

Abstract
Genome dynamics that allow pathogens to escape host immune responses are fundamental to our understanding of host-pathogen interactions. Here we present the first population-based study of the process of concerted evolution in the repetitive domain of a protein-coding gene. This gene, SOWgp, encodes the immunodominant protein in the parasitic phase of the human pathogenic fungi Coccidioides immitis and C. posadasii. We sequenced the entire gene from strains representing the geographic ranges of the two Coccidioides species. By using phylogenetic and genetic distance analyses we discovered that the repetitive part of SOWgp evolves by concerted evolution, predominantly by the mechanism of unequal crossing over. We implemented a mathematical model originally developed for multigene families to estimate the rate of homogenization and recombination of the repetitive array, and the results indicate that the pattern of concerted evolution is a result of homogenization of repeat units proceeding at a rate close to the nucleotide point mutation rate. The release of the SOWgp molecules by the pathogen during proliferation may mislead the host: we speculate that the pathogen benefits from concerted evolution of repeated domains in SOWgp by an enhanced ability to misdirect the host's immune system.
UNDERSTANDING the dynamic qualities of genomes that allow pathogens to escape immune response is vital to a deeper knowledge about host-pathogen interactions. We here present an empirical study of the concerted evolution of the repeated region within a gene encoding a protein that elicits a host response when the fungal pathogens Coccidioides immitis and C. posadasii cause human disease. Concerted evolution is manifested as the interdependent evolution of a repetitive region of the gene, resulting in a sequence similarity of repeat units greater within species than between them (Arnheim et al. 1980; Zimmer et al. 1980; Dover 1982). Concerted evolution can be driven by directional or stochastic processes, or both, and the two mechanisms that have been the principal explanations of concerted evolution in nuclear DNA are gene conversion and unequal crossing over (Smith 1976; Ohta 1980a; Hillis et al. 1991; Charlesworth et al. 1994). Studies of the underlying mechanisms of concerted evolution historically have relied on theoretical rather than empirical arguments (Smith 1974; Tartof 1974; Ohta 1980b; Szostak and Wu 1980; Coen et al. 1982), and the relative importance of these mechanisms remains controversial. Unequal crossing over currently is the favored mechanism to explain the observed sequence homogeneity among repeats in repetitive arrays (Kruger and Vogel 1975; Smith 1976; Stephan 1989; Charlesworth et al. 1994; Elder and Turner 1995). The increases and decreases in repeat number resulting from unequal crossing over lead to turnover among repeats and, in principle, stochastic fixation of a single repeat type. According to this model, unequal crossing over would most commonly occur in central regions of the array where repeats can mispair, and unique sequences flanking the repetitive array would inhibit exchanges involving the edge repeats, i.e., the repeats located at the termini. This bias toward homogenization of central repeats causes identity among repeats to decrease as spatial distance among repeats increases (Kimura and Ohta 1979; Ohta 1980a; Stephan 1989).
The Coccidioides spherule outer wall glycoprotein (SOWgp) single-copy gene contains tandemly repeated proline- and aspartic acid-rich motifs, ranging from 41 to 47 aa in size. It encodes a glycoprotein that is a component of the parasite cell surface. Not only is the coding part of each tandem repeat nearly identical in sequence among repeats, but also each repeat contains an intron nearly identical in sequence among repeats. Human infection by Coccidioides spp. possessing this gene leads to the respiratory disease coccidioidomycosis or San Joaquin Valley fever (Galgiani 1999). Two pathogenic species of Coccidioides cause infection, C. immitis and C. posadasii (Fisher et al. 2002), and both have a recombining genetic structure (Burt et al. 1996; Fisher et al. 2000b). The species are dimorphic, living as hyphal saprobes in the desert soil or as single cells that develop into multicellular spherules, which produce and release endospores in the mammalian host. SOWgp is expressed in the parasitic phase during growth of the first and second generation of presegmented and segmented spherules prior to endosporulation (Hung et al. 2002).
SOWgp appears to contribute to the virulence of Coccidioides spp. both by functioning as an adhesin and by modulating the host's immune response (Hung et al. 2000, 2002). The recombinant polypeptide (rSOWgp, expressed in Escherichia coli) has been shown to bind to mammalian extracellular matrix (ECM) proteins, and deletion of the gene in C. posadasii results in a significant reduction in virulence as well as in a partial loss of the ability of spherules to bind ECM proteins (Hung et al. 2002). The glycoprotein elicits both humoral and cellular immune response in patients with coccidioidal infection (Hung et al. 2000) and, intriguingly, the hydrophilic proline-rich repeat array of SOWgp is suggested to be the domain containing the B cell-dominant epitopes (Hung et al. 2000). Furthermore, the spherule outer wall fraction produced by a parasitic-phase culture of the Δsowgp82 mutant is not reactive with sera from patients with confirmed, disseminated coccidioidomycosis, suggesting that the SOWgp protein is the sole component of this parasitic cell surface layer that is recognized by patients' antibodies. Mice challenged with a lethal inoculum of spores derived from the Δsowgp82 knockout mutant mount a protective Th1 response to infection, while mice infected with the wild-type strain develop disseminated disease characterized by persistent inflammation and a dominant nonprotective Th2 pathway of host immune response (Cole and Hung 2003). These data suggest that, when spherules release endospores, the pathogen evades immune detection by releasing the immunodominant SOWgp from the outer cell wall of the endospores. This process may contribute further to the induction of the Th2 over the Th1 pathway of host immune response. This mechanism is thought to help mitigate the host immune response by misdirecting host defenses to released SOWgp and inducing the host to pointlessly damage itself through a fervid inflammatory response (Hung et al. 2002; Cole and Hung 2003).
