the larval arrest results from a failure to grow (Moorman and Plasterk 2002). In C. elegans, control of locomotion rate by the Gαs pathway is completely dependent on adenylyl cyclase (ACY-1) (Schade et al. 2005). Since the Gαs pathway (including acy-1) is expressed in both the muscle and the nervous system (Korswagen et al. 1997; Moorman and Plasterk 2002), we used a full-length acy-1 cDNA driven by ectopic promoters to investigate the extent to which the muscle and/or nervous system Gαs pathways contribute to the larval arrest and paralysis phenotypes of acy-1 nulls. Surprisingly, we found that the larval arrest of acy-1 nulls could be partially rescued by expressing the acy-1 cDNA in either the muscle or the nervous system, using the myo-3 or rab-3 promoters, respectively (Table 3). Muscle-specific acy-1 expression seemed to give the most robust rescue of larval arrest: one-half to two-thirds of acy-1 nulls expressing the myo-3::acy-1 transgene reached adulthood, although the percentage reaching adulthood did not increase significantly when we upped the gene dosage or used the acy-1(ce2) gain-of-function mutation (Table 3). The muscle-rescued acy-1 nulls that did reach adulthood seemed to exhibit near-normal growth rate and size (data not shown). In contrast, acy-1 nulls that expressed the acy-1(+) cDNA only in their nervous systems were slower growing and smaller than their muscle-rescued counterparts (data not shown). Nevertheless, about one-third of acy-1 nulls expressing rab-3::acy-1(+) from transgenes reached adulthood; however, upping rab-3::acy-1 doseage via the acy-1(ce2) gain-of-function mutation increased, rather than decreased, larval arrest (Table 3). These results suggest that both the muscle and the neuronal Gαs pathways promote progression from larval to adult stages by an unknown mechanism.
Lack of a neuronal Gαs pathway causes strong paralysis, but the muscle Gαs pathway also seems to contribute significantly to normal locomotion:
To determine the relative contributions of muscle and nervous system Gαs pathways to the paralysis of acy-1 nulls, we assayed the locomotion rates of transgenic strains in which acy-1 nulls were selectively rescued in the muscle or the nervous system. acy-1(pk1279) null mutants without any transgene moved at a rate of 0.75 body bends per minute, which is ∼3–4% of the wild-type rate (Figure 2A). However, acy-1 null strains that selectively expressed the acy-1(+) cDNA transgene in muscle had significantly slower locomotion rates than acy-1 null single mutants (<1% of wild type in three independent strains), even when the acy-1(+) cDNA transgene was expressed at relatively high levels, although overexpression of the acy-1 gain-of-function mutation in muscle did slightly improve locomotion rate (Figure 2A). In contrast, acy-1 null strains that selectively expressed either the acy-1(+) or the acy-1 gain-of-function cDNAs in the nervous system (using the rab-3 promoter) both had significantly higher locomotion rates than acy-1 null single mutants. For example, the neuronally expressed acy-1(gf) transgene improved the locomotion rate of the acy-1 null to ∼50% of wild type (Figure 2A). The inability of selective nervous system acy-1 expression to fully rescue the paralysis of the acy-1 null is unlikely to be due to insufficient expression from the rab-3 promoter, because this promoter drives strong GFP expression in all neurons (data not shown) and, when hooked to an unc-18 cDNA, fully rescues the paralysis of an unc-18 null mutant (Figure 2B); however, the lack of full rescue could still be due to inappropriate control of expression by the rab-3 promoter.
Figure 2.—
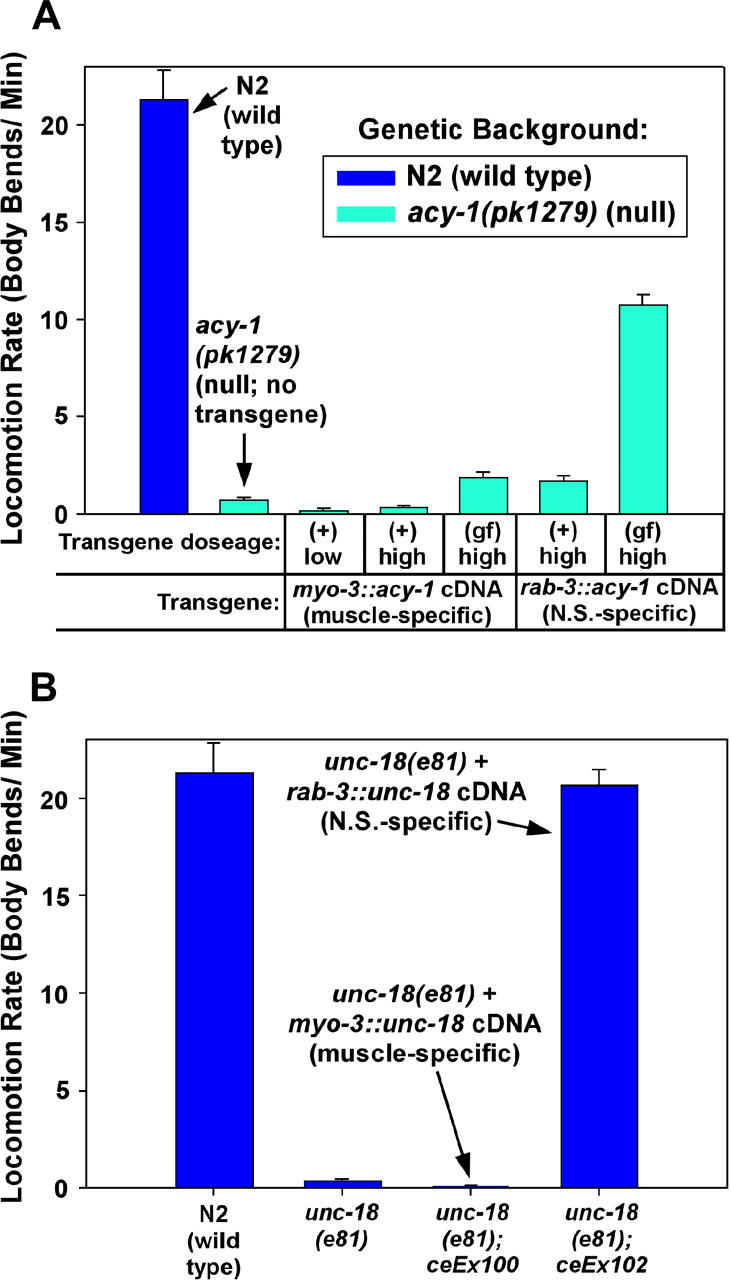
Lack of a neuronal Gαs pathway causes paralysis, but the muscle Gαs pathway also seems to contribute significantly to normal locomotion. (A) Neither muscle nor nervous system expression of acy-1 alone is sufficient to fully rescue the near-paralysis of acy-1 null mutants. The mean locomotion rates, expressed as body bends per minute, of transgenic strains that express the acy-1(+) or gain-of-function (gf) cDNA at various dosages in either the body-wall muscle or the nervous system in the acy-1(pk1279) null mutant background. Wild-type and the acy-1(pk1279) single mutant with no transgene are shown for comparison. Note that expressing the acy-1(+) cDNA in body-wall muscle, even at high doses, significantly reduces the locomotion rate of the acy-1(pk1279) null mutant (P = 0.0006 and 0.0057, respectively, for the low- and high-dose myo-3::acy-1(+) transgenes in A; see also the acy-1(pk1279); ceEx76 strain in Figure 3 (non-heat-shock conditions; P = 0.0001 for this strain). However, expressing the acy-1(gf) cDNA at high levels in either the body-wall muscle or the nervous system significantly improves the locomotion rate of the acy-1(pk1279) null mutant (P = 0.0025 and <0.0001 for (gf) expression in muscle or nervous system, respectively). Note, however, that none of the transgenic conditions restores wild-type levels of locomotion to the acy-1 null mutant, although nervous system expression has by far the greatest effect. Genotypes of the five transgenic strains, from left to right, are as follows: acy-1(pk1279); ceEx108, acy-1(pk1279); ceEx67, acy-1(pk1279); ceEx87, acy-1(pk1279); ceEx97, acy-1(pk1279); ceEx98. Error bars represent the standard error of the mean for populations of 10 animals. Statistical significance tests used the unpaired t-test with Welch correction. (B) Control experiments showing that the neuronal-specific rab-3 promoter driving the unc-18 cDNA can fully rescue the paralysis of an unc-18 null mutant and that the muscle-specific myo-3 promoter driving the unc-18 cDNA gives no rescue of the unc-18 null mutant. Locomotion rates are expressed as body bends per minute. Error bars represent the standard error of the mean for populations of 8–10 animals.
