

Abstract
Nuclear sequence variation and linkage disequilibrium (LD) were studied in 15 cold-hardiness- and 3 wood quality-related candidate genes in Douglas fir [Pseudotsuga menziesii (Mirb.) Franco]. This set of genes was selected on the basis of its function in other plants and collocation with cold-hardiness-related quantitative trait loci (QTL). The single-nucleotide polymorphism (SNP) discovery panel represented 24 different trees from six regions in Washington and Oregon plus parents of a segregating population used in the QTL study. The frequency of SNPs was one SNP per 46 bp across coding and noncoding regions on average. Haplotype and nucleotide diversities were also moderately high with Hd = 0.827 ± 0.043 and π = 0.00655 ± 0.00082 on average, respectively. The nonsynonymous (replacement) nucleotide substitutions were almost five times less frequent than synonymous ones and substitutions in noncoding regions. LD decayed relatively slowly but steadily within genes. Haploblock analysis was used to define haplotype tag SNPs (htSNPs). These data will help to select SNPs for association mapping, which is already in progress.
STUDIES of nuclear sequence variation and linkage disequilibrium (LD) across populations and genomic regions help to elucidate the evolutionary forces that shape patterns of variability (Nordborg and Innan 2002; Feder and Mitchell-Olds 2003; Luikart et al. 2003). Such studies are of general biological interest and are needed to design association mapping studies that will help us better understand the molecular basis of adaptation in plant populations (Goldstein and Weale 2001; Glazier et al. 2002; Schlötterer 2002; Borevitz and Nordborg 2003). However, with the exception of maize and Arabidopsis, little research has been conducted on LD in plants, although the mating system (selfing vs. outcrossing), population structure (continuous vs. isolated populations), life forms (annual vs. perennial), recombination rate, and other factors can strongly influence LD patterns (Flint-García et al. 2003). Douglas fir [Pseudotsuga menziesii (Mirb.) Franco] is found across a large and environmentally heterogeneous area in western North America and has evolved complex adaptive mechanisms (Campbell and Sugano 1975; Campbell and Sorensen 1978; Steiner 1979; Rehfeldt 1983, 1989; Li and Adams 1993; Aitken and Adams 1997; Anekonda et al. 2000). We are interested in the specific genes and alleles that underlie phenotypic variation in adaptive traits such as growth rate, bud set, bud flush, cold hardiness, and drought tolerance. Wood quality-related genes are also of great interest. Douglas fir is an excellent perennial plant species to use for studying these traits and genetic adaptation. It is evolutionarily old; phenotypically and genetically highly diverse; distributed in large, outcrossed, natural populations with high gene flow; and has relatively little within-population substructure (Merkle and Adams 1987; Moran and Adams 1989; Aagaard et al. 1998a,b; Viard et al. 2001). Douglas fir is also one of the most thoroughly studied trees in the United States and the most economically important tree in the Pacific Northwest.
Frost damage can negatively affect the annual growth of Douglas fir trees, particularly in the spring when new needle tissue is delicate and vulnerable. Fall frosts can damage actively elongating shoots in the autumn and adversely affect growth the following spring. Therefore, fall and spring cold hardiness are important adaptive traits in Douglas fir that show high genetic variation in common garden studies and vary among populations from environmentally diverse locations (reviewed in Wheeler et al. 2005).
Quantitative trait loci (QTL) mapping studies have confirmed these observations and have allowed us to begin dissecting these complex traits (Jermstad et al. 2001a,b, 2003; Wheeler et al. 2005). Several genomic regions responsible for genetic control of growth rhythm and cold-hardiness traits were found, but QTL mapping does not reveal which individual genes are responsible for these effects.
Association mapping is a powerful population genomic approach that unlike QTL mapping can identify individual genes and alleles that are responsible for phenotypic differences in adaptive traits (Neale and Savolainen 2004). However, limited genetic resources and the large genome of Douglas fir prevent a full genome scan. Instead, we plan to carry out a candidate gene-based association mapping using single-nucleotide polymorphisms (SNPs) (Rebbeck et al. 2004). SNPs are excellent markers for association mapping of genes controlling complex traits (e.g., Brookes 1999; Rafalski 2002; Carlson et al. 2004). However, to carry out association mapping it is necessary first to discover SNPs in candidate genes of interest and to study their variation. To achieve our goals we (1) developed a list of candidate genes for adaptive traits on the basis of data available from other plant species, (2) found their homologs or orthologs among Douglas fir genomic and EST sequences, (3) designed single-gene-specific primers to amplify single-gene PCR products, (4) sequenced them, (5) performed SNP discovery, (6) analyzed their diversity and LD, and (7) selected SNPs for association mapping. These steps are described in this article for a set of 18 genes that included late embryogenesis abundant protein genes, dehydrins, and other cold-induced and wood quality-related genes. The studied genes are mostly unlinked and represent a wide variety of protein-coding genes. Therefore, they are likely to reflect general genome variation in Douglas fir.
MATERIALS AND METHODS
Plant material and SNP discovery:
The SNP discovery panel consisted of 32 DNA samples isolated from 24 haploid (1N) seed megagametophytes collected from 24 unrelated trees from six regions in Washington and Oregon and 8 megagametophytes from the parents of a QTL mapping population—four megagametophytes from the maternal parent and four from the paternal parent (Figure 1) (Jermstad et al. 1998; see also supplemental Table 1S at http://www.genetics.org/supplemental/ for details). The eight samples from the mapping parents were used to test the PCR primers. If amplification and sequencing were successful, then the remaining 24 samples from the SNP discovery panel were used to amplify the DNA used for forward and reverse sequencing. The mapping parents segregated for a maximum of 2 alleles each; therefore, the maximum number of alleles studied and used for nucleotide diversity analysis was 28 (24 from the discovery panel and 4 from the mapping parents).
Figure 1.

Geographic location of Douglas fir trees used to collect single seed megagametophytes for SNP discovery and nucleotide diversity analysis.
Candidate gene selection:
Using published data on differential expression and physiological mechanisms involved in cold tolerance in plant species, we selected ∼500 genes and proteins that included cold acclimation, cold induced, cold resistant, chaperones, cryoprotectins, calmodulins, some dehydrins, LEA, and other cold-hardiness-related candidate genes and proteins (e.g., Close 1997; Palva and Heino 1998; Thomashow 1998, 1999, 2001; Wanner and Junttila 1999; Seki et al. 2001, 2002; Fowler and Thomashow 2002; Nogueira et al. 2003; Provart et al. 2003; Rabbani et al. 2003; Browse and Lange 2004; Cook et al. 2004). Then, using BLASTX, BLASTN, and TBLASTX tools they were compared with all available Douglas fir sequences submitted to GenBank, including most of the ∼11,700 ESTs obtained recently from Douglas fir seedlings in our laboratory (http://staff.vbi.vt.edu/estap). The highly homologous Douglas fir sequences that matched sequences in our database of cold-resistance-related genes and proteins were used to design PCR primers for sequencing. For this study we preferably selected those genes that were also positional candidates that collocated with cold-hardiness QTL in a previous study (Wheeler et al. 2005). A number of candidate genes were previously mapped by RFLP analysis using cDNAs as hybridization probes (Jermstad et al. 1998). Most of the positional candidates were good expressional and functional candidates. For comparison, we also included three wood quality-related genes that were recently studied in loblolly pine, Pinus taeda (Brown et al. 2004a). The details on the list of 18 candidate genes used in this study are presented in Table 1.
TABLE 1.
