

Abstract
Likelihood-based parentage inference depends on the distribution of a likelihood-ratio statistic, which, in most cases of interest, cannot be exactly determined, but only approximated by Monte Carlo simulation. We provide importance-sampling algorithms for efficiently approximating very small tail probabilities in the distribution of the likelihood-ratio statistic. These importance-sampling methods allow the estimation of small false-positive rates and hence permit likelihood-based inference of parentage in large studies involving a great number of potential parents and many potential offspring. We investigate the performance of these importance-sampling algorithms in the context of parentage inference using single-nucleotide polymorphism (SNP) data and find that they may accelerate the computation of tail probabilities >1 millionfold. We subsequently use the importance-sampling algorithms to calculate the power available with SNPs for large-scale parentage studies, paying particular attention to the effect of genotyping errors and the occurrence of related individuals among the members of the putative mother–father–offspring trios. These simulations show that 60–100 SNPs may allow accurate pedigree reconstruction, even in situations involving thousands of potential mothers, fathers, and offspring. In addition, we compare the power of exclusion-based parentage inference to that of the likelihood-based method. Likelihood-based inference is much more powerful under many conditions; exclusion-based inference would require 40% more SNP loci to achieve the same accuracy as the likelihood-based approach in one common scenario. Our results demonstrate that SNPs are a powerful tool for parentage inference in large managed and/or natural populations.
GENETIC markers have been used to infer parentage in applications across a range of fields from anthropology and ecology to forensics and law. Today the molecular markers of choice for parentage inference are highly polymorphic, repetitive loci such as the short tandem repeat loci commonly employed in human parentage testing (Hammond et al. 1994) and the microsatellites used in the field of molecular ecology (Queller et al. 1993). In contrast, single-nucleotide polymorphisms (SNPs) have not been widely employed for parentage inference and other forms of relationship estimation, because, possessing only two alleles, each SNP has lower resolving power per locus than most microsatellites (Glaubitz et al. 2003). However, SNPs have a number of features making them appropriate for large-scale genetic studies: they are abundant in most genomes surveyed (Brumfield et al. 2003); genotyping error rates are low (Ranade et al. 2001); scoring SNP genotypes requires minimal human interaction, making them amenable to high-throughput, low-cost genotyping; and SNP genotypes are easily standardized across laboratories. Indeed, because of these attractive features, SNPs have recently been employed for individual identification and paternity inference in large herds of cattle (Heaton et al. 2002; Werner et al. 2004) and for human forensic purposes (Lee et al. 2005).
Recently, several articles have reported specifically on the utility of SNPs for parentage inference, either for inferring parent–offspring pairs (Glaubitz et al. 2003) or for inferring paternity given an offspring, a known mother, and a candidate father (Krawczak 1999; Gill 2001). All of these studies measured the power for parentage inference in terms of the probability of exclusion (PE) (Chakraborty et al. 1988). The PE is an appropriate measure of power if the actual inference is done on the basis of exclusion—that is, if candidate parents are to be eliminated from consideration because of Mendelian incompatibilities with the candidate offspring. Although the method of exclusion is widely used, it has several important limitations. First, it uses only a portion of the information available in the data, and second, the method of exclusion is not easily adjusted to account for genotyping error. A more powerful method of parentage inference was introduced by Thompson (1976). This method, based on a likelihood-ratio statistic, is easily extended to allow for the possibility of genotyping error and has been implemented in various categorical assignment methods and computer programs (Meagher and Thompson 1986; SanCristobal and Chevalet 1997; Marshall et al. 1998; Gerber et al. 2000; Duchesne et al. 2002). For a recent review of the different approaches and methods available for inference of parentage, see Jones and Ardren (2003).
The level of confidence in inference from likelihood-based methods depends on the distribution of Λ, the log-likelihood ratio statistic, given the allele frequencies and other details of the sampling design. In general, the distribution of Λ is not analytically tractable, so its distribution is approximated by Monte Carlo simulation; however, none of the currently available Monte Carlo methods are suitable for estimating expected error rates in parentage studies involving large numbers of potential parents and offspring. This is a consequence of the large number of possible trios (putative mother, putative father, and putative offspring) that must be investigated in large studies. There can potentially be billions of trios in such a study, and therefore per-trio false-positive rates on the order of one in 1 billion are relevant. Standard, “naive” Monte Carlo estimates of such small probabilities are inaccurate and computationally impractical.
In this article, we develop importance-sampling methods (Hammersley and Handscomb 1964) that allow very small tail probabilities in the distribution of Λ to be estimated accurately and rapidly. These methods make likelihood-based inference in large-scale studies practical. We then assess the power of SNPs for likelihood-based parentage inference from trios. The results are presented in terms of per-trio false-positive and per-trio false-negative rates, from which it is straightforward to estimate expected studywide error rates (i.e., the expected absolute number of misassignments of offspring to parents). In addition, we incorporate the effects of genotyping error in the method by accounting for this error in both the likelihood-ratio and the Monte Carlo simulations, and we consider a wide range of possible nonparental relationships that can occur among three individuals. Our main interests are in situations where neither the true mother nor the true father of the child is known. However, cases where the true mother (or father) is known are handled easily within the same framework, and results are presented for such cases. Ultimately we show that with only a moderate number—∼60–100—of SNPs, enough power is achieved to accurately infer parentage in quite large populations.
In methods we describe likelihood inference of parentage and the importance-sampling methods. In results we provide calculations of per-trio error rates for a variety of different trio relationship categories, and we show that likelihood-based methods are more powerful than exclusion-based methods. We then show how such calculations can be used to provide expected studywide error rates, using a scenario from a hypothetical salmon population. In the discussion, we address several issues relevant to large-scale parentage studies with SNPs. Most importantly we point out that, for many scenarios, genetic linkage per se is not a great concern, although linkage disequilibrium between SNP markers can reduce power for parentage inference.
METHODS
The use of likelihood to infer relationships between individuals was proposed by Edwards (1967). Thompson (1976) developed likelihood-based methods for reconstructing human pedigrees by testing for parental relationships. While Thompson (1976) reported on methods for simultaneously inferring the parentage of large sibships, we focus on the inference of parentage in what Thompson refers to as “Q-triplets”—trios of individuals consisting of a putative offspring, a putative mother, and a putative father. We denote these three individuals by y, m, and f, respectively. Our methods section is organized as follows. First, we present some notation for SNP markers and briefly review likelihood inference of parentage. Then, we show how false-positive and false-negative rates can be computed. And finally, we present an efficient Monte Carlo method for estimating probabilities of incorrect parentage assignment.
We assume that the genetic data consist of L SNP markers. There are typically only two states (alleles) that each SNP locus takes in a population. For example, at one locus the two alleles may be A and G; at a different locus the two alleles may be C and A, and so forth. Instead of using the letter names of the DNA bases at each locus, we use 0 to denote the minor allele—the one at lowest frequency in the population—and 1 to denote the allele at higher frequency. We let q denote the frequency of the minor allele and p = 1 − q the frequency of the other allele. Since there are only two alleles, there are only three diploid genotypes possible at each locus. We name these genotypes according to the number of 1 alleles that they contain. Hence a genotype of 0 is homozygous for the 0 allele, a genotype of 1 is a heterozygote, and a genotype of 2 is homozygous for the 1 allele. The frequencies of these genotypes in the population, assuming Hardy–Weinberg equilibrium, are q2, 2pq, and p2, respectively. Throughout most of this article, we assume that the SNP markers are unlinked and are not in linkage disequilibrium (LD), but we briefly consider the effects of linkage in the discussion.
Parent–pair likelihood inference:
For a single trio of individuals, inferring whether m and f are both parents of y, an event that we denote by Q, or whether m, f, and y are three entirely unrelated individuals (U) is principally done using the log-likelihood-ratio statistic:
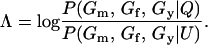 |
(1) |
P(Gm, Gf, Gy | Q) is the probability of the observed genotypes of m, f, and y at L loci under the assumption that m and f are the parents of y. P(Gm, Gf, Gy | U) is the probability of the observed genotypes under the assumption that m, f, and y are mutually unrelated individuals.
The above formulation of Thompson (1976) does not account for genotyping error, which can be problematic. For example, a single genotyping error among a true parental trio could lead to P(Gm, Gf, Gy | Q) being zero, which would make Λ = −∞, and one would wrongly reject the possibility that m and f were the parents of y. If independent estimates of the genotyping rates are known (or can be assumed), the analysis can be done conditional on those known (assumed) genotyping error rates. We assume that genotyping errors occur at a rate of μℓ per gene copy, and that they are independent between gene copies at a locus and between loci. This is a two-allele case of the error model considered by SanCristobal and Chevalet (1997). Because there are only two allelic types, the genotyping error model can be made simple and realistic—a genotyping error means that a 0 allele is observed as a 1 allele or that a 1 allele is observed as a 0 allele—without incurring a large computational burden as with markers having multiple alleles (Sieberts et al. 2002). It is possible that genotyping errors at the two gene copies at a locus would not occur independently of one another. In such a case the error model would have to be altered, which would not be difficult.
Assuming a genotyping error rate μℓ > 0 at the ℓth locus, the calculation of Λ is much like that in (1), except that P(Gm, Gf, Gy | Q) and P(Gm, Gf, Gy | U) are replaced by P(Gm, Gf, Gy | Q, μ) and P(Gm, Gf, Gy | U, μ), respectively, the latter two being probabilities computed conditional on the locus-specific genotyping error rates μ = (μ1,…, μL). Calculation of P(Gm, Gf, Gy | Q, μ) and P(Gm, Gf, Gy | U, μ) is straightforward. Details appear in the appendix.
In parentage inference, as typically pursued, if the statistic Λ is greater than some threshold value, Λc, then the trio is declared to be of type Q. If the trios being tested are all of either type Q or type U, then rejecting hypothesis U is equivalent to accepting hypothesis Q, and parentage inference of this sort is formally a hypothesis test. If the genotyping error rate is known exactly, then by the Neyman–Pearson lemma (Neyman and Pearson 1933), a test based on Λ is the most powerful test available (as noted by SanCristobal and Chevalet 1997). In other words, among all possible parentage tests, a test using the statistic in (1) will have the smallest type II error rate for any chosen type I error rate. The Neyman–Pearson lemma provides a theoretical justification for what has been noted by previous authors (Marshall et al. 1998) and is demonstrated later in this article—that likelihood-based methods can be more powerful than those based on parental exclusion.
If the trios being tested have some relationship other than Q or U, however, then such an analysis is not formally a hypothesis test, and the Neyman–Pearson lemma no longer applies. This means that without knowing the pattern of relatedness of the members of the trio—which is typically unknown—it is not possible to design a most powerful test for parentage. Nonetheless, the test statistic Λ, with the likelihood of U in the denominator, is still a reasonable choice of test statistic, even when the true relationship of the trio may be something other than Q or U (Meagher and Thompson 1986).