In summary, the paralysis of muscle-rescued acy-1 null mutants demonstrates that lack of acy-1 in the nervous system causes strong paralysis that sufficiently accounts for the paralysis of acy-1(pk1279) null mutants; however, the muscle Gαs pathway also seems to contribute significantly to normal locomotion.
Functional defects, rather than permanent developmental defects, cause paralysis in mutants lacking a neuronal Gαs pathway:
To determine whether the paralysis of the muscle-rescued acy-1 null mutant is due to functional or developmental defects, we transformed acy-1(pk1279) null mutants with a myo-3::acy-1(+) cDNA (to selectively rescue acy-1 in muscle) plus an acy-1 gain-of-function cDNA driven by a heat-shock-inducible promoter (to allow us to use a heat-shock treatment to induce acy-1 expression in the adult nervous system). Animals carrying transgenes with this mixture of cDNAs were often able to develop to adults of normal size and, in the absence of heat-shock treatment, were generally indistinguishable from the muscle-rescued acy-1 nulls described earlier, exhibiting locomotion rates ∼0.4% of wild type (Figure 3). However, when we subjected this strain to a 1-hr heat-shock treatment, followed by a 2-hr recovery period, its mean locomotion rate improved remarkably (almost 300-fold) to a level not significantly different from that of wild type (Figure 3 and Figure 3 supplemental movies at http://www.genetics.org/supplemental/). In contrast, heat-shock treatment of wild-type or muscle-rescued acy-1 null transgenic animals that did not have the heat-shock-promoted acy-1 cDNA did not significantly affect the locomotion rate of these strains at the 2-hr recovery time point. This suggests that the paralysis of acy-1 null mutants is largely due to neurons that cannot drive locomotion to any substantial extent without an intact Gαs pathway.
Knocking out the neuronal Gαs pathway appears to not significantly affect overall levels of neurotransmitter release:
Since the paralysis of mutants lacking a neuronal Gαs pathway is caused by neurons that cannot function properly, and since activating the C. elegans Gαs pathway increases overall steady-state levels of neurotransmitter release and causes hyperactive locomotion (Schade et al. 2005), we logically expected that knocking out the Gαs pathway in neurons might severely disrupt neurotransmitter release. To test this, we determined the aldicarb sensitivity of animals lacking a neuronal Gαs pathway (muscle-rescued acy-1 nulls). Surprisingly, we found that they exhibited approximately wild-type aldicarb sensitivity (Figure 4A). This is in striking contrast to the strong aldicarb resistance of similarly paralyzed Gαq reduction of function mutants (Figure 4A). Could the near-normal aldicarb sensitivity of animals lacking a neuronal Gαs pathway be caused by defective ACh receptor responses masking or compensating for a neurotransmitter release defect? This is unlikely because the neuronal Gαs pathway mutants showed a near-normal response to the ACh receptor agonist levamisole (Figure 4B). We conclude that knocking out the neuronal Gαs pathway via the acy-1 null mutation does not significantly affect steady-state levels of neurotransmitter release, despite the ostensibly incongruous fact that these mutants are strongly paralyzed for functional, nondevelopmental reasons. Furthermore, since synaptic vesicle priming is required for neurotransmitter release (Aravamudan et al. 1999; Augustin et al. 1999; Richmond et al. 1999, 2001), this result suggests that the Gαs pathway is not required for synaptic vesicle priming.
The Gαs pathway is largely dependent on the Gαq synaptic vesicle priming pathway to exert its effects on locomotion:
Since the Gαs pathway is not required for normal steady-state neurotransmitter release, why do mutants with a hyperactive Gαs pathway exhibit highly coordinated hyperactive locomotion and increased steady-state neurotransmitter release, and why does Gαs pathway activation suppress the paralysis and neurotransmitter release defect of a mutation that strongly reduces the function of the Gαq priming pathway (Schade et al. 2005)? To address this question, we investigated the relationship of the Gαs and Gαq pathways to each other by analyzing double mutants in which one pathway is completely knocked out and the other is strongly activated and visa versa. For reference, Table 1 summarizes the mutations that we used for this genetic epistasis analysis.
We first asked to what extent the Gαs pathway is dependent on the Gαq priming pathway to exert its effects on locomotion. To address this question, we analyzed double mutants containing an activated Gαs pathway in combination with the egl-30 (Gαq) null mutation ad810. We found that a strong acy-1 gain-of-function mutation, which hyperactivates the Gαs effector ACY-1 (adenylyl cyclase), caused only a small, although significant, suppression of the paralysis of the EGL-30 (Gαq) null (Figure 5A). Specifically, the strongly hyperactive locomotion rate of the acy-1 gain-of-function mutant was reduced by ∼98% by knocking out the Gαq pathway. This shows that only ∼2% of the effects of cAMP on locomotion occur independently of the Gαq pathway and thus that the neuronal Gαs pathway is largely dependent on the Gαq pathway to exert its effects. Our analysis of double mutants containing hyperactivated protein kinase A in combination with the egl-30 (Gαq) null again illustrated the strong dependence of the Gαs pathway on the Gαq pathway: the egl-30 null was completely unaffected by the strong kin-2 (regulatory subunit of protein kinase A, PKA) reduction-of-function mutation, which normally confers strongly hyperactive locomotion (Figure 5A and Figure 5 supplemental movies at http://www.genetics.org/supplemental/). This 100% block of activated protein kinase A caused by knocking out the Gαq pathway was not the result of the unexpected synthetic effects of combining the two mutations, because placing these double-mutant animals on plates containing phorbol esters quickly restored wild-type levels of locomotion (Figure 5A and Figure 5 supplemental movies at http://www.genetics.org/supplemental/). These results show that the ability of protein kinase A to stimulate locomotion is completely dependent on the Gαq pathway and thus that the Gαs and Gαq pathways must converge at some point in their joint regulation of locomotion rate.
