

Abstract
We develop a method for maximum-likelihood estimation of coalescence times in genealogical trees, based on population genetics data. For this purpose, a Viterbi-type algorithm is constructed to maximize the joint likelihood of the coalescence times. Marginal confidence intervals for the coalescence times based on the profile likelihoods are also computed. Our method of finding MLEs and calculating C.I.'s appears to be more accurate than alternative numerical maximization methods, and maximum-likelihood inference appears to be more accurate than other existing model-free approaches to estimating coalescent times. We demonstrate the method on two different data sets: human Y chromosome DNA data and fungus DNA data.
WE study the evolutionary history of a sample of nonrecombining DNA sequences, from a given population, which exhibits variation. The ancestral relationships of the individuals in the sample can be depicted by a rooted genealogical tree. Interest lies in estimating the coalescence times in the tree and, in particular, the time to the most recent common ancestor (TMRCA) of the sample.
Currently there are two distinct approaches for estimating coalescence times, which we call model-based and model-free. Both approaches use similar models for the mutation process, but differ in whether or not they assume a model for the genealogy.
Model-based approaches use coalescence models (Kingman 1982; Nordborg 2001) for the underlying genealogy of the sample. These approaches require specific assumptions about the demography (e.g., the presence of random mating, how population size has varied over time, etc.) to be made. Both Bayesian (Tavaré et al. 1997; Pritchard et al. 1999) and empirical Bayes (Griffiths and Tavaré 1994) methods for performing such inferences exist.
Model-free approaches do not assume any model for the genealogy. There are two such existing approaches for estimating the TMRCA, both based on method-of-moments estimators. Tang et al. (2002) partition the observed sequences into two subsets, corresponding to two clades that have diverged from the sample TMRCA, and use the number of nucleotide differences between pairs of sequences in different clades to obtain an estimator of the sample TMRCA. (This is an extension and significant improvement on an earlier approach by Templeton 1993). Alternatively Thomson et al. (2000) proposed a method, which uses the numbers of mutational differences between the observed DNA sequences and the sequence of the MRCA.
Which of the model-free and model-based approaches performs best will depend crucially on the validity of the demographic assumptions made in the model-based methods (for a comparative discussion, see Stumpf and Goldstein 2001). If they are correct, then model-based approaches will be more accurate; however, Tang et al. (2002) give evidence for the lack of robustness of model-based approaches to misspecification of the demographic model. One advantage of the current model-based approaches is that they enable inference for all coalescence times, whereas the current model-free approaches allow only for inference about the TMRCA.
We propose using maximum likelihood (ML) to perform inference about all coalescence times. We show how the Markovian structure of the mutation process on a genealogy can be used to calculate the maximum-likelihood estimates (MLEs) of the coalescence times, together with profile likelihoods for these times (from which confidence intervals can be calculated). The Markovian structure of the model enables us construct a Viterbi-type algorithm (Viterbi 1967) to maximize the joint likelihood of the coalescence times in a sequential manner. This algorithm is based on a recursion similar to that of the peeling algorithm (Felsenstein 1981) for the calculation of the joint likelihood of the allele types at the internal nodes of a tree. Our method assumes that the underlying genealogy, together with the number of mutations on each branch, can be obtained from the sequence data (which is the case for data where repeat mutations are rare—for example, data from the human Y chromosome). Usually there will be some uncertainty concerning the order of certain coalescence events in the genealogy and, therefore, there will be some multifurcating parts in the tree topology (which are referred to as ambiguities).
The problem of estimating coalescence times is an example of a regular inference problem where each observation is viewed as coming from a separate site, and as such MLEs will be asymptotically efficient and confidence intervals will be exact, in the limit, as the sequence length increases. Our approach lies within the model-free class and as such will produce valid inferences regardless of the underlying demographic model. The approach can be used to estimate the times of all coalescences (and not just the TMRCA as with previous model-free approaches) and can also be used to analyze data where the region sequenced differs among chromosomes.
Although the idea of estimating coalescence times by maximum likelihood in population genetics appears new, a number of maximum-likelihood methods exist (Felsenstein 1981) for phylogenetic analysis of a sample of DNA sequences, which could be applied to the population genetics data that we consider. These methods are based on an iterative numerical maximization scheme for maximizing the likelihood function over the unknown parameters, which can include the tree topology, the branch lengths, and the parameters that govern the evolutionary process along the tree. Furthermore, they provide standard errors (SEs) of the MLEs on the basis of the information matrices. The methods are employed in standard software for phylogenetic analysis, such as PAUP* (Swofford 1998), PAML (Yang 1997), and PHYLIP (Felsenstein 1993).
In a simulation study comparing our new Viterbi ML approach and the ML approach implemented in PAML, we found that the MLEs obtained by the two methods are the same when the tree topology is specified, but they differ if the tree topology is only partially known. The results concerning the accuracy of the two approaches suggest that PAML has worse performance than our method in the latter case (see Table 1). Furthermore, our approach to calculating confidence intervals is different from that of PAML: we use the profile likelihood rather than estimating standard errors from the information matrix. Theoretical considerations suggest that using profile likelihoods tends to be more robust (Pawitan 2001), and we found that confidence intervals for the TMRCA based on the profile likelihood were more accurate in our simulation study.
TABLE 1.
Comparison with PAML
| Measure | 0 < t < 0.25 | 0.25 < t < 0.5 | 0.5 < t < 0.75 | 0.75 < t < 1 | TMRCA = 1 |
|---|---|---|---|---|---|
| Viterbi ML | |||||
| Relative error | 1.1120 | 0.4704 | 0.4173 | 0.2734 | 0.2317 |
| Relative bias | −0.1657 | −0.1043 | −0.1124 | −0.0900 | 0.0067 |
| Coverage | 0.8795 | 0.8878 | 0.8571 | 0.9286 | 0.9300 |
| PAML | |||||
| Relative error | 0.9422 | 0.4913 | 0.4450 | 0.3388 | 0.2762 |
| Relative bias | −0.2737 | −0.1921 | −0.2582 | −0.1527 | 0.1023 |
| Coverage | 0.6406 | 0.6939 | 0.5714 | 0.7857 | 0.7700 |
Measures of accuracy in intervals of scaled time for simulations under demographic scenario i, population of constant size, are shown. We set m = 20 and ℓ = 5 × 105 bp. Partially known tree topologies were used.