We express the power for trio-based parentage assignment in terms of false-positive and false-negative error rates. These are analogs of type I and type II errors in hypothesis testing. A false-positive error occurs when we declare a trio to be of type Q when, in fact, it is not. A false-negative error occurs when we declare a trio to not be of type Q when, in fact, it is. The probability α of a false positive depends on the allele frequencies q = (q1,…, qℓ), the genotyping error rates μ, the true relationship, T ≠ Q, of the individuals within the trio, and the chosen value of Λc. It is the probability that a trio of type T yields Λ > Λc, which can be written as the expected value
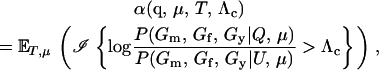 |
(2) |
where 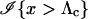 is the indicator function that takes the value 1 if x > Λc and 0 otherwise, and the subscript “T, μ” signifies that the expectation over all values of (Gm, Gf, Gy) is taken conditional on the trio being of type T and the genotyping error rates of the loci being μ. The probability of a false-negative error is the probability that a trio of type Q yields Λ < Λc, which is denoted by
is the indicator function that takes the value 1 if x > Λc and 0 otherwise, and the subscript “T, μ” signifies that the expectation over all values of (Gm, Gf, Gy) is taken conditional on the trio being of type T and the genotyping error rates of the loci being μ. The probability of a false-negative error is the probability that a trio of type Q yields Λ < Λc, which is denoted by
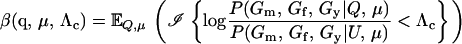 |
(3) |
The quantities α and β are per-trio error rates. β(q, μ, Λc), when multiplied by the number of parental trios compared in a study, gives the expected number of false negatives in the whole study. Likewise α(q, μ, T, Λc) multiplied by the number of type T trios for which Λ is evaluated gives the expected number of false positives involving trios of type T. If reasonable estimates (based, for example, on demography) can be made of the proportion of different trio types, T, in a sample, then a reasonable estimate of the total expected number of false positives and false negatives in the entire study (i.e., when all possible parent pairs are tested against all possible offspring) may be made. Being able to compute α and β makes it possible to choose a value of Λc that provides a suitable trade-off between false-positive and false-negative rates and to determine the number of loci necessary to reduce the total expected number of false positives and false negatives to an acceptable level.
Relatedness between putative parents and the youth:
There are many ways in which the putative parents m and f might be related to the youth y other than just as the true parents (Q) or as unrelated individuals (U). For example, in the forensics literature, there is considerable concern about situations in which a putative father is actually a brother or a cousin of the true father (e.g., Fung et al. 2002). Similarly, biologists have seen their attempts at pedigree reconstruction confounded by this “aunt and uncle effect” (Olsen et al. 2001). Because of age differences between parents and offspring in humans it is unlikely that a putative parent will actually be a sibling of the putative offspring. However, this situation may arise frequently in populations of plants and animals (Thompson and Meagher 1987), leading to difficulty in parentage assignment. Many studies have evaluated the effect of such relationships on parentage and paternity inference (Salmon and Brocteur 1978; Thompson and Meagher 1987; Goldgar and Thompson 1988; Double et al. 1997; Marshall et al. 1998; Heaton et al. 2002; Glaubitz et al. 2003; Sherman et al. 2004). Most of these have investigated the effect of relatedness on exclusion probabilities, and not on the distribution of Λ, or are concerned exclusively with paternity inference or the inference of parent–offspring pairs, and not with the analysis of general Q-triplets, thus ignoring many of the possible relationships among the members of a trio.
Here, we focus on 23 different relationships that cover many cases of interest. These 23 relationships are divided into eight basic types as shown in Figure 1. Figure 1a shows relationships in which the putative parents are related, via common descent from unobserved ancestors, to the true parents, M and F. The pairwise relationships between F and f, and M and m, are specified in terms of the coefficients κf and κm, respectively. In general, κ = (κ0, κ1, κ2), where κi is the probability that the pair shares i gene copies identical by descent at a locus and κ0 + κ1 + κ2 = 1 (Cotterman 1940; Thompson 1975). This type of trio relationship has been investigated in the context of parentage inference by Goldgar and Thompson (1988). We refer to these as C-type relationships. We consider 10 different variations of the C-type of relationship (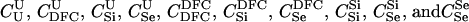 ) corresponding to four different degrees of pairwise relationship between F and f and M and m: self (Se), κ = (0, 0, 1); full-sibling (Si),
) corresponding to four different degrees of pairwise relationship between F and f and M and m: self (Se), κ = (0, 0, 1); full-sibling (Si), 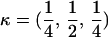 ; double first cousin (DFC),
; double first cousin (DFC), 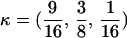 ; and unrelated (U), κ = (1, 0, 0). We denote these different C relationships using super- and subscripts for the relationships of f to F and of m to M, respectively. For example,
; and unrelated (U), κ = (1, 0, 0). We denote these different C relationships using super- and subscripts for the relationships of f to F and of m to M, respectively. For example,  denotes the trio in which f is a sibling of the true father (F) of y, and m is the true mother (M) of y. We follow the convention that, in a C-type relationship, m is always equally or more closely related to M as f is to F. Of course, for autosomal loci, the results are the same when the sex of the f and m individuals is reversed. Two special cases to note are
denotes the trio in which f is a sibling of the true father (F) of y, and m is the true mother (M) of y. We follow the convention that, in a C-type relationship, m is always equally or more closely related to M as f is to F. Of course, for autosomal loci, the results are the same when the sex of the f and m individuals is reversed. Two special cases to note are 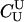 , which corresponds to an unrelated (U) trio, and
, which corresponds to an unrelated (U) trio, and 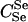 , which corresponds to a parental (Q) trio. The B-type relationships (Figure 1b) include cases in which one of the putative parents is a full-sibling of y and the other is not a descendant of M or F. The H-type relationships (Figure 1c) are similar to type B, except that one putative parent is a half sibling of y. We investigate four different variations of trio types B and H corresponding to the four different levels of κ investigated; for example, BSi denotes a trio in which the putative mother is a sibling of y and the putative father is a sibling of the true father. Once again, it should be noted that the fact that the mother is placed as the sibling of y in the B-type relationships or as the half-sibling of y in the H-type relationships is merely convention, and for an autosomal locus, the properties of the trio would be identical if the sexes of m and f were reversed. Finally, types D1–D5 (Figure 1, d–h) all include only a single case. They are situations in which both m and f are direct descendants of at least one of the true parents of y.
, which corresponds to a parental (Q) trio. The B-type relationships (Figure 1b) include cases in which one of the putative parents is a full-sibling of y and the other is not a descendant of M or F. The H-type relationships (Figure 1c) are similar to type B, except that one putative parent is a half sibling of y. We investigate four different variations of trio types B and H corresponding to the four different levels of κ investigated; for example, BSi denotes a trio in which the putative mother is a sibling of y and the putative father is a sibling of the true father. Once again, it should be noted that the fact that the mother is placed as the sibling of y in the B-type relationships or as the half-sibling of y in the H-type relationships is merely convention, and for an autosomal locus, the properties of the trio would be identical if the sexes of m and f were reversed. Finally, types D1–D5 (Figure 1, d–h) all include only a single case. They are situations in which both m and f are direct descendants of at least one of the true parents of y.
Figure 1.

Eight basic types of relationships in trios investigated in this article. The solid lines denote pedigree (i.e., parent–offspring) relationships and the dotted lines denote pairwise relatedness parameterized by κm and κf. Shaded nodes are the m, f, and y individuals whose genotype data are used to compute Λ. M and F represent the true mother and father of y. Unshaded nodes represent individuals that are not observed in the current m, f, and y trio (except in the case of “self” pairwise relationships—see text for more explanation).
Paternity inference:
Paternity inference is a special case of parentage inference in which the true mother of the child is known but the true father is unknown. We consider the version of the paternity inference problem in which all three members of a trio have been genotyped. Likelihood inference in this case proceeds much as before, except that the null hypothesis for each trio is no longer that they are all unrelated (U). Rather, the null hypothesis is that the putative mother is the true mother, but the putative father is unrelated to either of them. The likelihood-ratio statistic thus becomes
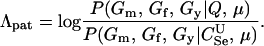 |
(4) |
The expressions for per-trio false-positive and false-negative rates in paternity inference can be computed for Λpat by making the appropriate modifications in (2) and (3). We consider the power for paternity assignment given five different scenarios that correspond to trio types 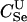 ,
, 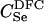 ,
, 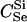 , BSe, and HSe. These paternity inference scenarios are denoted PU, PDFC, PSi, PF, and PH, respectively. The first three are cases where the true mother is known and the putative father is respectively either unrelated to the true father or a double first cousin or brother of the true father. The latter two are cases where the mother is known, and the putative father is a full-sibling or a half-sibling, respectively, of y.
, BSe, and HSe. These paternity inference scenarios are denoted PU, PDFC, PSi, PF, and PH, respectively. The first three are cases where the true mother is known and the putative father is respectively either unrelated to the true father or a double first cousin or brother of the true father. The latter two are cases where the mother is known, and the putative father is a full-sibling or a half-sibling, respectively, of y.
Importance sampling methods for α and β:
To simplify notation, we define G = (Gm, Gf, Gy). To estimate β by simulation, (3) suggests the Monte Carlo estimator
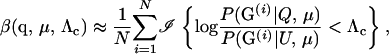 |
(5) |
where G(i) denotes the ith triplet of L-locus genotypes simulated from the distribution P(G | Q, μ) and N is the number simulated. As shown in results, the relationship between α and Λc is typically such that it may be impractical to reduce β below 0.1 or 0.05. Therefore, estimating β using (5) can usually be easily done.
The naive Monte Carlo estimator for α is
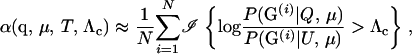 |
(6) |
where G(i) is simulated from the distribution P(G | T, μ). In many cases, this Monte Carlo estimator performs poorly because the desired value of α may be very small. For example, imagine that the potential parents include 3000 males and 3000 females and that 10000 offspring will be compared to all potential parent pairs. The total number of trios investigated in such a case is 90 billion. Therefore, to have few false positives in the whole study, α must be on the order of 10−10–10−8. Accurately estimating such small probabilities using (6) is virtually impossible: if α is 10−9, you might simulate 1 billion random trios from P(G | T, μ) and, using (6), still estimate α to be zero. In general, to estimate α with a Monte Carlo standard error of X% using (6) requires 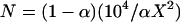 . For example, if α = 10−9, using (6) to achieve a Monte Carlo estimate of α with a standard error of 1% of its true value requires N ≈ 104/(10−9 × 1) = 1013, which would be computationally impractical.
. For example, if α = 10−9, using (6) to achieve a Monte Carlo estimate of α with a standard error of 1% of its true value requires N ≈ 104/(10−9 × 1) = 1013, which would be computationally impractical.
To accurately estimate small values of α, we use importance sampling (Hammersley and Handscomb 1964), simulating G(i) from a distribution P*(G) (note the superscript * to distinguish this distribution) called the importance-sampling distribution. In this case, so long as P*(G(i)) is nonzero for all possible values of G(i), and the values of G(i) are simulated from P*(G), α may be estimated using
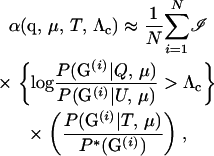 |
(7) |
for all Λc. If P*(G) is chosen well, the Monte Carlo variance of (7) can be much smaller than that of (6). Furthermore, it can be shown (e.g., Hammersley and Handscomb 1964) that the P*(G) that minimizes the Monte Carlo variance of the estimator of α satisfies
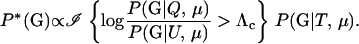 |
(8) |
In other words, P*(G) should be chosen such that it is zero for all values of G for which Λ ≤ Λc, and it should be proportional to P(G | T, μ) for all other values of G. In general, it is not possible both to simulate from such a distribution and to compute the probability of each realization from that distribution. However, a good importance-sampling distribution can still be constructed by trying to make P*(G) close to proportional to P(G | T, μ) for all values of G yielding Λ ≥ Λc or, at least, by ensuring that there are no values of G having aberrantly large values of the importance weights, 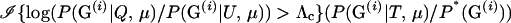 . Meeting the latter condition helps to avoid a situation that is a common downfall for importance-sampling schemes—infrequent realizations of G from P*(G) that contribute much larger-than-average terms to the sum in (7).
