

Abstract
We compared the sequence polymorphism of 12 genomic fragments in six geographically dispersed African populations to one European Drosophila melanogaster population. On the basis of one African and one European population half of these fragments have strongly reduced levels of variability outside of Africa. Despite this striking difference in European variation, we detected no significant difference in African variation between the two fragment classes. The joint analysis of all African populations indicated that all high-frequency European alleles are of African origin. We observed a negative Tajima's D in all African populations, with three populations deviating significantly from neutral equilibrium. Low, but statistically significant, population differentiation was observed among the African populations. Our results imply that the population structure and demographic past of African D. melanogaster populations need to be considered for the inference of footprints of selection in non-African populations.
IN the wake of a steadily growing number of sequenced genomes, hitchhiking mapping has become a popular approach for the identification of genomic regions, which were recently subjected to positive directional selection (Schlötterer 2003). The underlying idea is that the spread of a beneficial mutation is not limited to the target of selection alone, but also neutral flanking variation is affected [(hitchhiking) Kaplan et al. 1989; Maynard Smith and Haigh 1974]. Thus, any neutral genetic marker linked to a beneficial mutation could be used to identify the genomic region affected by such a selective sweep. Different approaches to the identification of nonneutral evolution in hitchhiking mapping studies have been suggested, such as the reduction in variability, increased level of linkage disequilibrium, and a skewed allele frequency spectrum (Kohn et al. 2000; Kim and Stephan 2002; Payseur et al. 2002; Schlötterer 2002, 2003). Apart from humans, Drosophila melanogaster is probably the organism for which most in-depth hitchhiking studies have been performed (Schlötterer et al. 1997; Harr et al. 2002; Glinka et al. 2003; Kauer et al. 2003a; Orengo and Aguadé 2004).
Biogeographical and genetic data suggest that D. melanogaster evolved in tropical Africa and colonized the rest of the world only recently (David and Capy 1988; Lachaise et al. 1988). Estimates for the colonization time range from ∼15,000 to 10,000 years ago for Europe and Asia, to some 100 years ago for the Americas and Australia (David and Capy 1988). The colonization of novel environments outside the ancestral species range probably imposed new selection pressure such as novel climatic conditions or food resources. Thus, it is likely that a burst of adaptations associated with the out-of-Africa habitat expansion occurred in the derived populations. Hence, the comparison of African and non-African D. melanogaster populations is a promising approach to gain insight into the genetic changes required for a habitat expansion from Africa to the rest of the world.
Consistent with previous microsatellite surveys (Kauer et al. 2003a), a genomic scan based on 105 X-linked intergenic and intronic fragments (Glinka et al. 2003) identified a large number of genomic regions deviating from neutral expectations. While microsatellite-based surveys focus mainly on the reduction of variability, the analysis of DNA sequences also provides information on the frequency spectrum of ancestral and derived sequence variants. Glinka et al. (2003) observed a pronounced difference between genomic regions putatively affected by a recent selective sweep and genomic regions, which showed no deviation from neutral expectations. Compared to neutrally evolving fragments, those with low levels of sequence polymorphism in a European population (i.e., putatively selected ones) displayed a pronounced excess of derived sites fixed in Europe but absent or rare in the African sample. A very similar pattern was described in a fine scale analysis of selective sweeps in non-African D. melanogaster, which combined microsatellite and DNA sequence polymorphism analysis. Two genes cramped and syntaxin4 were identified as putative targets of selection and both genes carried amino acid replacements that were fixed between African and non-African populations (Harr et al. 2002).
Given that fixed differences between African and non-African populations could be of central importance for the identification of the targets of selection, it is extremely important to gain more insight into how standing African variation is reflected in derived populations in genomic regions subjected to a recent selective sweep. Here we analyzed six X-linked noncoding fragments, identified as candidate regions affected by selection (Glinka et al. 2003), and six putatively neutrally evolving fragments selected from the same study in five additional sub-Saharan African populations. We detected considerable population structure among African populations and significant deviations from neutrality in three out of six African populations, strongly implying past demographic events in African D. melanogaster populations. Furthermore, all sites previously described to be fixed between African and non-African D. melanogaster were found to be segregating in our African samples.
MATERIALS AND METHODS
Fly stocks:
Sequence data previously published by Glinka et al. (2003) were acquired from the EMBL database (http://www.ebi.ac.uk) for 12 African lines originating from Lake Kariba, Zimbabwe (ZLK), 12 European lines originating from Leiden, The Netherlands (NL), and one D. simulans line (Davis, CA). Additional sequences were collected for 46 isofemale African D. melanogaster lines: 7 D. melanogaster lines were collected in Kampala, Uganda (KAM), 10 in Kisoro, Uganda (KIS), 10 lines in Kenya (KEN), 9 in Mali (MA), and 10 lines in the Sengwa Wildlife Reserve, Zimbabwe (ZS). Furthermore, one D. simulans from Kampala, Uganda, was included.
Loci sequenced:
A subset of 12 fragments was selected from 105 noncoding DNA fragments from both intronic and intergenic regions on the X chromosome (Glinka et al. 2003). When compared to an African sample, 6 of these fragments were previously shown to contain no or little variation in a European sample of 12 lines, whereas the other 6 fragments exhibited “normal” levels of polymorphism (Glinka et al. 2003). Primers were designed on the basis of release 3.2 of the complete D. melanogaster genome sequence. For two fragments (NV120 and LV375), our sequences were only partially overlapping with the published sequences. These fragments were also sequenced in one European population from Neumarkt (Germany) to obtain a European reference. The average length of the fragments was ∼400 bp (see Table 1 for details). Each fragment was sequenced in both directions. Sequence data have been submitted to GenBank under accession nos. AJ889255, AJ889565, AJ889618, AJ889823, and AJ889869, AJ889870, AJ889871, AJ889872, AJ889873, AJ889874, AJ889875, AJ889876, AJ889877, AJ889878, AJ889879, AJ889880, AJ889881, AJ889882, AJ889883, AJ889884, AJ889885, AJ889886, AJ889887, AJ889888, AJ889889, AJ889890, AJ889891, AJ889892, AJ889893, AJ889894, AJ889895, AJ889896, AJ889897, AJ889898, AJ889899, AJ889900, AJ889901, AJ889902, AJ889903, AJ889904, AJ889905, AJ889906, AJ889907, AJ889908, AJ889909, AJ889910, AJ889911, AJ889912, AJ889913, AJ889914. Primer sequences and amplification conditions are given in supplementary Table S1 (http://www.genetics.org/supplemental/).