MATERIALS AND METHODS
Model and assumptions:
We study the ancestral relationships of a sample of m DNA sequences from a nonrecombining chromosomal region, for example, the nonrecombining DNA region of the human Y chromosome. These can be depicted by a genealogical tree, or genealogy, with the root of the tree representing the MRCA of the sample. For ease of exposition we assume that the root of the genealogy is known. This assumption is not necessary, and later on we give the implementation details about how our method can be applied to the case where the root is unknown. The genealogical tree for a sample of m sequences has m tips, or leaf nodes, labeled 1, … , m, and (m − 1) internal nodes, labeled m + 1, … , 2m − 1, which correspond to (m − 1) coalescence events having occurred at times t = (tm+1, … , t2m−1) in the past. The pairs of branches in the tree that coalesce specify the topology of the genealogical tree. In the tree there are 2(m − 1) branches connecting two nodes or a tip and a node. The branch lengths, which are denoted by b = (b1, … , b2(m−1)), correspond to the times between the respective coalescence events; i.e., each branch length represents the time between the coalescence event or tip at the tipward end of the branch and the coalescence at the rootward end.
Our method for inferring the coalescence times in a genealogical tree is based on modeling the mutation process, without modeling the genealogy or making any assumptions about the population parameters and the population size and structure.
The DNA sequence data are modeled by assuming that each site evolves independently of all others and mutations occur as events in a Poisson process with rate 1. Defining the rate of the mutation process is equivalent to specifying the units in which time is measured. If the probability of mutation in the DNA sequence per meiosis is u, the 1 unit of time is 1/u generations. We assume the existence of a constant molecular clock; that is, we assume that the mutation rate is constant along the branches of the genealogical tree and that evolution is independent along different lineages.
We assume that the mutation rate is sufficiently low and, therefore, repeat mutations are rare enough that each historical mutation can be inferred from the data. This will occur when the data fit the infinitely many-sites assumption, where no repeat mutations are allowed. In situations where the infinitely many-sites assumption is invalid, it may still be possible to infer the sites at which repeat mutations have occurred and the specific mutations that have occurred at those sites. This is particularly true if repeat mutations are rare (for example, on the Y chromosome).
The tree topology can be obtained from the segregating data (see Gusfield 1991), with some uncertainty concerning the order in which certain pairs of a group of branches coalesce on a given lineage. An example of a partially known tree topology is shown in Figure 1, where the exact order of coalescence events 12, 13, and 14 is not known. Such ambiguous parts in the tree arise in the case where there are no mutations on a branch that connects two internal nodes. The ambiguities in the genealogy are represented by multifurcating parts in the tree topology. The order of coalescence events on different lineages like, for example, Figure 1, events 9 and 10, does not cause any ambiguity in the topology of the tree, since the pair of branches involved in each coalescence is known.
Figure 1.

Partially known tree topology for a sample of eight sequences. Mutations are depicted by solid circles, and the uncertainty about the order of coalescence events 12, 13, and 14 is represented by a multifurcation in the tree topology.
Assume that there are no ambiguities in the tree. Let ni denote the number of mutations on the ith branch that has length bi, i = 1, … , 2(m − 1). The probability distribution of ni|bi is a Poisson distribution with rate bi. Therefore, the log-likelihood function for the “observed” data n = (n1, … , n2(m−1)) and the unobserved variables b = (b1, … , b2(m−1)), up to an additive constant, is given by
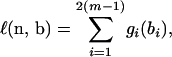 |
(1) |
where
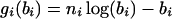 |
(2) |
is the contribution to the log-likelihood function of the data on the ith branch of the tree, and each bi is a function of at most two ti's. For maximum-likelihood estimation of coalescence times, the log-likelihood function in Equation 1 has to be maximized over b, subject to constraints that, according to the molecular clock hypothesis, the branch lengths from each internal node to all its descendant tips must be identical. If there are ambiguities over the tree topology, then this likelihood function needs to be maximized over all trees consistent with the partially known tree topology.
Sequential maximization of the log-likelihood:
Our aim is to maximize the log-likelihood function over the vector of branch lengths b to calculate the profile log-likelihoods and obtain MLEs and confidence intervals (C.I.'s) for the TMRCA and the coalescence times in the genealogy. The Markovian structure of the tree topology enables us to construct a Viterbi-type algorithm (Viterbi 1967) to maximize the log-likelihood in a sequential manner.
Initially we assume that the tree topology is known. The extension of the algorithm to the case of partially known tree topologies is detailed later. The first coalescence time is allowed to be arbitrarily close to zero, and the times of two subsequent coalescence events are allowed to be arbitrarily close to each other. For the construction of the Viterbi algorithm the Markovian structure of the genealogical tree is exploited: Given the numbers of mutations on each branch of the tree, the time at the ith node, i.e., the time of the ith coalescence event, depends only on the times at its immediately neighboring nodes.
Let fi(ti) be the logarithm of the joint probability of the coalescence time at the ith node of the genealogical tree and the numbers of mutations on the branches below the ith node, maximized over the coalescence times at and below its immediate descendant nodes. Let ℓi(ti) denote the profile log-likelihood of the coalescence time at the ith node. The Viterbi algorithm starts from pairs of tips that coalesce and in the forward part of the algorithm, going up the tree, recursively calculates the functions fi at subsequent nodes along different paths in the tree. At the final step of the recursion, the profile log-likelihood and the MLE of the TMRCA are obtained. Then, in the backward part of the algorithm, going down the tree, another recursion is built that calculates the profile log-likelihoods and the MLEs of the times of all coalescences.
The forward recursion:
The forward recursion of the algorithm (up the tree) is a procedure that sequentially maximizes the log-likelihood function in (1), starting from the tips and going toward the root of the tree. It is a recursion for calculating the function fi at an internal node i, i = m + 1, … , 2m − 1, given fj, fk at its immediate descendant nodes j and k, where each of the nodes j and k can be either a tip or an internal node of the tree (see Figure 2). Call the set of branches consisting of the branch connecting nodes i to j and all the branches below node j, 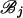 , and similarly for
, and similarly for 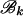 . Due to the independence of the mutation processes on
. Due to the independence of the mutation processes on 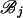 and
and 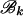 , conditional on ti, the function fi is given by the sum of two log-likelihood terms,
, conditional on ti, the function fi is given by the sum of two log-likelihood terms,
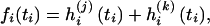 |
(3) |
where
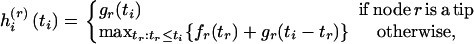 |
(4) |
for r = j, k. [Here hi(j)(ti) is the log-likelihood of the mutations on the branches in 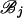 evaluated at ti and maximized over all other coalescence times.]
evaluated at ti and maximized over all other coalescence times.]