. Meeting the latter condition helps to avoid a situation that is a common downfall for importance-sampling schemes—infrequent realizations of G from P*(G) that contribute much larger-than-average terms to the sum in (7).
We consider two different forms for the importance-sampling distribution. In the first, which we refer to as I1, we use the distribution given that the trios are of type Q; that is, P*(G) ≡ P(G | Q, μ). This works remarkably well for cases where the members of the trio are mostly unrelated. The apparent reason for this can be understood by considering the case where the trio is truly unrelated (i.e., T = U) as follows: if G ∼ P(G | Q, μ) then a large proportion (1 − β) of realized values G(i) will yield values of Λ > Λc, and, in those cases, the importance weights will be 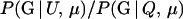 , which is precisely exp(−Λ). Since all values of G yielding importance weights greater than zero have Λ > Λc, the upper limit to the importance weights is exp(−Λc), and it turns out that a great many values of G simulated from P(G | Q, μ) yield values of exp(−Λ) near that limit. As a consequence, with T = U (and with other trio relationships with little relatedness between the members) this importance-sampling distribution does not generate any realizations of importance weights that are much larger than the average. Therefore, it provides a fairly stable importance-sampling distribution.
, which is precisely exp(−Λ). Since all values of G yielding importance weights greater than zero have Λ > Λc, the upper limit to the importance weights is exp(−Λc), and it turns out that a great many values of G simulated from P(G | Q, μ) yield values of exp(−Λ) near that limit. As a consequence, with T = U (and with other trio relationships with little relatedness between the members) this importance-sampling distribution does not generate any realizations of importance weights that are much larger than the average. Therefore, it provides a fairly stable importance-sampling distribution.
Unfortunately, for trios involving highly related members, the importance-sampling distribution I1 may perform poorly. For such relationships we develop a more ad hoc importance-sampling distribution, which we refer to as I2. To construct this distribution we adjust the probability of each of the 27 genotypic configurations at each locus according to a logistic weighting function. The goal is to increase the probability of the genotypic states yielding high values of Λ, so that the average value of Λ from the importance-sampling distribution is equal to that under the relationship Q (this ensures that a large proportion of the realizations from the importance-sampling distribution will have Λ > Λc). This procedure is applied independently to each of the L loci. To describe this at a single locus we let g denote the genotypic configuration of the trio at a single locus, and we denote the 27 configurations that g can take by the set 𝒢. At this single locus, the log-likelihood-ratio statistic is λ(g) = log[P(g | Q, μ)/P(g | U, μ)], and the expected value of λ under the hypothesis that the trio is of type Q is 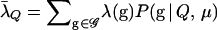 . The importance-sampling distribution P*(g) is formed by scaling P(g | T, μ) for each value of
. The importance-sampling distribution P*(g) is formed by scaling P(g | T, μ) for each value of  by a weight w(λ(g)) that depends on the value of λ that the particular g yields. For values of g that yield values of λ(g) >
by a weight w(λ(g)) that depends on the value of λ that the particular g yields. For values of g that yield values of λ(g) >  , the weight is >1. Otherwise it is ≤1 according to a logistic equation with two parameters: r > 0 and M (0 < M < 1). Mathematically,
, the weight is >1. Otherwise it is ≤1 according to a logistic equation with two parameters: r > 0 and M (0 < M < 1). Mathematically,
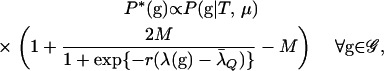 |
(9) |
and normalizing P*(g) is easily done.
From (9), we see that the weight w(g) is never >1 + M and never <1 − M. Since 0 < M < 1, this ensures that P*(g) > 0 whenever P(g | T, μ) > 0. For the relationships investigated in this article, a value of M = 0.85 was used, and r was adjusted so that the average value of λ(g) under the importance-sampling distribution was equal to (or, in practice, slightly larger than) 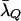 . Figure 2a plots w(λ(g)) as a function of λ when M = 0.85 and r = 0.72. Figure 2b shows the relationship between the importance-sampling distribution P*(g) obtained by weighting the original, true distribution—in this case P(g | BSe, μ)—using the weighting function depicted in Figure 2a. It makes it clear that the importance-sampling distribution is still highly correlated with the true distribution of g, as desired. This means the variance of the importance weights is not high. Also, as is clear from the absence of points above the y = x line in the left part of the graph, there are no values of g having very low probability under P*(g) but having very high importance-sampling weights. This is important since rare realizations of enormous importance weights often present difficulty for importance-sampling algorithms (Gelman et al. 1996).
. Figure 2a plots w(λ(g)) as a function of λ when M = 0.85 and r = 0.72. Figure 2b shows the relationship between the importance-sampling distribution P*(g) obtained by weighting the original, true distribution—in this case P(g | BSe, μ)—using the weighting function depicted in Figure 2a. It makes it clear that the importance-sampling distribution is still highly correlated with the true distribution of g, as desired. This means the variance of the importance weights is not high. Also, as is clear from the absence of points above the y = x line in the left part of the graph, there are no values of g having very low probability under P*(g) but having very high importance-sampling weights. This is important since rare realizations of enormous importance weights often present difficulty for importance-sampling algorithms (Gelman et al. 1996).
Figure 2.

The importance sampling scheme I2. (a) The logistic weighting function w(λ(g)) as a function of λ for M = 0.75 and r = 0.72. The dashed lines show the minimum and maximum possible values of w(λ(g)). (b) Plot of P(g | BSe, μ) for a single locus with q = 0.2 on the x-axis against P*(g), obtained by weighting values of P(g | BSe, μ) by the function in a, on the y-axis. The dashed line is the y = x line.
RESULTS
We begin by presenting the relationship between α, β, and Λc, and we use that relationship to portray the effect of genotyping error. Then we present three main results in three subsections: we assess the accuracy of the importance-sampling method for estimating false-positive rates, present the false-positive rates for a number of scenarios, and compare the power achieved by using a likelihood-based approach vs. relying solely on an exclusion criterion. Finally, we give a brief example in a hypothetical salmon population.
Figure 3 portrays the relationship between α and β for a hypothetical data set of 80 SNPs with minor allele frequencies of 0.2. The different curves show the relationship for different values of the genotyping error rate. The graph shows that α increases sigmoidally as a function of 1 − β, which is the probability of not making a false-negative error—also called the power. Between 1 − β values of ∼0.15 and 0.85, the logarithm of α increases roughly linearly with 1 − β. Toward the ends of the intervals, the increase of α is considerably steeper. Thus, although you may be able to identify the offspring of 95% of genotyped parent pairs with an acceptably low false-positive rate, the false-positive rate may climb quickly to unacceptable levels if you decrease Λc enough to expect to identify offspring of 99% of the genotyped parent pairs. The other noteworthy feature of Figure 3 is that, not unexpectedly, at any level of 1 − β, lowering the genotyping error rate reduces the false-positive rate. The solid lines in the graph show the curves for values of μ starting at 0.25 and decreasing in factors of 2 to 2−20 = 9.5 × 10−7. The curves are clearly converging to the limiting value of α that would be achieved if μ = 0. The dashed line in Figure 3 corresponds to μ = 0.005, which is at the high end of the range of μ (0.0001–0.005) estimated in recent, large, SNP genotyping studies (Gabriel et al. 2002; Kennedy et al. 2003; Barton et al. 2004; Hao et al. 2004; Mitra et al. 2004). Throughout the rest of this study, we assume μ = 0.005. In the following two sections, we focus our attention on values of α corresponding to 1 − β = 0.90 (Figure 3, vertical dotted line). In other words, when computing α, we set Λc so that β = 0.1.
Figure 3.

The relationship between false-positive rates, α, and power, 1 − β, for minor allele frequency, q = 0.2, and number of loci, L = 80, at different values of the genotyping error rate, μ. Note that the y-axis is on a log scale. Each solid curve represents the relationship for a value of μ = 2−n with n ∈ {2,…, 20}. For n = 2,…, 7, the value of μ is printed above the curve. The additional dashed curve plotted below the μ = 0.0078 curve corresponds to μ = 0.005, which is the value used for all subsequent analyses in this article. The dotted, vertical line at 1 − β = 0.90 shows the power used in several analyses later in this article.
Accuracy of the importance-sampling method:
It can be difficult to assess the performance of importance-sampling algorithms because the true distribution of the importance weights is typically unknown (Gelman et al. 1996). If the importance weights are highly variable, then the estimated variance of the Monte Carlo estimator may not represent the true variance. Here we assess how well the standard errors of estimates of α reflect the true uncertainty in the estimates. First, using a standard set of L = 60 loci with minor allele frequencies of q = 0.2, we make 100 independent importance-sampling estimates of α, corresponding to a false-negative rate of β = 0.10, using different random-number seeds. Then we compare the standard deviation of the 100 estimates of α to the Monte Carlo standard errors computed for each of the 100 estimates. If the importance-sampling method works well, then the standard error of each estimate of α will be small and will correspond well to the standard deviation of the estimates obtained by repeating the procedure 100 times. If it does not work well, then the standard errors will be large and/or they will not correspond well to the empirical distribution of the estimates.
We carried out this procedure for all 23 trio relationships (except the  case) and the 5 paternity inference relationships. For each relationship, we estimated α using both I1 and I2 with N = 100,000 Monte Carlo samples. The results are summarized in Table 1. For each relationship and each importance-sampling method we report the following:
case) and the 5 paternity inference relationships. For each relationship, we estimated α using both I1 and I2 with N = 100,000 Monte Carlo samples. The results are summarized in Table 1. For each relationship and each importance-sampling method we report the following:  , the mean α estimated over 100 replicate Monte Carlo simulations (this is equivalent to α estimated with N = 10 million); SD, the standard deviation of the 100 estimates of α calculated from their observed distribution; SD↓, the minimum, over the 100 replicate simulations, of the Monte Carlo standard errors of the estimates of α computed from the 100,000 Monte Carlo samples; SD↑, the maximum SD over the 100 replicate simulations; and ∉C.I., the number, out of 100 Monte Carlo simulations with N = 100,000, that failed to include
, the mean α estimated over 100 replicate Monte Carlo simulations (this is equivalent to α estimated with N = 10 million); SD, the standard deviation of the 100 estimates of α calculated from their observed distribution; SD↓, the minimum, over the 100 replicate simulations, of the Monte Carlo standard errors of the estimates of α computed from the 100,000 Monte Carlo samples; SD↑, the maximum SD over the 100 replicate simulations; and ∉C.I., the number, out of 100 Monte Carlo simulations with N = 100,000, that failed to include 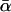 in the 90% confidence interval for the estimate of α. ∉C.I. should be close to 10.
in the 90% confidence interval for the estimate of α. ∉C.I. should be close to 10.
TABLE 1.