Description and map location by linkage group (LG) of 18 Douglas fir candidate genes used for SNP discovery sequencing and nucleotide diversity analysis
| Gene product | Abbreviated gene name | Potential adaptive role | Expression and position data | Gene sequence coverage | LG | Sequences | Total sites (bp) | Indels | Sites excluding alignment gaps | Coding sites | Exons | Noncoding (introns and UTRs) sites | Intron sites | Introns |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Translation elongation factor-1, α-subunit | EF1A | Translation enhancement | Cold induceda, colocated with QTLb | Partial | 1 | 27 | 1,072 | 0 | 1,072 | 743 | 1 | 329 | 0 | 0 |
| Thiazole biosynthetic enzyme | TBE | DNA damage tolerance | Colocated with QTLc | Complete | 1 | 28 | 2,954 | 15 | 2,380 | 1,029 | 2 | 1,614 | 1,430 | 1 |
| Flavanone-3- hydroxylase | F3H1 | Flavonoid pathway | Water-deficit inducedd, colocated with QTLb | Partial | 2 | 28 | 365 | 1 | 364 | 268 | 1 | 96 | 0 | 0 |
| Flavanone-3- hydroxylase | F3H2 | Flavonoid pathway | Water-deficit inducedd, colocated with QTLb | Partial | 2 | 28 | 647 | 1 | 640 | 441 | 2 | 206 | 88 | 1 |
| Formin-like protein AHF1 | Formin | Controls rearrange ments of the actin cytoskeleton | Colocated with QTLb | Partial | 2 | 28 | 337 | 0 | 337 | 337 | 1 | 0 | 0 | 0 |
| α-tubulin | AT1 | Microtubule cytoskeleton organization and bio genesis; cell motility | Colocated and associated with wood quality related QTL and traitse | Complete | 3 | 28 | 2,578 | 7 | 2,557 | 1,353 | 4 | 1,204 | 1,008 | 3 |
| Late embryo genesis abundant type 2 dehydrin- like protein | LEA2 | Stress response | Cold induceda, colocated with QTLb | Complete | 4 | 39 | 504 | 5 | 485 | 249 | 1 | 236 | 0 | 0 |
| Metallothionein- like protein | MT-like | Detoxification, leaf senes cence, fruit ripening | Stress induced; downregulated under the water deficitd, colo cated with QTLc | Complete | 5 | 28 | 579 | 2 | 564 | 204 | 3 | 360 | 202 | 2 |
| 60S ribosomal protein L31a | 60S-RPL31a | Protein biosynthesis | Cold inducedaf | Complete | 5 | 28 | 609 | 2 | 606 | 340 | 2 | 266 | 265 | 1 |
| Late embryo genesis abundant EMB11-like protein | LEA-EMB11 | Stress responsive LEA-3 proteins | Stress inducedg | Partial | 6 | 28 | 545 | 2 | 532 | 260 | 2 | 272 | 174 | 1 |
| 40S ribosomal protein S3a | 40S-RPS3a | Protein biosynthesis | Colocated with QTLc | Partial | 6 | 28 | 500 | 0 | 500 | 171 | 2 | 329 | 78 | 1 |
| Polyubiquitin | PolyUBQ | Protein degradation and proteolysis | Virus-inducedh, colocated with QTLc | Complete | 6 | 27 | 898 | 2 | 893 | 687 | 1 | 206 | 0 | 0 |
| Early response to dehydra tion protein | ERD15-like | Unknown | Dehydration inducedi, colocated with QTLc | Partial | 7 | 27 | 646 | 1 | 645 | 402 | 2 | 243 | 203 | 1 |
| Abscisic acid, water deficit stress and ripening- inducible protein | ABA-WDS | Dehydrin | Dehydration inducedj, colocated with QTLc | Partial | 7 | 28 | 344 | 0 | 344 | 344 | 1 | 0 | 0 | 0 |
| Water deficit- inducible protein | LP3-like | Dehydrin | Dehydration inducedk, colocated with QTLc | Partial | 7 | 38 | 481 | 3 | 449 | 372 | 1 | 109 | 109 | 1 |
| 4-coumarate: CoA ligase 1 | 4CL1 | Phenylpropa noid metabo lism, lignin and flavonoid biosynthesis | Differentially expressed in wood-forming tissuesa, colocated with wood quality- related QTLe | Partial | 11 | 32 | 628 | 0 | 628 | 566 | 1 | 62 | 62 | 1 |
| 4-coumarate: CoA ligase 2 | 4CL2 | Phenylpropanoid metabolism, lignin and flavonoid biosynthesis | Colocated with wood quality- related QTLe | Partial | 11 | 38 | 629 | 0 | 629 | 566 | 1 | 63 | 63 | 1 |
| Ascorbate peroxidase | APX | Essential for cell protection during oxidative stress | Cold inducedm | Partial | 13 | 28 | 867 | 7 | 847 | 274 | 3 | 573 | 311 | 2 |
| Mean: | 30 | 843.5 | 2.7 | 804.0 | 478.1 | 1.7 | 342.7 | 221.8 | 0.9 | |||||
| Total: | 536 | 15,183 | 48 | 14,472 | 8,606 | 31 | 6,168 | 3,993 | 16 |
DNA isolation, PCR primer design, amplification, and sequencing:
Genomic DNA was extracted from haploid megagametophytes after seed germination using the DNeasy plant mini kit (QIAGEN, Valencia, CA). The PCR amplification primers were based on the Douglas fir genomic, EST, or contig sequences (http://staff.vbi.vt.edu/estap) that were highly homologous to selected cold-resistance-related candidate genes described in other plants. The PCR primers were designed using the computer program GeneRunner v3.04 (Hastings Software, Hudson, NY; http://www.generunner.com) to amplify products 600–700 bp long. PCR amplifications were performed as described by Krutovsky et al. (2004) (see also supplemental Table 2S at http://www.genetics.org/supplemental/ for details on the primers). To obtain complete or almost complete gene sequences more than one primer pair was designed to amplify overlapping amplicons for the TBE, AT1, and APX genes (Table 1 and supplemental Table 2S). Nucleotide sequences were obtained directly by sequencing haploid PCR products using the ABI PRISM BigDye Primer Cycle sequencing kit v.3.1 (Applied Biosystems, Foster City, CA) and the ABI 3730 DNA analyzer at the College of Agricultural and Environmental Sciences Genomics Facility Center of the University of California (Davis, CA). All samples were sequenced in both directions. Raw sequences were base called by the PHRED program (Ewing and Green 1998; Ewing et al. 1998), assembled using the PHRAP program, and viewed through CONSED (Gordon et al. 1998, 2001). Multiple alleles of the same gene were aligned using the MACE program (B. Gilliland and C. Langley, University of California, Davis, CA). All chromatograms and SNPs were visually checked to exclude any sequencing errors.
Nucleotide diversity analysis:
Haplotypes were directly inferred from sequencing PCR products amplified in haploid megagametophytes from the SNP discovery panel. Multiple sequence alignments were analyzed using the DNA sequence polymorphism (DNASP) software version 4.0 (Rozas et al. 2003). Insertions and deletions (indels) were excluded from all estimates. Haplotype diversity (Hd) was computed using Equation 8.4 in Nei (1987), except that n was used instead of 2n. Nucleotide diversity was estimated by ΘW from the number of polymorphic segregating (S) sites (Watterson 1975, Equation 1.4a, but on a base pair basis; Nei 1987, Equation 10.3) and by π (Nei 1987, Equations 10.5 or 10.6, but on a per gene basis). Heterogeneity of ΘW among loci was assessed by using a likelihood-ratio test in which the probability of the observed number of segregating sites in a sample was calculated under the null hypothesis of a common, genomewide 4Neμ (Pg) and the alternative hypothesis of locus-specific values of 4Neμ (Pl), where an average for 18 genes ΘW was considered as a genomewide estimate. These probabilities were based on all (silent and nonsynonymous) segregating sites and were calculated for each gene by using the computer simulations that are implemented in DNASP. Simulations were based on the coalescent process for a neutral infinite-sites model and assumed a large constant population size (Hudson 1991). Then, the likelihood-ratio test statistic −2 ln(Pl/Pg) was calculated for each gene. Under certain assumptions this statistic is distributed as a χ2 with m − 1 d.f., where m is equal to the number of loci (see also Brown et al. 2004a).