We also found that activating the Gαs pathway did not significantly affect the larval arrest phenotype of the egl-30 (Gαq) null mutant. Consistent with the locomotion results described above, 100% of egl-30 null mutants arrested before reaching adulthood, regardless of whether or not they contained the strong kin-2 mutation that activates protein kinase A, and a strong acy-1 gain-of-function mutation allowed only 2 of 53 animals to reach adulthood (Table 2).
The neuronal Gαs pathway is required for the Gαq pathway to properly drive locomotion, and this is mediated by a pathway interaction that occurs downstream of DAG production:
As we showed earlier, animals that lack a neuronal Gαs pathway are strongly paralyzed for functional, nondevelopmental reasons, and yet steady-state neurotransmitter release, which is mediated by the Gαq pathway, is unimpaired in such animals. This suggests that the neuronal Gαs pathway is required for the wild-type Gαq priming pathway to properly drive locomotion. To further investigate the relationship of the Gαq and Gαs pathways to each other, we next did the complementary experiment to the one described in the last section: we knocked out the Gαs pathway, via the acy-1(pk1279) null mutation, and then asked to what extent we could bypass the resulting paralysis with mutations that strongly activate the Gαq pathway or with phorbol esters. The results of that experiment are shown in Figure 5, B and C. Whereas animals that lack a neuronal Gαs pathway are nearly paralyzed in response to wild-type levels of Gαq pathway activity, we found that hyperactivating the Gαq pathway, either via native mutations or by applying phorbol esters to activate the downstream priming part of the pathway, improved the locomotion rate of these animals to levels that were about one-third of the levels of egl-30 (Gαq) gain-of-function single mutants or phorbol-ester-treated control animals; however, we also observed that all of the double mutants containing a hyperactivated Gαq pathway (or phorbol ester treatment) in the absence of a neuronal Gαs pathway spend much of their time in a highly abnormal, tightly knotted, paralyzed posture (Figure 5, B and C, and Figure 5 supplemental movies at http://www.genetics.org/supplemental/). For example, in the absence of a neuronal Gαs pathway, phorbol esters induced a slow loopy movement interspersed with long periods of knotted paralysis, whereas hyperactivating Gαq in the absence of a neuronal Gαs pathway induced periods of surprisingly fluid movement interspersed with the tightly knotted phenotype, which dominated about two-thirds of the time (averaging 72% ± 2.9% of each assay interval; n = 12). These results, along with our observation that steady-state neurotransmitter release occurs normally in the absence of a neuronal Gαs pathway, suggest that the Gαq pathway can still exert its function independently of a neuronal Gαs pathway, but that these conditions do not support stable, coordinated locomotion. Furthermore, because phorbol esters confer the knotted phenotype on animals lacking a neuronal Gαs pathway (and only partially rescue their paralysis), these data suggest that the Gαs pathway regulates the locomotion response to DAG, and thus exerts its effects on the Gαq pathway downstream of DAG production. The phorbol ester results are all the more striking when compared to the effects of phorbol esters on the egl-30 (Gαq) null, which is significantly more paralyzed than animals lacking a neuronal Gαs pathway. egl-30 nulls are rescued to wild-type levels of locomotion by phorbol ester treatment and such animals do not exhibit the paralyzed, knotted phenotype. The larval arrest phenotype of animals that lack the Gαs pathway in both neurons and body-wall muscles was not mitigated by activating the Gαq pathway or by knocking out the inhibitory Gαo pathway (Table 2).
In summary of the last two sections, the first epistasis experiment (in which we knocked out the Gαq pathway and hyperactivated the Gαs pathway) suggests that the Gαs pathway is largely dependent on the Gαq pathway to exert its effects on locomotion. Therefore the Gαs pathway converges with the Gαq pathway and largely cannot bypass it to produce locomotion. In light of that result, the second epistasis experiment (in which we knocked out the neuronal Gαs pathway and hyperactivated the Gαq pathway) suggests that the Gαs pathway acts on the Gαq pathway to convert neurotransmitter release (a function of the Gαq priming pathway) into sustained, coordinated locomotion (a combined function of the Gαs and Gαq pathways).
The guanine nucleotide exchange factor RIC-8 (synembryn) appears necessary to maintain both the Gαq and the neuronal Gαs pathway in a functional state:
The above results suggests that coordinated locomotion requires appropriately balanced coactivation of both the Gαq and the Gαs pathways. Previous results suggested that maintaining a functional EGL-30 (Gαq) pathway requires RIC-8 (synembryn), originally identified in C. elegans as a novel, conserved protein that appears to function upstream of EGL-30 (Gαq) (Miller et al. 2000) and recently revealed by biochemical studies to be a GEF that helps monomeric Gα subunits (including, but not limited to, Gαq) attain the GTP-bound activated state independently of receptor stimulation (Tall et al. 2003). If RIC-8 is indeed essential to maintain a functional Gαq pathway, then, on the basis of the epistasis results in Figure 5A, we would predict that a ric-8 null mutant should not be suppressed by activating the Gαs pathway. To test this, we obtained the ric-8 null mutant ok98, a deletion mutant described in Figure 6A. About one-third of the animals homozygous for this deletion arrest as paralyzed larvae, although they can live for ∼1 week after hatching, while the rest eventually become paralyzed, sterile adults. The deletion also affects a second nonconserved gene of unknown function that is in an operon with ric-8; however, a fusion PCR fragment that does not contain this second gene provides nearly complete rescue of the paralysis of ric-8(ok98) mutants (Figure 6B).
The degree of paralysis of ric-8(ok98) (0.0139 ± 0.14 body bends/minute; Figure 6B) is not significantly different from that of a Gαq null mutant (0.024 ± 0.003 body bends/minute; Figure 1B), a result that is consistent with ric-8(ok98) being null or near null for the EGL-30 (Gαq) pathway. Consistent with our earlier results showing that the Gαs pathway is strongly dependent on the Gαq pathway to exert its effects, we found that the paralysis of ric-8(ok98) null mutants is not strongly suppressed by activating the Gαs pathway (Figure 6C and Figure 6 supplemental QuickTime movies at http://www.genetics.org/supplemental/). Of all of the mutations that most strongly activate the Gαs pathway, only the gsa-1(ce81) and gsa-1(ce94) gain-of-function mutations cause significant, albeit weak, suppression of the ric-8(ok98) null mutant; however, even the gsa-1(ce94); ric-8(ok98) double mutant has a locomotion rate that is only ∼2% of the gsa-1(ce94) single mutant (Figure 6C). Nevertheless, this degree of suppression, although small with respect to paralysis, was sufficient to rescue the ∼33% larval lethality, although these double mutants, like ric-8(ok98) single mutants, were completely sterile (data not shown).