Assessment of the importance-sampling methods
|
I1
|
I2
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| T | 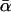 |
SD | SD↓ | SD↑ | ∉C.I. | 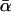 |
SD | SD↓ | SD↑ | ∉C.I. | ISI |
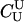 |
5.23e-08 | 0.89 | 0.92 | 0.95 | 9 | — | — | — | — | — | 2.4e+06 |
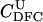 |
7.82e-07 | 1.27 | 1.09 | 1.35 | 13 | — | — | — | — | — | 7.9e+04 |
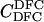 |
1.29e-05 | 1.20 | 1.06 | 1.24 | 11 | 7.76e-06 | 104.26 | 3.07 | 608.03 | 49 | 5.4e+03 |
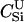 |
8.76e-06 | 2.20 | 1.80 | 4.47 | 6 | 4.39e-06 | 142.90 | 3.24 | 772.98 | 60 | 2.4e+03 |
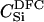 |
1.49e-04 | 1.57 | 1.34 | 2.47 | 9 | 1.47e-04 | 18.52 | 3.95 | 129.35 | 17 | 2.7e+02 |
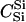 |
1.66e-03 | 1.46 | 1.29 | 2.08 | 6 | 1.67e-03 | 2.54 | 1.70 | 6.17 | 10 | 2.8e+01 |
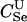 |
5.43e-04 | 14.72 | 4.34 | 67.70 | 24 | 5.53e-04 | 5.39 | 3.08 | 12.36 | 13 | 6.2e+00 |
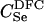 |
8.57e-03 | 6.02 | 2.76 | 30.61 | 12 | 8.59e-03 | 1.02 | 0.89 | 1.07 | 11 | 1.1e+01 |
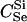 |
7.49e-02 | 3.25 | 1.89 | 9.92 | 9 | 7.48e-02 | 0.49 | 0.54 | 0.56 | 5 | 5.2e+00 |
| BU | 5.08e-04 | 24.40 | 4.82 | 137.21 | 27 | 5.58e-04 | 22.95 | 3.23 | 135.82 | 29 | — |
| BDFC | 2.76e-03 | 23.99 | 5.09 | 137.72 | 22 | 2.91e-03 | 2.10 | 1.52 | 4.83 | 10 | 7.8e+00 |
| BSi | 1.14e-02 | 54.21 | 4.28 | 508.81 | 33 | 1.21e-02 | 0.74 | 0.85 | 1.07 | 4 | 1.5e+01 |
| BSe | 1.04e-01 | 204.62 | 4.04 | 1859.42 | 58 | 1.18e-01 | 0.44 | 0.48 | 0.50 | 7 | 3.9e+00 |
| HU | 8.75e-06 | 2.03 | 1.72 | 3.53 | 8 | 6.64e-06 | 256.77 | 1.78 | 2439.39 | 61 | 2.8e+03 |
| HDFC | 1.48e-04 | 1.91 | 1.34 | 3.26 | 14 | 1.51e-04 | 50.34 | 4.45 | 472.74 | 38 | 1.9e+02 |
| HSi | 1.67e-03 | 1.54 | 1.29 | 3.19 | 8 | 1.66e-03 | 2.22 | 1.66 | 3.96 | 11 | 2.5e+01 |
| HSe | 7.45e-02 | 2.98 | 2.02 | 8.43 | 10 | 7.48e-02 | 0.54 | 0.54 | 0.57 | 8 | 4.2e+00 |
| D1 | 4.52e-02 | 79.70 | 3.89 | 468.64 | 41 | 7.27e-02 | 0.56 | 0.54 | 0.57 | 10 | 4.1e+00 |
| D2 | 1.42e-02 | 125.43 | 3.99 | 1052.07 | 59 | 1.17e-02 | 0.91 | 0.82 | 0.99 | 12 | 1.0e+01 |
| D3 | 1.93e-04 | 15.29 | 3.65 | 52.77 | 23 | 1.90e-04 | 20.17 | 4.13 | 161.01 | 23 | — |
| D4 | 1.67e-03 | 1.36 | 1.30 | 3.41 | 5 | 1.67e-03 | 2.96 | 1.68 | 15.78 | 8 | 3.2e+01 |
| D5 | 7.03e-05 | 162.84 | 4.72 | 1293.03 | 55 | 8.05e-05 | 12.33 | 3.94 | 58.29 | 19 | — |
| PU | 1.29e-09 | 1.33 | 1.26 | 1.31 | 9 | — | — | — | — | — | 4.4e+07 |
| PDFC | 8.79e-07 | 1.13 | 1.24 | 1.35 | 4 | 8.72e-07 | 2.46 | 2.15 | 4.84 | 9 | 8.9e+04 |
| PSi | 1.08e-04 | 1.42 | 1.24 | 1.44 | 14 | 1.08e-04 | 1.17 | 1.04 | 1.10 | 11 | 6.8e+02 |
| PF | 1.46e-04 | 285.69 | 3.04 | 2807.57 | 56 | 1.48e-04 | 1.21 | 1.06 | 1.11 | 14 | 4.6e+02 |
| PH | 6.88e-10 | 117.31 | 3.21 | 709.76 | 48 | 1.06e-09 | 30.62 | 4.68 | 166.84 | 27 | — |
For different relationships, given in the T column, 100 independent Monte Carlo estimates of α corresponding to β = 0.1 were made using both methods I1 and I2. Results for I1 appear on the left and those for I2 on the right. The description of the quantities given in the columns headed by 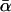 , SD, SD ↓, SD ↑, and ∉C.I. is given in the text. Values for the best-performing importance-sampling method are in italics. For four relationships—BU, D3, D5, and PH—neither I1 nor I2 provided an acceptable reduction in Monte Carlo error. The ISI column gives the factor by which the best importance-sampling algorithm speeds up the estimation of α relative to the naive Monte Carlo estimator of (6). All calculations were done assuming L = 60 loci with minor allele frequencies of q = 0.2.
, SD, SD ↓, SD ↑, and ∉C.I. is given in the text. Values for the best-performing importance-sampling method are in italics. For four relationships—BU, D3, D5, and PH—neither I1 nor I2 provided an acceptable reduction in Monte Carlo error. The ISI column gives the factor by which the best importance-sampling algorithm speeds up the estimation of α relative to the naive Monte Carlo estimator of (6). All calculations were done assuming L = 60 loci with minor allele frequencies of q = 0.2.
The results (Table 1) show that, for most relationships, at least one of either I1 or I2 performs well. For each true relationship, the results for the best-performing importance-sampling method appear in italics (i.e., for each relationship type, the importance-sampling method in italics is the one that should be used in practice). It is apparent that I1 performs best in trios in which the members are not highly related (i.e., when a putative parent was related no more closely than as a full-sibling to a true parent or as a half-sibling to y), and I2 outperforms I1 when members of the trios are more closely related (i.e., when only a single putative parent is a true parent or when a putative parent is a full-sibling of y). In most cases, the best importance-sampling method was able to estimate α with a standard error of between 0.5 and 2% using N = 100,000, which takes <10 sec of user time on a 1.25-GHz G4 Apple laptop. The only exceptions were relationship 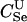 , which could only be estimated to a standard error of 5%, but could still be reliably estimated, and the relationships BU, D3, D5, and PH, for which α could not be accurately estimated using either I1 or I2. It is probable that a different importance-sampling scheme tailored to those relationships could be devised that would work better, but we do not pursue that here. In the rest of this article, we either exclude those relationships from subsequent analyses or note results for them with caution.
, which could only be estimated to a standard error of 5%, but could still be reliably estimated, and the relationships BU, D3, D5, and PH, for which α could not be accurately estimated using either I1 or I2. It is probable that a different importance-sampling scheme tailored to those relationships could be devised that would work better, but we do not pursue that here. In the rest of this article, we either exclude those relationships from subsequent analyses or note results for them with caution.
The “ISI” column in Table 1 gives the approximate factor by which importance sampling decreases computation time compared to the naive Monte Carlo estimator of Equation 6. This value is obtained by calculating the size of the Monte Carlo sample N that would be needed if using the naive Monte Carlo estimator to achieve the same Monte Carlo standard error for α and then dividing that by 100,000. For the 60-locus conditions we considered, the importance-sampling method is between 3.9 times and 44 million times faster, depending on the relationship. The speed improvement due to importance sampling is greater for smaller values of α and will hence be greater for larger numbers of loci, for values of α corresponding to higher values of β, and for more distantly related trio members. In many cases for which I2 is the best importance-sampling method, the improvement from importance sampling is marginal, primarily because the false-positive rates are high enough that estimating them with (6) is quite feasible.
Power of the likelihood-ratio method:
For the 19 trio relationships and four paternity inference scenarios for which either I1 or I2 works well, we computed α at β = 0.1, assuming μ = 0.005, using L loci with q = 0.2, where L ranged from 20 to 120 in steps of 20. The results appear in Figure 4, in which α is plotted against L for each relationship. It is immediately clear that log α decreases linearly with L, so α decreases exponentially with L. In practical terms this means that, as more SNP loci are available, it should be possible to perform accurate parentage inference in ever larger populations. The slope of the line for each relationship tells how much extra information is available from each locus. For example, for  trios, the slope of the line is −0.125, which means that an additional 10 loci will decrease the false-positive rate by a factor of 1010×0.125 = 17.8, and an additional 24 loci will decrease the false-positive rate by a factor of 1000. As can be seen in the plot, with L = 100, the false-positive rate for
trios, the slope of the line is −0.125, which means that an additional 10 loci will decrease the false-positive rate by a factor of 1010×0.125 = 17.8, and an additional 24 loci will decrease the false-positive rate by a factor of 1000. As can be seen in the plot, with L = 100, the false-positive rate for  trios is extremely low—only 4.6 × 10−13. By contrast, the slope of the line for trios of type BSe is only −0.0126, so even an additional 24 loci will decrease the false-positive rate for such relationships only by a factor of 2. And, even with 100 loci, α = 0.037 for a trio in which the putative father is the true father and the putative mother is a sister of the putative offspring. This difficulty of distinguishing siblings from parents is not unique to SNPs and has been investigated by Thompson and Meagher (1987).
trios is extremely low—only 4.6 × 10−13. By contrast, the slope of the line for trios of type BSe is only −0.0126, so even an additional 24 loci will decrease the false-positive rate for such relationships only by a factor of 2. And, even with 100 loci, α = 0.037 for a trio in which the putative father is the true father and the putative mother is a sister of the putative offspring. This difficulty of distinguishing siblings from parents is not unique to SNPs and has been investigated by Thompson and Meagher (1987).
Figure 4.

α as a function of number of loci. The x-axis plots L, the number of loci having minor allele frequency q = 0.2. The y-axis gives values of α at β = 0.1 for the different relationships. Genotyping error rate μ is assumed to be 0.005. α was computed by importance sampling, using N = 100,000, for values of L between 20 and 120 in steps of 20. Vertical bars at each value of L used show the 90% confidence interval around the estimated α. For most relationships these vertical lines are imperceptible because the importance-sampling algorithms work well (they are most apparent along the line for relationship 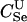 ). Note that the y-axis is on a log scale.
). Note that the y-axis is on a log scale.
In general, α increases as the degree of relatedness between the members of a trio increases (Figure 4), as expected. The pairs D1 and HSe, D2 and BSi, and D4 and HSi all have false-positive rates that are similar to one another. For the first two pairs, this happens to be coincidental, but for D4 and HSi, at all values of μ and q, the false-positive rates are identical. This is because, with unlinked markers, P(G | D4) = P(G | HSi) for all genotypic configurations G, although they are different relationships.
For a particular β the false-positive rate is minimized at a minor allele frequency of 0.5. This is a special case of the well-known result that, on average, a locus is most informative for relationship estimation when its alleles are equifrequent (Thompson 1975). Were we to replot Figure 4 using q = 0.5 we would find it to look much the same as before, but the slopes of all of the lines would be steeper. In Figure 5, the effect of allele frequency on α is presented for eight different relationships. It is interesting to note that the beneficial effect of increasing the minor allele frequency of the SNPs used is greater at low frequencies than at high frequencies. For example, little additional power is gained by increasing q from 0.4 to 0.5 for all relationships.