Neutrality tests:
Neutrality test statistics D (Tajima 1989, Equation 38), D*, and F* (Fu and Li 1993, pp. 700 and 702, respectively) were calculated and tested using 10,000 simulations to test the hypothesis that mutations in the gene are selectively neutral (Kimura 1983). If a population sample fits the infinite-sites model, π and ΘW have equal expectations. Tajima (1989) developed the D-test statistic, which is π − ΘW divided by the standard deviation of this difference. The difference between π and ΘW (Tajima's D) reflects the degree of nonequilibrium conditions in the genetic history of the population. The D*-test statistic is based on the differences between the number of singletons (mutations appearing only once among the sequences) and the total number of mutations. The F*-test statistic is based on the differences between the number of singletons and the average number of nucleotide differences between pairs of sequences. Significantly negative values for these statistics are consistent with negative (purifying) selection and can also indicate a recent selective sweep of a linked mutation, whereas significantly positive values are consistent with positive, balancing, or diversifying selection for two or more alleles (Kreitman 2000). To find regions under selection within genes the distributions of the D-, D*-, and F*-statistics were studied along the gene sequences using a sliding window with a window length and step size of 100 and 25 sites, respectively. Coalescence simulations without recombination were used to test deviations of the observed π- and ΘW-estimates from average values and the significance of the D-, D*-, and F*-statistics (Hudson 1991).
The nonsynonymous (dN; amino acid replacing) to synonymous (dS; no amino acid replacing) substitution ratio is a strong indicator of selection (Li 1997). The dN/dS ratio measures the magnitude and direction of selective pressure on a gene sequence, with ratios = 1, <1, and >1 indicating neutral evolution, negative selection, and positive selection, respectively. The average number of potentially nonsynonymous and synonymous substitution sites, estimates of the number of nonsynonymous (dN) and synonymous (dS) substitutions per site, variance and *standard errors, and Z-test [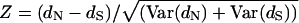 ] for neutrality (dN = dS) were computed using the molecular evolutionary genetics analysis (MEGA) software version 3.0 (http://www.megasoftware.net; Kumar et al. 2004) and the distance-based modified Nei-Gojobori method (Nei and Gojobori 1986) with the Jukes-Cantor model (Jukes and Cantor 1969) and bootstrap based on 1000 replicates (Nei and Kumar 2000).
] for neutrality (dN = dS) were computed using the molecular evolutionary genetics analysis (MEGA) software version 3.0 (http://www.megasoftware.net; Kumar et al. 2004) and the distance-based modified Nei-Gojobori method (Nei and Gojobori 1986) with the Jukes-Cantor model (Jukes and Cantor 1969) and bootstrap based on 1000 replicates (Nei and Kumar 2000).
Analysis of LD and haploblock structure within genes:
LD descriptive statistics r2 (Hill and Robertson 1968) and D′ (Lewontin 1964) were calculated using TASSEL (http://www.maizegenetics.net/bioinformatics/tasselindex.htm) and DNASP software. When more than two alleles were present at a locus, a weighted average of D′ or r2 was calculated (Farnir et al. 2000). If there were only two alleles at both loci, then a one-sided Fisher's exact test was calculated to determine the significance of LD. If there were more than two alleles, then permutations were used to calculate the proportion of permuted gamete distributions that are less probable than the observed gamete distribution under the null hypothesis of independence (Weir 1996). Only parsimony informative sites were included in the analysis of the LD decay within genes over distance. LD between genes was analyzed using alleles with a frequency of ≥15% for all genes, except the ERD15-like gene, for which alleles were less frequent.
Selection of htSNPs for association mapping:
To select haplotype tag SNPs (htSNPs) for association mapping, within-gene haploblock structure and haplotype coverage were studied using HaploblockFinder (Zhang and Jin 2003; http://cgi.uc.edu/∼kzhang), SNPtagger (Xiayi and Cardon 2003; http://www.well.ox.ac.uk/∼xiayi/haplotype/index.html), and SNPCherryPicker (Harris et al. 2003) software. These programs use different approaches and criteria to select htSNPs, and, therefore, we believe that they complement each other, and their combined use helps us select the consensus set of htSNPs.
Population structure:
Using MEGA a consensus neighbor-joining (NJ) tree (Saitou and Nei 1987) was reconstructed for all 28 Douglas fir samples on the basis of the 1000 Jukes-Cantor pairwise distance matrices (Jukes and Cantor 1969) calculated from the bootstrap-generated multiple-nucleotide alignments for all 18 genes combined. The FST statistic (Weir 1996), which measures the genetic variance among populations divided by the total genetic variance of the entire population, was used to quantify the degree of genetic differentiation between population samples from the six regions included in the SNP discovery panel using the ARLEQUIN ver. 2.0 software (Excoffier et al. 2004; http://lgb.unige.ch/arlequin). The analysis of molecular variance (AMOVA) approach implemented in Arlequin (Excoffier et al. 1992) is essentially similar to other approaches based on analyses of variance of gene frequencies, but it takes into account the number of mutations between haplotypes. The sample differentiation was also tested, using an exact test based on haplotype frequencies (Raymond and Rousset 1995; Goudet et al. 1996). The nearest-neighbor statistic (Snn) that measures how often the “nearest neighbors” (in sequence space) of sequences are from the same locality in geographic space was used to test for population differentiation among six regions and two states, from which samples were collected, as described in Hudson (2000).
RESULTS
Sequence analysis:
Thirty-two haploid DNA samples were sequenced for 18 candidate genes. The average size of a gene sequence was ∼843 bp (Table 1). In total, 15,183 bp of genomic DNA for 18 genes or 441,664 bp considering all samples were sequenced. Indels were found in 12 sequences, with the average number of 2.7 indels per sequence and the average length of 14.8 bp per indel. However, if the TBE gene with numerous large indels is excluded from analysis, the average numbers are 1.9 indels per sequence and 4.2 bp per indel. The average numbers of exons and introns were 1.7 and 0.9 per sequence, respectively, for all 18 partially and completely sequenced genes, or 2.2 and 1.2 per gene for 6 genes that were sequenced completely. The exon and intron sizes varied greatly, with the average lengths of 281 and 246 bp for all genes, respectively, or 292 and 402 bp for 6 genes that were sequenced completely. However, if the TBE gene, which had uncommonly large introns, is excluded from analysis, then the average lengths of exons and introns based on the five completely sequenced genes become 258 and 246 bp, respectively, which are very similar to the values based on all 18 genes.
Nucleotide diversity:
Four hundred SNPs were found in 18 genes, or 1 SNP for every 46 bp (Table 2). With the exception of 6 trinucleotide SNPs, all segregating sites had only two alternative nucleotides. Almost one-third of SNPs were singletons, and most SNPs (349) were either synonymous or in noncoding regions. Haplotype diversity was very high with Hd = 0.827 ± 0.043 and an average number of 11 different haplotypes per gene (Table 3). The total nucleotide diversities were also relatively high with π = 0.00655 ± 0.00082 and ΘW = 0.00702 ± 0.00269 on average. The estimates of nucleotide diversity, π and ΘW, varied significantly across loci (P < 0.00006 in the heterogeneity test) with values as low as π = 0.00237 in the 4CL2 gene and ΘW = 0.00229 in the formin-like gene and almost six to seven times higher in the LEA-EMB11-like gene, where π = 0.01378 and ΘW = 0.01594. Coalescence simulations also showed that the EF1A, TBE, AT1, and LEA-EMB11-like genes had statistically higher than average values for haplotype number (h) and diversity (Hd), while the formin-like gene had the lowest values for h, Hd, and ΘW. In general, π and ΘW were similar with a tendency for ΘW to be slightly higher than π, apparently as a result of an excess of low-frequency SNPs (supplemental Figure 1S at http://www.genetics.org/supplemental/). Due to this, the neutrality test statistics tended to be negative (Table 3).
TABLE 2.
