

Abstract
Until around 1990, most multigene families were thought to be subject to concerted evolution, in which all member genes of a family evolve as a unit in concert. However, phylogenetic analysis of MHC and other immune system genes showed a quite different evolutionary pattern, and a new model called birth-and-death evolution was proposed. In this model, new genes are created by gene duplication and some duplicate genes stay in the genome for a long time, whereas others are inactivated or deleted from the genome. Later investigations have shown that most non-rRNA genes including highly conserved histone or ubiquitin genes are subject to this type of evolution. However, the controversy over the two models is still continuing because the distinction between the two models becomes difficult when sequence differences are small. Unlike concerted evolution, the model of birth-and-death evolution can give some insights into the origins of new genetic systems or new phenotypic characters.
Keywords: birth-and-death evolution, concerted evolution, origins of new genetic systems, gene conversion, MHC genes, ribosomal RNA
INTRODUCTION
A multigene family is a group of genes that have descended from a common ancestral gene and therefore have similar functions and similar DNA sequences. A group of related multigene families is sometimes called a supergene family. A well-known example of a supergene family is the globin superfamily that is composed of the α-like, β-like, and some other gene families (134a). A gene family may also be subdivided into subfamilies whenever convenient.
The evolution of multigene families has been the subject of controversy for many years. The paradigm of evolution of multigene families before 1970 was that of hemoglobin α, β, γ, and δ chains and myoglobin (54). The genes encoding these polypeptides or proteins are phylogenetically related and have diverged gradually as the duplicate genes acquired new gene functions. This mode of evolution may be called “divergent evolution” (Figure 1a). Around 1970, however, a number of researchers showed that ribosomal RNAs (rRNAs) in Xenopus are encoded by a large number of tandemly repeated genes and that the nucleotide sequences of the intergenic regions of the genes are more similar within a species than between two related species (11).
Figure 1.
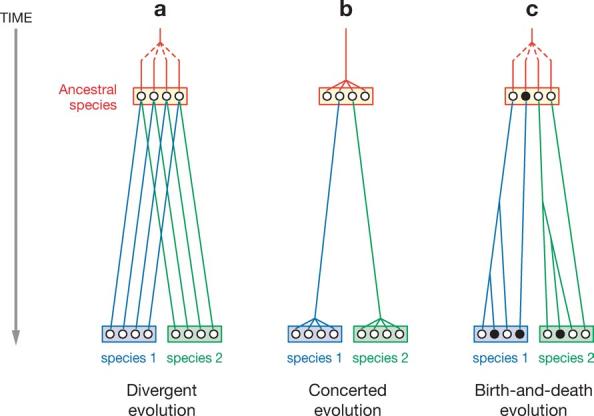
Three different models of evolution of multigene families. Open circles stand for functional genes and closed circles for pseudogenes.
These observations were difficult to explain by the model of divergent evolution, and a new model called “concerted evolution” was proposed (Figure 1b). In this model all the members of a gene family are assumed to evolve in a concerted manner rather than independently, and a mutation occurring in a repeat spreads through the entire member genes by repeated occurrence of unequal crossover or gene conversion. This model is capable of explaining previously puzzling observations about the evolution of rRNA genes.
This apparent success led many authors to believe that most multigene families evolve following the model of concerted evolution, and a number of authors investigated the evolutionary modes of various multigene families (48, 110, 178). Later, however, the applicability of concerted evolution to some gene families was questioned as both DNA and amino acid sequence data became available, and another model called birth-and-death evolution (98) was proposed. In this model new genes are created by gene duplication, and some duplicated genes are maintained in the genome for a long time, whereas others are deleted or become nonfunctional through deleterious mutations (Figure 1c). This model applies to most multigene families concerned with immune systems such as immunoglobulins and major histocompatibility complex (MHC) (53, 116) and disease-resistance genes (173). In recent years, even a highly conserved gene family such as the ubiquitin family was shown to be subject to birth-and-death evolution. Yet the controversy over the evolution of multigene families continues, partly because there are so many different types of gene families and partly because the general mechanism of gene conversion is still unclear.
In this paper we review recent studies of evolution of multigene families with some historical backgrounds.
CONCERTED EVOLUTION
Ribosomal RNA and Other RNA Genes
One of the best-known examples of concerted evolution comes from the study of rRNA genes. The rRNA gene family of the African toads Xenopus laevis and X. mulleri consists of about 450 repeats or members. Each member consists of the 18S, 5.8S, and 28S RNA genes, external transcribed spacers (ETS1 and ETS2), internal transcribed spacers (ITS1 and ITS2), and an intergenic spacer (IGS) (Figure 2) (15). Using DNA or RNA hybridization techniques, Brown et al. (11) showed that the nucleotide sequences of IGS are very similar among member genes of the same species but differ by about 10% between X. laevis and X. mulleri. This observation could not be explained by the then popular model of divergent evolution. According to this model, the differences in nucleotide sequence between different repeats of the same species are expected to be as large as those between repeats of different species. The explanation becomes more difficult to accept if we note that the nucleotide sequences of the 18S and 28S coding regions are virtually identical between X. laevis and X. mulleri. Actually, the 18S and 28S coding regions are very similar even among distantly related organisms such as animals and plants.
Figure 2.