Figure 5.

The effect of allele frequency on false-positive rates. L = 60 SNPs with minor allele frequency as given on the x-axis were simulated and α was computed for the eight relationships listed in the inset. The y-axis is the negative log to the base 10 of the ratio between α at q = 0.5 (the minimum value of α) and α with q as given on the x-axis. For example, for the 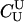 relationship, the false-positive rate for 60 loci is almost 100 times larger when all loci have q = 0.2 than when they have q = 0.5.
relationship, the false-positive rate for 60 loci is almost 100 times larger when all loci have q = 0.2 than when they have q = 0.5.
Comparison to exclusion-based methods:
We calculated the false-positive and false-negative rates achieved by the exclusion method with 100 loci and then calculated how many loci would be required to achieve the same α and β using the likelihood-based method. This procedure was performed assuming that μ = 0.005 and that the minor allele frequency of all loci was 0.20, 0.35, or 0.50. It was repeated for all 22 trio relationship types.
The exclusion criterion used was that of declaring a trio to be a parental trio only if <2 of 100 loci were observed to be incompatible with Mendelian inheritance in the trio. This method of allowing a small number of incompatible loci, to account for mutations or genotyping errors, is common practice in forensic paternity inference (Fung et al. 2002). The calculation of exclusion probabilities is standard, but is repeated here for explicitness. Letting X denote the number of loci, of L, exhibiting Mendelian incompatibilities in a trio of relationship T, it is apparent that, if the allele frequencies and genotyping error rates are constant across all loci, then X ∼ Binomial(L, v), where v is the probability that a locus is incompatible with Mendelian inheritance. In the notation of (A2) we have
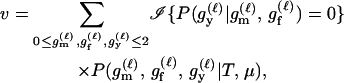 |
where 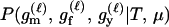 is computed assuming that μ = 0.005, and
is computed assuming that μ = 0.005, and 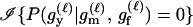 is computed assuming that μ = 0. Hence, it is straightforward to compute βExc = P(X > 1 | Q) and αExc(T) = P(X ≤ 1 | T). For μ = 0.005 and q = 0.20, 0.35, or 0.50, βExc with 100 loci is 0.80, 0.82, or 0.83, respectively.
is computed assuming that μ = 0. Hence, it is straightforward to compute βExc = P(X > 1 | Q) and αExc(T) = P(X ≤ 1 | T). For μ = 0.005 and q = 0.20, 0.35, or 0.50, βExc with 100 loci is 0.80, 0.82, or 0.83, respectively.
Table 2 contains the results of these comparisons. In all cases, the number of loci required of the likelihood method to achieve exactly the same α and β as the exclusion approach would be a noninteger value. This value was approximated by interpolating between the nearest consecutive integer values of the number of loci, and the results shown in Table 2 are rounded to the nearest integer. Two important trends are apparent. First, the likelihood method realizes greater improvements over the exclusion method for trios in which the individuals are not highly related to one another. Second, the benefit of conducting likelihood inference increases as the allele frequency decreases. For distinguishing 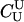 trios from parental trios, it requires <70 loci at q = 0.2 to achieve the same performance that the exclusion method achieves with 100 loci.
trios from parental trios, it requires <70 loci at q = 0.2 to achieve the same performance that the exclusion method achieves with 100 loci.
TABLE 2.
Number of loci required for the likelihood-ratio method to achieve the same α and β as 100 loci using an exclusion-based method
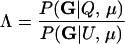
|
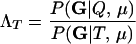
|
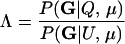
|
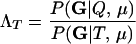
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T | 0.2 | 0.35 | 0.5 | 0.2 | 0.35 | 0.5 | T | 0.2 | 0.35 | 0.5 | 0.2 | 0.35 | 0.5 |
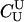 |
69 | 77 | 80 | 69 | 77 | 80 | BSi | 107 | 113 | 114 | 55 | 60 | 61 |
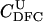 |
73 | 80 | 83 | 71 | 80 | 82 | BSe | 144 | 142 | 141 | 24 | 22 | 21 |
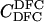 |
77 | 85 | 87 | 76 | 83 | 86 | HU | 77 | 84 | 86 | 72 | 81 | 84 |
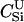 |
76 | 84 | 86 | 72 | 81 | 84 | HDFC | 82 | 89 | 91 | 78 | 86 | 88 |
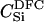 |
82 | 89 | 91 | 78 | 86 | 88 | HSi | 89 | 95 | 96 | 83 | 89 | 91 |
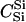 |
89 | 95 | 97 | 83 | 89 | 91 | HSe | 111 | 113 | 114 | 78 | 85 | 88 |
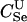 |
88 | 92 | 94 | 63 | 76 | 80 | D1 | 151 | 158 | 161 | 34 | 41 | 43 |
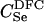 |
96 | 101 | 102 | 71 | 81 | 84 | D2 | 100 | 100 | 100 | 57 | 62 | 64 |
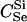 |
110 | 113 | 113 | 78 | 85 | 87 | D3 | 89a | 96a | 98a | 69 | 77 | 79 |
| BU | 85a | 92a | 95a | 58 | 68 | 71 | D4 | 89 | 95 | 97 | 83 | 90 | 91 |
| BDFC | 95 | 102 | 105 | 60 | 67 | 69 | D5 | 102a | 103a | 108a | 54 | 62 | 65 |
The T column denotes the true relationship of the trio. For each relationship, there are six columns. The first three columns give the number of loci required to have α and β comparable to the exclusion method when using the test statistic 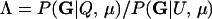 and when q = 0.2, 0.35, and 0.5 as indicated by the column headings. The fourth through sixth columns to the right of each relationship show the number of loci required for a test of relationship T vs. relationship Q, based on
and when q = 0.2, 0.35, and 0.5 as indicated by the column headings. The fourth through sixth columns to the right of each relationship show the number of loci required for a test of relationship T vs. relationship Q, based on 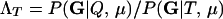 (the most powerful test statistic that could be used if all the trios were known a priori to be either of type Q or of type T), to have the same α and β as the exclusion-based method.
(the most powerful test statistic that could be used if all the trios were known a priori to be either of type Q or of type T), to have the same α and β as the exclusion-based method.
Values that must be treated cautiously because the accuracy of the importance-sampling methods is questionable for these relationships.
For 13 of the 22 relationships, the likelihood method using 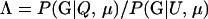 is clearly preferable to the exclusion method. For the remaining 9 relationships, however, the exclusion method performs as well or better than the likelihood method. The results for BSe and D1 are particularly striking—the likelihood method may require as many as 144 or 161 loci to achieve the same power as the exclusion method with 100 loci. This can occur, because, as mentioned in methods, when the true relationship is not U (or
is clearly preferable to the exclusion method. For the remaining 9 relationships, however, the exclusion method performs as well or better than the likelihood method. The results for BSe and D1 are particularly striking—the likelihood method may require as many as 144 or 161 loci to achieve the same power as the exclusion method with 100 loci. This can occur, because, as mentioned in methods, when the true relationship is not U (or 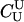 , as we have been calling it), then there is no guarantee that a likelihood-ratio test based on
, as we have been calling it), then there is no guarantee that a likelihood-ratio test based on 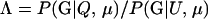 is the most powerful test available. This observation is related to the paradoxical phenomenon encountered in the estimation of pairwise genealogical relationships in which “bilateral relatives such as full-sibs may be more likely parents than the true parent individuals” (Thompson and Meagher 1987, p. 585).
is the most powerful test available. This observation is related to the paradoxical phenomenon encountered in the estimation of pairwise genealogical relationships in which “bilateral relatives such as full-sibs may be more likely parents than the true parent individuals” (Thompson and Meagher 1987, p. 585).
Thompson and Meagher (1987) showed that the discrimination of pairwise parent–offspring and sibling–sibling relationships can be improved by jointly considering the two likelihood ratios that arise by using either the likelihood of the parent pair or that of the sibling relationship in the numerator. In a similar way, it is possible to use a combination of different likelihood ratios to more efficiently discriminate parental trios from other trios with closely related members. The Neyman–Pearson lemma indicates that the most powerful statistic for distinguishing a trio of type T from a trio of type Q would be 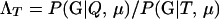 . Table 2 shows that for BSe and D1—the two relationships for which the likelihood method seems to work poorly relative to exclusion—using ΛT allows the likelihood method with as few as 21 and 34 loci, respectively, to perform as well as exclusion with 100 loci. In general, using ΛT seems most advantageous in situations in which one (or both) of the putative parents of the trio is a full sibling of the putative offspring. It thus seems likely that a test statistic that is a combination of both
. Table 2 shows that for BSe and D1—the two relationships for which the likelihood method seems to work poorly relative to exclusion—using ΛT allows the likelihood method with as few as 21 and 34 loci, respectively, to perform as well as exclusion with 100 loci. In general, using ΛT seems most advantageous in situations in which one (or both) of the putative parents of the trio is a full sibling of the putative offspring. It thus seems likely that a test statistic that is a combination of both 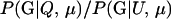 and ΛT for one or a variety of trio relationships, T, could offer a more powerful likelihood approach when some of the trios are expected to include highly related individuals such as full siblings. Of course, as pointed out by Thompson and Meagher (1987), the utility of such an approach depends on the correlation between Λ and ΛT.
and ΛT for one or a variety of trio relationships, T, could offer a more powerful likelihood approach when some of the trios are expected to include highly related individuals such as full siblings. Of course, as pointed out by Thompson and Meagher (1987), the utility of such an approach depends on the correlation between Λ and ΛT.
An example:
To illustrate the scale of study that is possible, we consider the prospects for large-scale parentage inference to infer the mothers and fathers of a cohort born in a large, hypothetical, chinook salmon population. Using the program spip (Anderson and Dunham 2005) we simulated a population of roughly constant size in which an average of 3820 females and 3540 males return to their natal stream each year to spawn and die. Of the male spawners, an average of 28, 57, and 15% were 3-, 4-, and 5-year-olds, respectively. Of the females, on average, 79% were 4-year-olds, and the rest were 5-year-olds. Female mate fidelity was set so that most females had fewer than four male mates (thus creating many more full-sibships than would occur if mating were at random) and the variance in reproductive success of males and females was set so that the effective number of spawners was roughly one-quarter of the census number of spawners (Waples et al. 1993). This serves to create larger families than expected under Wright–Fisher-like reproduction, thus increasing the degree of relatedness between individuals in the population. The population was simulated forward in time, starting at year −40. At year 0 we simulated the nonlethal collection of genetic data from all males and females returning to spawn (3825 females and 3450 males). Such sampling could occur, for example, if all the fish had to pass through a weir or fish ladder before spawning. At years 3, 4, and 5, we simulated genetic sampling of all spawning adults—21,819 in all. Of those fish, 7336 were offspring of the parents genotyped in year 0, and the rest were offspring of fish that spawned in years −2, −1, 1, or 2. We then imagined that parentage was to be inferred by comparing each of the 21,819 fish from years 3 to 5 to all possible pairs of the 3825 females and 3450 males that spawned in year 0—a total of 21,819 × 3825 × 3450 ≈ 2.9 × 1011 trio comparisons.