By examining variation in SOWgp within and between species, the evolutionary processes underlying this gene's formation, maintenance, and action may be revealed. Here we present the first population-based study of the evolution of a repetitive domain in a protein-coding gene, based on complete sequence of the entire gene from strains selected to represent populations from the whole geographic ranges of the two species that possess it. We used phylogenetic and genetic distance analyses to discover that the repetitive part of SOWgp evolves by concerted evolution, principally by the mechanism of unequal crossing over. Further, we used a mathematical model originally developed for multigene families by Ohta (1983), to estimate the rate of homogenization and recombination of the repetitive array. The adaptive significance for the pathogen of having repeated domains evolving in concert in SOWgp is speculated to be the enhancement of the immunomodulation such that the pathogen is able to efficiently evade the host immune system.
MATERIALS AND METHODS
Fungal material:
Eight strains of C. immitis and 16 strains of C. posadasii were used in this study (Table 1). All strains were previously genotyped and assigned to species by microsatellite markers (Fisher et al. 2002), with the exception of two additional strains that were identified as C. immitis by sequencing of diagnostic nuclear gene loci (GAPDH, glnA, and hxkA) (Johannesson et al. 2004).
TABLE 1.
Characteristics of fungal material used in the study
| Species | Source | Origina | RMSCCb | No. SOWgp repeat units |
|---|---|---|---|---|
| C. immitis | R. Talbot | Central California | 2012 | 4 |
| Unknown | — | 2018 | 4 | |
| T. Kirkland | Southern California | 2102 | 4 | |
| R. Talbot | Central California | 2271 | 4 | |
| R. Talbot | Central California | 2275 | 4 | |
| R. Talbot | Central California | 2278 | 3 | |
| I. Gutierrez | Mexico | 3505 | 5 | |
| Unknown | — | 1705 | 5 | |
| C. posadasii | J. Galgiani | Arizona | 1038 | 5 |
| J. Galgiani | Arizona | 1039 | 5 | |
| J. Galgiani | Arizona | 1040 | 4 | |
| J. Galgiani | Arizona | 1049 | 5 | |
| J. Galgiani | Arizona | 1444 | 4 | |
| T. Kirkland | Southern California | 2103 | 5 | |
| R. Diaz | Mexico | 2345 | 5 | |
| R. Diaz | Mexico | 2346 | 5 | |
| R. Diaz | Mexico | 2347 | 5 | |
| R. Diaz | Mexico | 2348 | 5 | |
| R. Negroni | South America | 2377 | 3 | |
| R. Negroni | South America | 2378 | 5 | |
| R. Negroni | South America | 2379 | 5 | |
| R. Negroni | South America | 3272 | 5 | |
| I. Gutierrez | Mexico | 3490 | 4 | |
| I. Gutierrez | Mexico | 3503 | 5 |
Origins of the strains of Coccidioides spp. follow the classification of populations made by Fisher et al. (2001).
Roche Molecular Systems Culture Collection, Alameda, California.
DNA manipulations:
Total genomic DNA was extracted from lyophilized material as in Burt et al. (1995). All primers were designed manually, on the basis of the GenBank sequence AF308873. The entire coding region of the SOWgp gene was amplified from all strains using the primers SOWgpF5 (GAAGCGCAAGAGAACTGTATG) and SOWgpR3 (CAAATCCATCTACCCAACTT). Each PCR reaction was performed using the Expand High-Fidelity PCR system (Roche Diagnostics, Mannheim, Germany) according to the manufacturer's recommendation, using an Eppendorf thermal cycler. The amplicons were cloned into pCR4-TOPO vector using the TOPO TA cloning kit (Invitrogen, Carlsbad, CA) to facilitate accurate sequencing of an ∼30-bp A-T rich segment of one intron in the region flanking the repetitive part of SOWgp. To obtain the complete sequence of the repeats, direct sequencing of the PCR products was performed with two additional internal sequencing primers, SOWgpF2 (ATGAGGAAACGAGGTGCTAC) and SOWgpR2 (TGGGCTCTGGCATAATGGTA), after purifying the products using the Qiaquick PCR purification kit (QIAGEN, Valencia, CA). Nucleotide sequences were determined using an Applied Biosystems (Foster City, CA) 3100 sequencer and the Taq DyeDeoxi Terminator cycle system (ABI). The repeat units of SOWgp were delimited as in Hung et al. (2002), and sequence alignment of flanking and repetitive parts was performed manually.
Sequence evolution of SOWgp:
Tests of neutral sequence evolution were performed on alignments of both the entire gene and individual repeat units, by using the D-statistic of Tajima (1989), as implemented in DNAsp3.52 (Rozas and Rozas 1999). Furthermore, we calculated the pairwise ratios of nonsynonymous substitutions per nonsynonymous site (Dn) to synonymous substitutions per synonymous site (Ds) for the aligned repeat units, by using maximum-likelihood estimates in PAML3.13d (Goldman and Yang 1994; Yang 1997). If positive Darwinian selection occurred, Dn would be greater than Ds. Only internal repeats were included in this analysis, and where repeat units were identical, only one representative was used.