Figure 2.

A step of the sequential maximization of the log-likelihood. The coalescence of two internal nodes is shown. Simpler cases arise if one or both nodes j and k are tips.
Clearly, the procedure starts from several pairs of tips and follows different paths (lineages) up the tree. The exact order that the nodes in different lineages are processed does not matter; it is only required that before a node is processed all nodes below it are processed.
At the final step of the sequential procedure, the profile log-likelihood of the time t2m−1 at the root of the tree, up to an additive constant, is obtained as
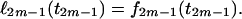 |
The maximum-likelihood estimate for the TMRCA will be the value of t2m−1 that maximizes ℓ2m−1(t2m−1). The function f2m−1 is convex and a unique maximum exists, and the MLE of the TMRCA is given by
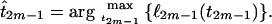 |
The backward recursion:
The MLEs of the coalescence times at the internal nodes of the tree can be obtained by tracing back the sequence of times that sequentially maximized the joint log-likelihood. Furthermore, starting from the root and going down the tree, we can calculate the profile log-likelihoods for the times at the internal nodes. If 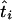 is the MLE of the coalescence time at node i, the MLE of the time at one of its immediate descendant nodes, j, is given by
is the MLE of the coalescence time at node i, the MLE of the time at one of its immediate descendant nodes, j, is given by
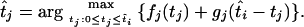 |
The first term on the right-hand side is the contribution to the log-likelihood from the number of mutations on all branches below node j, and the second term is the contribution to the log-likelihood from the branch above node j (conditional on ti, the number of mutations on all other branches in the tree is independent of tj).
The profile log-likelihood of tj is given (see appendix a) by the recursion
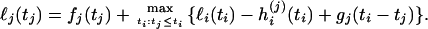 |
Calculating confidence intervals:
Marginal confidence intervals for a coalescence time can be obtained from its profile log-likelihood. Let ℓi* denote the maximum value of the profile log-likelihood of ti and define the deviance of ℓi(ti) from ℓi* by
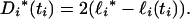 |
This deviance, evaluated at the true coalescent time, converges in distribution to the chi-square distribution with 1 d.f., denoted by 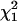 . Thus, an approximate 100(1 − α)% C.I. for ti will include all those values of ti for which the respective value of the deviance is less than the upper 100(1 − α)% point cα of the
. Thus, an approximate 100(1 − α)% C.I. for ti will include all those values of ti for which the respective value of the deviance is less than the upper 100(1 − α)% point cα of the 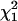 distribution, i.e., {ti : Di*(ti) ≤ cα}.
distribution, i.e., {ti : Di*(ti) ≤ cα}.
Dealing with ambiguities:
Ambiguous parts in the tree topology, for which the order of coalescence events on the same lineage is not known, demand a slightly more complicated procedure. One possibility would be to analyze all the subtrees arising from the different orderings of the coalescence times and maximize the likelihood over these topologies. However, this is a rather impracticable approach because the number of subtrees increases exponentially with the number of branches.
Figure 3a shows an example of an ambiguous region of a tree. The maximum-likelihood estimate of the topology for this region will be of the form shown in Figure 3b: Namely a pair of branches will coalesce first, and then all the remaining branches at the ambiguity will coalesce (in some order) to the parental branch of the branches at the ambiguity that have already coalesced. The reason for this is that this topology places no constraints on the intermediate coalescence times (tj for j = i − 4, … , i − 1 in Figure 3b), whereas any other topology would place some constraints on these times. It trivially follows that the unconstrained maximum will be larger.
Figure 3.

(a) Partially known tree topology with six branches involved. The exact order of the coalescence events at nodes i − 4, i − 3, i − 2, i − 1, and i is not known. (b) One possible topology that is consistent with the ambiguity.
It is possible to maximize over all such topologies by considering in turn all pairs of branches that coalesce first together with all possible branches that coalesce last. For simplicity we explain the method for the case of Figure 3; the extension to the general case is straightforward.
Denote by rj the immediate descendant node on the right of node j, j = i − k, … , i, where k = 4 in our example. For a given pair of branches that coalesce first, at time ti−k, and a branch that is involved in the last coalescence at time ti, introduce a function f*i(ti) that is given by
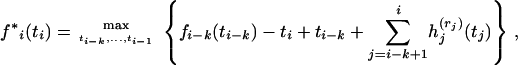 |
(5) |
where the functions 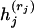 are defined in (4) and maximization over the coalescence times ti−k, … , ti−1 is subject to the constraints ti−k ≤ tj ≤ ti, j = i − k + 1, … , i − 1. This maximization allows for all possible orderings of intermediate coalescence times tj for j = i − k + 1, … , i − 1. The function fi(ti) is given by pointwise maximization of (5) over all possible pairs of branches that coalesce first and the branch that coalesces last.
are defined in (4) and maximization over the coalescence times ti−k, … , ti−1 is subject to the constraints ti−k ≤ tj ≤ ti, j = i − k + 1, … , i − 1. This maximization allows for all possible orderings of intermediate coalescence times tj for j = i − k + 1, … , i − 1. The function fi(ti) is given by pointwise maximization of (5) over all possible pairs of branches that coalesce first and the branch that coalesces last.
Implementation details:
We provide the implementation details of our method and we present some theoretical results that validate the sequential maximization of the log-likelihood function.
Time discretization:
To construct a Viterbi-type algorithm for maximum-likelihood estimation of coalescence times we need to discretize the continuous support of these variables. Each of the ti's will take values on a finite set of (T + 1) equally spaced points, i.e., S = {0, d, 2d, … , Td}, where d is the difference between any two subsequent points. Within the sequential procedure for maximizing the log-likelihood, the function hi(j)(ti) at each internal node i with immediate descendant node j is calculated by evaluating the function fj(tj) + gj(ti − tj) at a grid of values for ti and tj, with tj ≤ ti, to obtain a discrete approximation to the true continuous curve, and then maximizing over tj for each distinct value of ti. The computing time required for this procedure increases with the number of points used.
Sequences of different lengths:
The previous discussion was based on the assumption that the available chromosomal segment was the same for all individuals and, therefore, all the DNA sequences in the sample had the same length ℓ. However, in practice, the available sequences might be of different lengths, each of them consisting of a different set of nonoverlapping chromosomal segments.