We assumed 100 SNPs with q = 0.2 and μ = 0.005. Our goal was to estimate the total number of false-positive and false-negative errors expected in conducting such a study. To do this, we first had to calculate the number of trios of different relationship categories that would be encountered. This was achieved by enumeration of the relationships between the putative parents at year 0 and all the true parents of individuals spawning in years 3–5. This approach explicitly takes account of the effects of variation in family size on the distribution of such relationships. Any individuals sharing ancestors more than two generations apart were considered to be unrelated—a reasonable assumption given that the effect of such distant relationships on the distribution of Λ is minimal. Because of the semelparous nature of salmon, and the age structure of their populations, the only types of trios that will be encountered are of the C category. Enumerating the relationships between the true and the putative parents we found the vast majority, 99.8%, of trios to be of type  , with the remainder of the trio categories involving pairwise relationships of Se, Si, half-sib (HS), first cousin (FC), and half-cousin (HC). The latter three relationships have not been previously considered in this article, but are dealt with in a similar manner using their coefficients: for HS,
, with the remainder of the trio categories involving pairwise relationships of Se, Si, half-sib (HS), first cousin (FC), and half-cousin (HC). The latter three relationships have not been previously considered in this article, but are dealt with in a similar manner using their coefficients: for HS, 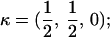 for FC,
for FC, 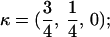 and for HC,
and for HC, 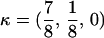 . A small proportion of trios included putative parents related in aunt–niece or other relationships. These relationships were not only rare, but they also did not contribute to the overall false-positive rate, so we do not include them in the results. We used (5) to choose Λc = 22.79 to yield a false-negative rate of β = 0.051.
. A small proportion of trios included putative parents related in aunt–niece or other relationships. These relationships were not only rare, but they also did not contribute to the overall false-positive rate, so we do not include them in the results. We used (5) to choose Λc = 22.79 to yield a false-negative rate of β = 0.051.
Overall, the prospects for parentage assignment in such a large population are promising (Table 3). Of the 7336 sampled offspring of the males and females genotyped at year 0, the expected number assigned to their parents is (1 − β) × 7336 = 6962. Of the 2.9 × 1011 trios that were not of type Q, 226 are expected to have Λ > Λc = 22.79. All but approximately one of these expected incorrect assignments will involve one correct parent. Hence, 94.9% of 225 expected, misassigned offspring are expected to also belong to correct Q trios having Λ > Λc = 22.79. To be conservative, all offspring associated with >1 trio having Λ > Λc could be discarded as having unidentified parents. This would leave 6748 offspring expected to be correctly assigned to their parents and ∼13 offspring expected to be incorrectly assigned; i.e., <2 of 1000 assignments of offspring to parents are expected to be incorrect.
TABLE 3.
Numbers of trios of different types, per-trio false-positive rates, and expected total numbers of false positives for the hypothetical chinook salmon study described in the text
| T | N | α | Err | αEXC | ErrEXC |
|---|---|---|---|---|---|
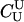 |
2.9e+11 | 2.6e-12 | 0.75 | 8.5e-09 | 2465 |
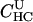 |
2.7e+08 | 8.9e-12 | <0.01 | 1.5e-08 | 4.05 |
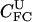 |
1.0e+08 | 2.7e-11 | <0.01 | 2.7e-08 | 2.70 |
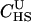 |
7.8e+07 | 2.4e-10 | 0.02 | 8.4e-08 | 6.55 |
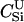 |
3.1e+07 | 1.3e-08 | 0.40 | 7.7e-07 | 23.9 |
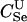 |
1.4e+07 | 1.2e-05 | 168 | 5.0e-05 | 700 |
 |
2.1e+05 | 3.0e-11 | <0.01 | 2.8e-08 | <0.01 |
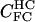 |
7.9e+04 | 8.4e-11 | <0.01 | 5.0e-08 | <0.01 |
 |
6.5e+04 | 7.9e-10 | <0.01 | 1.6e-07 | 0.01 |
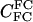 |
3.3e+04 | 2.8e-10 | <0.01 | 9.2e-08 | <0.01 |
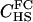 |
2.5e+04 | 2.6e-09 | <0.01 | 3.1e-07 | <0.01 |
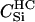 |
2.4e+04 | 4.1e-08 | <0.01 | 1.6e-06 | 0.04 |
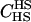 |
1.9e+04 | 2.3e-08 | <0.01 | 1.1e-06 | 0.02 |
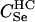 |
1.5e+04 | 4.1e-05 | 0.61 | 1.1e-04 | 1.65 |
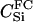 |
1.0e+04 | 1.3e-07 | <0.01 | 3.2e-06 | 0.03 |
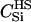 |
7.3e+03 | 1.3e-06 | <0.01 | 1.2e-05 | 0.09 |
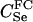 |
5.8e+03 | 1.2e-04 | 0.70 | 2.5e-04 | 1.45 |
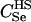 |
4.1e+03 | 9.9e-04 | 4.06 | 1.2e-03 | 4.92 |
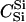 |
3.0e+03 | 6.8e-05 | 0.20 | 1.7e-04 | 0.51 |
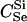 |
1.7e+03 | 3.0e-02 | 51.0 | 2.0e-02 | 34.0 |
| Totals | 2.9e+11 | — | 226 | — | 3245 |
The T column gives the relationship of the trio. The N column gives the number of such trios among the 2.9 × 1011 trios compared in the study. The α column gives the false-positive rates, and the “Err” column gives the total expected number of parental misassignments expected from each trio category when using a likelihood-based assignment method. The value in the Err column is the product of the values in the N and α columns. The αEXC and ErrEXC columns show the results when using an exclusion-based method.
Table 3 also provides the numbers of misassigned offspring expected in the study if an exclusion-based criterion is used. In this case, excluding trios if they carry more than two loci with Mendelian incompatibilities leads to a false-negative rate of βEXC = 0.051—identical to the false-negative rate used above in estimating error rates using the likelihood-based method. Using this exclusion-based criterion, 2465 of the 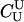 trios are expected to be incorrectly classified as parental trios. The other expected misassignments include 38 trios that do not include either correct parent and 742 trios in which there is at least one correct parent. If one were to apply the same conservative rule of excluding offspring that are nonexcluded from more than one parent pair, then parentage would be assigned to ∼2465 + 38 + 6962 − 742 × 0.949 = 8761 offspring. Of these, 742 × 0.051 + 2465 + 38 = 2541 are expected to be incorrect. Thus, ∼2541/8761 ≈ 29% of assigned offspring would be assigned to an incorrect parent pair using an exclusion-based method. This number overestimates the true expected number, somewhat, because it does not account for the fact that some fraction of the 2541 incorrectly assigned offspring is expected to be assigned to more than one incorrect parent pair. Nonetheless, it clearly makes the point that the likelihood-based approach is far more powerful than the exclusion-based approach in a population where most parents are unrelated.
trios are expected to be incorrectly classified as parental trios. The other expected misassignments include 38 trios that do not include either correct parent and 742 trios in which there is at least one correct parent. If one were to apply the same conservative rule of excluding offspring that are nonexcluded from more than one parent pair, then parentage would be assigned to ∼2465 + 38 + 6962 − 742 × 0.949 = 8761 offspring. Of these, 742 × 0.051 + 2465 + 38 = 2541 are expected to be incorrect. Thus, ∼2541/8761 ≈ 29% of assigned offspring would be assigned to an incorrect parent pair using an exclusion-based method. This number overestimates the true expected number, somewhat, because it does not account for the fact that some fraction of the 2541 incorrectly assigned offspring is expected to be assigned to more than one incorrect parent pair. Nonetheless, it clearly makes the point that the likelihood-based approach is far more powerful than the exclusion-based approach in a population where most parents are unrelated.
DISCUSSION
We predict that SNPs will quickly become the marker of choice for parentage inference in populations of heavily managed species, as well as for large populations, because SNPs are well suited to the high-throughput genotyping required of large studies and because SNP genotyping error rates are low. The advantages of SNPs are particularly apparent in situations where multiple laboratories collaborate on the genotyping effort, and standardization of microsatellite allele calls across all the labs would be costly or infeasible. In this article we provide several advances that allow the application of likelihood-based methods to large-scale parentage inference using SNPs. We describe two importance-sampling algorithms that make the calculation of small false-positive rates, in the presence of genotyping error, computationally feasible. We show that the importance-sampling methods work well for a range of trio types, but do not work well for some cases involving putative parents that are full- or half-siblings of the youth. Developing an efficient importance-sampling algorithm for those cases (BU, D3, D5, and PH) remains an open problem. For trios involving unrelated individuals, the importance-sampling method is millions of times faster than a naive Monte Carlo estimator, even with as few as 60 loci. Although we have focused on SNPs, both importance-sampling algorithms could be modified to handle cases involving other, multiallelic loci.
We present simulations demonstrating that likelihood-based inference of parentage may be considerably more efficient than a method based on the exclusion of trios with an excess of Mendelian incompatibilities. In the case of totally unrelated trios, the likelihood method can achieve the same power and accuracy as the exclusion method with 30% fewer loci. Another way of stating this result is that, for distinguishing unrelated trios from parental trios, the method of exclusion could require up to 143 loci to perform as well as the likelihood-based method with 100 loci. Since most of the trios compared in a large study will likely be unrelated (as shown in the salmon example), this greater efficiency of the likelihood method is particularly germane. However, for trios involving one correct parent and a sibling of the other parent, as well as for trios in which one putative parent is a full-sibling of the youth itself, the method of exclusion performs better than the likelihood method. This argues for the application, when such situations are likely, of a hybrid approach in which trios are initially compared on the basis of the standard likelihood ratio for parentage [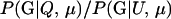 ], and all those having Λ greater than the critical value, Λc, should be investigated further, perhaps by applying an exclusion-based test or perhaps by using a statistic like ΛT described in this article. The latter would be a sort of sequential version of the method recommended in Thompson and Meagher (1987) for dealing with the case where full-siblings of the youth are putative parents. Such a sequential procedure would have to be designed carefully so that the overall false-positive and false-negative rates could still be reliably calculated.
], and all those having Λ greater than the critical value, Λc, should be investigated further, perhaps by applying an exclusion-based test or perhaps by using a statistic like ΛT described in this article. The latter would be a sort of sequential version of the method recommended in Thompson and Meagher (1987) for dealing with the case where full-siblings of the youth are putative parents. Such a sequential procedure would have to be designed carefully so that the overall false-positive and false-negative rates could still be reliably calculated.
We have given a brief summary of the false-positive rates that can be expected using different numbers of SNP loci. We show that false-positive rates decrease exponentially with the number of loci. The consequence of this is that one typically requires only a modest increase in the number of loci to accommodate even a rather large increase in the number of potential parents and offspring in a study. This feature, combined with the fact that SNPs are abundant in the genome of most organisms (Brumfield et al. 2003), is encouraging. Our calculations show that false-positive rates for unrelated trios can be extremely small with a moderate and feasible number of SNP loci. Unfortunately, for closely related trios, particularly those in which a full-sibling of the offspring is a putative parent, the false-positive rates, even with a large number of loci, can be high, especially if one of the putative parents is, indeed, the correct one. This problem is not unique to parentage inference with SNPs, but, in fact, exists for all genetic marker systems (see, for example, Thompson and Meagher 1987). Fortunately, in some contexts, occurrence of such closely related trios will be quite rare. This is particularly true in studies of large populations, as our salmon population example demonstrates. However, nonparental trios containing highly related members may be a substantial problem in some situations, such as small populations, species with extremely high variance in reproductive success, or populations that have recently experienced a reduction in effective size.