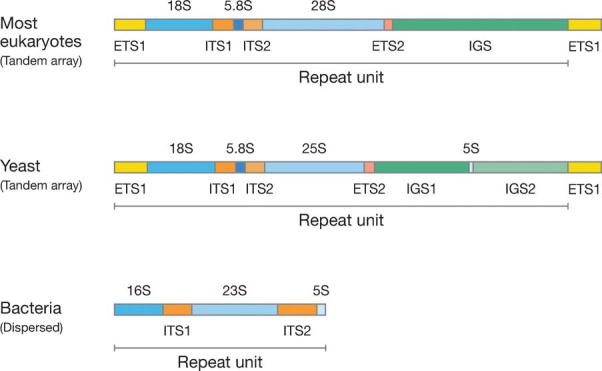
Molecular structures of rRNA gene repeats in different organisms.
Concerted evolution: a form of multigene family evolution in which all the member genes are assumed to evolve as a unit in concert and a mutation occurring in a repeat spreads through the entire member genes by repeated occurrence of unequal crossover or gene conversion
Gene conversion: a form of nonreciprocal recombination in which a DNA segment of a recipient gene is copied from a donor gene
This puzzling observation can be explained by the model of horizontal or concerted evolution originally proposed by Brown et al. (11). According to this model, unequal crossover occurs randomly among members of a gene family, and repeated occurrence of unequal crossover has an effect of homogenizing the member genes, as mentioned above. In this case, the number of member genes may increase or decrease by chance, but a certain range of the number of genes is maintained because of the functional requirement. In the absence of mutation, this process will eventually lead to the homogeneity of all member genes of a family. In reality, of course, mutation always occurs, so that a gene family is expected to have some variant genes. It should now be clear that when a species diverges into two species and the gene cluster in each descendant species evolves independently, the cluster within each species tends to have similar gene copies because of unequal crossover, whereas the genes belonging to the cluster of the two species gradually diverge by mutation (Figure 1b). This is exactly what we observe with the IGS regions of rRNA genes in Xenopus. Later, Smith (144, 145) conducted computer simulations to show that concerted evolution indeed can explain the observation about the evolutionary change of the IGS region. As mentioned above, the 18S and 28S coding regions are virtually identical between X. laevis and X. mulleri as well as between different copies of the same species. This identity has apparently been maintained by strong purifying selection that operates for the coding regions. Thus, we can explain the entire set of observations about the rRNA gene family of Xenopus in terms of unequal crossover, mutation, and purifying selection.
In addition to these factors, gene conversion (55, 143) was proposed as a mechanism of homogenizing the member genes of multigene families. The role of gene conversion is similar to that of unequal crossover. The only difference is that the latter may increase or decrease the number of genes, whereas the former does not. The idea of gene conversion later became popular among theoretical population geneticists, partly because it is easy to develop mathematical models of concerted evolution (8, 90-92, 111-113, 161). In reality, the molecular mechanism of gene conversion in multigene families is not well understood, particularly when sequence identity is patchy, though gene conversion in yeast can be explained by a DNA breakage followed by invasive DNA replication (120). Furthermore, in most mathematical formulations the effect of purifying selection operating in the coding regions of 18S and 28S genes has been neglected. Therefore, caution should be exercised in the application of the mathematical formulas to real data. The gene conversion theory has also become popular among researchers of MHC polymorphism, as is discussed below.
Note that the relative contributions of unequal crossover (or gene conversion) and purifying selection to the homogenization of the rRNA genes have rarely been discussed. For this reason, the homogeneity of the rRNA-coding regions (18S and 28S) was often attributed to unequal crossover rather than to purifying selection. Actually, even the IGS regions appear to be subject to purifying selection in Xenopus because this region contains elements of promoters and enhancers (15, 132). Furthermore, the ITS regions have a level of variability as low as that of the rRNA-coding regions in the fungus Fusarium graminearum (K. O'Donnell, personal communication). This has probably occurred because they are closely linked to the highly conserved rRNA-coding regions or because they have some important functions. It is therefore necessary to keep in mind that concerted evolution applies primarily to the IGS region, and even in this region a substantial proportion of mutations may be eliminated by purifying selection.
In recent years, the so-called complete genome sequence has been published for many different organisms. Unfortunately, the rRNA gene regions are usually excluded from the sequence, mainly because of the difficulty of sequencing a large number of similar genes. In humans, chimpanzees, gorillas, and orangutans, however, some sequence data are available for a small portion of the rRNA gene regions (35). The hominoid genome has about 500 rRNA gene repeats, and the molecular structure of each repeat unit is similar to that of Xenopus (15). However, hominoid rRNA genes are clustered in the telomeric regions of five different chromosomes. The pattern of sequence similarity among the IGS, 18S, and 28S regions of each chromosome is very similar to that of Xenopus genes. The 18S and 28S gene regions are virtually identical within and between hominoid species, and the IGS regions from the same chromosomes are also similar and show only about 0.5% nucleotide differences.
The rRNA gene clusters closely located at the chromosomal ends are also very similar in each hominoid species, but the IGS regions distal to the telomere are somewhat differentiated (35). This observation suggests that genes are exchanged between different chromosomes through unequal crossover or gene conversion that occurs primarily in the chromosomal region proximal to the telomere. In fact, the distal IGS regions often showed a substantial amount of sequence difference (3 ∼ 7% per site).