Phylogenetic analyses of sequences of each repeat unit:
The DNA sequences of all repeats for all strains were used to create a phylogeny for the repeats using maximum-parsimony analyses in PAUP 4.0b (Swofford 2001). Searches were performed using the heuristic search option and support for the branching arrangements was evaluated by bootstrap analyses of 1000 resampled data sets.
Genetic distance and diversity of the repeats:
A series of likelihood-ratio tests of nested models of DNA substitutions of the data set was completed using Modeltest3.04 (Posada and Crandall 1998). These tests revealed that the best model of substitutions for this data set was a transversional model of substitution with equal base frequencies (TVMef + I + G) (Posada and Crandall 1998). PAUP 4.0b was used to generate distance matrices from the multiple alignments using both this parameter-rich model of substitutions and the uncorrected measurement of distance implemented in the program. Indels were treated as single characters.
Two comparisons of pairwise distances were performed on strains with the same number of repeats for each species. To reveal whether the repeat units evolve in concert, we compared the mean distance between the sequences of different repeat units within each species to the mean distance between the sequences of the corresponding repeat units between the species. To reveal the mechanism of concerted evolution, we determined if the genetic distance of repeat sequences increased with physical distance in the primary sequence for each species. To avoid pseudo-replication in the statistical analyses, the genetic distances of repeat units at different physical distances were analyzed in separate tests, each including only comparisons within strains, and separately for each repeat position; e.g., for each strain with five repeats we calculated separately the genetic distance between 1, 2, 3, 4, and 5 and all other repeats. One-way analyses of variance were performed to test for a significant increase in genetic distance with physical distance, using JMP IN version 4 (SAS 2001).
Diversity of the repeats for each species was calculated as 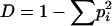 , where the summation is over all repeat sequence types, and pi is the frequency in the data set of the ith repeat sequence type.
, where the summation is over all repeat sequence types, and pi is the frequency in the data set of the ith repeat sequence type.
Detection of gene conversion events:
The alignments of the entire repetitive array, from strains of each of the two species harboring the same number of repeats, were analyzed for evidence of gene conversion by using GENECONV version 1.81 (Sawyer 2000). This method detects pairs of sequences that share unusually long stretches of similarity given their overall polymorphism (Sawyer 1989) and is unbiased with regard to individual repeat unit boundaries. We searched for evidence of significant gene conversion events between ancestors of two sequences in the alignments by using global permutation P-values (Sawyer 2000).
Estimation of population size and rates of homogenization and recombination:
Ohta (1983) developed an infinite-alleles model for the concerted evolution of a multigene family. Equations 7, 8, and 9 of this model predict the equilibrium values of three summary statistics for population data on DNA containing n repeated domains: f, the observed average pairwise identity of corresponding repeats on separate chromosomes; C1, the observed average pairwise identity of all repeats within chromosomes; and C2, the observed average pairwise identity of all noncorresponding repeats between chromosomes. Ohta's model predicts these statistics given values for v, the allelic mutation rate; N, the effective population size; β, the rate at which equal interchromosomal crossover occurs between repeated domains; and λ, the rate at which a repeat domain is converted to be identical to another of the (n − 1) homologous domains.
In this study, we implemented Ohta's model to estimate the rate of homogenization and recombination of the repetitive array of SOWgp. The summary statistics f, C1, and C2 were calculated from the population sample of repeat sequence data for each species, by weighted averaging over groups of strains having the same number of repeats. Statistics f, C1, and C2 were set equal to the equilibrium Equations 7, 8, and 9 of Ohta (1983). All sites with INDELs were excluded from the analysis. We assumed an allelic mutation rate, v, based on the estimated nucleotide mutation rate for the filamentous fungus Neurospora crassa (Drake 1991), with a number of neutrally evolving sites of either 16 (the number of polymorphic sites within species in this data set, v = 7.2 × 10−10) or 197 (all sites in the repeat domain, v = 8.9 × 10−9). The resulting set of three nonlinear equations could not be algebraically solved. To estimate the parameters N, β, and λ from our data, we found approximate solutions to these equations by numerical exploration of the parameter solution space and minimization of the square difference between the predicted and observed values of f, C1, and C2. Random perturbations of the proposed parameter values were drawn from triangular distributions that, at the extremes, halved or doubled each parameter in the current approximate solution, and perturbations were accepted in each generation of the search whenever a uniform variate between zero and one was less than the ratio of the proposed to the current square difference. Each numerical solution was the best result from 5000 iterations of this algorithm. Solutions were examined by eye for fit to observed values and by further manual perturbation to ensure that the solution found was a global rather than a local best fit.
RESULTS
PCR amplification and sequencing of the SOWgp gene from the 24 strains from the two Coccidioides species revealed from three to five tandem repeats. In the eight strains of C. immitis, 1 strain had three repeats, 5 had four, and 2 had five. In the 16 strains of C. posadasii, 1 strain had three repeats, 3 had four, and 12 had five repeats (Table 1).
Sequence variability in regions flanking the repetitive array:
Sequence variability flanking the repeat array was low. All polymorphisms are presented by using GenBank sequence AF308873 as a reference for C. posadasii. In the 447-bp exon sequence, two characters were fixed between the species: one nucleotide replacement (T → A at position 1315) and one 9-bp deletion (between positions 1573 and 1581) were found in C. immitis. In the 135-bp intron, four substitutions were fixed between the species: in C. immitis we found a T at position 1445, an A at position 1459, a C at position 1618, and a T at position 1632. Intraspecific variation was confined to nucleotide polymorphisms in exons, with three polymorphic sites in C. immitis (strain 2018, G → A synonymous substitution at position 1512; strain 2278, G → A synonymous substitution at position 1524; strain 2278, T → C replacement substitution at position 1516) as well as in C. posadasii (strain 2345, T → C synonymous substitution at position 1382 and C → T replacement substitution at position 1525; strains 1040, 1049, and 2103, C → T replacement substitution at position 1667). Tajima's D-test of neutral sequence evolution for the flanking regions failed to reject neutrality in the region of SOWgp flanking the repetitive part.