If the only, or major, function of RIC-8 at the synapse is to mediate Gαq nucleotide exchange and thereby to maintain activation of the Gαq pathway, then it should be possible to bypass the paralysis of ric-8(ok98) null mutants by incubating them on plates containing phorbol esters. Recall that incubating Gαq null mutants on plates containing phorbol esters for only 2 hr improves their locomotion rate ∼900-fold, to levels similar to that of the wild-type strain on drug-free control plates (Figure 1C). In strong contrast, we found that ric-8(ok98) mutants are only partially suppressed by incubation on phorbol ester plates (Figure 6D and Figure 6 supplemental QuickTime movies at http://www.genetics.org/supplemental/). Although the degree of suppression is highly significant, the peak locomotion rate induced by phorbol treatment of ric-8(ok98) is still only ∼20% of that seen in phorbol ester-treated egl-30 null mutants (compare Figure 6C to Figure 1C). This strongly suggests that Gαq nucleotide exchange is not RIC-8's only function. We therefore considered the possibility that RIC-8 is also required to maintain activation of the neuronal GSA-1 (Gαs) pathway. Consistent with this idea, phorbol-ester-treated ric-8(ok98) null mutants have locomotion rates similar to those seen in phorbol-ester-treated animals that lack the neuronal Gαs pathway (compare Figure 6D to Figure 5C), and gsa-1(ce94); ric-8(ok98) double mutants have locomotion rates similar to those seen when the Gαs pathway is activated in an EGL-30 (Gαq) null background (compare Figure 6D to Figure 5A). If ric-8(ok98) null mutants are effectively knocked out for both the Gαq and the Gαs pathways, then appropriate coactivation of both pathways might rescue their paralysis, and this indeed is the case. Incubating gsa-1(ce94); ric-8(ok98) double mutants on plates containing 5 μm phorbol esters for only 2 hr improves their locomotion rate to levels slightly greater than that of the wild-type strain on the drug-free control plates and, remarkably, restores beautifully coordinated locomotion (Figure 6D and Figure 6 supplemental movies at http://www.genetics.org/supplemental/). However, we also observed a late inhibitory effect of unknown origin that began dominating ∼30–90 min after the 2-hr peak locomotion point. After 4 hr of exposure to the phorbol ester plates, the gsa-1(ce94); ric-8(ok98) double mutants were more paralyzed than they had been before exposure to phorbol esters (0.0 ± 0 body bends/minute; n = 8), whereas the wild-type strain N2 continued to exhibit hyperactive locomotion at the 4-hr time point (33.2 ± 1.9 body bends/minute; n = 8), although even its locomotion rate was reduced from its 2-hr peak. We have not further investigated the cause of the strong inhibitory influence that we observed at later time points in phorbol-ester-exposed gsa-1(ce94); ric-8(ok98) double mutants.
In summary, because ric-8 null mutants have phenotypes consistent with lacking both the Gαq and the neuronal Gαs pathways; because singly activating each pathway in the ric-8 null mutant results in phenotypes consistent with lacking the cognate pathway; and because simultaneous activation of both pathways restores wild-type levels of coordinated locomotion to the ric-8 null mutant, we conclude that the RIC-8 guanine nucleotide exchange factor is likely to be required to maintain both the Gαq and the neuronal Gαs pathways in a functional state.
DISCUSSION
The Gαq pathway is the core pathway for priming synaptic vesicles:
In our investigation of the relationship of the Gαq pathway to synaptic vesicle priming, we showed that we can completely block the Gαq pathway by a near-null mutation in UNC-13 that specifically blocks synaptic vesicle priming, that we can completely rescue the paralysis and neurotransmitter release defect of a Gαq null by applying an appropriate concentration of a synaptic vesicle priming stimulator (phorbol ester), and that phorbol ester is largely, if not completely, dependent on the UNC-13 priming protein to exert its effects on locomotion rate. These results suggest that the presynaptic Gαq pathway is equivalent to the core synaptic vesicle priming pathway.
The fact that Gαq nulls are even more paralyzed than strong reduction-of-function unc-13(s69) mutants is interesting because unc-13(s69) mutants exhibit almost no detectable spontaneous or evoked neurotransmitter release (Richmond et al. 1999). This finding, combined with the results presented here, strongly suggests that the Gαq pathway is essential for neurotransmitter release and that its purpose is to activate UNC-13 and possibly other molecules associated with synaptic vesicle priming.
Our results show that we can completely suppress the paralysis and neurotransmitter release defect of a Gαq null by flooding the mutant's nervous system with an appropriate concentration of phorbol ester and that this effect is strongly UNC-13 dependent. However, this does not necessarily mean that the effects of phorbol esters are mediated entirely by direct interactions with UNC-13. That phorbol esters have an effect on UNC-13 by direct interactions seems evident from a prior study showing that a mutation that eliminates the ability of UNC-13 to bind DAG affects the dose-response relationship between phorbol esters and aldicarb sensitivity (Lackner et al. 1999). Furthermore, a more recent study used the same mutation to demonstrate that DAG binding to UNC-13 is essential for use-dependent alterations of synaptic efficacy (Rhee et al. 2002). However, that same study also showed that UNC-13-mediated synaptic vesicle priming is not dependent on DAG binding to UNC-13, because eliminating the ability of UNC-13 to bind DAG does not significantly affect evoked or spontaneous release at synapses that had not been previously manipulated (Rhee et al. 2002). Our analysis of egl-30(tg26); unc-13(s69) double mutants suggests that all effectors of the presynaptic Gαq pathway (including DAG) must ultimately channel their effects through UNC-13 or a through an UNC-13-dependent process. Although our results also show that nearly all of the effects of phorbol esters ultimately require UNC-13, in light of the study by Rhee et al. (2002) we believe that much of this requirement must be indirect (e.g., through protein kinase C phosphorylating UNC-13 or UNC-13 interacting proteins). Indeed, we and others have found that the DAG/phorbol-ester-binding protein TPA-1 (PKC δ/θ) is required for the hyperactive locomotion response induced by phorbol esters (Tabuse et al. 1989; K. G. Miller, unpublished observations).
The Gαq and Gαs pathways converge via pathway interactions downstream of DAG production:
Our genetic experiments clearly show that the neuronal Gαs pathway is largely dependent on the Gαq synaptic vesicle priming pathway to exert its effects on locomotion and that activated protein kinase A (a key downstream effector of the Gαs pathway) is completely dependent on the Gαq pathway. This result shows that the Gαs and Gαq pathways must converge at some point. Despite this convergence, the Gαs and Gαq pathways are functionally distinct, because, as we show in this article, knocking out either pathway in neurons causes strong paralysis that is the result of neurons that cannot function properly (i.e., the two pathways are not redundant). Furthermore, the Gαq pathway appears to be the major determinant of steady-state neurotransmitter release, whereas the Gαs pathway clearly is not. In a second set of epistasis experiments, we discovered that the Gαs pathway is involved in the locomotion response of the activated Gαq pathway and, importantly, our phorbol ester studies suggest that the Gαs pathway is involved in the locomotion response to the messenger molecule DAG. Our results therefore support a model in which the Gαs pathway interacts with the Gαq priming pathway at a point downstream of DAG production (Figure 7).