Suppose that there are J different nonoverlapping chromosomal segments (for example, genes or combinations of genes) present in the sample. Let ℓj, μj, j = 1, … , J be the sequence length and the average mutation rate per site, respectively, for the jth segment and let uj = μjℓj be the (total) mutation rate for that segment. Each sequence in the sample will consist of a subset of those segments and its mutation rate u(i), i = 1, … , m will be given by the sum of the corresponding mutation rates. If two sequences of different lengths coalesce, then the length and the mutation rate of the ancestral sequence u(i), i = m + 1, … , 2m − 1 will be that of the union of the two sequences. The mutation rate for the sequence carried by the MRCA will be the total mutation rate for the chromosomal region consisting of all chromosomal segments; i.e., 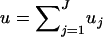 .
.
For example, Figure 5 shows the genealogical tree for a sample of human Y chromosomes where the DNA sequence sampled differs among individuals. Four nonoverlapping chromosomal segments are present in the sample, namely the four genes SMCY, DFFRY, DBY, and UTY1, which have different mutation rates. The branches in the tree are colored according to which genes are present in the sequence or ancestral sequence represented by that branch; red branches correspond to SMCY (the mutation rate on these branches is 6.11 × 10−5), blue branches correspond to the region consisting of DFFRY, DBY, and UTY1 (the mutation rate on these branches is 4.81 × 10−5), and black branches correspond to all four genes being present (the mutation rate on these branches is 1.092 × 10−4).
Figure 5.

The genealogical tree for a worldwide sample of 82 individuals from the human male population, for which a segment of the Y chromosome is sequenced. Each distinct sequence in the sample is represented at the tips of the tree by its multiplicity in the sample. Mutations are represented by black circles on the branches of the tree. The different colors of the branches correspond to sequences of different lengths: Black corresponds to the sequences consisting of the four genes; blue is for those consisting of the three genes DFFRY, DBY, and UTY1; red is for SMCY; and green is for UTY1. The MLEs of the coalescence times, in thousands of years, are given on the vertical timescale on the right of the tree.
Again we define time by assuming that if the probability of mutation in the whole chromosomal region is u, the 1 unit of time is 1/u generations. This amounts to assuming that mutations occur at the ith branch of the tree with relative rate u(i)/u, where u(i) is the mutation rate of the sequence at the bottom of the ith branch. Therefore, the distribution of ni|bi will be Poisson with rate u(i)bi/u. This affects the contribution to the log-likelihood of the data on each branch of the tree, which now becomes
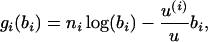 |
for i = 1, … , 2(m − 1). If 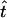 is the MLE for the time of an event in the past obtained by this generalized approach, then the respective MLE of the time in years is
is the MLE for the time of an event in the past obtained by this generalized approach, then the respective MLE of the time in years is 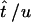 .
.
Unknown root:
Our method is also applicable for the estimation of coalescence times in genealogical trees where the root is assumed to be unknown. When the root of the genealogy is not known, we need to consider all the possible locations of the root that are consistent with the DNA sequence data, maximize the log-likelihood function for each of the corresponding trees, and then maximize over all the possible tree topologies arising from the different locations of the root.
In practice, we may search among the genealogies that are consistent with the data, starting from an arbitrarily chosen tree topology and then iteratively placing the root at neighboring positions. The arising genealogical trees will be analyzed using our method and the maximum-likelihood tree will be used for inference. Not all of the topologies that are consistent with the data need to be analyzed. The likelihood of a topology with the root placed near the initial position will often be greater than the likelihood of the starting topology; however, it starts reducing as the root is placed at more distant positions. Therefore, we may stop our search if the decrease of the likelihood, after placing the root at a more distant position, exceeds some prespecified value.
Monotonicity of search:
The problem we deal with is a regular-likelihood problem. In appendix b we prove that if i and r are two internal nodes in the tree and r is an immediate descendant of i, as the value of ti increases the value of tr that maximizes the function fr(tr) + gr(ti − tr) also increases. This result enables speeding up the computations needed for sequential maximization of the likelihood. If tr* is the value of tr that maximizes the function for a given ti = ti*, then for ti = ti* + d, only the values tr: tr* ≤ tr ≤ ti* + d need to be considered while searching for the new maximum.
RESULTS
Simulation study:
We consider the properties of the maximum-likelihood estimates of the TMRCA and the coalescence times obtained by using both our Viterbi-type algorithm and the iterative numerical maximization algorithm for phylogenetic analysis (implemented by PAML). For the TMRCA we also compare our method with the two existing model-free methods. The first (Thomson et al. 2000) provides a simple estimate of the TMRCA that is based on the numbers of mutational differences between each sequence in the sample and the MRCA, while the second (Tang et al. 2002) provides a more sophisticated model-free estimate that is based on a method-of-moments approach.
Our simulation study is based on data simulated under three different demographic scenarios: (i) population of constant effective size Ne; (ii) exponentially growing population of effective size at present Ne; and (iii) structured population of total effective size Ne, which consists of two equally sized subpopulations migrating to each other.
The data were simulated under the coalescent (Hudson 1991, 1992). We assumed that the root of the genealogies was known (this is often the case, as outgroup data can be used to infer the root). We assumed a finitely many-sites mutation model with two alleles per site and a small mutation rate per site per meiosis, μ = 10−9, similar to that of Y chromosome DNA data. In practice, repeat mutations were very rare in these simulations.
For our simulations we varied the sample size m and the length of the simulated sequences ℓ, which corresponds to varying the mutation rate. We measured the accuracy of our method and of the other approaches on the basis of the relative error and the bias of the point estimates and on the 95% coverage probability of the C.I.'s. If t is a quantity to be estimated, such as the sample TMRCA or a coalescence time, the relative error and bias of an estimate 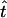 are defined as
are defined as
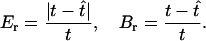 |
The relative error and bias represent the deviation of the estimate from the true value relative to the magnitude of the true value; these appear to be more appropriate measures of error and bias than the absolute error and usual bias in the simulation study as they avoid giving extra weight to simulated data sets with old TMRCAs.
To implement our method we needed to discretize time. We used d = 0.05, 0.1, depending on the values of m and ℓ, which provides a sufficiently good approximation to the log-likelihood curve.
The method of Tang et al. (2002), after partitioning the sample of sequences into two subsets that correspond to the two clades having diverged from the MRCA of the sample, estimates the TMRCA by using the average pairwise nucleotide differences between the two clades. In our simulation study we have used programs available from the authors to implement this method, using the partition that corresponds to the true root.
The iterative numerical maximization algorithm implemented by PAML performs ML analysis of nucleotide sequences under the assumption of some nucleotide substitution model. To make a fair comparison we adopted the simple Jukes and Cantor (1969) nucleotide substitution model in PAML, which assumes equal equilibrium frequencies for the four nucleotides and equal rates for all types of substitution.