Divergence of repeat unit sequences:
Alignment of the 108 repeat units from all strains of Coccidioides revealed 34 distinct repeat types (see supplementary Figure S1 at http://www.genetics.org/supplemental/). Thirty-one of 197 nucleotide positions were found to be polymorphic (27 SNPs and two 3-bp deletions; at 2 positions both a SNP and a deletion were found). Nine positions were found to be polymorphic in edge and internal repeats in both species, but only 1 of them was polymorphic in the internal repeats of both species. In the coding sequence, 10 SNPs resulted in synonymous substitutions, while 6 resulted in replacement substitutions. Diversity (D) of repeats was 0.83 for C. immitis and 0.94 for C. posadasii. When excluding edge repeats, D was 0.75 for C. immitis and 0.90 for C. posadasii. The evolutionary analyses of repeat unit divergence indicate that the sequence of the repeat units evolves under purifying selection. Tajima's D-test of neutral sequence evolution for the aligned repeat units failed to reject neutrality. Furthermore, Dn/Ds ratios for pairwise sequence comparisons (cf. Swanson and Vacquier 1998) were found to be very low. In 84% of the comparisons the ratio was <0.5. Dn/Ds was <1 for all comparisons except for five cases for which the only polymorphism in the coding region was one replacement substitution between intraspecific sequence types (data not shown).
Phylogenetic relationship of repeat sequences:
The unrooted maximum-parsimony consensus tree (of 100 retained most parsimonious trees) of the repeat sequences is shown in Figure 1. The 3′ edge repeats of the two species cluster together and are each other's closest relatives. The same is true for the 5′ edge repeats, with the inclusion of a small cluster of a few C. posadasii internal repeats. However, the internal repeat units from the two Coccidioides species do not group together by corresponding position. Rather, they group on the basis of species.
Figure 1.

Unrooted consensus tree of repeat sequences from strains of C. immitis and C. posadasii. Bootstrap values of >50% are given by the branches. Repeats are named as follows: i, immitis; p, posadasii; RMSCC strain number; repeat x out of (y). Edge repeats are encircled with green for C. immitis and red for C. posadasii. C. immitis internal repeats are encircled with yellow.
Genetic distance of repeats between and within species:
Using the parameter-rich model of DNA substitution, the mean genetic distance of corresponding repeat units between the species is 6.4%, smaller than the mean genetic distance between sequences of all the different repeat units within C. immitis (7.4%), and identical to that in C. posadasii. When excluding the two edge repeats, the distance between the corresponding repeats between species is 4.7%, this time higher than the mean genetic distance between repeat units within C. immitis (1.8%) and again equal to that within C. posadasii (Table 2).
TABLE 2.
Mean pairwise genetic distance between sequential SOWgp repeats within C. immitis (C.i.) and C. posadasii (C.p.) and between corresponding repeat sequences between the two species
| Mean pairwise genetic distance (SD)
|
||||
|---|---|---|---|---|
| Different repeats within species
|
Corresponding repeats between species
|
|||
| Repeats included | C.i. | C.p. | Grouped | |
| All | 0.074 (0.055) | 0.064 (0.032) | 0.065 (0.036) | 0.064 (0.037) |
| Internala | 0.018 (0.012) | 0.047 (0.016) | 0.045 (0.018) | 0.047 (0.023) |
The edge repeats are excluded.
The average genetic distance between sequences of repeats at different physical distances in the array is shown in Figure 2. The tests of genetic distance of repeat units at different positions revealed that the physical distance in the array significantly affects the genetic distances of the repeats (one-way ANOVAs, P < 0.005). However, when the edge repeats were excluded from the analyses, no significant difference was found in any of the data sets. The same results were obtained when using the uncorrected measurement of distance in PAUP 4.0b.
Figure 2.

Average genetic distance between sequences of repeats at different physical distances in the array. 0, corresponding position; 1, adjacent position; 2–4, separated by one to three repeats. (A) All repeats included; (B) only internal repeats included. The bars represent mean values ±SE for all genetic distances at each position.
Gene conversion:
By using global permutation P-values obtained from GENCONV (Sawyer 2000), we detected one fragment of DNA that had been converted between ancestors of sequences in the alignment. This 311-bp-long fragment was found between the C. posadasii strain 2103 and four other strains: 2345, 2346, 2348, and 3503 (P = 0.0034). SOWgp of all these strains harbors 5 repeat units and the converted fragment spans the last 63 bp on repeat number 3, the entire repeat 4, and the first 51 bp on the last repeat.
Population size and rate of homogenization and recombination:
The summary statistics of f, C1, and C2 and parameter estimates of λ, β, and N are given in Table 3 (using a weighted average of n = 3, 4, and 5 and v = 7.2 × 10−10). Estimates for individual parameters when n = 3, 4, or 5 and when v is assigned a value of either 7.2 × 10−10 or 8.9 × 10−9 are shown in supplementary Table S1 (http://www.genetics.org/supplemental/). The observed identity of both corresponding (f) and noncorresponding (C1 and C2) repeat units is greater for C. immitis than for C. posadasii. For both species, the identity between corresponding repeats decreases when excluding edge repeats from the data set, while the identity between noncorresponding repeats increases.