Figure 7.—
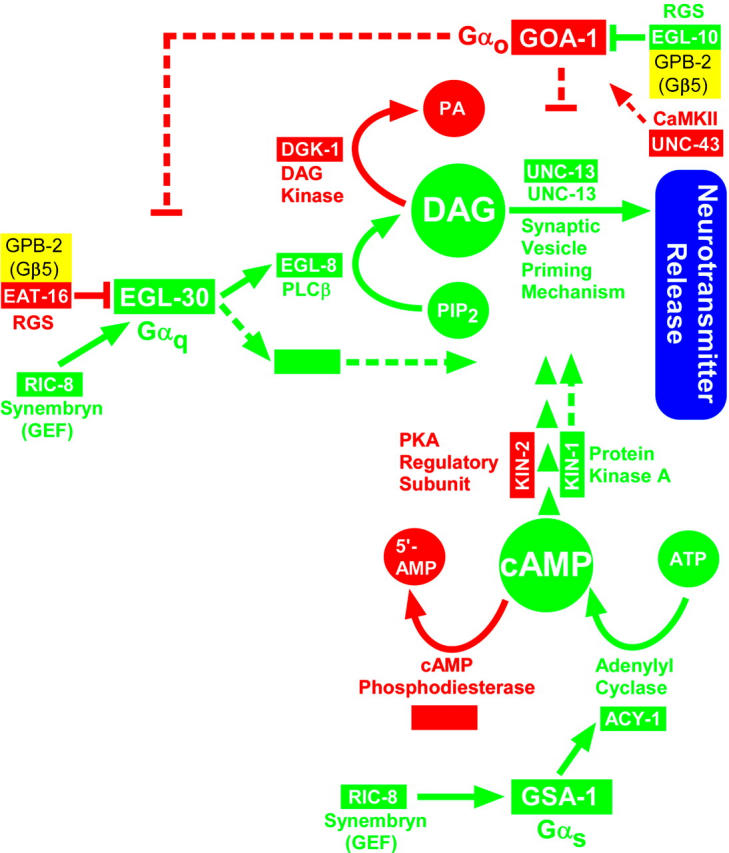
Molecular circuit model of the synaptic signaling network. Solid lines indicate that direct interactions are known or likely, while dashed lines and/or large gaps between line endpoints and downstream effectors indicate poorly understood interactions or missing components. Proteins that promote locomotion or neurotransmitter release are green, while proteins that inhibit locomotion or neurotransmitter release are red. The core pathway of the network is the EGL-30 (Gαq) synaptic vesicle priming pathway described in Figure 1. The network also includes another G protein, GOA-1 (Gαo), that negatively regulates the EGL-30 (Gαq) pathway by one or more unknown mechanisms, a DAG kinase that antagonizes the EGL-30 pathway, and two RGS proteins that negatively regulate each G protein. Results presented in the current study suggest that the neuronal Gαq and Gαs pathways converge downstream of DAG production, possibly at or near the synaptic vesicle priming mechanism. Even though the neuronal Gαs pathway is functionally distinct from the Gαq pathway and appears to not be required for synaptic vesicle priming, it is largely dependent on the Gαq synaptic vesicle priming pathway to exert its effects in producing locomotion. Further linking the Gαq and Gαs pathways, the Gα guanine nucleotide exchange factor RIC-8 (synembryn) appears essential to maintain both pathways in a functional state (this study) and could, we hypothesize, possibly function to maintain extended coactivation of the Gαq and Gαs pathways at optimal synapses for locomotion. Other studies, in addition to this study, have contributed to this model (Maruyama and Brenner 1991; Mendel et al. 1995; Segalat et al. 1995; Brundage et al. 1996; Koelle and Horvitz 1996; Hajdu-Cronin et al. 1999; Lackner et al. 1999; Miller et al. 1999, 2000; Nurrish et al. 1999; Richmond et al. 1999, 2001; Robatzek and Thomas 2000; Chase et al. 2001; Robatzek et al. 2001; van der Linden et al. 2001; Tall et al. 2003; Schade et al. 2005).
Previous studies have demonstrated that the function of a third Gα pathway, the Gαo pathway, is also closely linked to the core Gαq priming pathway (Hajdu-Cronin et al. 1999; Miller et al. 1999, 2000; Nurrish et al. 1999; Robatzek and Thomas 2000; Chase et al. 2001; Robatzek et al. 2001; van der Linden et al. 2001; Figure 7). The major purpose of the Gαo pathway appears to be to negatively regulate the core Gαq priming pathway (Hajdu-Cronin et al. 1999; Miller et al. 1999). However, despite the fact that knocking out either the Gαo or Gαs pathways has opposite effects on locomotion rate, and despite the fact that both pathways are dependent on the Gαq priming pathway to exert their effects on locomotion, the Gαo pathway and Gαs pathways do not simply act in directly opposing ways on the Gαq pathway. This is evident from the observation that too much Gαo pathway activity, caused by knocking out the EGL-10 RGS protein (Koelle and Horvitz 1996), confers aldicarb resistance (Miller et al. 1996; K. G. Miller; data not shown), which indicates decreased steady-state neurotransmitter release, whereas knocking out the neuronal Gαs pathway does not significantly affect aldicarb sensitivity (this study). Each of the three Gα pathways in the synaptic signaling network is therefore functionally distinct, despite their ultimate convergence.
Since the synaptic vesicle priming mechanism is a known downstream target of DAG/phorbol esters, it seems reasonable to hypothesize that the Gαq and Gαs pathways converge at or near the synaptic vesicle priming mechanism. Although our experiments do not address how these two G protein pathways are linked at the molecular level, recent studies provide evidence for at least one physical connection between the Gαq and Gαs pathways. Studies of mouse RIM1α, a synaptic molecule that directly interacts with mouse UNC-13 (Betz et al. 2001), found that protein kinase A, which is activated by the Gαs pathway, directly phosphorylates RIM1α and that this event triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses (Lonart et al. 2003). In light of our findings that the Gαq pathway is completely dependent on the UNC-13 priming protein to exert its effects on locomotion, the mouse RIM1α studies provide evidence of a physical link between the two pathways and the synaptic vesicle priming mechanism. However, RIM1α cannot be the only major target of the Gαs pathway, at least in C. elegans, because C. elegans strains that lack UNC-10 (RIM1α) are much more active than the neuronal Gαs pathway nulls that we describe here (Koushika et al. 2001; K. G. Miller, data not shown).
The Gαs pathway is not required for the core synaptic vesicle priming function:
Our study shows that knocking out the neuronal Gαs pathway via the acy-1 null mutation does not significantly affect overall steady-state levels of neurotransmitter release, as measured by aldicarb sensitivity. Although surprising at first glance, this finding is in agreement with recent Drosophila studies showing that evoked release remains unchanged, or even increases, at low rates of artificial stimulation in Gαs null synapses (Hou et al. 2003; Renden and Broadie 2003). Since synaptic vesicle priming appears to be required for neurotransmitter release (Aravamudan et al. 1999; Augustin et al. 1999; Richmond et al. 1999, 2001), these results also demonstrate that the Gαs pathway is not required for the core priming function. However, previous electrophysiological studies comparing gain-of-function manipulations of the Gαs and Gαq pathways have found that both pathways can affect synaptic vesicle and/or synapse priming (reviewed in the Introduction). So what exactly is the difference between the functions of the Gαq and Gαs pathways?