The 5S RNA genes form separate gene clusters and are located on a different genomic region. This gene family includes 9000∼24,000 member genes in Xenopus (10) and about 500 members in humans (35). These 5S rRNA genes are also known to undergo concerted evolution (10). Furthermore, the gene families of small nuclear RNAs (snRNA) involved in intron-splicing and other important cellular metabolisms apparently undergo concerted evolution. An extensive study of concerted evolution of U2 snRNA genes in primates has been conducted by Weiner and his group (70, 122, 123), and these authors have shown that the coding regions of U2 snRNA gene members are very similar to one another but the intergenic regions are heterogeneous within each species. These results again demonstrate the importance of purifying selection in the coding regions.
In yeast, the basic unit of rRNA gene repeats includes an additional RNA gene (5S gene) in the middle of the IGS region, but IGS is again subject to concerted evolution (125, 146, 151, 170). In bacterial genomes there are only a few rRNA gene repeats, and they are generally dispersed in the genome (69). For example, Escherichia coli has 7 copies of the rRNA operon, which is composed of 16S, 23S, and 5S rRNA genes (Figure 2). The spacer (ITS1) between the 16S and 23S genes usually contains one or two tRNA genes, and these tRNA genes are not necessarily the same among different copies of the operon.
The rRNA-coding regions are again very similar among different operons. The sequence difference of the 16S gene among repeats is 0.0055 per site in E. coli and 0 in Haemophilus influenzae. However, the ITS1 is quite heterogeneous. This region often contains unique nucleotide sequences shared by only a few operons. These sequences are patchy and could represent traces of gene conversion events that occurred in the past. On the basis of these observations, Liao (69) concluded that the rRNA genes in bacteria are generally homogenized by gene conversion. However, these observations can also be explained by strong purifying selection and occasional unequal crossover. If unequal crossover occurs in the ITS1 regions as well as outside the rRNA operons, a unique nucleotide sequence in an ITS can be transferred to other ITSs. In this case, we would expect that the sequence length of ITS varies from operon to operon. In fact, this is the case in E. coli, and the length of the ITS1 between the 16S and 23S genes varies from 500 bp to 800 bp.
The above literature survey indicates that most RNA genes in both prokaryotes and eukaryotes are subject to concerted evolution. However, there are exceptions to this rule. The most conspicious is the rRNA genes in species of the malaria parasite plasmodia. In these species the number of rRNA gene is low (a few to a dozen copies), and they are dispersed in different chromosomes (68a, 86a). These genes are grouped into a few different classes in terms of function and structure. These different classes of rRNA genes are used in different stages of the life cycle of plasmodia, which infest both insects and vertebrates (68a). The gene sequences are similar within each class of rRNA genes but are different between different classes. The inter-class differences are often substantial and amount to about 10 percent. Conducting a phylogenetic analysis of the sequences from several closely related species, Rooney (135) concluded that the gene family in plasmodia actually evolve following the birth-and-death model. The existence of different classes of rRNA genes in the same genome has been reported in several other organism such as flat-worms (14a) and oak tree (89a).
The 5S RNA gene family is also heterogeneous in some species. The best known example is that of filamentus fungi. The number of copies of 5S RNA genes in these species is 50 ∼ 100, and they are dispersed in the genome rather than organized as a tandem array. They can also be grouped into a few different classes by means of sequence similarity. Studying the evolutionary pattern of the sequences from four species of this group of fungi, Rooney & Ward (137) concluded that this 5S RNA gene family is subject to birth-and-death evolution. They found that 18% to 83% of genes are pseudogenes. There are several other species in which heterogeneous 5S genes exists. Examples are Xenopus laevis (162a) and wheat (58a).
Tandemly Arrayed Histone Genes
Not long after the evolution of rRNA gene families was explained by the model of concerted evolution, many researchers began to assume that this model applies to various other multigene families (17, 110, 115, 144, 145). The general view then was that a gene family that produces a large amount of gene products is subject to concerted evolution to homogenize the genes. One such family was the histone gene family of sea urchins (20, 21, 44, 47, 57). This is a large multigene family with several hundred members that are divided into four classes on the basis of developmental and tissue-specific expression patterns (Figure 3): (a) “early histone genes” that are active during late oogenesis through the blastula stage of embryogenesis, (b) “cleavage stage histone genes” that encode the first histones expressed after fertilization, (c) “late histone genes” that are active from the late blastula stage onwards, and (d) “sperm histone genes” that are expressed only during spermatogenesis (19, 78, 83, 101, 102).
Figure 3.
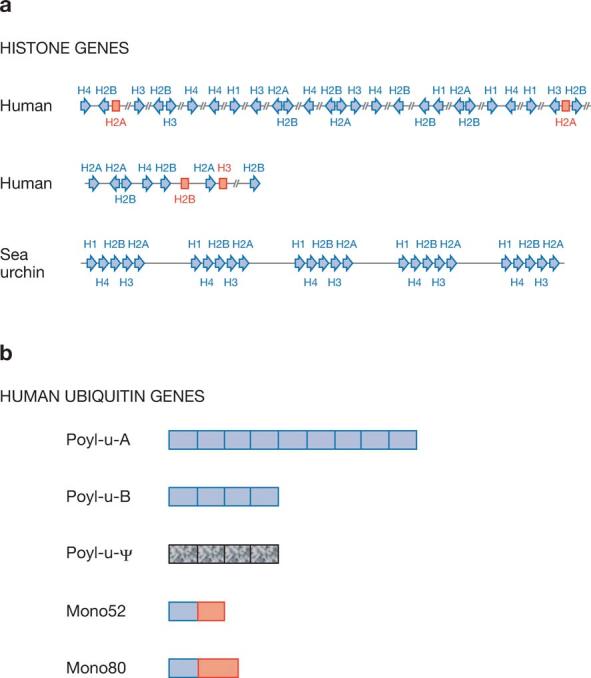
Genomic structures of histone gene (a) and ubiquitin gene repeats (b). The red square box in human histone gene repeats represents a pseudogene. Expressed histone genes are shown in blue, and an arrow indicates the direction of transcription when known. Each box in the poly-u gene complex represents one copy of ubiquitin gene. Poly-uΨ shows a poly-u pseudogene. A monomeric ubiquitin locus is composed of a ubiquitin gene and a ribosomal protein gene with 52 codons (Mono52) or 80 codons (Mono80).