TABLE 3.
Observed identity between repeat units and parameter estimates for the concerted evolution of SOWgp in C. immitis (C.i.) and C. posadasii (C.p.)
| All repeats
|
Internal repeatsa
|
|||
|---|---|---|---|---|
| C.i. | C.p. | C.i. | C.p. | |
| f | 0.78 | 0.30 | 0.61 | 0.25 |
| C1 | 0.020 | 0.0071 | 0.091 | 0.026 |
| C2 | 0.014 | 0.0022 | 0.077 | 0.0075 |
| λ | 4.3 × 10−11 | 2.6 × 10−11 | 1.1 × 10−10 | 5.3 × 10−11 |
| β | 6.8 × 10−8 | 6.5 × 10−9 | 2.8 × 10−8 | 7.6 × 10−9 |
| N | 9.3 × 107 | 7.9 × 108 | 2.0 × 108 | 9.9 × 108 |
Weighted averages of estimates are given for n = 3, 4, and 5 repeated domains, assuming an allelic mutation rate of 7.2 × 10−10.
The edge repeats are excluded.
The estimated rates of homogenization (λ) and recombination (β) among repeat units were higher for C. immitis than for C. posadasii, while the estimated population size is smaller for C. immitis than for C. posadasii. The observed degree of repeat unit identity can be explained by a rate of homogenization events of 10−10–10−11/generation in both C. immitis and C. posadasii. The recombination rate between adjacent repeat units is estimated to be 10−7–10−8/generation in C. immitis and 10−8–10−9/generation in C. posadasii.
DISCUSSION
Our data show that the repetitive part of the SOWgp gene evolves under concerted evolution. Corresponding internal repeat units of the two Coccidioides species do not group together in the phylogenetic tree. Instead, the internal repeats group by species. Furthermore, the mean genetic distance between internal repeats within a species is never larger than the genetic distance of corresponding repeats between the two species and, in C. immitis, it is smaller. Thus, internal repeats of SOWgp are as similar as, or more similar than, corresponding repeats between the species and do not evolve as independent units. Analysis using the theoretical predictions of Ohta (1983) indicates that the pattern of concerted evolution is a result of homogenization of repeat units proceeding at a rate close to the nucleotide point mutation rate (Table 3).
Our results suggest the rate of homogenization is higher in C. immitis than in C. posadasii. One plausible explanation for this is that C. immitis has a smaller population size than C. posadasii (Table 3). This finding is in accordance with what is expected based on the geographic distribution of the two species; while C. immitis is confined to northern Mexico, central and southern California, C. posadasii has a wider distribution, ranging from southern California and Texas, through Arizona to Argentina. Furthermore, the genetic diversity of microsatellites found in C. immitis is much smaller than that found in C. posadasii (Fisher et al. 2001), again consistent with a smaller population size for C. immitis.
When comparing the parameters estimated from our data with the parameters estimated from the major histocompatibility complex (MHC) (Ohta 1982), we find that the rate at which repetitive units are made identical to each other is much lower for the repeat units in SOWgp in Coccidioides (λ between 10−10 and 10−11) than for the MHC genes in humans (λ between 10−5 and 10−6 (Ohta 1982)). The difference can be explained in part by considering the role of unequal crossing over in the homogenization process of SOWgp repeat units. The estimated recombination rate is substantially higher between the MHC repeats (β = 10−3) than between the SOWgp repeats (β between 10−7 and 10−9). A lower rate of recombination internal to SOWgp compared to the rate of recombination internal to MHC is expected, because recombination in SOWgp is over a shorter physical distance than recombination in MHC, i.e., between domains of a gene rather than between genes in a multigene family.
We suggest that the most important mechanism of concerted evolution in the SOWgp gene is unequal crossing over. By examining the sequences for extended regions of homology, we found evidence for possible gene conversion of only one fragment, a 311-bp-long fragment that has been converted between strains of C. posadasii. This fragment corresponds to the fourth repeat unit out of five, with some additional sequence upstream and downstream. However, the test (Sawyer 2000) does not explicitly distinguish between gene conversion and unequal crossing over, and the statistical significance of this test could be a byproduct of homogenization by unequal crossing over of repeat units. Unequal crossing over as a mechanism of concerted evolution in SOWgp is supported by the overall high diversity of repeat sequence types in the gene. Furthermore, a positive correlation between genetic distance of repeats and physical distance in the primary sequence was found in the SOWgp data set, and the first and last repeat in the SOWgp array appear to evolve independently of the internal repeats. Homogenization by unequal crossing over occurs in regions of the array that are free to recombine, while unique sequences flanking a repetitive array inhibit exchanges of edge repeats (Ohta 1980a; Stephan 1989). However, these peripheral repeats also are influenced by the presence of unequal exchange, which causes identity among repeats to decrease as physical distance among repeats increases (Ohta 1980a). This pattern has been documented in other systems, such as the α-satellite arrays in the human genome (Cooper et al. 1993), in an array of repeats on the paternal sex ratio chromosome in the parasitic wasp Nasonia vitripennis (McAllister and Werren 1999), and in the egg vitelline envelope receptor for abalone sperm lysin (Galindo et al. 2002). This observed positive correlation of genetic distance and spatial distance in the array of SOWgp repeat units provides further support for the idea that concerted evolution is predominantly by the stochastic mechanism of unequal crossing over. A final line of evidence for unequal crossing over is the observed polymorphism in repeat number in SOWgp. It is widely understood that a mechanism such as unequal crossing over is required to generate variation in the number of copies of a repeat (Hood et al. 1975; Tartof 1975; Smith 1976).