TABLE 1.
List of loci analyzed
| Locus | Absolute position (bp) |
r | Relative distance (kb) |
Sequence type | Gene |
|---|---|---|---|---|---|
| NV57 | 3,338,549 | 3.138 | — | Intron | AlstR |
| NV120 | 6,811,021 | 2.178 | 3472.5 | Exon (289 nt)-intron | CG4607 |
| LV122 | 6,902,001 | 2.178 | 91.0 | Intergenic | — |
| LV125 | 7,029,642 | 1.926 | 127.6 | Intron | Unc119 |
| LV130 | 7,257,235 | 1.601 | 227.6 | Intergenic | — |
| NV139 | 7,762,275 | 1.486 | 505.0 | Exon (100 nt)-intron | Tbh |
| LV157 | 8,708,919 | 2.725 | 946.6 | Intron | rdgA |
| LV203 | 10,846,533 | 2.545 | 2137.6 | Exon (184 nt)-intron | CG1961 |
| NV216 | 11,404,294 | 3.44 | 557.8 | Exon (99 nt)-intron | Ptp10D |
| NV278 | 13,215,206 | 4.925 | 1810.9 | Intron | CG32635 |
| LV375 | 14,458,023 | 4.934 | 1242.8 | Intron | NetB |
| NV287 | 15,423,083 | 4.33 | 965.1 | Intergenic | — |
For each locus we provide absolute position on the X chromosome in base pairs (based on the D. melanogaster genome release 3.2); recombination rate (r) expressed as recombination events per site per generation × 10−8 (Glinka et al. 2003); relative distance between consecutive loci in kilobases; sequence type (for each locus partly covering an exonic region, the number of coding nucleotides is provided on the basis of release 3.2 of the D. melanogaster genome); and genes where the intronic loci are located. NV, normal variability; LV, low variability.
DNA preparation and sequencing:
DNA was extracted from a single male fly from each strain using a high-salt extraction protocol (Miller et al. 1988). DNA amplification was carried out in a 20-μl reaction volume. A typical cycling profile consisted of 3 min denaturation at 94° followed by 35 cycles of 94° for 40 sec, annealing temperature for 50 sec, and extension at 72° for 1 min. PCR products were sequenced for both strands using BigDye Terminators v1.1 cycle sequencing chemistry. The extension products were purified with Sephadex G-50 fine (Amersham Biosciences, Sweden) and separated on a MegaBACE 500 automated capillary sequencer. Forward and reverse strands were assembled using the Autoassembler 2.1 software and checked manually. Sequences were aligned with ClustalX (Thompson et al. 1997) and adjusted manually using the BioEdit sequence alignment editor (Hall 1999).
Statistical analysis:
Basic population genetic parameters were calculated using the DnaSP 4.00 software (Rozas et al. 2003). To assess whether the observed allele frequency spectrum for the African sample was in accordance with the expectations from the neutral model we calculated Tajima's D (Tajima 1989). To test the significance of the average Tajima's D across loci, we applied a multilocus version of Tajima's D-test using the HKA software provided by J. Hey (http://www.lifesci.rutgers.edu/~heylab/). This method evaluates whether the average observed value of Tajima's D is consistent with the equilibrium model by comparing it to its simulated equilibrium distribution (Kliman et al. 2000). We performed 10,000 independent standard coalescent simulations for each locus and population, conditioned on the observed number of segregating sites (S). We assumed no intragenic recombination, but free recombination between fragments. For each simulation, we noted whether it produced a D-value smaller or greater than the observed one.
Genetic differentiation among the African populations was analyzed by means of the haplotype statistic HST (Hudson et al. 1992) and the nearest-neighbor statistic Snn (Hudson 2000). HST is based on the haplotype frequencies in the sample alone and does not utilize the information on the number of differences between haplotypes. Snn is a measure of how often the nearest neighbors of sequences in sequence space are from the same locality. This method has been shown to combine the advantages of haplotype-based and sequence-based methods over a wide range of sample sizes and haplotype diversities (Hudson 2000). We calculated global and pairwise HST and Snn values for each fragment separately. To assess levels of significance, we permuted genotypes among populations 10,000 times. In cases where the sample size differed among populations the weighting factors recommended in Hudson et al. (1992) were used. Populations were tested for all pairwise combinations (including the ZLK population from Glinka et al. 2003). The differentiation probabilities P from pairwise population comparisons for all fragments were combined to the χ2-distributed quantity −2∑ln P with 2k d.f. (k being the number of fragments). This method of combining probabilities allows us to create an overall test for significance from a series of separate significance tests on different sets of data (Sokal and Rohlf 1995). The validity of this test depends on the assumption of free recombination for all fragments. In Drosophila, linkage disequilibrium dissipates within a few kilobases (Miyashita and Langley 1988; Langley et al. 2000). As the minimum physical distance between fragments analyzed was 91 kb (average ∼1.1 Mb, Table 1), we do not consider nonindependence of the fragments to be of major effect for our analyses.