A close functional relationship between the presynaptic Gαq and Gαs pathways:
Synthesizing our findings with previous knowledge of the Gαs and Gαq pathways, we hypothesize that the purpose of the neuronal Gαs pathway may be to transduce critical positional information onto the Gαq pathway to stabilize the priming of selected synapses that are optimal for producing locomotion, whereas the Gαq pathway globally generates the core, obligatory signals for synaptic vesicle priming and has only limited abilities to interpret positional cues. Information on which synapses need to be primed may come, at least in part, from retrograde signals from the muscle (for regulating movements) or from other neurons (Davis et al. 1998; Tao and Poo 2001; Doi and Iwasaki 2002). According to this hypothesis, in a mutant with an overactive Gαs pathway, hyperstabilization of “optimal” release sites for locomotion results in increased neurotransmitter release at those sites (aldicarb hypersensitivity) and strongly hyperactive, highly coordinated locomotion as observed by Schade et al. (2005). The hypothesis further predicts that knocking out the Gαs pathway in the nervous system prevents transduction of positional information and thus prevents neurons from determining which synapses need to be optimally primed for locomotion. Neurotransmitter release, mediated by the global Gαq priming pathway, still occurs at near-normal levels, but in the absence of proper positional information, it is uncoupled from driving locomotion, thus explaining the paralysis of these animals (e.g., Figure 3). Forcing release in the absence of a neuronal Gαs pathway by hyperactivation of the Gαq pathway restores movement for brief periods (perhaps due to a limited ability of the Gαq pathway to interpret positional cues that do not depend on the Gαs pathway) but most of the time results in a knotted, paralyzed phenotype (because the nervous system cannot determine which synapses are optimal for locomotion; e.g., Figure 5C. Finally, activating the Gαs pathway in a Gαq null background has little effect because the Gαs pathway is largely dependent on the Gαq pathway to exert its synapse-activating effects (e.g., Figure 5A). We note, however, that although this interpretation of our data seems consistent with an “optimal synapse priming” role for the Gαs pathway, we consider this hypothesis speculative and in need of further investigation.
A role for RIC-8 (synembryn) in maintaining functional Gαq and neuronal Gαs pathways:
Our epistasis results using the ric-8 null mutant strongly suggest that, at least in neurons, RIC-8 is required to maintain both the Gαq and the Gαs pathways in a functional state. This concept initially surprised us. Vertebrate forms of RIC-8 have been found to interact with both Gαq and Gαs (Klattenhoff et al. 2003; Tall et al. 2003); however, RIC-8's nucleotide exchange activity has been demonstrated only for Gαq, Gαo/i, and Gα13 (Tall et al. 2003). One potentially trivial explanation is that the vertebrate RIC-8A and RIC-8B isoforms are specialized for different Gα proteins, whereas the one isoform in C. elegans retains a broad Gα specificity. Indeed, due to problems purifying nonaggregated RIC-8B, only the RIC-8A isoform was tested and found to have no effect on Gαs nucleotide exchange (Tall et al. 2003). Since only the RIC-8B isoform significantly interacts with Gαs by yeast two-hybrid, we think it is quite possible that the vertebrate RIC-8B isoform mediates receptor-independent Gαs nucleotide exchange.
On the basis of their findings that RIC-8 interacts preferentially with monomeric GDP-bound Gα proteins and does not stimulate nucleotide exchange when Gα is complexed to βγ, Tall et al. (2003) proposed that RIC-8 functions to amplify the duration of a signal that comes from an individual G protein. That is, G-protein-coupled receptors function as the initial GEF to activate Gαβγ heterotrimers and to produce free active GTP-bound Gα. Then, after the Gα hydrolyzes its GTP to convert to the inactive GDP-bound form, and before it reassociates with βγ, RIC-8 reactivates it by exchanging GTP for GDP. RIC-8 therefore has the ability, at least in vitro, to maintain a G-protein pathway in its active state for an indefinite period of time in the absence of continuous receptor stimulation.
Although little or nothing is known about how long the Gαq pathway can remain active at a synapse, it is interesting to note that, in C. elegans, one can induce strongly hyperactive locomotion by flooding the animal's nervous system with an appropriate concentration of phorbol esters (Miller et al. 2000; this study). This suggests that synapses in intact animals have a high tolerance for a continuously activated Gαq pathway. In addition, our finding that RIC-8 null mutants are just as paralyzed as EGL-30 (Gαq) null mutants suggests that RIC-8-mediated activation of the Gαq pathway in neurons is critically important and that receptor-mediated activation of the Gαq pathway is not sufficient to maintain a functional pathway, at least with respect to producing locomotion (likewise for the neuronal Gαs pathway since RIC-8 also appears necessary to maintain it in a functional state).
Coactivation of the Gαq and Gαs pathways drives C. elegans locomotion:
The knotted phenotype that results from hyperactivating the Gαq pathway in the absence of a neuronal Gαs pathway (as well as the paralysis that results from wild-type levels of Gαq pathway activity in the absence of a neuronal Gαs pathway) suggests that the activity of both pathways must be appropriately controlled or coordinated (at least at selected synapses) to achieve stable, coordinated locomotion. We have not yet investigated whether or not RIC-8, by virtue of its essential role in maintaining both pathways, might play a role in coordinating Gαq and Gαs pathway activation.
Although ours is the first study to investigate the relationship of the Gαq and Gαs pathways in intact, behaving, animals, previous studies using cultured neurons seem consistent with the possibility that coactivation of the Gαq and Gαs pathways mediates synapse activation. Studies of Aplysia sensory neurons in culture have shown that application of serotonin, which can activate both the Gαs and Gαq pathways in those neurons (Byrne and Kandel 1996), induces rapid synapse maturation and that the resulting synaptic facilitation occurs by means of an “all-or-none” switching on of synapses (Coulson and Klein 1997; Royer et al. 2000). A study of serotonin's effects on the crayfish neuromuscular junction proposed a similar conclusion as a possibility (Wang and Zucker 1998). Although these studies do not reveal the relationship of the Gαq and Gαs pathways to each other or even the relative contributions of each pathway to the measured output, they are consistent with coactivation of both pathways being a major determinant of whether or not a synapse is turned on. In other intriguing studies in which the activity state of the Gαq pathway was totally unknown and not controlled, manipulations that increase cAMP in primary cultures of vertebrate neurons activated previously “silent” synapses (Tong et al. 1996; Chavis et al. 1998; Ma et al. 1999).
Acknowledgments
The authors are grateful to the C. elegans Gene Knockout Consortium for providing the ric-8(ok98) allele, to Celine Moorman and Ron Plasterk for providing the acy-1(pk1276) mutant, to Ann Rose for providing unc-13(s69), and to Kouichi Iwasaki for providing his egl-30(tg26) mutant ahead of publication. All DNA sequences were obtained from the Core DNA Sequencing Facility at Oklahoma Medical Research Foundation. Some of the strains used here were provided by the C. elegans Genetics Center. This work was supported by grants from the National Institute of Mental Health (MH62400) and the Oklahoma Center for the Advancement of Science and Technology (HR02-093).