Comparison with phylogenetic analysis by maximum likelihood:
We have considered simulation experiments with samples of size m = 20, mutation rate μ = 10−9, and sequence lengths ℓ = 5 × 105, 106, and 2 × 106 bp for scenario i, with effective population size Ne = 5000. For each different experiment our results are based on 100 simulated data sets.
The MLEs obtained by the two methods are the same when the tree topology is specified. However, when only a partially known tree topology can be inferred from the data (which is almost always the case in practice) the answers of the two methods are different. If a partially known tree topology is specified in PAML, it is assumed that all the branches involved in each ambiguity coalesce simultaneously. This leads to less accurate estimation of coalescence times and usually underestimation of the TMRCA.
Results concerning the estimation of the TMRCA and the coalescence times for a simulation experiment where the partially known topologies obtained by the sequence data were used can be found in Table 1. For each simulated genealogy we scaled time by setting the TMRCA to be 1. Then we divided time into disjoint intervals, each of which covers 25% of the unit of scaled time. The coalescences were grouped according to which interval the respective coalescence time fell in and the measures of accuracy were then calculated within each interval. It can be seen that our ML estimates are more accurate than those obtained by PAML in terms of the relative error. An alternative implementation of PAML is to not specify a tree and allow PAML to search for the best one. The results in this case are similar to those of Table 1. For the TMRCA the average relative error is 0.2752, the relative bias is 0.0808, and the coverage is 0.81.
The coverage properties of our C.I.'s are in all cases better than those of PAML's C.I.'s, especially in cases where the mutation rate is small. Table 2 shows results concerning the coverage of the C.I.'s for the TMRCA and for the other coalescence times averaged in intervals for different simulation experiments. In these experiments we assumed that the tree topologies were known, so both methods give the same MLEs. The C.I.'s of the two methods differ as our approach calculates C.I.'s on the basis of the profile likelihood while PAML use asymptotic normality of the MLEs and estimates the variance of the MLEs from the information matrix. It can be seen that our C.I.'s are better in terms of coverage than PAML's C.I.'s. As the mutation rate increases both methods become accurate and asymptotically identical. The likelihood-based C.I.'s are on average 96% of the length of the information-based C.I.'s. The difference in coverage of the C.I.'s between the two methods is even larger for these examples if the partially known trees are used, especially for the TMRCA and coalescence times close to the TMRCA (see, for example, Table 1).
TABLE 2.
Comparison with PAML
| ℓ (bp) | 0 < t < 0.25 | 0.25 < t < 0.5 | 0.5 < t < 0.75 | 0.75 < t < 1 | TMRCA = 1 |
|---|---|---|---|---|---|
| Viterbi ML | |||||
| 5 × 105 | 0.9395 | 0.9432 | 0.9157 | 0.9121 | 0.9200 |
| 106 | 0.9508 | 0.9326 | 0.9717 | 0.9191 | 0.9700 |
| 2 × 106 | 0.9203 | 0.9157 | 1.0000 | 0.9231 | 0.9600 |
| PAML | |||||
| 5 × 105 | 0.7014 | 0.8352 | 0.9043 | 0.9091 | 0.9000 |
| 106 | 0.7964 | 0.8764 | 0.9245 | 0.8485 | 0.9500 |
| 2 × 106 | 0.9124 | 0.8675 | 0.9744 | 0.9231 | 0.9600 |
Coverage in intervals of scaled time for simulations under demographic scenario i, population of constant size, is shown. We set m = 20 and we considered different values of ℓ. The tree topologies were assumed to be known.
Comparison with model-free methods:
We considered simulation experiments with samples of size m = 20, mutation rate μ = 10−9, and sequence lengths ℓ = 106, 2 × 106, and 3 × 106 bp for the three demographic scenarios, in accordance with what is known for the human Y chromosome. We chose the effective population size to be Ne = 5000. For scenario ii we used a model according to which the population halves backward in time in Ne generations (see Harding et al. 1997), and for scenario iii we assumed that each individual migrates (backward in time) independently at rate Ne/2, which is common across the two subpopulations. For scenario i we have also considered simulation experiments with m = 50 and ℓ = 106 and 2 × 106 bp and m = 100 and ℓ = 106 bp. For each different experiment our results are based on 500 simulated data sets. The CPU times per average run varied between 5 and 35 sec on a desktop PC.
The results of the comparative study can be found in Tables 3, 4, and 5 for the three simulation scenarios, respectively. In almost all cases our estimates are more accurate than those obtained by the existing methods in terms of the relative error, though the difference between our approach and the method of Tang et al. (2002) is small. All methods are, in general, unbiased, while their accuracy increases with ℓ. The C.I.'s obtained by the method of Thomson et al. (2000) are inaccurate and, being very wide, give high coverage probabilities. Both our method and the method of Tang et al. (2002) have coverage probabilities close to 95%. Our C.I.'s are on average 94% of the length of those of Tang et al. and 60% of those of Thomson et al.
TABLE 3.
Comparison with model-free methods
| m | ℓ (bp) | Measure | Viterbi ML | Tang et al. (2002) | Thomson et al. (2000) |
|---|---|---|---|---|---|
| 20 | 106 | Relative error | 0.1784 | 0.1841 | 0.1910 |
| Relative bias | 0.0071 | −0.0028 | 0.0110 | ||
| Coverage | 0.9360 | 0.9240 | 0.9960 | ||
| 20 | 2 × 106 | Relative error | 0.1111 | 0.1208 | 0.1293 |
| Relative bias | 0.0042 | −0.0068 | 0.0009 | ||
| Coverage | 0.9500 | 0.9540 | 0.9980 | ||
| 20 | 3 × 106 | Relative error | 0.1018 | 0.1081 | 0.1149 |
| Relative bias | −0.0006 | −0.0140 | −0.0097 | ||
| Coverage | 0.9440 | 0.9480 | 0.9960 | ||
| 50 | 106 | Relative error | 0.1674 | 0.1736 | 0.1898 |
| Relative bias | 0.0095 | −0.0172 | −0.0094 | ||
| Coverage | 0.9220 | 0.9120 | 0.9940 | ||
| 50 | 2 × 106 | Relative error | 0.1179 | 0.1214 | 0.1335 |
| Relative bias | −0.0077 | −0.0128 | −0.0098 | ||
| Coverage | 0.9500 | 0.9540 | 0.9980 | ||
| 100 | 106 | Relative error | 0.1696 | 0.1750 | 0.1895 |
| Relative bias | 0.0273 | 0.0056 | 0.0159 | ||
| Coverage | 0.9277 | 0.9404 | 0.9894 |
Measures of accuracy for simulations under demographic scenario i, population of constant size, are shown.