We determined the time to the most recent common ancestor of the European fragments, by estimating the number of putatively new mutations in the derived populations that arose after the split from the ancestral populations: first, we identified all alleles that were present only in the European sample. Second, we excluded those variants that were found in the same individual, reasoning that they more likely represent a rare haplotype rather than two novel mutations occurring on the same chromosome. Third, we dated the colonization event on the basis of a silent mutation rate of 15.4 × 10−9 substitutions/year/bp (Li 1997). Finally, we used the genetree software (Bahlo and Griffiths 2000) to determine the distribution of the time to the most recent common ancestor (TMRCA) of the European population scaled in Ne generations. We circumvented the potential problem of ancestral African variation segregating in the European population by using only those mutations for which we inferred a European origin. The coalescent simulations implemented in genetree assumed a constant population size and θ values identical to the estimates for the African population [mean θ for low variability (LV) fragments = 0.0142; mean θ for normal variability (NV) fragments = 0.0191].
RESULTS
DNA sequence variation:
We sequenced 12 noncoding fragments from predominantly intronic and intergenic regions on the X chromosome in five African populations from Uganda (Kampala and Kisoro), Kenya, Mali, and Zimbabwe (ZS) (see Table 1 for details). The number of segregating sites, haplotype diversity, nucleotide diversity, levels of divergence, Tajima's D, and Fay and Wu's H for the combined African sample are given in Table 2 (see supplementary Table S2 at http://www.genetics.org/supplemental/ for a breakdown for each locus by population). To account for different mutation rates in the genomic regions sequenced, we followed an approach previously suggested (e.g., Schlenke and Begun 2003) and standardized the population variation estimators by the divergence between D. simulans and D. melanogaster. Similar levels of variability were detected in all African populations, with average silent heterozygosity-to-divergence ratios ranging from 0.18 to 0.26. Silent heterozygosity-to-divergence ratios (averaged across populations) differed among loci and ranged from 0.13 to 0.37. Our set of fragments contained six regions, which were previously shown to have low levels of polymorphism in a European population (LV fragments), while the remaining six fragments harbored normal levels of variability (NV fragments) (Glinka et al.'s 2003 supplementary Table S2 at http://www.genetics.org/supplemental/). In our sample of African populations no significant difference in variability (silent heterozygosity-to-divergence ratios) was detected between LV (0.21 ± 0.08) and NV (0.26 ± 0.09) fragments (Mann-Whitney U-test, P = 0.31).
TABLE 2.
Summary statistics for 12 X-chromosomal fragments in African and European population samples
| Total African sample
|
European sample
|
||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fragment | Length (bp) |
N | S | H | HD | π | θW |
D (%) |
DT | HFW | N | S | H | HD | π | θW |
D (%) |
DT | HFW |
| Low variation in Europe | |||||||||||||||||||
| LV122 | 513 | 55 | 20 | 21 | 0.76 | 0.0026 | 0.0085 | 1.29 | −2.25** | −0.72 | 11 | 0 | 1 | 0.00 | 0.0000 | 0.0000 | 1.71 | — | — |
| LV125 | 240 | 58 | 19 | 27 | 0.92 | 0.0098 | 0.0171 | 7.68 | −1.42 | −0.82 | 12 | 0 | 1 | 0.00 | 0.0000 | 0.0000 | 7.81 | — | — |
| LV130 | 553 | 52 | 36 | 34 | 0.97 | 0.0108 | 0.0144 | 5.78 | −0.97 | −0.80 | 12 | 1 | 2 | 0.17 | 0.0003 | 0.0006 | 5.98 | −1.14 | 0.15 |
| LV157 | 304 | 57 | 24 | 30 | 0.95 | 0.0083 | 0.0171 | 3.09 | −1.64 | −0.11 | 12 | 1 | 2 | 0.17 | 0.0005 | 0.0010 | 5.14 | −1.14 | 0.15 |
| LV203 | 525 | 56 | 40 | 45 | 0.99 | 0.0177 | 0.0166 | 5.29 | −0.03 | −1.46 | 12 | 1 | 2 | 0.17 | 0.0003 | 0.0006 | 5.16 | −1.14 | 0.15 |
| LV375a | 446 | 46 | 16 | 15 | 0.76 | 0.0047 | 0.0082 | 3.24 | −1.33 | −4.86* | 11 | 0 | 1 | 0.00 | 0.0000 | 0.0000 | 3.37 | — | — |
| Normal variation in Europe | |||||||||||||||||||
| NV57 | 536 | 54 | 31 | 47 | 0.99 | 0.0085 | 0.0127 | 3.43 | −1.22 | 2.71 | 12 | 7 | 6 | 0.76 | 0.0042 | 0.0041 | 3.88 | 0.05 | −2.39 |
| NV120a | 486 | 46 | 42 | 40 | 0.99 | 0.0124 | 0.0197 | 3.5 | −1.27 | −0.08 | 11 | 13 | 7 | 0.89 | 0.0114 | 0.0091 | 3.78 | 1.07 | −0.55 |
| NV139 | 332 | 57 | 31 | 34 | 0.97 | 0.0145 | 0.0203 | 9.53 | −0.92 | 0.17 | 12 | 9 | 5 | 0.67 | 0.0101 | 0.0086 | 9.62 | 0.73 | −0.27 |
| NV216 | 513 | 56 | 54 | 42 | 0.98 | 0.0190 | 0.0229 | 5.03 | −0.69 | −0.91 | 12 | 16 | 6 | 0.82 | 0.0112 | 0.0087 | 4.96 | 1.23 | −0.03 |
| NV278 | 552 | 55 | 53 | 41 | 0.98 | 0.0127 | 0.0210 | 5.29 | −1.46 | −7.04 | 12 | 19 | 6 | 0.85 | 0.0157 | 0.0114 | 5.73 | 1.68 | 0.39 |
| NV287 | 426 | 57 | 30 | 35 | 0.96 | 0.0081 | 0.0153 | 4.05 | −1.54 | −5.14 | 12 | 9 | 5 | 0.79 | 0.0046 | 0.0059 | 4.4 | −0.92 | −1.64 |
Length, excluding sites with gaps/missing data; N, number of lines; S, number of segregating sites; H, number of haplotypes; HD, haplotype diversity; π, nucleotide diversity; θ, the neutral parameter θ = 4Neμ estimated from the number of segregating sites; D, divergence between D. simulans and D. melanogaster; DT, Tajima's D; HFW, Fay and Wu's H. Significance levels for HFW were estimated from 1000 standard coalescent simulations assuming no recombination.