Our results suggest that the evolutionary process generating length variation at the SOWgp locus is occurring more rapidly than that generating sequence variation among repeats within the species. In both species, we found identical SOWgp repeat sequences in strains originating from the outskirts of the species geographical range, as well as from repeat arrays of different sizes (data not shown). These results suggest that these repeat sequences were moved across this geographical range very recently and imply that recombination associated with the observed diversity of SOWgp must also have been very recent.
The results presented in this study suggest that the sequence of SOWgp evolves under purifying selection. However, the persistence of concerted evolution of the repetitive structure of SOWgp in both species of Coccidioides suggests that there may be a selective advantage to maintaining a protein comprising multiple identical domains. We suggest that the possible adaptive significance of SOWgp consisting of repeat units evolving in concert is to allow the pathogen to generate multiple identical epitopes. The pathogen is believed to misdirect the host's immune system by inducing high titers of antibody production and stimulating a nonprotective Th2 pathway of immune response. The immunodominant SOWgp is expressed at the surface of spherules and is released from the outer cell wall of endospores at sporulation. The individual repeat unit of SOWgp is the primary antigen-binding site of the antibody and, by having numerous identical epitopes in SOWgp, a nonprotective response of the immune system would be multiplied, allowing the pathogen to more efficiently divert the efforts of the immune system. This proposed model for the function of the repeat units of SOWgp suggests a selective advantage in having numerous repeat units. However, there appears to be a functional upper limit of SOWgp repeat number in Coccidioides, possibly related to its biochemical activity as an adhesin.
Although intriguing, our conjecture of adaptive significance of SOWgp consisting of repeat units evolving in concert is speculative. In spite of the two Coccidioides species being deeply divergent, with estimations of the time of genetic isolation ranging from 11 to 12.8 million years (Koufopanou et al. 1997; Fisher et al. 2000a), it could reasonably be argued that concerted evolution of the repetitive structure of SOWgp in both species has been retained as a result of chance alone. The likelihood of losing copy number variation through stochastic processes will depend not only on the divergence time but also on other factors such as the population rate of unequal crossing over and the degree to which the allelic unequal crossover rate increases with copy number (e.g., Townsend and Rand 2004). Furthermore, lethal threshold selection on copy number, as in the above model, suggests a selective disadvantage to both low and high copy number, and unequal crossing over will tend to destabilize the number of repeats. However, the increases and decreases in repeat number resulting from unequal crossing over could result only in a selection coefficient at the threshold length that is equal to or lesser than the already low rate of unequal crossover. One way to test the proposed model of adaptive significance of multiple SOWgp repeat units would be to use a murine model to investigate the virulence of individuals of Coccidioides manipulated to possess a different number of repeat units in the array.
Concerted evolution has previously been suggested to be of biological importance in a diverse range of systems. For example, genes evolving by concerted evolution have been hypothesized to produce a species-specific, selective force on the gene of their cognate interacting protein (Dover and Flavell 1984; Dover 1993) and to underlie species-specific gamete interaction in free-spawning abalones (Swanson and Vacquier 1998). Concerted evolution is thought to be the mechanism behind species-specific transcription of tandemly repeated ribosomal DNA by RNA polymerase I (Grummt et al. 1982; Miesfeld and Arnheim 1984; Rudloff et al. 1994; Heix et al. 1997), and variation in the number of copies in an array by unequal crossing over can also be a means for varying the rate of synthesis of the product encoded by that gene (Schimke et al. 1978; Anderson and Roth 1979; Zimmer et al. 1980). This article provides the first evidence that repeat units proposed to be important for immunomodulation by a pathogen are subjected to concerted evolution and suggests that concerted evolution is important for pathogenicity. The presence in other fungal species of adhesins that are polymorphic for internal repeat number (Verstrepen et al. 2004) suggests that the concerted evolution seen in Coccidioides may be of general significance.
Acknowledgments
We thank Georgiana May, Anne Pringle, and Willie Swanson for advice and useful discussions. Financial support from the Fulbright Commission and Carl Tryggers Stiftelse för Vetenskaplig Forskning to H.J., from the Miller Institute for Basic Research in Science to J.P.T., and from the National Institutes of Health (AI37232) and the National Science Foundation (0316710) to J.W.T. is gratefully acknowledged.
References
- Anderson, R. P., and J. R. Roth, 1979. Gene duplication in bacteria: alteration of gene dosage by sister-chromosome exchanges. Cold Spring Harbor Symp. Quant. Biol. 43(2): 1083–1087. [DOI] [PubMed] [Google Scholar]
- Arnheim, N., M. Krystal, R. Schmickel, G. Wilson, O. Ryder et al., 1980. Molecular evidence for genetic exchanges among ribosomal genes on nonhomologous chromosomes in man and apes. Proc. Natl. Acad. Sci. USA 77: 7323–7327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt, A., D. A. Carter, G. L. Koenig, T. J. White and J. T. Taylor, 1995. A safe method of extracting DNA from Coccidioides immitis. Fungal Genet. Newsl. 42: 23. [Google Scholar]
- Burt, A., D. A. Carter, G. L. Koenig, T. J. White and J. W. Taylor, 1996. Molecular markers reveal cryptic sex in the human pathogen Coccidioides immitis. Proc. Natl. Acad. Sci. USA 93: 770–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, B., P. Sniegowski and W. Stephan, 1994. The evolutionary dynamics of repetitive DNA in eukaryotes. Nature 371: 215–220. [DOI] [PubMed] [Google Scholar]
- Coen, E. S., J. M. Thoday and G. Dover, 1982. Rate of turnover of structural variants in the rDNA gene family of Drosophila melanogaster. Nature 295: 564–568. [DOI] [PubMed] [Google Scholar]
- Cole, G. T., and C.-Y. Hung, 2003. A GPI-anchored cell surface antigen contributes to the virulence of Coccidioides, pp. 123–124 in Presentation Summaries and Abstracts of the 15th Congress of the International Society for Human and Animal Mycology, edited by M. G. Rinaldi and J. R. Graybill. Imedex, Alpharetta, GA.