TABLE 4.
Comparison with model-free methods
| m | ℓ (bp) | Measure | Viterbi ML | Tang et al. (2002) | Thomson et al. (2000) |
|---|---|---|---|---|---|
| 20 | 106 | Relative error | 0.2090 | 0.2034 | 0.2197 |
| Relative bias | 0.0235 | −0.0007 | 0.0106 | ||
| Coverage | 0.9380 | 0.9340 | 0.9800 | ||
| 20 | 2 × 106 | Relative error | 0.1499 | 0.1547 | 0.1636 |
| Relative bias | 0.0008 | −0.0067 | 0.0045 | ||
| Coverage | 0.9280 | 0.9400 | 1.000 | ||
| 20 | 3 × 106 | Relative error | 0.1173 | 0.1222 | 0.1285 |
| Relative bias | 0.0012 | −0.0105 | −0.0099 | ||
| Coverage | 0.9320 | 0.9540 | 0.9960 |
Measures of accuracy for simulations under demographic scenario ii, exponentially growing population, are shown.
TABLE 5.
Comparison with model-free methods
| m | ℓ (bp) | Measure | Viterbi ML | Tang et al. (2002) | Thomson et al. (2000) |
|---|---|---|---|---|---|
| 20 | 106 | Relative error | 0.1369 | 0.1400 | 0.1555 |
| Relative bias | 0.0050 | −0.0018 | 0.0101 | ||
| Coverage | 0.9440 | 0.9500 | 0.9860 | ||
| 20 | 2 × 106 | Relative error | 0.1025 | 0.1064 | 0.1101 |
| Relative bias | 0.0031 | 0.0036 | 0.0014 | ||
| Coverage | 0.9400 | 0.9480 | 0.9960 | ||
| 20 | 3 × 106 | Relative error | 0.0810 | 0.0846 | 0.0906 |
| Relative bias | 0.0074 | 0.0071 | 0.0053 | ||
| Coverage | 0.9418 | 0.9398 | 0.9940 |
Measures of accuracy for simulations under demographic scenario iii, structured population, are shown.
Coalescence times:
The existing model-free methods are not able to estimate the coalescence times at the internal nodes of the genealogy. We conducted a simulation study to assess the performance of our method for estimating the coalescence times. We considered three simulation experiments with samples of size m = 40, one for each of the scenarios described in the previous paragraph. The mutation rates were chosen so that the average number of mutations was equal to 50 for all scenarios. Again 500 genealogies were simulated for each scenario. The CPU times per average run were: 7 sec for scenario i, 14 sec for scenario ii, and 8 sec for scenario iii.
The accuracy of the estimate of a coalescence time depends on the relative age of the coalescence with respect to the TMRCA of the sample. For each simulated genealogy we scaled time by setting the TMRCA to be 1 and we divided time into disjoint intervals, each of which covers 5% of the unit of scaled time. The coalescences were again grouped according to which interval the respective coalescence time fell in and the measures of accuracy were calculated within each interval. Our method estimates accurately the coalescence times that are closer to the TMRCA while it estimates poorly the ones that are closer to the present. The results are shown in Figure 4.
Figure 4.

Measures of accuracy in intervals for the simulation experiments for scenarios i, population of constant size; ii, exponentially growing population; and iii, structured population. Estimated (a) relative errors, (b) bias, and (c) coverage probabilities are shown. The straight line in subplot b corresponds to zero bias, while the one in subplot c corresponds to 95% coverage. The vertical error bars in subplots b and c represent 95% C.I.'s for the corresponding quantities, while the dotted lines are smoothed versions of the respective estimates.
Application I: Y chromosome DNA data:
We consider a data set consisting of a worldwide sample of 82 DNA sequences from the human Y chromosome. These data were taken from Shen et al. (2000) and they consist of four genes (SMCY, DFFRY, DBY, and UTY1) of the nonrecombining Y chromosome region. Not all of the four genes are sequenced for all individuals, and thus the available sequences are of different lengths: The length of the SMCY gene is 39,931 bp, whereas that of the segment consisting of DFFRY, DBY, and UTY1 is 38,468 bp. Ninety-eight segregating sites are in the sample and the estimate by Shen et al. (2000) of the mutation rate per site for SMCY was 1.53 × 10−9, whereas the estimate of the mutation rate for the region consisting of DFFRY, DBY, and UTY1 was 1.25 × 10−9.
The genealogical tree obtained for these data is shown in Figure 5. Apart from the MRCA, two important mutations are indicated in the tree. Mutation 1 defines a clade separate from the deep African lineages, while mutation 2 corresponds to a hypothetical bottleneck before the global expansion (see Thomson et al. 2000).
A subset of these data was previously analyzed by Thomson et al. (2000), using genetree under the assumption of constant population and of exponential growth, as well as using their simple model-free estimator for the TMRCA. In their study they considered only the sequences for which the chromosomal region consisting of the genes SMCY, DFFRY, and DBY was available. For reasons of comparison, we analyzed the same reduced data set consisting of 43 sequences, with 55 segregating sites present in the sample. We also used the method of Tang et al. (2002) to estimate the TMRCA of this sample. We report our results together with the previous results for the TMRCA in Table 6 and those for the ages of mutations in Table 7. The ages of mutations 1 and 2 were estimated by Thomson et al. (2000) using genetree, under the assumption of constant population size and also using the number of segregating sites within the sequences that contained each mutation (Griffiths and Tavaré 1998). Our estimates are for the coalescence times and hence the numbers in Table 7 are not directly comparable.
TABLE 6.
Point estimates and C.I.s for the TMRCA of the reduced sample
| Method | TMRCA (yr) | C.I. |
|---|---|---|
| Maximum likelihood | 63,000 | (40,000–100,000) |
| Tang et al. (2002) | 63,000 | (29,000–98,000) |
| Thomson et al. (2000) | 70,000 | (10,000–130,000) |
| genetree (constant population) | 84,000 | (55,000–149,000) |
| genetree (exponential growth) | 59,000 | (40,000–140,000) |
TABLE 7.