0.01 < P ≤ 0.05;
P ≤ 0.01.
European sample from Neumarkt, Germany; Lake Kariba, Zimbabwe, is missing from the African sample.
Deviation from mutation-drift equilibrium:
We did not observe an excess of high-frequency-derived sites. Of 70 population-locus pairs, only 5 had a significantly negative H-value (Fay and Wu 2000). After a Bonferroni correction, no population-locus pair was found to be significant.
We used Tajimas's D to test if the analyzed sub-Saharan D. melanogaster populations were in mutation-drift equilibrium. In the absence of selection, populations in mutation-drift equilibrium will have Tajima's D-values close to zero. However, demographic events, such as population expansion, bottlenecks, and admixture, will result in a genome-wide deviation from zero. We used a multilocus test based on coalescent simulations to compare the observed Tajima's D-values at all loci jointly against the neutral expectation for mutation-drift equilibrium (Kliman et al. 2000). For three populations (Kampala, Mali, ZS) no significant deviation from neutrality was observed across fragments, while for the three remaining ones (Kenya, Kisoro, ZLK), a significantly negative Tajima's D was observed (Table 3).
TABLE 3.
Tajima'sD across 12 unlinked loci [10 loci for Zimbabwe (Lake Kariba)]
| Population |
|
|
|
Dsim < Dobs (%)d | P | |||
|---|---|---|---|---|---|---|---|---|
| Zimbabwe (Lake Kariba) | 11.7 | −0.668 | −0.069 | 1.86 | * | |||
| Zimbabwe (Sengwa) | 9.4 | −0.381 | −0.068 | 11.55 | NS | |||
| Uganda (Kampala) | 6.9 | −0.173 | −0.047 | 32.32 | NS | |||
| Uganda (Kisoro) | 9.6 | −0.740 | −0.063 | 0.35 | ** | |||
| Kenya | 9.8 | −0.581 | −0.070 | 2.40 | * | |||
| Mali | 8.7 | −0.176 | −0.058 | 33.39 | NS |
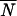
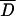
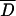
NS, not significant; *0.01 < P ≤ 0.05; **P ≤ 0.01.
Mean number of lines for each population across 12 loci.
Observed mean value of Tajima's D across 12 loci.
Simulated mean value of Tajima's D across 12 loci.
Percentage of 10,000 independent standard coalescent simulations that generated a more extreme mean Tajima's D.
Could this overall pattern have been generated by one or two loci strongly deviating from neutral expectations rather than reflect a genome-wide deviation from neutrality? Upon visual inspection we found that the significantly negative overall Tajima's D-values in three populations were due to a moderately negative Tajima's D of most loci (see supplementary Table S3 at http://www.genetics.org/supplemental/). Only one population-locus pair had a significantly negative Tajima's D, indicating that multiple loci contributed to the significant deviation from neutral expectations across all loci. We further substantiated this observation by performing a sign test for each population to evaluate if more loci with a negative Tajima's D were observed than expected by chance. Significant deviations from expectations (P < 0.05, paired sign tests) were found for each of the three populations that showed significantly negative Tajima's D, whereas no significant trend in Tajima's D-values was detected for the other three populations (P > 0.39, paired sign tests). This confirms that the overall significant deviation from neutrality for three populations is likely to result from the joint effect of all loci rather than a few outliers.
The three populations that report significant negative overall Tajima's D have on average the largest sample sizes. To rule out that the difference among the populations reflects different statistical power rather than a biologically significant result, we repeated the multilocus analysis with equal sample sizes for all populations (N = 7) after randomly discarding individuals from the larger populations. Two populations (Kenya and Kisoro) remained significant, whereas ZLK increased only marginally beyond the threshold with 5.97% Dsim < Dobs. Hence, the observed heterogeneity among the sub-Saharan D. melanogaster populations is explained better by biological differences among the populations than by differences in statistical power.
Genetic differentiation among populations:
To test whether our African population samples were drawn from a single panmictic population, we estimated genetic differentiation among populations using the haplotype statistic HST (Hudson et al. 1992) and the nearest-neighbor statistic (Snn) (Hudson 2000). A separate analysis of all 12 loci indicated a statistically significant differentiation (after applying a sequential Bonferroni correction) among the African populations for 6 loci (NV57, NV120, NV139, NV216, NV278, and LV203) using HST and for 5 loci (NV57, NV139, NV216, NV278, and LV203) using Snn (supplementary Table S4 at http://www.genetics.org/supplemental/). A meta-analysis combining the probabilities over all 12 loci (Sokal and Rohlf 1995) indicated a highly significant differentiation of African D. melanogaster populations. The meta-analysis of population pairs showed a highly significant differentiation among some pairs, but no significant differentiation among others. Most important, the pattern of differentiation did not follow a geographic pattern; i.e., more distantly located population pairs were not always highly differentiated and vice versa (Table 4). In general, for population pairs showing a highly significant differentiation, both measures of differentiation provided congruent results, and among less-differentiated population pairs we observed some inconsistencies for the HST and Snn statistic (Table 4).
TABLE 4.