Number of SNPs discovered in 18 Douglas fir candidate genes
| Gene | Total sites (bp) | Total SNP sites (S) | Average frequency of SNPs (bp/SNP) | Singleton SNPs | Trinucleotide SNPs | Parsimony informative SNPs | SNPs in coding regions | SNPs in noncoding regions | Silent (synonymous and noncoding) SNPs |
|---|---|---|---|---|---|---|---|---|---|
| EF1A | 1,072 | 14 | 77 | 5 | 0 | 9 | 7 | 7 | 14 |
| TBE | 2,954 | 58 | 51 | 22 | 1 | 36 | 20 | 38 | 51 |
| F3H1 | 365 | 14 | 26 | 10 | 0 | 4 | 8 | 6 | 8 |
| F3H2 | 647 | 14 | 46 | 2 | 1 | 12 | 6 | 8 | 11 |
| Formin-like | 337 | 3 | 112 | 0 | 1 | 3 | 3 | 0 | 1 |
| AT | 2,578 | 93 | 28 | 27 | 1 | 66 | 30 | 63 | 93 |
| LEA2 | 504 | 18 | 28 | 5 | 0 | 13 | 7 | 11 | 15 |
| MT-like | 579 | 20 | 29 | 0 | 0 | 20 | 4 | 16 | 18 |
| 60S-RPL31a | 609 | 21 | 29 | 3 | 0 | 18 | 6 | 15 | 21 |
| LEA-EMB11 | 545 | 33 | 17 | 7 | 1 | 26 | 13 | 20 | 27 |
| 40S-RPS3a | 500 | 12 | 42 | 2 | 1 | 10 | 1 | 11 | 12 |
| PolyUBQ | 898 | 17 | 53 | 2 | 0 | 15 | 9 | 8 | 17 |
| ERD15-like | 646 | 14 | 46 | 2 | 0 | 12 | 7 | 7 | 10 |
| ABA-WDS | 344 | 9 | 38 | 4 | 0 | 5 | 9 | n/a | 5 |
| LP3-like | 481 | 16 | 30 | 3 | 0 | 13 | 12 | 4 | 12 |
| 4CL1 | 628 | 8 | 79 | 5 | 0 | 3 | 5 | 3 | 4 |
| 4CL2 | 629 | 10 | 63 | 3 | 0 | 7 | 7 | 3 | 6 |
| APX | 867 | 26 | 33 | 9 | 0 | 17 | 3 | 23 | 24 |
| Mean | 843.5 | 22.2 | 46 | 6.2 | 0.3 | 16.1 | 8.7 | 14.2 | 19.2 |
| Total | 15,183 | 400 | 111 | 6 | 289 | 157 | 243 | 349 |
TABLE 3.
Total haplotype (Hd) and nucleotide (π and ΘW per site) diversity and neutrality test statistics (D, D*, and F*) in 18 Douglas fir candidate genes
| Gene | Total haplotypes | Haplotypes excluding indels (h) | Hd ± SD | π ± SD | ΘW ± SD | Tajima's D | Fu and Li's D* | Fu and Li's F* |
|---|---|---|---|---|---|---|---|---|
| EF1A | 17 | 17*** | 0.940 ± 0.031* | 0.00274 ± 0.00020 | 0.00339 ± 0.00136* | −0.656 | −0.493 | −0.636 |
| TBE | 24 | 21*** | 0.963 ± 0.024** | 0.00516 ± 0.00063 | 0.00626 ± 0.00209 | −0.723 | −0.847 | −0.951 |
| F3H1 | 7 | 6* | 0.690 ± 0.061* | 0.00528 ± 0.00179 | 0.00988 ± 0.00396 | −1.576* | −2.550* | −2.633* |
| F3H2 | 8 | 8 | 0.828 ± 0.042 | 0.00629 ± 0.00075 | 0.00562 ± 0.00225 | 0.150 | 0.384 | 0.365 |
| Formin-like | 3 | 3*** | 0.585 ± 0.045** | 0.00480 ± 0.00023 | 0.00229 ± 0.00144* | 1.498 | 1.066 | 1.381 |
| AT1 | 18 | 18*** | 0.966 ± 0.017*** | 0.00936 ± 0.00055 | 0.00935 ± 0.00304 | −0.037 | −0.146 | −0.131 |
| LEA2 | 14 | 12 | 0.884 ± 0.027 | 0.00647 ± 0.00066 | 0.00878 ± 0.00321 | −0.862 | −0.234 | −0.522 |
| MT-like | 13 | 12 | 0.907 ± 0.027 | 0.01334 ± 0.00098* | 0.00911 ± 0.00342 | 1.639* | 1.621** | 1.909** |
| 60S-RPL31a | 8 | 8 | 0.701 ± 0.088* | 0.01011 ± 0.00126 | 0.00891 ± 0.00332 | 0.479 | 0.758 | 0.786 |
| LEA-EMB11 | 18 | 17*** | 0.950 ± 0.024** | 0.01378 ± 0.00163* | 0.01594 ± 0.00560** | −0.593 | 0.396 | 0.093 |
| 40S-RPS3a | 8 | 8 | 0.810 ± 0.046 | 0.00601 ± 0.0011 | 0.00617 ± 0.00255 | −0.336 | 0.632 | 0.390 |
| PolyUBQ | 9 | 9 | 0.840 ± 0.047 | 0.00544 ± 0.00049 | 0.00494 ± 0.00192 | 0.357 | 0.886 | 0.845 |
| ERD15-like | 8 | 7 | 0.598 ± 0.105* | 0.00438 ± 0.00130 | 0.00563 ± 0.00227 | −0.757 | 0.712 | 0.304 |
| ABA-WDS | 9 | 9 | 0.825 ± 0.045 | 0.00662 ± 0.00097 | 0.00672 ± 0.00298 | −0.048 | −0.914 | −0.761 |
| LP3-like | 14 | 12 | 0.866 ± 0.033 | 0.00662 ± 0.00085 | 0.00848 ± 0.00319 | −0.713 | 0.368 | 0.017 |
| 4CL1 | 9 | 9 | 0.841 ± 0.038 | 0.00268 ± 0.00026 | 0.00316 ± 0.00143* | −0.460 | −1.900* | −1.705 |
| 4CL2 | 10 | 10 | 0.814 ± 0.041 | 0.00237 ± 0.00033* | 0.00378 ± 0.00159* | −1.128 | −0.319 | −0.676 |
| APX | 12 | 12 | 0.884 ± 0.041 | 0.00636 ± 0.00080 | 0.00789 ± 0.00285 | −0.700 | −0.504 | −0.665 |
| Mean | 11.6 | 11.0 | 0.827 ± 0.043 | 0.00655 ± 0.00082 | 0.00702 ± 0.00269 | −0.248 | −0.060 | −0.144 |
Italic entries are statistically significant values for test statistics (D, D*, and F*) and values for haplotype number (h), haplotype diversity (Hd), and nucleotide diversity (π and ΘW) that were statistically different from average values based on coalescence simulations (*P < 0.05; **P < 0.01; ***P < 0.001).
A detailed description of the nucleotide diversity in different nucleotide sequence sites and regions is presented in Table 4. The nonsynonymous substitutions were almost five times less frequent than silent substitutions (synonymous substitutions and substitutions in noncoding regions), where π = 0.00210 vs. π = 0.01055 and ΘW = 0.00261 vs. ΘW = 0.01132 for nonsynonymous vs. silent substitutions, respectively.
TABLE 4.
Mean nucleotide diversity (π and Θ per site) in different nucleotide sites or gene regions for 18 Douglas fir candidate genes
| Sites | π | ΘW |
|---|---|---|
| All (coding + noncoding) | 0.00655 | 0.00702 |
| Coding | ||
| All coding | 0.00456 | 0.00491 |
| Nonsynonymous | 0.00210 | 0.00261 |
| Synonymous | 0.01275 | 0.01300 |
| Noncoding | 0.01000 | 0.01107 |
| Silent (synonymous + noncoding) | 0.01055 | 0.01132 |
Neutrality tests:
Thirteen of the 18 genes had negative values of the neutrality test statistics D, D*, and F*, but they were significant only for the F3H1 gene (Table 3). The 4CL1 gene was also possibly under negative selection, but only D* was statistically significant (although the F*-value was almost significant with P = 0.065). Unlike F3H1 and 4CL1, the MT-like gene was possibly under positive selection. The sliding-window analysis revealed statistically significant values of D, D*, and F* in a few regions within the TBE, formin, AT1, and APX genes that were possibly under selection (see, for instance, APX in supplemental Figure 2S at http://www.genetics.org/supplemental/).
The neutrality of sequence polymorphism was also assessed using the ratio of nonsynonymous (dN) to synonymous (dS) nucleotide substitutions. Six genes had a dN/dS ratio significantly <1 (supplemental Table 3S at http://www.genetics.org/supplemental/). Only the 4CL1 gene had dN/dS > 1, but it was not statistically significant.