Point estimates and C.I.s for the ages of coalescences (mutations) 1 and 2 for the reduced sample
| Method | T1 (yr) | C.I. | T2 (yr) | C.I. |
|---|---|---|---|---|
| Maximum likelihood | 45,000 | (32,000–74,000) | 37,000 | (27,000–67,000) |
| genetree | 47,000 | (35,000–89,000) | 40,000 | (31,000–79,000) |
| Segregating sites | 43,000 | (37,000–111,000) | 42,000 | (36,000–109,000) |
We also analyzed the full data by using our Viterbi ML method. Ninety-eight segregating sites were present in the full sample. The analysis of these data using our ML program took ∼10 min. The results are given in Table 8. The estimates obtained by analyzing the full data are smaller than those obtained by analyzing the reduced data set. The extra information contained in this larger data set has reduced the width of the confidence intervals by ∼50%.
TABLE 8.
MLEs and likelihood-based C.I.s for the TMRCA and the coalescence times 1 and 2 for the full data
| MLE (yr) | C.I. | |
|---|---|---|
| TMRCA | 56,000 | (39,000–83,000) |
| T1 | 36,000 | (27,000–50,000) |
| T2 | 32,000 | (23,000–41,000) |
Application II: fungus DNA data:
In this application we consider the fungal data set of Carbone and Kohn (2001). This data set consists of DNA data from multiple nuclear loci in 385 isolates of the haploid, plant parasitic ascomycete fungus Sclerotinia. Two genealogies were constructed from the nonrecombinant haplotypes in these data: one that describes the ancestry of the population Sclerotinia sclerotiorum (Figure 6) and another that describes the ancestry of the three different species present in the data, namely S. sclerotiorum, S. minor, and S. trifoliorum (Figure 7). There were three sampling areas for these species: temperate, subtropical, and wild. Nested analysis of the population S. sclerotiorum has shown that it was subdivided into nine subpopulations. For further details of the data see Carbone and Kohn (2001).
Figure 6.

The genealogical tree for a sample from the population S. sclerotiorum. The 19 distinct haplogroups in the sample are represented at the tips of the tree by their multiplicities in the sample. Mutations are represented by solid circles on the branches of the tree. The MLEs of the coalescence times, in units of Ne generations, are given on the vertical timescale on the right of the tree. The Carbone and Kohn estimates are also given in parentheses and substantially different estimates are marked with asterisks. Likelihood-based C.I.'s for the oldest coalescence times are given in brackets below the respective point estimates.
Figure 7.

The genealogical tree for a sample from the three species S. sclerotiorum, S. minor, and S. trifoliorum. The 11 distinct haplogroups in the sample are represented at the tips of the tree by their multiplicities in the sample. Mutations are represented by solid circles on the branches of the tree. The MLEs of the coalescence times, in units of Ne generations, are given on the vertical timescale on the right of the tree. The Carbone and Kohn estimates are also given in parentheses. Likelihood-based C.I.'s for the oldest coalescence times are given in brackets below the respective point estimates.
Both genealogies have been analyzed by Carbone and Kohn, using a combination of parsimony, maximum-likelihood, and coalescent methods to distinguish divergence times of populations and species. Their analysis on the population scale was based on a coalescent model, assuming no recombination and selection, which included migration among the nine subpopulations. We analyzed the genealogical tree of Figure 6, using our ML method. The analysis took 70 sec. To obtain estimates in units of Ne generations (to be comparable with the estimates in Carbone and Kohn 2001) we scaled our estimates by a factor of u = θ/2, using the estimate of the mutation parameter θ = 9.9 from Carbone and Kohn (2001).
Our estimates are shown in Figure 6 (the vertical timescale on the right), where we also give the Carbone and Kohn estimates in parentheses. Substantial differences between the two estimates of some coalescence times were observed (see Figure 6). The large estimates of Carbone and Kohn are due to the fact that the respective coalescent events involve multiple sequences from different subpopulations that migrate to each other. Under the assumptions of a specific migration model and of constant migration rate the estimated coalescence times are pushed to be higher. Their analysis is based on assuming a specific migration model and that the inferred migration rates are exact. If those assumptions are not valid the resulting estimates will be inaccurate.
For example, the estimate of the coalescence time marked with two asterisks in Figure 6 is surprisingly large. The length of the branch below that node is 8% of the total length of the tree, with only one mutation having occurred on this part of the tree. The expected number of mutations according to the Poisson mutation process is 5. The P-value for observing one or fewer mutations on this part of the tree is 0.04.
The coalescent analysis of Carbone and Kohn (2001) on the species scale was based on an estimated migration matrix for the three sampling areas. Our estimates, in units of Ne generations, scaled according to the estimate θ = 11.8 from Carbone and Kohn (2001), are shown in Figure 7 (the vertical timescale on the right), where we also give the Carbone and Kohn estimates in parentheses. The analysis of this tree took 45 sec. This genealogy has also been analyzed by DeIorio and Griffiths (2004) under a coalescent model with migration, using an importance-sampling technique for computing the likelihood of samples of genes in subdivided populations. Their estimate of the TMRCA was 3.0 coalescent units.
DISCUSSION
In this article we have developed a method for maximum-likelihood estimation of coalescence times in genealogical trees, based on the Viterbi algorithm. Inference is based on a sample of DNA sequences from a population and it is assumed that the genealogy of the sample and the numbers of mutations on the branches of the tree can be obtained from these data. We have used a model-free approach to estimating coalescence times in a genealogy, assuming a Poisson mutation process and no recombination, and we have shown how the Markovian structure of the mutation process on the genealogy can be exploited to calculate MLEs and C.I.'s on the basis of the profile likelihoods of the coalescence times.
Our approach to calculating MLEs appears to be more accurate than other numerical maximization approaches in the cases where only partially known tree topologies can be inferred from the data. Furthermore, our Viterbi ML approach enables confidence intervals to be obtained from profile likelihoods rather than from approximate standard errors estimated from the information matrix. Both the theoretical considerations (Pawitan 2001) and the results presented here suggest that confidence intervals calculated from the profile likelihood will be more accurate.
Our method has the advantage of the optimal asymptotic properties (as the sequence length increases) of maximum-likelihood estimation and in simulation studies was shown to give slight improvements over the method of Tang et al. (2002). However, the main advantages of our method over existing model-free approaches are that it can naturally cope with data where different chromosomes have been sampled in different regions and that it provides estimates and confidence intervals for all coalescent times, not just for the TMRCA.
The limitation of our method is that it assumes that the genealogy is known together with the number of mutations on each branch. Such data arise under an infinite-sites mutation model for nonrecombining regions. In the analysis of the fungus DNA data, our method was applied to a recombining region by first removing recombinant haplotypes (which is a common approach to such data, see Harding et al. 1997; Carbone and Kohn 2001).