Pairwise genetic differentiation for six African populations
| Kampala | Kisoro | Kenya | Mali | ZS | ZLK | |
|---|---|---|---|---|---|---|
| A | ||||||
| Kampala | 77.99 | 77.04 | 57.32 | 69.81 | 91.21 | |
| Kisoro | <0.0001** | 27.71 | 26.47 | 47.25 | 39.24 | |
| Kenya | <0.0001** | 0.27 | 33.53 | 19.24 | 25.38 | |
| Mali | 0.0002** | 0.33 | 0.093 | 28.17 | 56.54 | |
| ZS | <0.0001** | 0.0031* | 0.74 | 0.25 | 33.10 | |
| ZLK | <0.0001** | 0.0062* | 0.19 | <0.0001** | 0.033 (NS) | |
| B | ||||||
| Kampala | 77.95 | 72.32 | 64.51 | 78.35 | 89.32 | |
| Kisoro | <0.0001** | 22.03 | 39.43 | 31.81 | 24.22 | |
| Kenya | <0.0001** | 0.58 | 31.67 | 18.05 | 17.42 | |
| Mali | <0.0001** | 0.027 (NS) | 0.15 | 37.06 | 41.49 | |
| ZS | <0.0001** | 0.15 | 0.80 | 0.04 (NS) | 34.21 | |
| ZLK | <0.0001** | 0.24 | 0.65 | 0.0037* | 0.026 (NS) |
The values above the diagonal give the combined probabilities (−2∑ln P) from the significance tests of pairwise differentiation at 12 unlinked loci [10 loci in comparisons involving Zimbabwe (Lake Kariba)] for (A) Snn and (B) HST. The values below the diagonal give the significance levels in the meta-analysis (see materials and methods for details). NS, not significant; *0.01 < P ≤ 0.05; **P ≤ 0.01 after sequential Bonferroni correction.
African origin of European variation:
Fragments with low variability in a non-African population were previously found to harbor a disproportionately high number of fixed or high-frequency-derived mutations in non-African D. melanogaster (Glinka et al. 2003). Given the evidence for population substructure in African D. melanogaster, we were interested in using a large set of African D. melanogaster populations to determine the proportion of European variation that was already segregating in Africa. In particular, we tested whether the proportion of mutations absent in our Africa sample differed between LV fragments and NV fragments. As outlined by Glinka et al. (2003), we used the D. simulans sequence to distinguish between ancestral and derived variants.
On the basis of the original comparison between one European and one African population (Glinka et al. 2003) we identified a total of 15 fixed differences (European alleles) in the six LV fragments. Twelve of these alleles were derived and 3 were ancestral with respect to the D. simulans sequence. No fixed difference was identified for the six NV fragments. The joint analysis of all African populations indicated that all 15 sites identified as fixed differences between one African and one European D. melanogaster population could be detected in the extended African sample (Table 5). The mean frequency of those alleles that were misclassified as European was 0.16 ± 0.10 (n = 15) in the extended African sample. For most sites the misclassified alleles were present at a low frequency (0.05–0.18). At three sites, however, the misclassified allele was present at a moderate to high frequency (0.27, 0.28, and 0.45) in at least some African populations. Interestingly, two of these high-frequency sites were categorized as a derived site in Europe in the data set of Glinka et al. (2003).
TABLE 5.
Polymorphism for those sites that were identified as fixed differences between one African (Lake Kariba) and one EuropeanD. melanogaster population for six “low variability” fragments
| LV fragment
|
||||||
|---|---|---|---|---|---|---|
| 122 | 125 | 130 | 157 | 203 | 375a | |
| 4 | 02 | 44555 | 01 | 023 | 22 | |
| 7 | 36 | 36378 | 24 | 304 | 56 | |
| 5 | 46 | 76192 | 85 | 632 | 97 | |
| Africa | ||||||
| Kampala | C | GC | ATTTC | TC | GAG | CT |
| Kampala | . | .. | … | .. | TG. | .. |
| Kampala | . | .. | … | .. | TG. | .. |
| Kampala | . | .. | … | .. | … | .. |
| Kampala | . | .. | … | .. | … | .. |
| Kampala | . | .. | CCCAT | .. | .G. | .. |
| Kampala | G | .. | CCCAT | .. | … | .. |
| Kisoro | . | .. | … | .. | .G. | .. |
| Kisoro | . | .. | … | .. | TG. | .C |
| Kisoro | . | .. | … | .. | .G. | .. |
| Kisoro | . | .. | … | .. | … | .C |
| Kisoro | . | .. | CC… | .. | … | .. |
| Kisoro | G | .. | T… | .. | TG. | .C |
| Kisoro | G | A. | … | .. | .G. | .. |
| Kisoro | . | .. | … | .. | … | .. |
| Kisoro | . | .. | .C… | .. | .G. | .. |
| Kisoro | . | .. | .. | |||
| Kenya | G | TA | CCCAT | .. | .GC | .. |
| Kenya | . | .. | … | .. | ..T | .C |
| Kenya | . | .. | … | .. | … | .C |
| Kenya | . | T. | … | .. | .G. | .C |
| Kenya | . | .. | … | CT | TGC | .. |
| Kenya | . | .. | … | .. | TGC | TC |
| Kenya | . | .. | T… | .. | TG. | .. |
| Kenya | . | .. | T… | .. | … | .. |
| Kenya | . . | … | .T | TG. | TC | |
| Kenya | .. | .. | … | TC | ||
| Mali | . | .. | … | .. | .G. | .. |
| Mali | . | .. | … | .. | TGC | .. |
| Mali | . | .. | … | .. | TG . | .. |
| Mali | G | .. | CCCAT | .. | … | .. |
| Mali | . | A. | … | .. | TG. | .. |
| Mali | G | .. | T… | .. | … | .. |
| Mali | . | .. | … | CT | TG. | .. |
| Mali | G | .. | CCCAT | .. | TGC | .. |
| Mali | G | TA | CT | |||
| Sengwa | G | T. | … | .. | … | .. |
| Sengwa | . | .. | T… | .. | TGC | .. |
| Sengwa | . | TA | … | .. | .G. | .. |
| Sengwa | . | .. | T… | .. | … | .C |
| Sengwa | G | .. | CCCAT | .. | .G. | TC |
| Sengwa | . | TA | … | .. | T.. | .. |
| Sengwa | . | .. | CCCAT | .. | … | .C |
| Sengwa | . | .. | .T | … | .. | |
| Sengwa | . | .. | .. | … | .. | |
| Sengwa | .. | .. | .G. | .C | ||
| Lake Kariba | . | .. | … | .. | … | |
| Lake Kariba | . | A. | … | .. | … | |