The early histone genes are present in about 300–500 repeat units in most sea urchin species. In the sea urchin Lytechinus pictus, they are arranged in 3 tandem arrays that consist of virtually identical repeating units of the 5 histone genes (H1, H2A, H2B, H3, and H4), each of which is separated by noncoding IGS regions (21, 47) (Figure 3). In this species, only the early genes are arranged in tandem array, whereas the other three classes of genes appear to be dispersed throughout the genome and present in significantly fewer copy numbers (16, 78). Using DNA heteroduplex and restriction mapping analyses, it was demonstrated that the IGS regions of the early gene tandem arrays in L. pictus show a considerable amount of variation, whereas the protein-encoding regions are highly conserved (20, 21, 47). This was taken as evidence for concerted evolution in the early histone genes of this species. Not long afterwards, researchers studying the sequence divergence of late histone genes of L. pictus claimed that these genes also undergo concerted evolution (130), as did another study on the late genes of the sea urchin Strongylocentrotus purpuratus (83). However, it was later shown that histone genes generally evolve following a birth-and-death process with strong purifying selection (127, 136), as discussed below.
Like the sea urchin genes, the 5 histone genes in Drosophila melanogaster are arranged in a repeating unit (71). This unit is repeated about 110 times in a tandem array found on chromosome 2L (121). Two types of repeat units that differ with respect to length (5.0 kb and 4.8 kb) have apparently arisen owing to differences in the noncoding region (71). In addition to the tandem array, so-called variant histone genes are located in other parts of the genome (119). On the basis of restriction fragment patterns, Coen et al. (18) argued that the histone genes of D. melanogaster and its close relatives underwent concerted evolution. According to these authors, the absence of different banding patterns within a species was evidence for concerted evolution, because a restriction site must have spread to all other repeat units after it had arisen subsequent to the divergence of Drosophila species. However, this line of inference based on negative data should not be used as support for concerted evolution.
Matsuo & Yamazaki (82) later obtained nucleotide sequences of several different histone H3 genes and their flanking regions from Drosophila. They obtained data from 10 clones of a single chromosome (a single individual), 10 clones from different chromosomes (a population sample) of D. melanogaster, and a single clone from a single chromosome of a sibling species, D. simulans. They found that variability within an individual was 1.7 times less than that of a population sample and 14 times less than the interspecific variation. Using complex mathematical models, they argued that these observations can be explained by concerted evolution. However, their sequence data indicate that the number of polymorphic sites is quite small and is not substantially different between the protein coding and flanking regions.
Pseudogenes: nonfunctional genes generated by nonsense mutation, frameshift mutation, or partial nucleotide deletion
By the end of the 1980s, most researchers in the field had concluded that virtually all multigene families evolve in a concerted fashion. Therefore, the studies with sea urchin and Drosophila histone genes mentioned above were received as confirmation of the general view. No one considered the possibility that purifying selection is an alternative explanation for the similarity of histone sequences. This happened because the above studies, which were often performed using restriction fragment analysis, did not have sufficient power to distinguish between intralocus and interlocus variation of histone genes. Recently, this view has changed, as discussed below.
Other Genes
There are many other genes for which concerted evolution has been reported. A well-known example is the heatshock protein gene (hsp70 genes) in Drosophila. D. melanogaster has a relatively large family of hsp genes, of which two, hsp70Aa and hsp70Ab, are a pair of inverted tandem repeats, and the nucleotide sequences of the two genes are virtually identical. D. simulans, a sibling species of D. melanogaster, also has a similar tandem pair of nearly identical hsp70 genes. Finding these pairs of genes, Leigh Brown & Ish-Horowicz (68) and Bettencourt & Feder (7) proposed that the within-species identity of the two genes is caused by gene conversion. This is certainly a plausible explanation, but if we consider the evolution of the entire members of this gene family, the evolutionary pattern does not necessarily conform to concerted evolution (see below).
There are many reports about possible gene conversion in small multigene families. One example is that between the two loci of Rh blood group genes in hominoids (14b, 60). Conducting a detailed statistical analysis, Kitano & Saitou (60) reported that several gene conversions occurred in each of humans, chimpanzees, and gorillas. However, it is very difficult to distinguish between gene conversion and independent nucleotide substitution in their case (65a). Their results are also dependent on the phylogenetic tree of the genes used. Therefore, their conclusion is questionable. Furthermore, the sequence identity of the two genes was rather low compared with the average identity of genes from humans and chimpanzees. A more detailed study is necessary about this gene family.