Acknowledgments
This work was supported by Engineering and Physical Sciences Research Council grant GR/R91724.
APPENDIX A: PROFILE LOG-LIKELIHOODS OF COALESCENCE TIMES
The profile log-likelihood of tj, ℓj, is the log-likelihood function of the vector of coalescence times maximized over the times at all the nodes in the tree except for the jth. This is obtained by a recursion. If ℓi is the profile log-likelihood of the time at node i, which is the parent of node j, then the function ℓi(ti) − hi(j)(ti) + gj(ti − tj), maximized over ti, is the contribution to ℓj from all the branches in the tree that are not parts of the subtree below node j. The contribution to ℓj from this subtree is simply the function fj(tj), that is, the log-likelihood up to node j maximized over the times at the nodes below node j. Therefore,
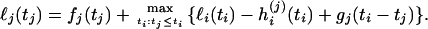 |
APPENDIX B: MONOTONICITY OF MLES OF COALESCENCE TIMES
We show that in (4),
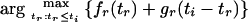 |
is a monotonically increasing function of ti.
Theorem 1: Let gr, fr be as defined in (2) and (3), respectively. If i and r are two internal nodes in the tree and r is an immediate descendant of i, as the value of ti increases the value of tr that maximizes the function fr(tr) + gr(ti − tr) also increases.
Proof: Let
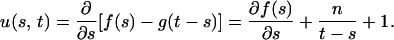 |
For fixed s, the partial derivative of u(s, t) with respect to t is
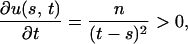 |
so that u(s, t) is monotonically increasing with respect to t. For fixed t, the partial derivative of u(s, t) with respect to s is
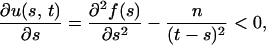 |
so that u(s, t) is monotonically decreasing with respect to s.
For a given value of t, let s* be the value of s that maximizes f(s) − g(t − s), that is, u(s*, t) = 0. Since u(s, t) is monotonically increasing with respect to t, if t′ > t, then u(s*, t′) > u(s*, t) = 0. For fixed t = t′, s′* > s* exists such that u(s′*, t′) = 0 < u(s*, t′), since u(s, t) is monotonically decreasing with respect to s, where s′* is the unique value of s that maximizes f(s) − g(t′ − s).
References
- Carbone, I., and L. M. Kohn, 2001. A microbial population-species interface: nested cladistic and coalescent inference with multilocus data. Mol. Ecol. 10: 947–964. [DOI] [PubMed] [Google Scholar]
- DeIorio, M., and R. C. Griffiths, 2004. Importance sampling on coalescent histories. ii: subdivided population models. Adv. Appl. Probab. 36: 434–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein, J., 1981. Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17: 368–376. [DOI] [PubMed] [Google Scholar]
- Felsenstein, J., 1993. Phylogenetic Inference Package (PHYLIP). University of Washington, Seattle.
- Griffiths, R. C., and S. Tavaré, 1994. Ancestral inference in population genetics. Stat. Sci. 9: 307–319. [Google Scholar]
- Griffiths, R. C., and S. Tavaré, 1998. The age of a mutation in a general coalescent tree. Stoch. Models 14: 273–295. [Google Scholar]
- Gusfield, D., 1991. Efficient algorithms for inferring evolutionary trees. Networks 21: 19–28. [Google Scholar]
- Harding, R. M., S. M. Fullerton, R. C. Griffiths, J. Bond, M. J. Cox et al., 1997. Archaic African and Asian lineages in the genetic ancestry of modern humans. Am. J. Hum. Genet. 60: 772–789. [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., 1991. Gene genealogies and the coalescent process, pp. 1–44 in Oxford Surveys in Evolutionary Biology, Vol. 7, edited by D. Futuyma and J. Antonovics. Oxford University Press, London/New York/Oxford.
- Hudson, R. R., 1992. Gene trees, species trees and the segregation of ancestral alleles. Genetics 131: 509–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jukes, T. H., and C. R. Cantor, 1969. Evolution of protein molecules, pp. 21–132 in Mammalian Protein Metabolism, edited by H. N. Munro. Academic Press, New York.
- Kingman, J. F. C., 1982. The coalescent. Stoch. Proc. Appl. 13: 235–248. [Google Scholar]
- Nordborg, M., 2001. Handbook of Statistical Genetics, pp. 179–212. John Wiley & Sons, London.
- Pawitan, Y., 2001. In All Likelihood Statistical Modelling and Inference Using Likelihood. Oxford University Press, Oxford.
- Pritchard, J. K., M. T. Seielstad, A. Perez-Lezaun and M. W. Feldman, 1999. Population growth of human Y chromosomes: a study of Y chromosome microsatellites. Mol. Biol. Evol. 16: 1791–1798. [DOI] [PubMed] [Google Scholar]
- Shen, P., F. Wang, P. A. Underhill, C. Franco, W. Yang et al., 2000. Population genetic implications from sequence variation in four y chromosome genes. Proc. Natl. Acad. Sci. USA 97: 7354–7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf, M., and D. B. Goldstein, 2001. Genealogical and evolutionary inference with the human Y chromosome. Science 291: 1738–1742. [DOI] [PubMed] [Google Scholar]
- Swofford, D. L., 1998. PAUP*: Phylogenetic Analysis Using Parsimony and Other Methods. Sinauer Associates, Sunderland, MA.
- Tang, H., D. O. Siegmund, P. Shen, P. J. Oefner and M. W. Feldman, 2002. Frequentist estimation of coalescence times from nucleotide sequence data using a tree-based partition. Genetics 161: 447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavaré, S., D. J. Balding, R. C. Griffiths and P. Donnelly, 1997. Inferring coalescent times from DNA sequence data. Genetics 145: 505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Templeton, A. A., 1993. The “Eve” hypothesis: a genetic critique and reanalysis. Am. Anthropol. 95: 51–72. [Google Scholar]
- Thomson, R., J. K. Pritchard, P. Shen, P. J. Oefner and M. W. Feldman, 2000. Recent common ancestry of human Y chromosomes: evidence from DNA sequence data. Proc. Natl. Acad. Sci. USA 97: 7360–7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viterbi, A. J., 1967. Error bounds for convolutional codes and an asymptotically optimal decoding algorithm. IEEE Trans. Inf. Theory 13: 260–269. [Google Scholar]
- Yang, Z., 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13: 555–556 (http://abacus.gene.ucl.ac.uk/software/paml.html). [DOI] [PubMed] [Google Scholar]