LD, haploblock structure within genes, and selection of htSNPs for association mapping:
A considerable amount of LD was found within sequences. A total of 4349 pairwise comparisons were estimated for parsimony informative sites among pairs of sites within 18 genes. Almost one-third of them (1316) showed LD statistically significant by a Fisher's exact test, which remained significant for 326 pairs even after Bonferroni correction. Figure 2 shows LD estimates (r2) plotted against the pairwise distances between parsimony SNPs within all 18 genes. The LD declined linearly as distances between sites increased, but a fair amount of LD remained, even for pairs separated by >500 bp. There were a few significant LDs for tightly linked or even for unlinked genes (supplemental Figure 3S at http://www.genetics.org/supplemental/), but none of them remained significant after Bonferroni correction. Depending on the threshold values used to define a block, from 1 up to 58 haploblocks per gene were revealed (supplemental Table 4S at http://www.genetics.org/supplemental/). These thresholds included a minimal LD value, minimal frequency of the SNP allele to be included, minimal chromosome and haplotype coverage, and htSNP coverage. However, using reasonable thresholds, there were approximately ∼2–3 haploblocks per gene (except very long genes such as TBE) that could be genotyped with approximately four to five SNPs per gene on average.
Figure 2.

Scatterplot of pairwise distances and linkage disequilibrium (LD) estimates (r2) between all parsimony informative Douglas fir SNPs in 18 genes with a second-order polynomial best-fit curve (y = 5E-08x2 − 0.0002x + 0.28).
Population structure:
NJ trees revealed no significant clustering or obvious geographic structure among samples (supplemental Figure 4S at http://www.genetics.org/supplemental/). The nucleotide site data gave a low estimate of FST = 0.028 among six regions that was not statistically different from zero on the basis of 1000 permutations (Weir 1996). The exact differentiation test also did not reveal any differentiation (P = 1) between regions. The nearest-neighbor statistic revealed no significant differentiation between populations either in the pairwise tests or globally (Snn = 0.071, P = 0.848 on the basis of 1000 permutations).
DISCUSSION
This is the first extensive study of nucleotide diversity and LD for candidate genes in Douglas fir. It provides important data on nucleotide diversity and haplotype structure in Douglas fir natural populations, and, together with similar studies in pines, it lays a foundation for association mapping in conifers. An efficient strategy for selecting the most informative SNPs for association mapping was also developed.
Average nucleotide diversity in Douglas fir was higher than that in human and soybean, but lower than that in maize, and similar to that in Drosophila (Table 5). The similarity to Drosophila is not completely surprising given that both Douglas fir and Drosophila have large population sizes and high outcrossing rates. Compared with other conifers, Douglas fir has higher levels of diversity than do loblolly pine (Brown et al. 2004a; Neale and Savolainen 2004; S. C. González-Martínez, E. Ersoz, G. R. Brown, N. C. Wheeler and D. B. Neale, unpublished results), Scots pine (P. sylvestris) (Dvornyk et al. 2002; García-Gil et al. 2003), and sugi (Kado et al. 2003). Potentially, a difference in mutation rates and/or in historic effective population sizes (Ne) between Douglas fir and pines could explain the difference in the level of nucleotide diversity observed in Douglas fir and loblolly pine. Unfortunately, we are unaware of any direct estimates of mutation rate at the nucleotide level in pines vs. Douglas fir. Indirect estimates that are inferred from observed nucleotide differentiation between closely related pine species are based on assumptions of the neutral model as well as on the rough assumptions of divergence time and Ne. These estimates can be highly biased and produce a circular argument. Unfortunately, paleobotanical data are also very incomplete and highly inconclusive. Therefore, there are no unambiguous data that would suggest that Douglas firs have maintained large Ne during the Holocene or Pleistocene, while pines have not. However, more importantly, our study, as well as other conifer studies cited above, revealed manyfold difference in estimates of π and ΘW between different genes, which highlights the problems of comparing variation among species when estimates are based on one or a few loci (e.g., Dvornyk et al. 2002; García-Gil et al. 2003; Ingvarsson 2005). Comparisons among species should be either based on many loci or, ideally, restricted to orthologous loci.
TABLE 5.
Nucleotide diversity (Θ per site) across different regions and species
| Species | Loci | Total | Coding regions | Noncoding, including introns and untranscribed regions | Synonymous SNPs | Nonsynonymous SNPs | Reference |
|---|---|---|---|---|---|---|---|
| Humana | 75 | 8.3 ± 1.9 | 8.0 ± 1.9 | 8.5 ± 2.0 | 15.1 ± 3.6 | 5.7 ± 1.4 | Halushka et al. (1999) |
| 106 | 5.3 ± 1.3 | 5.4 ± 1.3 | 5.2 ± 1.3 | 11.7 ± 2.9 | 3.4 ± 0.9 | Cargill et al. (1999) | |
| Soybean | 143 | 5.3 ± 1.9 | 10.0 ± 3.9 | 3.8 ± 1.5 | Zhu et al. (2003) | ||
| Sugib | 7 | 20 | 35c | 34 | 7 | Kado et al. (2003) | |
| Maritime pine | 8 | 21 | 21 | 17 | 4.6 | 1.5 | Pot et al. (2005) |
| Monterey pine | 8 | 19 | 8d | 8 | 8d | 1.5 | Pot et al. (2005) |
| Loblolly pine | 19 | 41 | 66c | 11 | Brown et al. (2004a) | ||
| 18 | 50 ± 29 | 86 ± 84 | 23 ± 21 | S. C. González-Martínez, E. Ersoz, G. R. Brown, N. C. Wheeler and D. B. Neale (unpublished results) | |||
| Douglas fir | 18 | 70 ± 27 | 49 ± 23 | 113 ± 46c | 130 ± 60 | 26 ± 17 | This study |
| Drosophilaa | 24 | 70 ± 58 | 40 ± 31 | 105 ± 80 | 130 ± 92 | 15 ± 14 | Moriyama and Powell (1996) |
| Arabidopsis | 357e | 70 | 80 | 100 | 10 | Schmid et al. (2005) | |
| Maize | 21 | 96 ± 32 | 72 ± 25 | 111 ± 37 | 173 ± 61 | 39 ± 14 | Tenaillon et al. (2001) |
All Θ-values are ×104.
Compiled in Zwick et al. (2000).
Unweighed average Θ calculated from Table 3 in Kado et al. (2003).
Based on silent (synonymous plus noncoding regions) substitutions.
Corrected values (D. Pot, personal communication).
Based on 12 accessions including 5 accessions previously used in genetic mapping (Col-0, Cvi-0, Ler, Nd-0, and Ws-0) and an additional 7 accessions (Ei-2, CS22491, Gü-0, Lz-0, Wei-0, Ws-0, and Yo-0) with a high average genetic distance to other accessions.
The average π- and ΘW-values were similar in this study. Both π- and ΘW-values estimate the equilibrium neutral parameter Θ = 4Neμ for autosomal loci, a central parameter in population genetic models for the balance between mutation and random genetic drift, where Ne is the effective population size and μ is the neutral mutation rate per nucleotide site. This parameter summarizes the rate at which processes of mutation and random genetic drift generate and maintain variation within a gene, assuming that natural selection has not been operating. Although the number of segregating sites does not represent all the information in the sample, under the neutral infinite-sites model the frequency spectrum of sites is determined by Θ, which in turn is estimated by S. Violations of the assumptions of the infinite-sites model will lead to biases in the estimate of Θ. The similarity of π- and ΘW-values shows that those violations were not significant. Nevertheless, negative values of the neutrality test statistics (Tajima's D, D*, and F*) and dN/dS < 1 in most studied genes suggested that they are mainly under negative selection or reflect a recent population expansion. However, it is difficult, if not impossible, to distinguish between population growth and selection, if only intraspecific polymorphism is studied. The frequency spectrum can be different for different genes, depending on the combined effect of many factors, such as mutation, population size, recombination rate, gene conversion, and selection intensity. Comparison of intraspecific and interspecific polymorphism in orthologous genes between closely related species can help to detect or confirm genes under selection (Hudson et al. 1987; McDonald and Kreitman 1991; Kreitman 2000).