References
- Aravamudan, B., and K. Broadie, 2003. Synaptic Drosophila UNC-13 is regulated by antagonistic G-protein pathways via a proteasome-dependent degradation mechanism. J. Neurobiol. 54: 417–438. [DOI] [PubMed] [Google Scholar]
- Aravamudan, B., T. Fergestad, W. S. Davis, C. K. Rodesch and K. Broadie, 1999. Drosophila Unc-13 is essential for synaptic transmission. Nat. Neurosci. 2(11): 965–971. [DOI] [PubMed] [Google Scholar]
- Augustin, I., C. Rosenmund, T. C. Südhof and N. Brose, 1999. Munc13–1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature 400: 457–461. [DOI] [PubMed] [Google Scholar]
- Avery, L., and S. Wasserman, 1992. Ordering gene function: the interpretation of epistasis in regulatory hierarchies. Trends Genet. 8: 312–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz, A., M. Okamoto, F. Benseler and N. Brose, 1997. Direct interaction of the rat unc-13 homologue Munc13–1 with the N terminus of syntaxin. J. Biol. Chem. 272(4): 2520–2526. [DOI] [PubMed] [Google Scholar]
- Betz, A., P. Thakur, H. J. Junge, U. Ashery, J. S. Rhee et al., 2001. Functional interaction of the active zone proteins Munc13–1 and RIM1 in synaptic vesicle priming. Neuron 30: 183–196. [DOI] [PubMed] [Google Scholar]
- Brenner, S., 1974. The genetics of C. elegans. Genetics 77: 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundage, L., L. Avery, A. Katz, U. Kim, J. E. Mendel et al., 1996. Mutations in a C. elegans Gqα gene disrupt movement, egg laying, and viability. Neuron 16: 999–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne, J. H., and E. R. Kandel, 1996. Presynaptic facilitation revisited: state and time dependence. J. Neurosci. 16: 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase, D. L., G. Patikoglou and M. R. Koelle, 2001. Two RGS proteins that inhibit Gαo and Gαq signaling in C. elegans neurons require a Gβ5-like subunit for function. Curr. Biol. 11: 222–231. [DOI] [PubMed] [Google Scholar]
- Chavis, P., P. Mollard, J. Bockaert and O. Manzoni, 1998. Visualization of cyclic AMP-regulated presynaptic activity at cerebellar granule cells. Neuron 20: 773–781. [DOI] [PubMed] [Google Scholar]
- Chen, C., and W. G. Regehr, 1997. The mechanism of cAMP-mediated enhancement at a cerebellar synapse. J. Neurosci. 17: 8687–8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulson, R. L., and M. Klein, 1997. Rapid development of synaptic connections and plasticity between sensory neurons and motor neurons of Aplysia in cell culture: implications for learning and regulation of synaptic strength. J. Neurophysiol. 77: 2316–2327. [DOI] [PubMed] [Google Scholar]
- Davis, G. W., A. DiAntonio, S. A. Petersen and C. S. Goodman, 1998. Postsynaptic PKA controls quantal size and reveals a retrograde signal that regulates presynaptic transmitter release in Drosophila. Neuron 20: 305–315. [DOI] [PubMed] [Google Scholar]
- Doi, M., and K. Iwasaki, 2002. Regulation of retrograde signaling at neuromuscular junctions by the novel C2 domain protein AEX-1. Neuron 33: 249–259. [DOI] [PubMed] [Google Scholar]
- Hajdu-Cronin, Y. M., W. J. Chen, G. Patikoglou, M. R. Koelle and P. W. Sternberg, 1999. Antagonism between Goα and Gqα in C. elegans: the RGS protein EAT-16 is necessary for Goα signaling and regulates Gqα activity. Genes Dev. 13: 1780–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert, O., 2002. PCR fusion-based approach to create reporter gene constructs for expression analysis in transgenic C. elegans. BioTechniques 32: 728–730. [DOI] [PubMed] [Google Scholar]
- Hou, D., K. Suzuki, W. J. Wolfgang, C. Clay, M. Forte et al., 2003. Presynaptic impairment of synaptic transmission in Drosophila embryos lacking Gsα. J. Neurosci. 23: 5897–5905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, L. S., and P. W. Sternberg, 1995 Genetic dissection of developmental pathways, pp. 97–122 in Caenorhabditis elegans: Modern Biological Analysis of an Organism, edited by H. F. Epstein and D. C. Shakes. Academic Press, San Diego.
- Kandel, E. R., and C. Pittenger, 1999. The past, the future and the biology of memory storage. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354: 2027–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klattenhoff, C., M. Montecino, X. Soto, L. Guzman, X. Romo et al., 2003. Human brain synembryn interacts with Gsα and Gqα and is translocated to the plasma membrane in response to isoproterenol and carbachol. J. Cell. Physiol. 195: 151–157. [DOI] [PubMed] [Google Scholar]
- Koelle, M. R., and H. R. Horvitz, 1996. EGL-10 regulates G protein signaling in the C. elegans nervous system and shares a conserved domain with many mammalian proteins. Cell 84: 112–125. [DOI] [PubMed] [Google Scholar]
- Kohn, R. E., J. S. Duerr, J. McManus, A. Duke, T. L. Rakow et al., 2000. Expression of multiple UNC-13 proteins in the Caenorhabditis elegans nervous system. Mol. Biol. Cell 11: 3441–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korswagen, H. C., J. Park, Y. Ohshima and R. H. Plasterk, 1997. An activating mutation in a Caenorhabditis elegans Gs protein induces neural degeneration. Genes Dev. 11: 1493–1503. [DOI] [PubMed] [Google Scholar]
- Koushika, S. P., J. E. Richmond, G. Hadwiger, R. M. Weimer, E. M. Jorgensen et al., 2001. A post-docking role for active zone protein Rim. Nat. Neurosci. 4: 997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuromi, H., and Y. Kidokoro, 2000. Tetanic stimulation recruits vesicles from reserve pool via a cAMP-mediated process in Drosophila synapses. Neuron 27: 133–143. [DOI] [PubMed] [Google Scholar]
- Lackner, M. R., S. J. Nurrish and J. M. Kaplan, 1999. Facilitation of synaptic transmission by EGL-30 Gqα and EGL-8 PLCβ: DAG binding to UNC-13 is required to stimulate acetylcholine release. Neuron 24: 335–346. [DOI] [PubMed] [Google Scholar]
- Lin, R. C., and R. H. Scheller, 2000. Mechanisms of synaptic vesicle exocytosis. Annu. Rev. Cell Dev. Biol. 16: 19–49. [DOI] [PubMed] [Google Scholar]
- Lonart, G., S. Schoch, P. S. Kaeser, C. J. Larkin, T. C. Sudhof et al., 2003. Phosphorylation of RIM1α by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell 115: 49–60. [DOI] [PubMed] [Google Scholar]
- Ma, L., L. Zablow, E. R. Kandel and S. A. Siegelbaum, 1999. Cyclic AMP induces functional presynaptic boutons in hippocampal CA3–CA1 neuronal cultures. Nat. Neurosci. 2: 24–30. [DOI] [PubMed] [Google Scholar]