| Lake Kariba | . | .. | … | .. | … | |
| Lake Kariba | . | A. | … | .. | … | |
| Lake Kariba | . | .. | … | .. | … | |
| Lake Kariba | . | .. | T… | .. | … | |
| Lake Kariba | . | .. | … | .. | … | |
| Lake Kariba | . | .. | … | .. | … | |
| Lake Kariba | . | .. | … | .. | … | |
| Lake Kariba | . | .. | … | .. | … | |
| Lake Kariba | . | .. | … | .. | … | |
| Lake Kariba | . | .. | T… | … | ||
| Europe | ||||||
| Leiden | G | TA | CCCAT | CT | TGC | TC |
| Leiden | G | TA | CCCAT | CT | TGC | TC |
| Leiden | G | TA | CCCAT | CT | TGC | TC |
| Leiden | G | TA | CCCAT | CT | TGC | TC |
| Leiden | G | TA | CCCAT | CT | TGC | TC |
| Leiden | G | TA | CCCAT | CT | TGC | TC |
| Leiden | G | TA | CCCAT | CT | TGC | TC |
| Leiden | G | TA | CCCAT | CT | TGC | TC |
| Leiden | G | TA | CCCAT | CT | TGC | TC |
| Leiden | G | TA | CCCAT | CT | TGC | TC |
| Leiden | G | TA | CCCAT | CT | TGC | TC |
| Leiden | TA | CCCAT | CT | TGC | ||
| D. simulans | . | .. | .A.A. | .. | .G. | .C |
The data for the Leiden and Lake Kariba populations were taken from Glinka et al. (2003).
The European sample for fragment 375 derives from Neumarkt, Germany.
Given the significant population structure in African D. melanogaster, we were interested in the number of African populations that need to be sampled to correctly identify ancestral African variants. To do so, we selected sites for which the European allele was absent in at least one African population. As such sites could be erroneously scored as novel European variants if only one of the African populations is taken for comparison, we used them to determine the probability of misclassification for a given number of African populations (Figure 1). If only a single African population was considered, on average only 57% of the alleles were correctly identified to be of African origin. For NV fragments the probability to falsely classify variants to be of European origin was higher than that for LV fragments (Figure 1). When more than a single African population was considered, the probability of misclassification decreased with an increasing number of African populations. However, for NV fragments even after sampling four African populations almost 10% (8.3%) of the alleles were missed in the African populations and consequently incorrectly classified as Europe specific (Figure 1).
Figure 1.—

The probability of misclassifying an African allele detected in a European population as Europe specific (and thus of outside-African origin). We considered only those African alleles that were not detected in at least one African population. The probability was calculated as
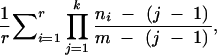 |
DISCUSSION
Demography:
Given that D. melanogaster originated in sub-Saharan Africa, it has been widely assumed that African D. melanogaster populations are close to equilibrium. Nevertheless, recent multilocus sequence polymorphism analyses reported a negative Tajima's D-value in African populations (Harr et al. 2002; Glinka et al. 2003; Baudry et al. 2004). Consistent with these results, we also found negative mean Tajima's D-values for all populations. However, for only three of the populations analyzed, Tajima's D was significantly different from neutral expectations. This distinction between the African populations was due to neither individual outlier loci nor an artifact of sample size differences. The combined evidence from our study and the previous reports strongly suggests that at least some African D. melanogaster populations are not in equilibrium and their allele frequencies are strongly influenced by past demographic events. While it is generally assumed that negative Tajima's D-values indicate a population expansion, it needs to be stressed that other demographic events, such as admixture from a diverged population, could also result in a similar pattern. Interestingly, a recent microsatellite analysis of different African D. melanogaster populations also indicated a significant deviation from stable equilibrium populations (Dieringer et al. 2005).
Population structure and inference of fixed derived differences:
Consistent with previous reports (Baudry et al. 2004; Dieringer et al. 2005), our analysis of 12 DNA fragments indicated significant population differentiation among African D. melanogaster populations. While microsatellite data indicated that temporal heterogeneity may be at least as important as geographic location (Dieringer et al. 2005), our study included too few populations to address this question systematically. Nevertheless, it is interesting that the two populations from Zimbabwe (Sengwa and Lake Kariba) were significantly differentiated, while more distantly located populations (e.g., Sengwa and Mali) were not.
The significant differences among African populations considerably complicate the inference of fixed differences among African and non-African D. melanogaster populations. Some alleles may be scored as absent in Africa (and thus of putative non-African origin) while other African population samples may contain this allele. Our analysis, therefore, strongly suggests that multiple African populations need to be analyzed before firm conclusions can be drawn about their origin. As our population sample was rather small, we were not able to test to what extent larger population samples could compensate for the analysis of multiple populations.