The rate of decay of LD with distance is a critical factor that affects the success of association mapping on the basis of SNPs in candidate genes. If LD affects large regions or genomic blocks, then association with phenotypic traits would be easier to detect, but it would be more difficult to assign it to the particular candidate gene or quantitative trait nucleotide (QTN). If LD decays quickly, then the associations found between a particular SNP and phenotypic trait would be more likely to be causative rather than due to linkage with other unknown genes. LD is a result of the interplay of many factors, such as mutation and recombination rates, mating system, selection, population size, structure, and history. The intragenic recombination that affects LD within genes was estimated in this study, but not presented and discussed here because we believe that the limited sample size was insufficient for its reliable estimation. The estimation of recombination requires that considerably larger segments of contiguous DNA be sequenced, and more data should be collected to fully address this problem. LD varies greatly in different species ranging from 200–1500 bp in maize up to 50–100 kb in Arabidopsis (see Rafalski and Morgante 2004, Table 2, for review). Our data indicate that LD decayed >50% over relatively short segments (from r2 = ∼0.25 to ∼0.10 within 2000 bp, Figure 2). These data confirmed recent studies in loblolly pine and suggest that conifers may have LD at the lower end of the spectrum (Brown et al. 2004a; S. C. González-Martínez, E. Ersoz, G. R. Brown, N. C. Wheeler and D. B. Neale, unpublished results), making these species potentially very amenable for candidate gene vs. genomewide-based studies (Neale and Savolainen 2004). Unlike candidate gene-based association studies the genomewide scans depend more on strong LD over long regions in the genome. However, it should be noted that this study was not specifically designed to address LD in the genome, but rather within genes, and many distal pairwise comparisons are underrepresented because studied sequences were relatively short.
A few significant LDs were found between tightly linked or even between unlinked genes (supplemental Figure 3S at http://www.genetics.org/supplemental/), although none of them remained significant after Bonferroni correction. Nevertheless, these associations could be a sign of either population substructure or strong epistatic interactions between genes. The latter one is especially likely for the EF1 and 60SRPL31a genes, because both are involved in ribosomal biosynthesis, and for the F3H1 and F3H2 genes that are apparently involved in the same metabolic pathway.
Association mapping requires careful selection of SNPs for genotyping. Our data will help us select the most informative and potentially useful htSNPs in 18 candidate genes for association mapping. We developed a complex approach that takes into account all available data to increase the likelihood of detecting associations. The polymorphic SNPs that were discovered in this study in coding regions, which cause nonsynonymous substitutions, mark haploblocks, and are under positive selection, are the best candidates for the association mapping study that is now in progress.
Connecting phenotype with genotype is the fundamental aim of genetics (Botstein and Risch 2003). The candidate gene-based association studies are considered as one of the best approaches to connect complex phenotypes with genotypes (Pflieger et al. 2001; Botstein and Risch 2003; Neale and Savolainen 2004; Rebbeck et al. 2004). This study proved that Douglas fir meets the most important conditions for candidate gene-based association studies such as high phenotypic variation, high SNP polymorphism in candidate genes, and moderate LD. The lack of population subdivision observed in the SNP discovery panel will also facilitate association mapping. However, it is too early to make a conclusion about population structure. The further study of a much larger sample of ∼1300 trees from an association study will provide more data on population structure.
Acknowledgments
We thank the members of the Douglas fir Genome Project for their participation and contributions (http://dendrome.ucdavis.edu/dfgp/collaborators.html), Nicholas Wheeler (Molecular Tree Breeding Services, Centralia, WA) for providing samples for the SNP discovery panel, Glenn T. Howe (Department of Forest Science, Oregon State University, Corvallis, OR) and Santiago C. González-Martínez (Unit of Forest Genetics, Center of Forest Research, Madrid, Spain) for reviewing the manuscript and helpful recommendations, and Jennifer Manares (University of California, Davis, CA) for technical assistance with PCR and gel electrophoresis. Funding for this project was provided by the Pacific Southwest Research Station, U.S. Department of Agriculture (USDA) Forest Service within the American Forest & Paper Association Agenda 2020 program. Trade names and commercial products or enterprises are mentioned solely for information and no endorsement by the USDA is implied.
References
- Aagaard, J. E., K. V. Krutovskii and S. H. Strauss, 1998. a RAPDs and allozymes exhibit similar levels of diversity and differentiation among populations and races of Douglas-fir. Heredity 81: 69–78. [Google Scholar]
- Aagaard, J. E., K. V. Krutovskii and S. H. Strauss, 1998. b RAPD markers of mitochondrial origin exhibit lower population diversity and higher differentiation than RAPDs of nuclear origin in Douglas fir. Mol. Ecol. 7: 801–812. [Google Scholar]
- Aitken, S. N., and W. T. Adams, 1997. Spring cold hardiness under strong genetic control in Oregon populations of coastal Douglas-fir. Can J. For. Res. 27: 1773–1778. [Google Scholar]
- Anekonda, T. S., W. T. Adams, S. N. Aitken, D. B. Neale, K. D. Jermstad et al., 2000. Genetics of cold-hardiness in a cloned full-sib family of coastal Douglas-fir. Can. J. For. Res. 30: 837–840. [Google Scholar]
- Aranda, M. A., M. Escaler, D. Wang and A. J. Maule, 1996. Induction of HSP70 and polyubiquitin expression associated with plant virus replication. Proc. Natl. Acad. Sci. USA 93: 15289–15293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borevitz, J. O., and M. Nordborg, 2003. The impact of genomics on the study of natural variation in Arabidopsis. Plant Physiol. 132: 718–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botstein, D., and N. Risch, 2003. Discovering genotypes underlying human phenotypes: past successes for Mendelian disease, future approaches for complex disease. Nat. Genet. 33(Suppl.): 228–237. [DOI] [PubMed] [Google Scholar]
- Brookes, A. J., 1999. The essence of SNPs. Gene 234: 177–186. [DOI] [PubMed] [Google Scholar]
- Brown, G. R., D. L. Bassoni, G. P. Gill, J. R. Fontana, N. C. Wheeler et al., 2003. Identification of quantitative trait loci influencing wood property traits in loblolly pine (Pinus taeda L.). III. QTL verification and candidate gene mapping. Genetics 164: 1537–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, G. R., G. P. Gill, R. J. Kuntz, C. H. Langley and D. B. Neale, 2004. a Nucleotide diversity and linkage disequilibrium in loblolly pine. Proc. Natl. Acad. Sci. USA 101: 15255–15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, G. R., G. P. Gill, R. J. Kuntz, J. A. Beal, D. Nelson et al., 2004. b Associations of candidate gene single nucleotide polymorphisms with wood property phenotypes in loblolly pine. Plant & Animal Genome XII Conference, San Diego, January 10–14, 2004 (http://www.intl-pag.org/pag/12/abstracts/W22_PAG12_98.html).