Many authors have claimed that the genetic variability of MHC loci is caused by gene conversion, and this was thought to be a source of genetic variability within loci rather than a homogenizing factor (36, 73, 86, 112, 164). Similar hypotheses were presented to explain the high degree of antibody diversity (110). These views remained popular until a more realistic model of maintenance of polymorphism was proposed.
BIRTH-AND-DEATH EVOLUTION
MHC Genes
The birth-and-death model of evolution of multigene families was first proposed to explain the unusual pattern of evolution of MHC genes in mammals (51, 65, 96, 98). The function of MHC genes is to bind self or foreign peptides and present them to T lymphocytes, thereby triggering an immune response (63). MHC genes can be divided into class I and class II genes on the basis of molecular structure and function of the polypeptide encoded. Class I genes can be further divided into classical and nonclassical genes. The classical class I (Ia) genes are highly polymorphic, and the number of alleles per locus sometimes exceeds 100. This high degree of polymorphism is important for protecting the host from attack by various types of parasites (viruses, bacteria, fungi, and others), which are always changing with time. By contrast, the nonclassical class I (Ib) genes are less polymorphic and their functions may be quite different from those of Ia genes.
In the 1970s and 1980s when most investigators believed that multigene families were generally subject to concerted evolution, the MHC gene family was no exception. Therefore, some authors attempted to explain the polymorphism of MHC genes by means of unequal crossover or gene conversion (74, 111, 112, 164). In particular, Ohta (112) and Weiss et al. (164) proposed that the high degree of polymorphism at Ia loci could be explained by gene conversion. This view is based on the idea that if some parts of a sequence at a monomorphic locus are converted by another nucleotide sequence from another locus, polymorphism is generated at the first locus. Enhancement of polymorphism would occur even if both loci are polymorphic to some extent as long as the nucleotide sequences between the two loci are sufficiently different and gene conversion occurs in both ways between the two loci. The problem with this idea is that it does not explain why gene conversion starts to occur between two previously differentiated loci suddenly at an evolutionary time. The coexistence of Ia and Ib genes in the same DNA region is also difficult to explain. If gene conversion occurs continuously between the two loci, the extent of polymorphism should be essentially the same for the two loci. Furthermore, if phylogenetic analysis is conducted for the alleles from different loci, there would be no monophyletic clades formed for each locus. In reality, this is not the case. Klein & Figueroa (62) and Kriener et al. (65a) critically examined data that seemingly supported the idea of concerted evolution and concluded that the evidence is weak. They argued that some of the data showing the identical gene segments between paralogous pairs of genes can be explained by co-ancestry of the segments or even clustered mutations (65a, 169).
The idea of gene conversion was weakened considerably when Hughes & Nei (50, 52) showed that MHC polymorphism is primarily caused by overdominant selection that operates at the peptide-binding region of MHC molecules. This finding made it unnecessary to invoke gene conversion as an explanation for MHC polymorphism. It also provided a theoretical basis for the concept of trans-specific (long-term) polymorphism previously discovered (5, 28, 62, 66, 85). In a phylogenetic analysis of MHC class I and class II genes, Hughes & Nei (52, 53, 97) showed that the evolutionary pattern of these genes was very different from what would be expected under concerted evolution. The tree for class I genes from a number of vertebrate species is presented in Figure 4a. It indicates that different orders or families of mammals often have different genes or genetic loci. For example, the classical loci A, B, and C are shared only by hominoid species (e.g., human, gorilla, and orangutan), but the New World monkeys (e.g., tamarin) and nonprimate mammals do not have the genes. Similarly, cats and mice have different Ia loci. In other words, different families or orders of mammals do not have truly orthologous genes. This evolutionary pattern indicates that some genes were generated by gene duplication and some duplicate genes were lost after the divergence of mammalian orders (51, 97). Actually, the genomic regions of human and mouse MHC genes contain a large number of pseudogenes (58), exactly as would be expected under the birth-and-death model (Figure 1c). Also, this conclusion makes sense biologically because the genetic variability at MHC loci is generated to defend the host from many new types of parasites. Gene conversion and unequal crossover are not essential for this purpose, though they may occur.
Figure 4.
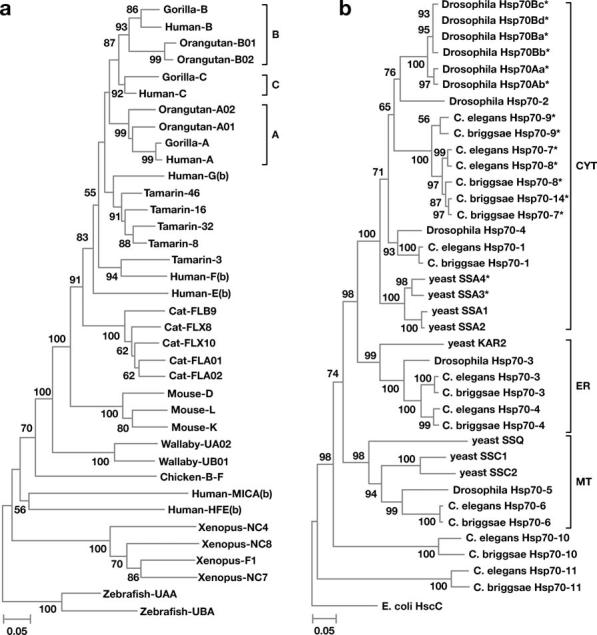
(a) Phylogenetic tree of MHC class I genes from vertebrates. (b) Phylogenetic tree of heat shock protein genes (hsp70) from two species of nematodes, Drosophila melanogaster, and yeast. CYT, ER, and MT stand for the genes expressed in cytoplasm, endoplasmic reticulum, and mitochondria, respectively. The * sign indicates a highly heat-inducible gene. Modified from Nei et al. (96) and Nikolaidis & Nei (109).