Back migration:
Our analysis indicated that all high-frequency alleles of a European population were also detected in at least one African population. Two alternative explanations can be put forward for this observation. Either the alleles are of African origin or they originated in Europe and were brought to Africa by back migration. Back migration of European alleles has previously been suggested for several African D. melanogaster populations (Bénassi and Veuille 1995; Capy et al. 2000; Kauer et al. 2003b). If recent back migration to Africa were responsible for shared variation between African and European populations, the European haplotype should be conserved in Africa. To test this hypothesis we focused on those sites that were at a high frequency (fixed) in the European population, but at a low frequency (<0.2) in Africa. Five LV fragments were analyzed. Each putative European allele resided in at least three different haplotypes in the African sample (haplotype diversity h ≥ 0.54). Given that we analyzed only short fragments (<600 bp), sufficient time was required to generate the high haplotype diversity in African flies carrying the putative European allele. Thus, we propose that either these alleles are of African origin and reached a high frequency in the European population or they are derived from an old admixture event. Due to the low number of putative European mutations we do not have enough power to distinguish between these two scenarios. Considering that two additional putative European alleles had a high frequency in most African populations, we favor the ancestral African variation hypothesis.
Selective sweeps coincide with the out-of-Africa habitat expansion:
A recent series of publications found strong evidence for a significant number of selective sweeps in non-African D. melanogaster populations (Harr et al. 2002; Glinka et al. 2003; Kauer et al. 2003a; Sáez et al. 2003; Orengo and Aguadé 2004). The timing of the selective sweeps is, however, not clear. One possibility would be that beneficial mutations occurred sequentially after D. melanogaster expanded its habitat. Alternatively, beneficial mutations may have all occurred within a very narrow time interval and only the presence of the majority of the mutations made the successful colonization possible. Finally, most of the mutations may already have been segregating in Africa, but became beneficial only in the European context (Orr and Betancourt 2001; Catania and Schlötterer 2005). In an attempt to shed some light on this important question, we compared LV and NV fragments. We estimated the number of new mutations that have putatively arisen in the derived populations after the habitat expansion and after the selective sweeps, respectively (see materials and methods). All identified mutations were singletons and derived with respect to D. simulans. In total, we identified three mutations in the LV fragments and five mutations in the NV fragments. Using the approximate calculation proposed by Baudry et al. (2004) and a mean silent mutation rate of 15.4 × 10−9 substitutions/year/bp (Li 1997), we obtained an estimated age of 6102 years for the LV fragments, and 8678 years for the NV fragments. Both estimates are very similar, and the value for the LV fragments is almost identical to the 6415 years obtained by Baudry et al. (2004). We inferred confidence intervals for these estimates using the genetree software (Bahlo and Griffiths 2000) to calculate the TMRCA for the putative novel mutations of the LV and NV fragments. The genetree analysis was based on the inferred European alleles only to avoid the confounding effect of ancestral African variation. We ran three replicas of a set of 10,000,000 coalescent simulations conditioned on θ-estimates for the African samples for the two classes of fragments, respectively (mean θ for LV fragments, 0.0142; mean θ for NV fragments, 0.0191). The mean TMRCA estimates did not differ significantly for the two classes of fragments (Table 6). When we conditioned the simulations on mean African θ of all fragments the estimates were similar (results not shown). Thus, our data do not show a significant difference for the minimum age estimate of the colonization event and the sweeps putatively associated with it. Nevertheless, due to the small number of mutations our test does not have enough power to determine the age of the beneficial mutations, in particular, if some heterogeneity among the LV fragments is assumed. A larger set of fragments may provide more insight into this question.
TABLE 6.
Estimates of the time to the most recent common ancestor (TMRCA) of putative novel (postsweep and postbottleneck) mutations scaled in coalescent units
| Fragments | No. of simulations |
θafr | Mean TMRCA |
SD |
|---|---|---|---|---|
| LV | 10,000,000 | 0.0142 | 2.1363 | 1.1040 |
| LV | 10,000,000 | 0.0142 | 2.1148 | 1.1175 |
| LV | 10,000,000 | 0.0142 | 2.1460 | 1.1992 |
| NV | 10,000,000 | 0.0191 | 1.7576 | 1.0908 |
| NV | 10,000,000 | 0.0191 | 2.7724 | 0.9590 |
| NV | 10,000,000 | 0.0191 | 3.1506 | 1.3901 |
Simulations were conditioned on the mean θ-estimates of the African sample for the two classes of fragments (LV, NV), respectively. Three replicates were performed assuming a constant population size.
Implications for polymorphism analysis in non-African populations:
Our analysis indicated that almost all variation detected in non-African D. melanogaster flies is most likely of African origin. This result is consistent with a previous comparison of African and non-African D. melanogaster populations (Schlötterer and Harr 2002). The implication of this observation is that all estimates of θ or the population recombination parameter in non-African populations are heavily influenced by ancestral African variation. Thus, African variation and the past demographic history of non-African populations are pertinent to the interpretation of neutrality tests based on allele frequency data.
Acknowledgments
We thank the members of the C.S. lab for constructive suggestions and critical discussion. B. Charlesworth provided helpful comments on an earlier version of the article. Two anonymous reviewers provided helpful criticism. This work was supported by Fonds zur Förderung der Wissenschaftlichen Forschung grants to C.S.