- Browse, J., and B. M. Lange, 2004. Counting the cost of a cold-blooded life: metabolomics of cold acclimation. Proc. Natl. Acad. Sci. USA 101: 14996–14997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, R. K., and F. C. Sorensen, 1978. Effect of test environment on expression of clines and on delimitation of seed zones in Douglas-fir. Theor. Appl. Genet. 51: 233–246. [DOI] [PubMed] [Google Scholar]
- Campbell, R. K., and A. I. Sugano, 1975. Phenology of bud burst in Douglas-fir related to provenance, photoperiod, chilling and flushing temperature. Bot. Gaz. 136: 290–298. [Google Scholar]
- Cargill, M., D. Altshuler, J. Ireland, P. Sklar, K. Ardlie et al., 1999. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat. Genet. 22: 231–238. [DOI] [PubMed] [Google Scholar]
- Carlson, C. S., M. A. Eberle, L. Kruglyak and D. A. Nickerson, 2004. Mapping complex disease loci in whole-genome association studies. Nature 429: 446–452. [DOI] [PubMed] [Google Scholar]
- Close, T. J., 1997. Dehydrins: a commonality in the response of plants to dehydration and low temperature. Physiol. Plant. 100: 291–296. [Google Scholar]
- Cook, D., S. Fowler, O. Fiehn and M. F. Thomashow, 2004. A prominent role for the CBF cold response pathway in configuring the low-temperature metabolome of Arabidopsis. Proc. Natl. Acad. Sci. USA 101: 15243–15248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, J.-Z., and D. I. Dunstan, 1996. Expression of abundant mRNAs during somatic embryogenesis of white spruce [Picea glauca (Moench) Voss]. Planta 199: 459–466. [DOI] [PubMed] [Google Scholar]
- Dvornyk, V., A. Sirviö, M. Mikkonen and O. Savolainen, 2002. Low nucleotide diversity at the pal1 locus in the widely distributed Pinus sylvestris. Mol. Biol. Evol. 19: 179–188. [DOI] [PubMed] [Google Scholar]
- Dubos, C., G. Le Provost, D. Pot, F. Salin, C. Lalane et al., 2003. Identification and characterization of water-stress-responsive genes in hydroponically grown maritime pine (Pinus pinaster) seedlings. Tree Physiol. 23: 169–179. [DOI] [PubMed] [Google Scholar]
- Ewing, B., and P. Green, 1998. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8: 186–194. [PubMed] [Google Scholar]
- Ewing, B., L. Hillier, M. Wendl and P. Green, 1998. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8: 175–185. [DOI] [PubMed] [Google Scholar]
- Excoffier, L., P. E Smouse and J. M. Quattro, 1992. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131: 479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier, L., S. Schneider and D. Roessli, 2004. ARLEQUIN ver 2.0: a software for population genetics data analysis (http://lgb.unige.ch/arlequin). [PMC free article] [PubMed]
- Farnir, F., W. Coppieters, J.-J. Arranz, P. Berzi, N. Cambisano et al., 2000. Extensive genome-wide linkage disequilibrium in cattle. Genome Res. 10: 220–227. [DOI] [PubMed] [Google Scholar]
- Feder, M. E., and T. Mitchell-Olds, 2003. Evolutionary and ecological functional genomics. Nat. Rev. Genet. 4: 649–655. [DOI] [PubMed] [Google Scholar]
- Flint-García, S. A., J. M. Thornsberry and E. S. Buckler, IV, 2003. Structure of linkage disequilibrium in plants. Annu. Rev. Plant. Biol. 54: 357–374. [DOI] [PubMed] [Google Scholar]
- Fowler, S., and M. F. Thomashow, 2002. Arabidopsis transcriptome profiling indicates that multiple regulatory pathways are activated during cold acclimation in addition to the CBF cold response pathway. Plant Cell 14: 1675–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y.-X., and W.-H. Li, 1993. Statistical tests of neutrality of mutations. Genetics 133: 693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Gil, M. R., M. Mikkonen and O. Savolainen, 2003. Nucleotide diversity at two phytochrome loci along a latitudinal cline in Pinus sylvestris. Mol. Ecol. 12: 1195–1206. [DOI] [PubMed] [Google Scholar]
- Glazier, A. M., J. H. Nadeau and T. J. Aitman, 2002. Finding genes that underlie complex traits. Science 298: 2345–2349. [DOI] [PubMed] [Google Scholar]
- Goldstein, D. B., and M. E. Weale, 2001. Population genomics: linkage disequilibrium holds the key. Curr. Biol. 11: R576–R579. [DOI] [PubMed] [Google Scholar]
- Gordon, D., C. Abajian and P. Green, 1998. Consed: a graphical tool for sequence finishing. Genome Res. 8: 195–202. [DOI] [PubMed] [Google Scholar]
- Gordon, D., C. Desmarais and P. Green, 2001. Automated finishing with autofinish. Genome Res. 11: 614–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudet, J., M. Raymond, T. de Meeüs and F. Rousset, 1996. Testing differentiation in diploid populations. Genetics 144: 1933–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halushka, M. K., J. B. Tan, K. Bentley, L. Hsie, N. P. Shen et al., 1999. Patterns of single-nucleotide polymorphisms in candidate genes for blood-pressure homeostasis. Nat. Genet. 22: 239–247. [DOI] [PubMed] [Google Scholar]
- Harris, M., J. M. Martin, J. F. Peden and C. J. Rawlings, 2003. SNP cherry picker: maximizing the chance of finding an association with a disease SNP. Bioinformatics 19: 2141–2143. [DOI] [PubMed] [Google Scholar]
- Hertzberg, M., H. Aspeborg, J. Schrader, A. Andersson, R. Erlandsson et al., 2001. A transcriptional roadmap to wood formation. Proc. Natl. Acad. Sci. USA 98: 14732–14737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, W. G., and A. Robertson, 1968. Linkage disequilibrium in finite populations. Theor. Appl. Genet. 38: 226–231. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 1991. Gene genealogies and the coalescent process. Oxf. Surv. Evol. Biol. 7: 1–44. [Google Scholar]
- Hudson, R. R., 2000. A new statistic for detecting genetic differentiation. Genetics 155: 2011–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., M. Kreitman and M. Aguade, 1987. A test of neutral molecular evolution based on nucleotide data. Genetics 116: 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingvarsson, P. K., 2005. Nucleotide polymorphism and linkage disequilibrium within and among natural populations of European aspen (Populus tremula L., Salicaceae). Genetics 169: 945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jermstad, K. D., D. L. Bassoni, N. C. Wheeler and D. B. Neale, 1998. A sex-averaged linkage map in coastal Douglas-fir (Pseudotsuga menziesii [Mirb.] Franco) based on RFLP and RAPD markers. Theor. Appl. Genet. 97: 762–770. [Google Scholar]
- Jermstad, K. D., D. L. Bassoni, K. S. Jech, N. C. Wheeler and D. B. Neale, 2001. a Mapping of quantitative trait loci controlling adaptive traits in coastal Douglas-fir. I. Timing of vegetative bud flush. Theor. Appl. Genet. 102: 1142–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jermstad, K. D., D. L. Bassoni, N. C. Wheeler, T. S. Anekonda, S. N. Aitken et al., 2001. b Mapping of quantitative trait loci controlling adaptive traits in coastal Douglas-fir. II. Spring and fall cold-hardiness. Theor. Appl. Genet. 102: 1152–1158. [Google Scholar]
- Jermstad, K. D., D. L. Bassoni, K. S. Jech, G. A. Ritchie, N. C. Wheeler et al., 2003. Mapping of quantitative trait loci controlling adaptive traits in coastal Douglas fir. III. QTL by environment interactions. Genetics 165: 1489–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jukes, T. H, and C. R. Cantor, 1969. Evolution of protein molecules, pp. 21–132 in Mammalian Protein Metabolism, edited by H. N. Munro. Academic Press, New York.
- Kado, T., H. Yoshimaru, Y. Tsumura and H. Tachida, 2003. DNA variation in a conifer, Cryptomeria japonica (Cupressaceae sensu lato). Genetics 164: 1547–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M., 1983. The Neutral Theory of Molecular Evolution. Cambridge University Press, Cambridge, UK.
- Kiyosue, T., K. Yamaguchi-Shinozaki and K. Shinozaki, 1994. ERD15, a cDNA for a dehydration-induced gene from Arabidopsis thaliana. Plant Physiol. 106: 1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitman, M., 2000. Methods to detect selection in populations with applications to the human. Annu. Rev. Genomics Hum. Genet. 1: 539–559. [DOI] [PubMed] [Google Scholar]
- Krutovsky, K. V., M. Troggio, G. R. Brown, K. D. Jermstad and D. B. Neale, 2004. Comparative mapping in the Pinaceae. Genetics 168: 447–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S., K. Tamura and M. Nei, 2004. MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief. Bioinform. 5: 150–163. [DOI] [PubMed] [Google Scholar]
- Kuwabara, C., D. Takezawa, T. Shimada, T. Hamada, S. Fujikawa et al., 2002. Abscisic acid- and cold-induced thaumatin-like protein in winter wheat has an antifungal activity against snow mould, Microdochium nivale. Physiol. Plant. 115: 101–110. [DOI] [PubMed] [Google Scholar]
- Lewontin, R. C., 1964. The interaction of selection and linkage. I. General considerations: heterotic models. Genetics 49: 49–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, P., and W. T. Adams, 1993. Genetic control of bud phenology in pole-size trees and seedlings of coastal Douglas-fir. Can. J. For. Res. 23: 1043–1051. [Google Scholar]
- Li, W.-H., 1997. Molecular Evolution. Sinauer, Sunderland, MA.