The method of parentage inference described here requires that independent estimates of the genotyping error rate be available for all loci or that some reasonable genotyping error rates can be assumed. In the absence of any prior knowledge about true parental relationships, it would not be possible to jointly estimate the genotyping error rate and the relationships. Decreasing the genotyping error rate decreases the false-positive rate at a given false-negative rate. The power analyses described here were done assuming a per-gene-copy genotyping error rate of 0.5%. This value is at the very upper end of reported genotyping error rates for SNPs, and it still provides ample power for parentage inference. Also, SNPs with a minor allele frequency of 0.5 provide the most power for parentage inference, although little additional power is gained above q = 0.4. In many of the simulations, we used q = 0.2, so it should be kept in mind that comparable power could be achieved with fewer loci if they are selected such that q > 0.2.
Throughout our simulations we have assumed that the allele frequencies among the parents are known without error. For large studies, involving thousands of parents, this is a reasonable assumption because, unless they are all descended from a small number of individuals, the large sample of parents should provide an excellent estimate of the allele frequencies. It should also be pointed out that, since parentage inference is not concerned with the inference of evolutionary history, the ascertainment of SNPs through discovery in particular populations or genomic regions (Wakeley et al. 2001) does not bias the results of parentage inference in any way. In fact, SNP ascertainment leads to an advantage in parentage inference because ascertainment typically leads to an overrepresentation of SNPs at intermediate allele frequencies—exactly the type of loci that are most powerful for parentage.
In addition to being conditional on genotyping error rates and allele frequencies, our estimates of false-positive rates have also been made conditional on the true, but unobserved, relationship of the members of the trio. To obtain an estimate of the false-positive rate for a trio randomly drawn from a population, it would be necessary to know the distribution of all trio types in that population. Since this distribution depends on a number of demographic and life-history features, including age at first and last reproduction, age structure, and distribution of family sizes, it will typically be unknown and inference must be made by assuming the distribution of trio types. For this reason, large-scale parentage inference may initially be useful in heavily monitored or managed species. Our example of a large salmon population reflects this—given good information about age structure and family size variance, a reasonable approximation of the distribution of trio relationships in a population can be obtained. We reiterate here that salmon populations are special in that all trio relationships will be of a 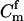 type. To efficiently compute false-positive rates of randomly drawn trios from more generally structured populations may require devising specialized importance-sampling algorithms for the BU, D3, D5, and PH relationships. This remains an open problem.
type. To efficiently compute false-positive rates of randomly drawn trios from more generally structured populations may require devising specialized importance-sampling algorithms for the BU, D3, D5, and PH relationships. This remains an open problem.
An argument that has been made against the use of SNPs in the estimation of relationships is that, since so many SNPs are required, there is high probability that some of them will be physically linked, and that genetic linkage may not even be recognized because the markers may not be in linkage disequilibrium (see, for example, Glaubitz et al. 2003). There are two important points to be made with regard to the effect of linkage and LD on parentage inference. First, as has been pointed out previously (Chakraborty and Hedrick 1983; Jones and Ardren 2003), LD will always decrease the per-locus power for parentage inference, because each locus no longer provides independent information. Consequently, whenever possible, SNPs used for parentage inference should be chosen to have no significant LD. This should not be difficult, even for physically linked SNPs, since LD has been observed to drop to low values over physical distances of <200 kb (Pritchard and Przeworski 2001). Second, in the absence of LD, the effect of physical linkage on parentage inference depends on the true relationship of the individuals in the trio. Most importantly, for all trios of the 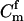 type, physical linkage without LD does not affect the distribution of Λ. Therefore, for example, physical linkage (with no LD) is irrelevant in the analysis of mother–father–youth trios in a salmon population, where only trios of the
type, physical linkage without LD does not affect the distribution of Λ. Therefore, for example, physical linkage (with no LD) is irrelevant in the analysis of mother–father–youth trios in a salmon population, where only trios of the 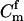 type are possible. This is true because, with no LD, the probability P(G |
type are possible. This is true because, with no LD, the probability P(G | 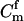 ) is the same whether or not alleles occur together on the same haplotype, because there is no information available in
) is the same whether or not alleles occur together on the same haplotype, because there is no information available in 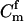 -type trios to infer the haplotypic phase of alleles that are heterozygous in all trio members.
-type trios to infer the haplotypic phase of alleles that are heterozygous in all trio members.
Physical linkage does, however, affect the distribution of Λ for trios in which two members may have each inherited genetic material from a single founder of the pedigree that connects the trio members (i.e., the B, H, and D trio types). Although the mean of the distribution of Λ remains unchanged, physical linkage increases the variance of the distribution for such trios. Accordingly, false-positive rates calculated for such trios under the assumption of no linkage (as examined here) will underestimate the true false-positive rate in the presence of linkage. One solution to this problem would be to simulate the genetic data using a method that explicitly takes account of the linkage. However, developing an importance-sampling scheme to make this type of simulation efficient with many linked markers might be difficult. Additionally, for trio types in which physical linkage affects the distribution of Λ, a more powerful test statistic than Λ could be derived that took account of the linkage. Such a method could build upon the framework of Sieberts et al. (2002), but requires that estimates of the recombination fraction between markers are available.
We have focused on the estimation of false-positive rates and their use in calculating expected studywide error rates. Such calculations are useful for guiding study design and determining the number of loci required to achieve a certain degree of reliability. They do not, however, address the actual practice of carrying out the trio comparisons. As pointed out by Meagher and Thompson (1986), comparing all offspring to all possible parent pairs could be computationally prohibitive—performing 1011 trio comparisons is extremely time consuming. Fortunately, this computational burden can be reduced by a number of strategies. We will provide details in a separate article, but we note here that the number of trios for which the likelihood must be evaluated can be significantly reduced by first excluding individual males and females from consideration by using a nonstringent (i.e., having a low false-negative rate) exclusion criterion based on large numbers of Mendelian incompatibilities. This is computationally advantageous with SNPs because assessing Mendelian incompatibility for many loci at once can be done rapidly by employing bitwise logic operations. Furthermore, for searches of large databases of parents, a suffix tree (McCreight 1976) representation of the genotypes of males and females would allow rapid identification of nonexcluded parents, and the problem of identifying parents or parent pairs sharing zero, or a small number, of Mendelian incompatibilities with any individual can be translated into a special case of the approximate keyword search problem, for which fast algorithms are known (Myers 1994).
We also note that we have not addressed several other improvements to the practice of large-scale parentage inference that could be made. For example, in large studies, it is likely that some males or females will have more than one offspring assigned to them. After a preliminary inquiry based on trios, larger family groups could be analyzed as a unit to provide sharper parentage inferences, reducing the false-positive rate. However, it should be noted that many such comparisons (i.e., those involving multiple potential children of the same parent or parent pair) will be affected by physical linkage, even in the absence of LD.
We distribute two computer programs written in C implementing the calculations presented in this article. snpSumPed calculates probabilities of the 27 configurations of genotypes that m, f, and y might carry, given the minor allele frequency, the genotyping error rate, and the true relationship between members of the trio. The user specifies the trio relationship as a combination of pedigree relationships and pairwise relationships. TrioTests implements the importance-sampling schemes I1 and I2, given the joint probabilities of trio configurations computed by snpSumPed. Both programs are available for download from santacruz.nmfs.noaa.gov/staff/eric_anderson/.
Acknowledgments
We thank Elizabeth Thompson, Robin Waples, and David Hankin for discussion of statistical issues; Paul Moran and Tasha Belfiore for discussion of genotyping issues; Kevin Dunham for assistance in running large numbers of simulations; and three anonymous referees for comments that considerably improved the manuscript.
APPENDIX
For L loci that are not in LD, Λ is found by taking a product over loci:
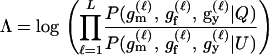 |
(A1) |
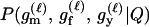 is the joint probability that, at the ℓth locus, m carries genotype
is the joint probability that, at the ℓth locus, m carries genotype 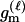 , f carries genotype
, f carries genotype 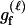 , and their offspring y carries genotype
, and their offspring y carries genotype 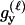 . This is equal to
. This is equal to 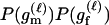
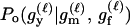 , where
, where 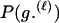 is the frequency of genotype
is the frequency of genotype 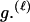 in the population and
in the population and 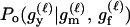 is the probability that a child of parents with genotypes
is the probability that a child of parents with genotypes 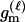 and
and 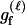 at locus ℓ has the genotype
at locus ℓ has the genotype 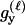 .
. 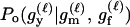 takes values of 0,
takes values of 0, 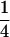 , or
, or 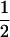 and is easily computed as a consequence of Mendel's laws (see, for example, Marshall et al. 1998 or Neff et al. 2001).
and is easily computed as a consequence of Mendel's laws (see, for example, Marshall et al. 1998 or Neff et al. 2001). 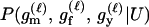 is merely
is merely 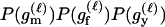 .
.
The probability 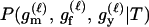 for a trio relationship T, like those in Figure 1, is computed as a sum over genotypes of unobserved individuals. Writing
for a trio relationship T, like those in Figure 1, is computed as a sum over genotypes of unobserved individuals. Writing  for the genotypes at locus ℓ of the individuals in the pedigree that are not m, f, or y, we have
for the genotypes at locus ℓ of the individuals in the pedigree that are not m, f, or y, we have 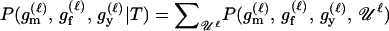 . The joint probability
. The joint probability 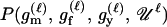 can be expressed as a product over the genotypes of each member of the pedigree (Thompson 2000): founders of the pedigree contribute a factor that is just the frequency of their genotype in the population; an individual with genotype a in the pedigree that is the offspring of individuals with genotypes b and c contributes the factor Po(a | b, c) to the product; and an individual with genotype d that shares a pairwise relationship with coefficients κ (see text) with an individual e contributes the factor P(d | e, κ)—the probability that an individual has genotype d given that he is related via identity coefficients κ to an individual with genotype e (Li and Sacks 1954). Since pairwise relationships parameterized by κ define valid conditional probabilities only for noninbred pairs of individuals, they may be correctly included in the joint probability of the genotypes of individuals on pedigrees if individuals in specified pairwise relationships are: not inbred, not the offspring of any others in the pedigree, and not in specified pairwise relationships to any others in the pedigree. As an example, referring to Figure 1a, letting M and F denote the genotypes at locus ℓ of the true mother and father of y, respectively,
can be expressed as a product over the genotypes of each member of the pedigree (Thompson 2000): founders of the pedigree contribute a factor that is just the frequency of their genotype in the population; an individual with genotype a in the pedigree that is the offspring of individuals with genotypes b and c contributes the factor Po(a | b, c) to the product; and an individual with genotype d that shares a pairwise relationship with coefficients κ (see text) with an individual e contributes the factor P(d | e, κ)—the probability that an individual has genotype d given that he is related via identity coefficients κ to an individual with genotype e (Li and Sacks 1954). Since pairwise relationships parameterized by κ define valid conditional probabilities only for noninbred pairs of individuals, they may be correctly included in the joint probability of the genotypes of individuals on pedigrees if individuals in specified pairwise relationships are: not inbred, not the offspring of any others in the pedigree, and not in specified pairwise relationships to any others in the pedigree. As an example, referring to Figure 1a, letting M and F denote the genotypes at locus ℓ of the true mother and father of y, respectively,
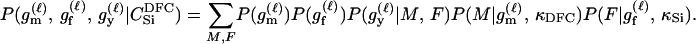 |
If loci are physically unlinked and not in LD, then  for all trio relationships, T. In the special case of
for all trio relationships, T. In the special case of  -type relationships, the above relation holds if the loci are not in LD, even if they are physically linked.