- Cooper, K. F., R. B. Fisher and C. Tyler-Smith, 1993. Structure of the sequences adjacent to the centromeric alphoid satellite DNA array on the human Y chromosome. J. Mol. Biol. 230: 787–799. [DOI] [PubMed] [Google Scholar]
- Dover, G., 1982. Molecular drive: a cohesive mode of species evolution. Nature 299: 111–117. [DOI] [PubMed] [Google Scholar]
- Dover, G. A., 1993. Evolution of genetic redundancy for advanced players. Curr. Opin. Genet. Dev. 3: 902–910. [DOI] [PubMed] [Google Scholar]
- Dover, G. A., and R. B. Flavell, 1984. Molecular coevolution: DNA divergence and the maintenance of function. Cell 38: 622–623. [DOI] [PubMed] [Google Scholar]
- Drake, J. W., 1991. A constant rate of spontaneous mutation in DNA-based microbes. Proc. Natl. Acad. Sci. USA 88: 7160–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elder, Jr., J. F., and B. J. Turner, 1995. Concerted evolution of repetitive DNA sequences in eukaryotes. Q. Rev. Biol. 70: 297–320. [DOI] [PubMed] [Google Scholar]
- Fisher, M. C., G. Koenig, T. J. White and J. W. Taylor, 2000. a A test for concordance between the multilocus genealogies of genes and microsatellites in the pathogenic fungus Coccidioides immitis. Mol. Biol. Evol. 17: 1164–1174. [DOI] [PubMed] [Google Scholar]
- Fisher, M. C., G. L. Koenig, T. J. White and J. W. Taylor, 2000. b Pathogenic clones versus environmentally driven population increase: analysis of an epidemic of the human fungal pathogen Coccidioides immitis. J. Clin. Microbiol. 38: 807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher, M. C., G. L. Koenig, T. J. White, G. San-Blas, R. Negroni et al., 2001. Biogeographic range expansion into South America by Coccidioides immitis mirrors New World patterns of human migration. Proc. Natl. Acad. Sci. USA 98: 4558–4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher, M. C., G. L. Koenig, T. J. White and J. T. Taylor, 2002. Molecular and phenotypic description of Coccidioides posadasii sp nov., previously recognized as the non-California population of Coccidioides immitis. Mycologia 94: 73–84. [PubMed] [Google Scholar]
- Galgiani, J. N., 1999. Coccidioidomycosis: a regional disease of national importance—rethinking approaches for control. Ann. Intern. Med. 130: 293–300. [DOI] [PubMed] [Google Scholar]
- Galindo, B. E., G. W. Moy, W. J. Swanson and V. D. Vacquier, 2002. Full-length sequence of VERL, the egg vitelline envelope receptor for abalone sperm lysin. Gene 288: 111–117. [DOI] [PubMed] [Google Scholar]
- Goldman, N., and Z. H. Yang, 1994. Codon-based model of nucleotide substitution for protein-coding DNA-sequences. Mol. Biol. Evol. 11: 725–736. [DOI] [PubMed] [Google Scholar]
- Grummt, I., E. Roth and M. R. Paule, 1982. Ribosomal RNA transcription in vitro is species specific. Nature 296: 173–174. [DOI] [PubMed] [Google Scholar]
- Heix, J., J. C. Zomerdijk, A. Ravanpay, R. Tjian and I. Grummt, 1997. Cloning of murine RNA polymerase I-specific TAF factors: conserved interactions between the subunits of the species-specific transcription initiation factor TIF-IB/SL1. Proc. Natl. Acad. Sci. USA 94: 1733–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillis, D. M., C. Moritz, C. A. Porter and R. J. Baker, 1991. Evidence for biased gene conversion in concerted evolution of ribosomal DNA. Science 251: 308–310. [DOI] [PubMed] [Google Scholar]
- Hood, L., J. H. Campbell and S. C. Elgin, 1975. The organization, expression, and evolution of antibody genes and other multigene families. Annu. Rev. Genet. 9: 305–353. [DOI] [PubMed] [Google Scholar]
- Hung, C. Y., N. M. Ampel, L. Christian, K. R. Seshan and G. T. Cole, 2000. A major cell surface antigen of Coccidioides immitis which elicits both humoral and cellular immune responses. Infect. Immun. 68: 584–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung, C. Y., J. J. Yu, K. R. Seshan, U. Reichard and G. T. Cole, 2002. A parasitic phase-specific adhesin of Coccidioides immitis contributes to the virulence of this respiratory fungal pathogen. Infect. Immun. 70: 3443–3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannesson, H., P. Vidal, J. Guarro, R. A. Herr, G. T. Cole et al., 2004. Positive directional selection in the proline-rich antigen (PRA) gene among the human pathogenic fungi Coccidioides immitis, C. posadasii and their closest relatives. Mol. Biol. Evol. 21: 1134–1145. [DOI] [PubMed] [Google Scholar]
- Kimura, M., and T. Ohta, 1979. Population genetics of multigene family with special reference to decrease of genetic correlation with distance between gene members on a chromosome. Proc. Natl. Acad. Sci. USA 76: 4001–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koufopanou, V., A. Burt and J. W. Taylor, 1997. Concordance of gene genealogies reveals reproductive isolation in the pathogenic fungus Coccidioides immitis. Proc. Natl. Acad. Sci. USA 94: 5478–5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger, J., and F. Vogel, 1975. Population genetics of unequal crossing over. J. Mol. Evol. 4: 201–247. [Google Scholar]
- McAllister, B. F., and J. H. Werren, 1999. Evolution of tandemly repeated sequences: What happens at the end of an array? J. Mol. Evol. 48: 469–481. [DOI] [PubMed] [Google Scholar]
- Miesfeld, R., and N. Arnheim, 1984. Species-specific rDNA transcription is due to promoter-specific binding factors. Mol. Cell Biol. 4: 221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta, T., 1980. a Evolution and Variation of Multigene Families. Springer-Verlag, New York.