Figure 4a also shows that a few nonclassical loci as indicated by (b) diverged from Ia loci a long time ago and now have different functions, as discussed below. This type of acquisition of new functions by duplicate genes is also an important feature of birth-and-death evolution that is not shared by concerted evolution. Phylogenetic analysis of class II region genes showed essentially the same evolutionary pattern as that of class I genes (53). However, the rate of gene birth and gene death is much lower in the class II gene family than in the class I gene family (97, 126, 152).
Despite these findings, a substantial number of authors maintain that gene conversion or unequal crossover is an important factor for generating polymorphism (12, 114, 124). Some investigators invoked gene conversion to explain the identity of a single or a few nucleotides between different alleles at the same locus or different paralogous sequences (24, 162, 167). Actually, there is evidence that intralocus gene conversion or recombination occurs occasionally (6, 46, 79, 84, 138, 162, 172). Exon exchange between different loci has also been documented (40, 51). Furthermore, there is some molecular evidence that interlocus gene conversion occurs at MHC loci (2, 33, 41, 45, 46, 56, 168). Hogstrand & Bohme (45) reported that the frequency of occurence of gene conversion between the Ab and Eb loci in mice is about 2 × 10−6 per sperm per generation. If this estimate is right, it is much higher than the rate of nucleotide substitution (of the order of 10−9 per generation). Therefore, the nucleotide sequences of the two loci, Ab and Eb, would become nearly identical in the long run. In reality, however, a phylogenetic analysis of allelic sequences from loci Ab and Eb has shown that alleles from each locus form a monophyletic clade and the alleles from different loci do not intermingle (40). This is also generally true with the genes from different organisms (40), indicating that the effect of gene conversion on MHC polymophism is quite minor. These results suggest that either the current estimate of gene conversion frequency is too high or many gene conversions observed in the mouse experiment are not fixed in the natural population because of selective disadvantage compared with wild-type alleles (40). Martinsohn et al. (80) conducted a critical examination of papers claiming gene conversions and concluded that gene conversions or interlocus recombinations do occur but those that enhance polymorphism are not proven. This conclusion agrees with that of Gu & Nei (40). Therefore, MHC polymorphism appears to be primarily generated by nucleotide substitution and positive selection.
Immunoglobulin and Related Gene Families
Immunoglobulins are composed of heavy (H) chains and light chains, and the latter chains can be further divided into λ and κ chains. Each of the three groups of chains consists of constant and variable regions (63), and the polypeptides of variable regions in each category are encoded by a genomic cluster, called a variable region gene family. There are about 50 ∼ 100 genes in each of the H, λ, and κ variable region gene families in human. About 50% of these variable region genes are pseudogenes (81, 118). All of these multigene families are subject to birth-and-death evolution (116-118, 141, 142, 148). The molecular structure and the genomic organization of T-cell receptors are similar to those of immunoglobulins, and the variable region gene families for different classes of T-cell receptor genes are also known to be subject to birth-and-death evolutions (147, 149, 150). Most of these gene families include many pseudogenes. Therefore, the death rate of these genes is quite high. In both immunoglobulin and T-cell receptor families, individual functional genes apparently maintain their own identity without much effect from interlocus recombination or gene conversion, because the branch lengths of individual genes are usually very long and sometimes correspond to tens of millions of years. In the case of immunoglobulin κ variable region genes, the human genome contains about two times as many genes as does the chimpanzee because of a DNA block duplication that occurred in the human lineage about 2 mya. In this case, the orthologous genes between the original DNA segment and the new duplicated segment can easily be identified (141). This supports the idea that intergenic recombination and gene conversion have little effect on sequence evolution. Note also that the extent of polymorphism in these genes is much smaller than that of MHC genes.
In addition to the above multigene families, the gene families concerned with innate immunity have also been shown to undergo birth-and-death evolution (42, 43, 107, 108). The evolutionary pattern of these genes is more complex than those of the adaptive immune system. For example, the natural killer (NK) cell receptors of humans [KIR: killer cell immunoglobulin-like receptor (KIR genes)] are composed of immunoglobulin-like domains, but those of rodent receptors (Ly49) genes are of lectin-type, and the molecular structures of these two groups of genes are very different (63). It is unclear how these different types of NK cell receptors originated in two different orders of mammals. Both KIR and Ly49 gene families are known to be subject to birth-and-death evolution (42, 43). KIR genes are also subject to domain shuffling as well as to mutational changes (9, 42, 129, 156, 166). Furthermore, the number of member genes of these gene families has expanded very rapidly by gene duplication during the past 20–30 million years (42, 43, 59). Yet, about a half of these genes are apparently nonfunctional. For example, the rat genome contains 33 Ly49 genes, but 16 of them are pseudogenes (42).
Many other immune systems gene families undergo birth-and-death evolution (Table 1). In fact, almost all immune systems genes except solitary or small-sized gene families appear to be subject to this model of evolution, and the rate of gene turnover is generally quite high, as expected from their function.
Table 1.