- Luikart, G., P. R. England, D. Tallmon, S. Jordan and P. Taberlet, 2003. The power and promise of population genomics: from genotyping to genome typing. Nat. Rev. Genet. 4: 981–994. [DOI] [PubMed] [Google Scholar]
- McDonald, J. H., and M. Kreitman, 1991. Adaptive protein evolution at the Adh locus in Drosophila. Nature 351: 652–654. [DOI] [PubMed] [Google Scholar]
- Merkle, S. A., and W. T. Adams, 1987. Pattern of allozyme variation within and among Douglas-fir breeding zones in southwest Oregon. Can. J. For. Res. 17: 402–407. [Google Scholar]
- Moran, G. F., and W. T. Adams, 1989. Microgeographical patterns of allozyme differentiation in Douglas-fir from southwest Oregon. For. Sci. 35: 3–15. [Google Scholar]
- Moriyama, E. N., and J. R. Powell, 1996. Intraspecific nuclear DNA variation in Drosophila. Mol. Biol. Evol. 13: 261–277. [DOI] [PubMed] [Google Scholar]
- Neale, D. B., and O. Savolainen, 2004. Association genetics of complex traits in conifers. Trends Plant Sci. 9: 325–330. [DOI] [PubMed] [Google Scholar]
- Nei, M., 1987. Molecular Evolutionary Genetics. Columbia University Press, New York.
- Nei, M., and T. Gojobori, 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3: 418–426. [DOI] [PubMed] [Google Scholar]
- Nei, M., and S. Kumar, 2000. Molecular Evolution and Phylogenetics. Oxford University Press, New York.
- Nogueira, F. T. S., V. E. De Rosa, Jr., M. Menossi, E. C. Ulian and P. Arruda, 2003. RNA expression profiles and data mining of sugarcane response to low temperature. Plant Physiol. 132: 1811–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg, M., and H. Innan, 2002. Molecular population genetics. Curr. Opin. Plant Biol. 5: 69–73. [DOI] [PubMed] [Google Scholar]
- Palva, E. T., and P. Heino, 1998. Molecular mechanism of plant cold acclimation and freezing tolerance, pp. 3–14 in Plant Cold Hardiness, edited by P. H. Li and T. H. H. Chen. Plenum, New York.
- Pflieger, S., V. Lefebvre and M. Causse, 2001. The candidate gene approach in plant genetics: a review. Mol. Breed. 7: 275–291. [Google Scholar]
- Pot, D., L. McMillan, C. Echt, G. Le Provost, P. Garnier-Gere et al., 2005. Nucleotide variation in genes involved in wood formation in two pine species. New Phytol. 167: 101–112. [DOI] [PubMed] [Google Scholar]
- Provart, N. J., P. Gil, W. Chen, B. Han, H.-S. Chang et al., 2003. Gene expression phenotypes of Arabidopsis associated with sensitivity to low temperatures. Plant Physiol. 132: 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabbani, M. A., K. Maruyama, H. Abe, M. A. Khan, K. Katsura et al., 2003. Monitoring expression profiles of rice genes under cold, drought, and high-salinity stresses and abscisic acid application using cDNA microarray and RNA gel-blot analyses. Plant Physiol. 133: 1755–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafalski, A. J., 2002. Novel genetic mapping tools in plants: SNPs and LD-based approaches. Plant Sci. 162: 329–333. [Google Scholar]
- Rafalski, A., and M. Morgante, 2004. Corn and humans: recombination and linkage disequilibrium in two genomes of similar size. Trends Genet. 20: 103–111. [DOI] [PubMed] [Google Scholar]
- Raymond, M., and F. Rousset, 1995. An exact test for population differentiation. Evolution 49: 1280–1283. [DOI] [PubMed] [Google Scholar]
- Rebbeck, T. R., M. Spitz and X. Wu, 2004. Assessing the function of genetic variants in candidate gene association studies. Nat. Rev. Genet. 5: 589–597. [DOI] [PubMed] [Google Scholar]
- Rehfeldt, G. E., 1983. Genetic variability within Douglas-fir populations: implications for tree improvement. Silvae Genet. 32: 9–14. [Google Scholar]
- Rehfeldt, G. E., 1989. Ecological adaptations in Douglas-fir (Pseudotsuga menziesii var. glauca): a synthesis. For. Ecol. Manage. 28: 203–215. [Google Scholar]
- Rozas, J., J. C. Sánchez-DelBarrio, X. Messeguer and R. Rozas, 2003. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19: 2496–2497. [DOI] [PubMed] [Google Scholar]
- Saitou, N., and M. Nei, 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4: 406–425. [DOI] [PubMed] [Google Scholar]
- Schlötterer, C., 2002. Towards a molecular characterization of adaptation in local populations. Curr. Opin. Genet. Dev. 12: 683–687. [DOI] [PubMed] [Google Scholar]
- Schmid, K. J., S. Ramos-Onsins, H. Ringys-Beckstein, B. Weisshaar and T. Mitchell-Olds, 2005. A multilocus sequence survey in Arabidopsis thaliana reveals a genome-wide departure from a neutral model of DNA sequence polymorphism. Genetics 169: 1601–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki, M., M. Narusaka, H. Abe, M. Kasuga, K. Yamaguchi-Shinozaki et al., 2001. Monitoring the expression pattern of 1300 Arabidopsis genes under drought and cold stresses by using a full-length cDNA microarray. Plant Cell 13: 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki, M., M. Narusaka, J. Ishida, T. Nanjo, M. Fujita et al., 2002. Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. Plant J. 31: 279–292. [DOI] [PubMed] [Google Scholar]
- Steiner, K. C., 1979. Variation in bud-burst timing among populations of interior Douglas-fir. Silvae Genet. 28: 76–79. [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon, M. I., M. C. Sawkins, A. D. Long, R. L. Gaut, J. F. Doebley et al., 2001. Patterns of DNA sequence polymorphism along chromosome 1 of maize (Zea mays ssp mays L.). Proc. Natl. Acad. Sci. USA 98: 9161–9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomashow, M. F., 1998. Role of cold-responsive genes in plant freezing tolerance. Plant Physiol. 118: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomashow, M. F., 1999. Plant cold acclimation: freezing tolerance genes and regulatory mechanisms. Annu. Rev. Plant Physiol. Plant Mol. Biol. 50: 571–599. [DOI] [PubMed] [Google Scholar]
- Thomashow, M. F., 2001. So what's new in the field of plant cold acclimation? Lots! Plant Physiol. 125: 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viard, F., Y. A. El-Kassaby and K. Ritland, 2001. Diversity and genetic structure in populations of Pseudotsuga menziesii (Pinaceae) at chloroplast microsatellite loci. Genome 44: 336–344. [DOI] [PubMed] [Google Scholar]
- Wanner, L. A., and O. Junttila, 1999. Cold-induced freezing tolerance in Arabidopsis. Plant Physiol. 120: 391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson, G. A., 1975. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7: 256–276. [DOI] [PubMed] [Google Scholar]
- Weir, B. S., 1996. Genetic Data Analysis II. Sinauer Associates, Sunderland, MA.
- Wheeler, N. C., K. D. Jermstad, K. V. Krutovsky, S. N. Aitken, G. T. Howe et al., 2005. Mapping of quantitative trait loci controlling adaptive traits in coastal Douglas-Fir. IV. Cold-hardiness QTL verification and candidate gene mapping. Mol. Breed. 15: 145–156. [Google Scholar]
- Xiayi, K., and L. R. Cardon, 2003. Efficient selective screening of haplotype tag SNPs. Bioinformatics 19: 287–288. [DOI] [PubMed] [Google Scholar]
- Zhang, K., and L. Jin, 2003. HaploBlockFinder: haplotype block analyses. Bioinformatics 19: 1300–1301. [DOI] [PubMed] [Google Scholar]
- Zhu, Y. L., Q. J. Song, D. L. Hyten, C. P. Van Tassell, L. K. Matukumalli et al., 2003. Single-nucleotide polymorphisms in soybean. Genetics 163: 1123–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwick, M. E., D. J. Cutler and A. Chakravarti, 2000. Patterns of genetic variation in Mendelian and complex traits. Annu. Rev. Genomics Hum. Genet. 1: 387–407. [DOI] [PubMed] [Google Scholar]