- Ohta, T., 1980. b Linkage disequilibrium between amino acid sites in immunoglobulin genes and other multigene families. Genet. Res. 36: 181–197. [DOI] [PubMed] [Google Scholar]
- Ohta, T., 1982. Allelic and nonallelic homology of a supergene family. Proc. Natl. Acad. Sci. USA 79: 3251–3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta, T., 1983. On the evolution of multigene families. Theor. Popul. Biol. 23: 216–240. [DOI] [PubMed] [Google Scholar]
- Posada, D., and K. A. Crandall, 1998. MODELTEST: testing the model of DNA substitution. Bioinformatics 14: 817–818. [DOI] [PubMed] [Google Scholar]
- Rozas, J., and R. Rozas, 1999. DnaSP version 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics 15: 174–175. [DOI] [PubMed] [Google Scholar]
- Rudloff, U., D. Eberhard, L. Tora, H. Stunnenberg and I. Grummt, 1994. TBP-associated factors interact with DNA and govern species specificity of RNA polymerase I transcription. EMBO J. 13: 2611–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAS, 2001. JMP IN. SAS Institute, Cary, NC.
- Sawyer, S., 1989. Statistical tests for detecting gene conversion. Mol. Biol. Evol. 6: 526–538. [DOI] [PubMed] [Google Scholar]
- Sawyer, S., 2000. GENECONV, Version 1.80: a computer package for the statistical detection of gene conversion. Department of Mathematics, Washington University, St. Louis.
- Schimke, R. T., R. J. Kaufman, F. W. Alt and R. F. Kellems, 1978. Gene amplification and drug resistance in cultured murine cells. Science 202: 1051–1055. [DOI] [PubMed] [Google Scholar]
- Smith, G. P., 1974. Unequal crossover and the evolution of multigene families. Cold Spring Harbor Symp. Quant. Biol. 38: 507–513. [DOI] [PubMed] [Google Scholar]
- Smith, G. P., 1976. Evolution of repeated DNA sequences by unequal crossover. Science 191: 528–535. [DOI] [PubMed] [Google Scholar]
- Stephan, W., 1989. Tandem-repetitive noncoding DNA: forms and forces. Mol. Biol. Evol. 6: 198–212. [DOI] [PubMed] [Google Scholar]
- Swanson, W. J., and V. D. Vacquier, 1998. Concerted evolution in an egg receptor for a rapidly evolving abalone sperm protein. Science 281: 710–712. [DOI] [PubMed] [Google Scholar]
- Swofford, D. L., 2001. PAUP: Phylogenetic Analysis Using Parsimony, Version 3.1. Illinois Natural History Survey, Champaign, IL.
- Szostak, J. W., and R. Wu, 1980. Unequal crossing over in the ribosomal DNA of Saccharomyces cerevisiae. Nature 284: 426–430. [DOI] [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical methods for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartof, K. D., 1974. Unequal mitotic sister chromatid exchange and disproportionate replication as mechanisms regulating ribosomal RNA gene redundancy. Cold Spring Harbor Symp. Quant. Biol. 38: 491–500. [DOI] [PubMed] [Google Scholar]
- Tartof, K. D., 1975. Redundant genes. Annu. Rev. Genet. 9: 355–385. [DOI] [PubMed] [Google Scholar]
- Townsend, J. P., and D. M. Rand, 2004. Mitochondrial genome size variation in New World and Old World populations of Drosophila melanogaster. Heredity 93: 98–103. [DOI] [PubMed] [Google Scholar]
- Verstrepen, K. J., T. B. Reynolds and G. R. Fink, 2004. Origins of variation in the fungal cell surface. Nat. Rev. Microbiol. 2: 533–540. [DOI] [PubMed] [Google Scholar]
- Yang, Z., 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13: 555–556. [DOI] [PubMed] [Google Scholar]
- Zimmer, E. A., S. L. Martin, S. M. Beverley, Y. W. Kan and A. C. Wilson, 1980. Rapid duplication and loss of genes coding for the alpha chains of hemoglobin. Proc. Natl. Acad. Sci. USA 77: 2158–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]