References
- Bahlo, M., and R. C. Griffiths, 2000. Inference from gene trees in a subdivided population. Theor. Popul. Biol. 57: 79–95. [DOI] [PubMed] [Google Scholar]
- Baudry, E., B. Viginier and M. Veuille, 2004. Non-African populations of D. melanogaster have a unique origin. Mol. Biol. Evol. 21: 1482–1491. [DOI] [PubMed] [Google Scholar]
- Bénassi, V., and M. Veuille, 1995. Comparative population structuring of molecular and allozyme variation of Drosophila melanogaster Adh between Europe, West Africa and East Africa. Genet. Res. 65: 95–103. [DOI] [PubMed] [Google Scholar]
- Capy, P., M. Veuille, M. Paillette, J. M. Jallon, J. Vouidibio et al., 2000. Sexual isolation of genetically differentiated sympatric populations of Drosophila melanogaster in Brazzaville, Congo: The first step towards speciation? Heredity 84: 468–475. [DOI] [PubMed] [Google Scholar]
- Catania, F., and C. Schlötterer, 2005. Non-African origin of a local beneficial mutation in D. melanogaster. Mol. Biol. Evol. 22: 265–272. [DOI] [PubMed] [Google Scholar]
- David, J. R., and P. Capy, 1988. Genetic variation of Drosophila melanogaster natural populations. Trends Genet. 4: 106–111. [DOI] [PubMed] [Google Scholar]
- Dieringer, D., V. Nolte and C. Schlötterer, 2005. Population structure in African Drosophila melanogaster revealed by microsatellite analysis. Mol. Ecol. 14: 563–573. [DOI] [PubMed] [Google Scholar]
- Fay, J. C., and C.-I Wu, 2000. Hitchhiking under positive Darwinian selection. Genetics 155: 1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinka, S., L. Ometto, S. Mousset, W. Stephan and D. DeLorenzo, 2003. Demography and natural selection have shaped genetic variation in Drosophila melanogaster: a multilocus approach. Genetics 165: 1269–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, T. A., 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41: 95–98. [Google Scholar]
- Harr, B., M. Kauer and C. Schlötterer, 2002. Hitchhiking mapping: a population-based fine-mapping strategy for adaptive mutations in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 99: 12949–12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., 2000. A new statistic for detecting genetic differentiation. Genetics 155: 2011–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., D. D. Boos and N. L. Kaplan, 1992. A statistical test for detecting geographic subdivision. Mol. Biol. Evol. 9: 138–151. [DOI] [PubMed] [Google Scholar]
- Kaplan, N. L., R. R. Hudson and C. H. Langley, 1989. The “hitchhiking effect” revisited. Genetics 123: 887–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer, M. O., D. Dieringer and C. Schlötterer, 2003. a A microsatellite variability screen for positive selection associated with the “out of Africa” habitat expansion of Drosophila melanogaster. Genetics 165: 1137–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer, M. O., D. Dieringer and C. Schlötterer, 2003. b Nonneutral admixture of immigrant genotypes in African Drosophila melanogaster populations from Zimbabwe. Mol. Biol. Evol. 20: 1329–1337. [DOI] [PubMed] [Google Scholar]
- Kim, Y., and W. Stephan, 2002. Detecting a local signature of genetic hitchhiking along a recombining chromosome. Genetics 160: 765–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliman, R. M., P. Andolfatto, J. A. Coyne, F. Depaulis, M. Kreitman et al., 2000. The population genetics of the origin and divergence of the Drosophila simulans complex species. Genetics 156: 1913–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn, M. H., H. J. Pelz and R. K. Wayne, 2000. Natural selection mapping of the warfarin-resistance gene. Proc. Natl. Acad. Sci. USA 97: 7911–7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachaise, D., M.-L. Cariou, J. R. David, F. Lemeunier, L. Tsacas et al., 1988. Historical biogeography of the Drosophila melanogaster species subgroup. Evol. Biol. 22: 159–225. [Google Scholar]
- Langley, C. H., B. P. Lazzaro, W. Phillips, E. Heikkinen and J. M. Braverman, 2000. Linkage disequilibria and the site frequency spectra in the su(s) and su(w(a)) regions of the Drosophila melanogaster X chromosome. Genetics 156: 1837–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. H., 1997 Molecular Evolution. Sinauer Associates, Sunderland, MA.
- Maynard Smith, J., and J. Haigh, 1974. The hitch-hiking effect of a favourable gene. Genet. Res. 23: 23–56. [PubMed] [Google Scholar]
- Miller, S. A., D. D. Dykes and H. F. Polesky, 1988. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 16: 1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita, N., and C. H. Langley, 1988. Molecular and phenotypic variation of the white locus region in Drosophila melanogaster. Genetics 120: 199–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orengo, D. J., and M. Aguadé, 2004. Detecting the footprint of positive selection in a European population of Drosophila melanogaster: multilocus pattern of variation and distance to coding regions. Genetics 167: 1759–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr, H. A., and A. J. Betancourt, 2001. Haldane's sieve and adaptation from the standing genetic variation. Genetics 157: 875–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payseur, B. A., A. D. Cutter and M. W. Nachman, 2002. Searching for evidence of positive selection in the human genome using patterns of microsatellite variability. Mol. Biol. Evol. 19: 1143–1153. [DOI] [PubMed] [Google Scholar]
- Rozas, J., J. C. Sánchez-DelBarrio, X. Messeguer and R. Rozas, 2003. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19: 2496–2497. [DOI] [PubMed] [Google Scholar]
- Sáez, A. G., A. Tatarenkov, E. Barrio, N. H. Becerra and F. J. Ayala, 2003. Patterns of DNA sequence polymorphism at Sod vicinities in Drosophila melanogaster: unraveling the footprint of a recent selective sweep. Proc. Natl. Acad. Sci. USA 100: 1793–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlenke, T. A., and D. J. Begun, 2003. Natural selection drives Drosophila immune system evolution. Genetics 164: 1471–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlötterer, C., 2002. A microsatellite-based multilocus screen for the identification of local selective sweeps. Genetics 160: 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlötterer, C., 2003. Hitchhiking mapping—functional genomics from the population genetics perspective. Trends Genet. 19: 32–38. [DOI] [PubMed] [Google Scholar]
- Schlötterer, C., and B. Harr, 2002. Single nucleotide polymorphisms derived from ancestral populations show no evidence for biased diversity estimates in Drosophila melanogaster. Mol. Ecol. 11: 947–950. [DOI] [PubMed] [Google Scholar]
- Schlötterer, C., C. Vogl and D. Tautz, 1997. Polymorphism and locus-specific effects on polymorphism at microsatellite loci in natural Drosophila melanogaster populations. Genetics 146: 309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokal, R. R., and F. J. Rohlf, 1995 Biometry: The Principles and Practice of Statistics in Biological Research. W. H. Freeman, New York.
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J. D., T. J. Gibson, F. Plewniak and F. Jeanmougin, 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25: 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]