Some examples of gene families that undergo birth-and-death evolution
| Multigene family | Organism | Reference |
|---|---|---|
| (A) Immune system | ||
| MHC | Vertebrates | (51, 96, 98) |
| Immunoglobulins | Vertebrates | (96, 116) |
| T-cell receptors | Vertebrates | (149) |
| Natural killer cell receptors | Mammals | (42, 43) |
| Eosinophilic RNases | Rodents | (173) |
| Disease resistance (R) loci | Plants | (87) |
| Cecropins | Drosophila | (128) |
| α-Defensins | Mammals | (75) |
| β-Defensins |
Mammals |
(89) |
| (B) Sensory system | ||
| Chemoreceptors | Nematodes | (131) |
| Taste receptors | Mammals | (22) |
| Sex pheromone desaturases | Insects | (72, 133) |
| Olfactory receptors |
Mammals |
(103, 165) |
| (C) Development | ||
| Homeobox genes | Animals | (160) |
| MADS-box | Plants | (94) |
| WAK-like kinase |
Arabidopsis |
(158) |
| (D) Highly conserved | ||
| Histones | Eukaryotes | (26, 127, 136) |
| Amylases | Drosophila | (177) |
| Peroxidases | All kingdoms | (171) |
| Ubiquitins | Eukaryotes | (99) |
| Nuclear ribosomal RNA |
Protists, Fungi |
(135, 137) |
| (E) Miscellaneous | ||
| DUP240 genes | Yeast | (67) |
| Polygalacturonases | Fungi | (37) |
| 3-Finger venom toxins | Snakes | (30) |
| Replication proteins | Nanoviruses | (49) |
| ABC transporters | Eukaryotes | (3, 4, 23) |
Olfactory Receptor Genes
The MHC and immunoglobulin variable gene families are quite large, but the largest gene family in mammals is the olfactory receptor (OR) gene family. Olfactory (odor molecules) receptors are G-protein–coupled receptors that contain 7 α-helical transmembrane regions. OR genes are about 310 codons long and have no introns in the coding regions. These genes are expressed in sensory neurons of olfactory epithelia in nasal cavities. The human and mouse genomes contain about 800 and 1400 genes, respectively; over 60% of human OR genes are pseudogenes whereas only about 25% of mouse OR genes are pseudogenes (Table 2) (32, 34, 103, 106, 168, 175, 176, 179). These genes are scattered over many different locations of almost all chromosomes, and they are generally arranged as a tandem array in each genomic location. It is relatively easy to identify the orthologous gene pairs between humans and mouse by conducting phylogenetic analysis. This suggests that gene conversion or unequal crossover has not occurred frequently and that the number of OR genes apparently has increased by tandem gene duplication and chromosomal rearrangement (Figure 5). There are not many traces of transposition of genes mediated by ransposons.
Table 2.
Numbers of 9 group (α, β, …, κ) OR genes in 6 different species of vertebrates
| Zebrafish |
Pufferfish |
X. tropicalis |
Chicken |
Mouse |
Human |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Func | All | Func | All | Func | All | Func | All | Func | All | Func | All | |
| α (I) | 0 | 0 | 0 | 0 | 2 | 6 | 9 | 14 | 115 | 163 | 57 | 102 |
| β (I) | 1 | 2 | 1 | 1 | 5 | 19 | 0 | 0 | 0 | 0 | 0 | 0 |
| γ (II) | 1 | 1 | 0 | 0 | 370 | 802 | 72 | 543 | 922 | 1228 | 331 | 700 |
| δ (I) | 44 | 55 | 28 | 61 | 22 | 36 | 0 | 0 | 0 | 0 | 0 | 0 |
| ε (I) | 11 | 14 | 2 | 2 | 6 | 17 | 0 | 0 | 0 | 0 | 0 | 0 |
| ζ (I) | 27 | 40 | 6 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| η | 16 | 23 | 5 | 24 | 3 | 6 | 0 | 0 | 0 | 0 | 0 | 0 |
| θ | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 |
| κ |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
0 |
0 |
0 |
0 |
0 |
| Total | 102 | 137 | 44 | 98 | 410 | 888 | 82 | 558 | 1037 | 1391 | 388 | 802 |
I; class I genes. II; class II genes. Func; functional genes. All; functional genes plus pseudogenes. From Niimura & Nei (106).
Figure 5.
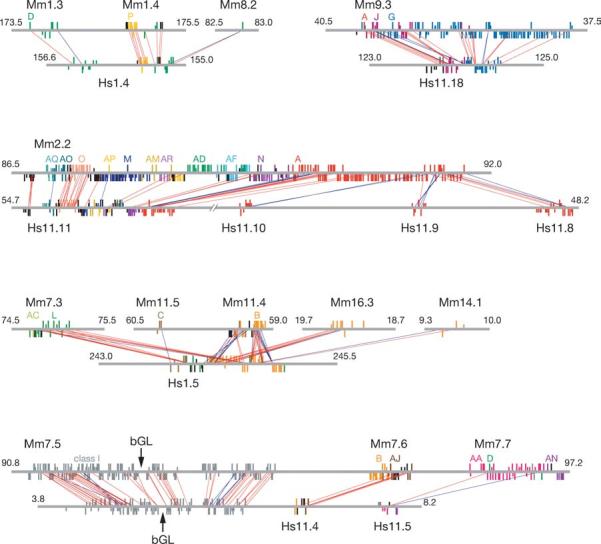
Orthologous relationships of class II OR genes between mouse (Mm) and human (Hs) genomic clusters. OR gene genomic clusters are indicated by chromosome number and Giemsa-stained band number in each species. Only a few chromosomes are presented. Long and short vertical bars show the locations of functional and nonfunctional OR genes, respectively. A vertical bar above a horizontal line indicates the transcriptional direction opposite to that below a horizontal line. Different colors represent different phylogenetic clades. Red and blue lines connecting mouse and human OR genes represent orthologous gene pairs. A red line indicates that transcriptional directions of orthologous genes are conserved between mice and humans, whereas a blue line indicates that they are inverted. Alphabetical letters such as A, J, G, and AQ represent different phylogenetic clades. Arrows show the location of the β-globin gene cluster (βGL). From Niimura & Nei (103).