- Maruyama, I. N., and S. Brenner, 1991. A phorbol ester/diacylglycerol-binding protein encoded by the unc-13 gene of Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 88: 5729–5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello, C. C., J. M. Kramer, D. Stinchcomb and V. Ambros, 1991. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 10(12): 3959–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendel, J. E., H. C. Korswagen, K. S. Liu, Y. M. Hajdu-Cronin, M. I. Simon et al., 1995. Participation of the protein Go in multiple aspects of behavior in C. elegans. Nature 267: 1652–1655. [DOI] [PubMed] [Google Scholar]
- Miller, K. G., A. Alfonso, M. Nguyen, J. A. Crowell, C. D. Johnson et al., 1996. A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc. Natl. Acad. Sci. USA 93: 12593–12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, K. G., M. D. Emerson and J. B. Rand, 1999. Goα and diacylglycerol kinase negatively regulate the Gqα pathway in C. elegans. Neuron 24: 323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, K. G., M. D. Emerson, J. McManus and J. B. Rand, 2000. RIC-8 (Synembryn): a novel conserved protein that is required for Gqα signaling in the C. elegans nervous system. Neuron 27: 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorman, C., and R. H. Plasterk, 2002. Functional characterization of the adenylyl cyclase gene sgs-1 by analysis of a mutational spectrum in Caenorhabditis elegans. Genetics 161: 133–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurrish, S., L. Segalat and J. M. Kaplan, 1999. Serotonin inhibition of synaptic transmission: Gαo decreases the abundance of UNC-13 at release sites. Neuron 24: 231–242. [DOI] [PubMed] [Google Scholar]
- Rand, J. B., and M. L. Nonet, 1997 Synaptic transmission, pp. 611–643 in C. elegans II, edited by D. H. Riddle, T. Blumenthal, B. J. Meyer and J. R. Priess. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [PubMed]
- Renden, R. B., and K. Broadie, 2003. Mutation and activation of Gαs similarly alters pre- and postsynaptic mechanisms modulating neurotransmission. J. Neurophysiol. 89: 2620–2638. [DOI] [PubMed] [Google Scholar]
- Rhee, J. S., A. Betz, S. Pyott, K. Reim, F. Varoqueaux et al., 2002. β phorbol ester- and diacylglycerol-induced augmentation of transmitter release is mediated by Munc13s and not by PKCs. Cell 108: 121–133. [DOI] [PubMed] [Google Scholar]
- Richmond, J. E., W. S. Davis and E. M. Jorgensen, 1999. UNC-13 is required for synaptic vesicle fusion in C. elegans. Nat. Neurosci. 2(11): 959–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond, J. E., R. M. Weimer and E. M. Jorgensen, 2001. An open form of syntaxin bypasses the requirement for UNC-13 in vesicle priming. Nature 412: 338–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robatzek, M., and J. H. Thomas, 2000. Calcium/calmodulin-dependent protein kinase II regulates Caenorhabditis elegans locomotion in concert with a Go/Gq signaling network. Genetics 156: 1069–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robatzek, M., T. Niacaris, K. Steger, L. Avery and J. H. Thomas, 2001. eat-11 encodes GPB-2, a Gβ5 ortholog that interacts with Goα and Gqα to regulate C. elegans behavior. Curr. Biol. 11: 288–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer, S., R. L. Coulson and M. Klein, 2000. Switching off and on of synaptic sites at aplysia sensorimotor synapses. J. Neurosci. 20: 626–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassa, T., S. Harada, H. Ogawa, J. B. Rand, I. N. Maruyama et al., 1999. Regulation of the UNC-18-Caenorhabditis elegans syntaxin complex by UNC-13. J. Neurosci. 19: 4772–4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schade, M. A., N. K. Reynolds, C. M. Dollins and K. G. Miller, 2005. Mutations that rescue the paralysis of Caenorhabditis elegans ric-8 (synembryn) mutants activate the Gαs pathway and define a third major branch of the synaptic signaling network. Genetics 169: 631–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segalat, L., D. A. Elkes and J. M. Kaplan, 1995. Modulation of serotonin-controlled behaviors by Go in Caenhorabditis elegans. Nature 267: 1648–1651. [DOI] [PubMed] [Google Scholar]
- Speese, S. D., N. Trotta, C. K. Rodesch, B. Aravamudan and K. Broadie, 2003. The ubiquitin proteasome system acutely regulates presynaptic protein turnover and synaptic efficacy. Curr. Biol. 13: 899–910. [DOI] [PubMed] [Google Scholar]
- Stevens, C. F., and J. M. Sullivan, 1998. Regulation of the readily releasable vesicle pool by protein kinase C. Neuron 21: 885–893. [DOI] [PubMed] [Google Scholar]
- Sudhof, T. C., 2000. The synaptic vesicle cycle revisited. Neuron 28: 317–320. [DOI] [PubMed] [Google Scholar]
- Tabuse, Y., K. Nishiwaki and J. Miwa, 1989. Mutations in a protein kinase C homolog confer phorbol ester resistance on Caenorhabditis elegans. Science 243: 1713–1716. [DOI] [PubMed] [Google Scholar]
- Tall, G. G., A. M. Krumins and A. G. Gilman, 2003. Mammalian Ric-8A (synembryn) is a heterotrimeric Gα protein guanine nucleotide exchange factor. J. Biol. Chem. 278: 8356–8362. [DOI] [PubMed] [Google Scholar]
- Tao, H. W., and M. Poo, 2001. Retrograde signaling at central synapses. Proc. Natl. Acad. Sci. USA 98: 11009–11015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, G., R. C. Malenka and R. A. Nicoll, 1996. Long-term potentiation in cultures of single hippocampal granule cells: a presynaptic form of plasticity. Neuron 16: 1147–1157. [DOI] [PubMed] [Google Scholar]
- Trudeau, L. E., D. G. Emery and P. G. Haydon, 1996. Direct modulation of the secretory machinery underlies PKA-dependent synaptic facilitation in hippocampal neurons. Neuron 17: 789–797. [DOI] [PubMed] [Google Scholar]
- van der Linden, A. M., F. Simmer, E. Cuppen and R. H. Plasterk, 2001. The G-protein β-subunit GPB-2 in Caenorhabditis elegans regulates the Goα-Gqα signaling network through interactions with the regulator of G-protein signaling proteins EGL-10 and EAT-16. Genetics 158: 221–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C., and R. S. Zucker, 1998. Regulation of synaptic vesicle recycling by calcium and serotonin. Neuron 21: 155–167. [DOI] [PubMed] [Google Scholar]
- Waters, J., and S. J. Smith, 2000. Phorbol esters potentiate evoked and spontaneous release by different presynaptic mechanisms. J. Neurosci. 20: 7863–7870. [DOI] [PMC free article] [PubMed] [Google Scholar]