-type relationships, the above relation holds if the loci are not in LD, even if they are physically linked.
Regardless of the true relationship, accounting for genotyping error of rate μℓ at locus ℓ is done by summing the trio probabilities over all possible values of the true underlying genotypes, g.*(ℓ), of m, f, and y, each weighted by P(g.(ℓ) | g.*(ℓ))—the probability of the observed genotypes, g.(ℓ), given the underlying, true genotypes. That is,
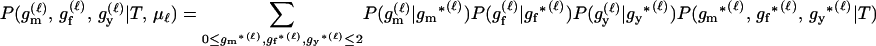 |
(A2) |
for all T. The sum involves only 27 terms, so it is easily computed. The probabilities 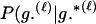 ) may be specified to accommodate any model of genotyping error in which genotyping errors are independent between individuals and loci. Values of
) may be specified to accommodate any model of genotyping error in which genotyping errors are independent between individuals and loci. Values of 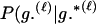 ) from the error model assumed in this article can be derived by standard probability arguments. For example,
) from the error model assumed in this article can be derived by standard probability arguments. For example, 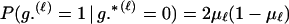 and
and 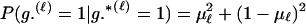 .
.
References
- Anderson, E. C., and K. K. Dunham, 2005. spip 1.0: a program for simulating pedigrees and genetic data in age-structured populations. Mol. Ecol. Notes 5: 459–461. [Google Scholar]
- Barton, A., J. A. Woolmore, D. Ward, S. Eyre, A. Hinks et al., 2004. Association of protein kinase C alpha (PRKCA) gene with multiple sclerosis in a UK population. Brain 127: 1717–1722. [DOI] [PubMed] [Google Scholar]
- Brumfield, R. T., P. Beerli, D. A. Nickerson and S. V. Edwards, 2003. The utility of single nucleotide polymorphisms in inferences of population history. Trends Ecol. Evol. 18: 249–256. [Google Scholar]
- Chakraborty, R., and P. W. Hedrick, 1983. Paternity exclusion and the paternity index for two linked loci. Hum. Hered. 33: 12–23. [DOI] [PubMed] [Google Scholar]
- Chakraborty, R., T. R. Meagher and P. E. Smouse, 1988. Parentage analysis with genetic markers in natural populations. I. The expected proportion of offspring with unambiguous paternity. Genetics 118: 327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotterman, C. W., 1940. A calculus for statistico-genetics. Ph.D. Thesis, Ohio State University, Columbus, OH.
- Double, M. C., A. Cockburn, S. C. Barry and P. E. Smouse, 1997. Exclusion probabilities for single locus paternity analysis when related males compete for matings. Mol. Ecol. 6: 1155–1166. [Google Scholar]
- Duchesne, P., M. H. Godbout and L. Bernatchez, 2002. PAPA (package for the analysis of parental allocation): a computer program for simulated and real parental allocation. Mol. Ecol. Notes 2: 191–193. [Google Scholar]
- Edwards, A. W. F., 1967. Automatic construction of genealogies from phenotypic information. Bull. Eur. Soc. Hum. Genet. 1: 42–43. [Google Scholar]
- Fung, W. K., Y. Chung and D. Wong, 2002. Power of exclusion revisited: probability of excluding relatives of the true father from paternity. Int. J. Leg. Med. 116: 64–67. [DOI] [PubMed] [Google Scholar]
- Gabriel, S. B., S. F. Schaffner, H. Nguyen, J. M. Moore, J. Roy et al., 2002. The structure of haplotype blocks in the human genome. Science 296: 2225–2229. [DOI] [PubMed] [Google Scholar]
- Gelman, A., J. B. Carlin, H. S. Stern and D. B. Rubin, 1996. Bayesian Data Analysis. Chapman & Hall, New York.
- Gerber, S., S. Mariette, R. Streiff, C. Bodenes and A. Kremer, 2000. Comparison of microsatellites and amplified fragment length polymorphism markers for parentage analysis. Mol. Ecol. 9: 1037–1048. [DOI] [PubMed] [Google Scholar]
- Gill, P., 2001. An assessment of the utility of single nucleotide polymorphisms (SNPs) for forensic purposes. Int. J. Leg. Med. 114: 204–210. [DOI] [PubMed] [Google Scholar]
- Glaubitz, J. C., O. E. Rhodes and J. A. Dewoody, 2003. Prospects for inferring pairwise relationships with single nucleotide polymorphisms. Mol. Ecol. 12: 1039–1047. [DOI] [PubMed] [Google Scholar]
- Goldgar, D. E., and E. A. Thompson, 1988. Bayesian interval estimation of genetic relationships: application to paternity testing. Am. J. Hum. Genet. 42: 135–142. [PMC free article] [PubMed] [Google Scholar]
- Hammersley, J. M., and D. C. Handscomb, 1964. Monte Carlo Methods. Methuen, London.
- Hammond, H. A., L. Jin, Y. Zhong, C. T. Caskey and R. Chakraborty, 1994. Evaluation of 13 short tandem repeat loci for use in personal identification applications. Am. J. Hum. Genet. 56: 1005–1006. [PMC free article] [PubMed] [Google Scholar]
- Hao, K., C. Li, C. Rosenow and W. Hung Wong, 2004. Estimation of genotype error rate using samples with pedigree information—an application on the GeneChip Mapping 10K array. Genomics 84: 623–630. [DOI] [PubMed] [Google Scholar]
- Heaton, M. P., G. P. Harhay, G. L. Bennett, R. T. Stone, W. M. Grosse et al., 2002. Selection and use of SNP markers for animal identification and paternity analysis in U.S. beef cattle. Mamm. Genome 13: 272–281. [DOI] [PubMed] [Google Scholar]
- Jones, A. G., and W. R. Ardren, 2003. Methods of parentage analysis in natural populations. Mol. Ecol. 12: 2511–2523. [DOI] [PubMed] [Google Scholar]
- Kennedy, G. C., H. Matsuzaki, S. Dong, W. M. Liu, J. Huang et al., 2003. Large-scale genotyping of complex DNA. Nat. Biotechnol. 21: 1233–1237. [DOI] [PubMed] [Google Scholar]
- Krawczak, M., 1999. Informativity assessment for biallelic single nucleotide polymorphisms. Electrophoresis 20: 1676–1681. [DOI] [PubMed] [Google Scholar]
- Lee, H. Y., M. J. Park, J.-E. Yoo, U. Chung, G.-R. Han et al., 2005. Selection of twenty-four highly informative SNP markers for human identification and paternity analysis in Koreans. Forensic Sci. Int. 148: 107–112. [DOI] [PubMed] [Google Scholar]
- Li, C. C., and L. Sacks, 1954. The derivation of joint distribution and correlation between relatives by the use of stochastic matrices. Biometrics 10: 347–360. [Google Scholar]
- Marshall, T. C., J. Slate, L. E. B. Kruuk and J. M. Pemberton, 1998. Statistical confidence for likelihood-based paternity inference in natural populations. Mol. Ecol. 7: 639–655. [DOI] [PubMed] [Google Scholar]
- McCreight, E. M., 1976. A space-economical suffix tree construction algorithm. J. ACM 23: 262–272. [Google Scholar]
- Meagher, T. R., and E. A. Thompson, 1986. The relationship between single parent and parent pair likelihoods in genealogy reconstruction. Theor. Popul. Biol. 29: 87–106. [Google Scholar]
- Mitra, N., T. Z. Ye, A. Smith, S. Chuai, T. Kirchhoff et al., 2004. Localization of cancer susceptibility genes by genome-wide single-nucleotide polymorphism linkage-disequilibrium mapping. Cancer Res. 64: 8116–8125. [DOI] [PubMed] [Google Scholar]
- Myers, E., 1994. A sub-linear algorithm for approximate keyword matching. Algorithmica 12: 345–374. [Google Scholar]
- Neff, B. D., J. Repka and M. R. Gross, 2001. A Bayesian framework for parentage analysis: the value of genetic and other biological data. Theor. Popul. Biol. 59: 315–331. [DOI] [PubMed] [Google Scholar]
- Neyman, J., and E. S. Pearson, 1933. On the problem of the most efficient tests of statistical hypotheses. Philos. Trans. R. Soc. Ser. A 231: 289–337. [Google Scholar]
- Olsen, J. B., C. Busack, J. Britt and P. Bentzen, 2001. The aunt and uncle effect: an empirical evaluation of the confounding influence of full sibs of parents on pedigree reconstruction. Heredity 92: 243–247. [DOI] [PubMed] [Google Scholar]
- Pritchard, J. K., and M. Przeworski, 2001. Linkage disquilibrium in humans: models and data. Am. J. Hum. Genet. 69: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queller, D. C., J. E. Strassmann and C. R. Hughes, 1993. Microsatellites and kinship. Trends Ecol. Evol. 8: 285–288. [DOI] [PubMed] [Google Scholar]
- Ranade, K., M. S. Chang, C. T. Ting, D. Pei, C. F. Hsiao et al., 2001. High-throughput genotyping with single nucleotide polymorphisms. Genome Res. 11: 1262–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon, D. B., and J. Brocteur, 1978. Probability of paternity exclusion when relatives are involved. Am. J. Hum. Genet. 30: 65–75. [PMC free article] [PubMed] [Google Scholar]
- SanCristobal, M., and C. Chevalet, 1997. Error tolerant parent identification from a finite set of individuals. Genet. Res. 70: 53–62. [Google Scholar]
- Sherman, G. B., S. D. Kachman, L. L. Hungerford, G. P. Rupp, C. P. Fox et al., 2004. Impact of candidate sire number and sire relatedness on DNA polymorphism-based measures of exclusion probability and probability of unambiguous parentage. Anim. Genet. 35: 220–226. [DOI] [PubMed] [Google Scholar]
- Sieberts, S. K., E. M. Wijsman and E. A. Thompson, 2002. Relationship inference from trios of individuals, in the presence of typing error. Am. J. Hum. Genet. 70: 170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, E. A., 1975. The estimation of pairwise relationships. Ann. Hum. Genet. 39: 173–188. [DOI] [PubMed] [Google Scholar]
- Thompson, E. A., 1976. Inference of genealogical structure. III. The reconstruction of genealogies. Soc. Sci. Inf. Sci. Soc. 15: 507–526. [Google Scholar]
- Thompson, E. A., 2000. Statistical Inference From Genetic Data on Pedigrees. Institute of Mathematical Statistics, Beachwood, OH.
- Thompson, E. A., and T. R. Meagher, 1987. Parental and sib likelihoods in genealogy reconstruction. Biometrics 43: 585–600. [PubMed] [Google Scholar]
- Wakeley, J., R. Nielsen, S. N. Liu-Cordero and K. Ardlie, 2001. The discovery of single-nucleotide polymorphisms—and inferences about human demographic history. Am. J. Hum. Genet. 69: 1332–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waples, R. S., O. W. Johnson, P. B. Aebersold, C. K. Shiflett, D. M. VanDoornik et al., 1993. A genetic monitoring and evaluation program for supplemented populations of salmon and steelhead in the Snake River basin. Technical report, Coastal Zone and Estuarine Studies Division, Northwest Fisheries Science Center, National Marine Fisheries Service, NOAA, Seattle.
- Werner, F. A. O., G. Durstewitz, F. A. Habermann, G. Thaller, W. Kramer et al., 2004. Detection and characterization of SNPs useful for identity control and parentage testing in major European dairy breeds. Anim. Genet. 35: 44–49. [DOI] [PubMed] [Google Scholar]