As is shown in Table 2, the human and mouse genomes have about 390 and 1040 functional genes, respectively. To examine whether this is due to the loss of OR genes in the human lineage or the gain of genes in the mouse lineage, Niimura & Nei (105) estimated the number of functional OR genes that existed in the most recent common ancestor (MRCA) of humans and mice. The number obtained was approximately 754 genes. This indicates that the human lineage lost many functional OR genes, whereas the mouse lineage gained a substantial number of genes (Figure 6a). Some authors have suggested that mice require a larger number of OR genes because they are nocturnal and heavily dependent on olfaction, whereas humans do not need so many genes because they often use the visual sense for finding mates and food.
Figure 6.
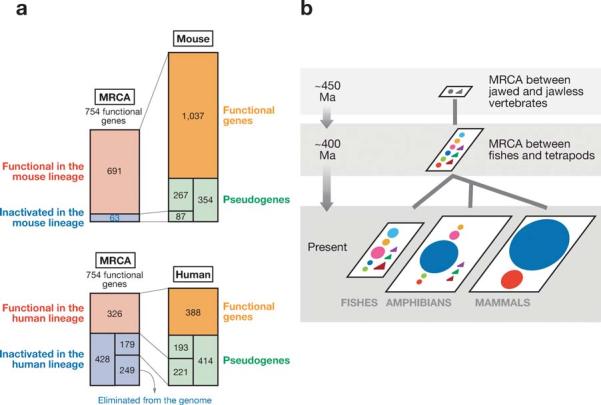
Evolutionary changes of functional OR genes. (a) The most recent common ancestor (MRCA) between humans and mice had approximately 754 functional OR genes. The number of functional OR genes increased to 1037 in the mouse lineage, whereas it declined to 388 in the human lineage. (b) The MRCA between jawed and jawless fishes had at least two functional genes, whereas the MRCA between fishes and tetrapods had at least 9 functional OR genes. Class II genes (blue circle) expanded enormously in amphibians and mammals.
In reality, the ability to smell is controlled not only by the number of OR genes but also by the brain function for odor recognition, and the human brain possibly has a higher power of distinguishing between subtle differences of odor molecules than the mouse brain (140). At present, however, the mechanism of odor recognition in the brain is virtually unknown, and therefore we do not consider this factor in this paper.
As indicated in Table 2, mammalian OR genes can be divided into class I and class II genes. Previously, class I genes were thought to be for aquatic odorants, and class II genes for airborne odorants (29). In mammals and chickens, most genes belong to class II genes, whereas class I genes make up only 2.5% ∼ 13%. It was later found that the zebrafish has one class II OR genes, although the search for OR genes in fish is still incomplete. Conducting phylogenetic analysis of OR genes from fish, X. tropicalis, chickens, and humans, Niimura & Nei (106) estimated that the MRCA of these species had at least 9 genes and that one of them generated mammalian class I genes and another generated class II genes (Figure 6b). Amphibians also have additional class I and class II genes, as well as five other groups of genes. Fish appear to have eight different groups of genes, but none of them appears to be very common. These results indicate that the currently dominant class I and class II genes in mammals are relatively recent products of multiple gene duplication events.
Pheromone Receptor and Synthesis Genes
Pheromones are water-soluble chemicals emitted and are sensed by individuals of the same species to elicit reproductive behaviors or physiological character changes. In terrestrial vertebrates they are perceived by the vomeronasal organ (VNO), which is located at the base of the nasal cavity and is separated from the main olfactory epithelium. One supergene family that controls the VNO receptors is called the V1R (vomeronasal receptor 1) gene family, a member of which consists of about 330 codons without introns (25). The pheromone receptors are G-protein–coupled receptors as in the case of olfactory receptors, but there is little sequence similarity between the two proteins.
The mouse genome has about 350 genes, but the number of functional genes is 187. The rat genome has 102 functional genes and about 50 pseudogenes (39). Similarly, opossum and cow have a substantial number of functional genes. By contrast, the human has only 4 functional genes and nearly 200 pseudogenes. The number of functional genes in dogs is also quite small. The difference between the mouse and human genomes apparently occurred by massive pseudogenization of V1R genes in the human. In fact, some primate species including humans do not have functional VNOs and therefore are thought to have no perception of vomeronasal pheromone. The small number of functional V1R genes in humans seem to be relics of V1R gene pseudogenization. This pseudogenization in humans apparently occurred because humans use visual and auditory senses for sexual and physiological behavior. This example indicates that the number of copies of a multigene family can vary enormously even among different orders of mammals.
In insects a more advanced pheromone recognition system has developed. Mate finding in most moth species is achieved by female-emitted sex pheromones that are dispersed in a wide area. The key enzymes responsible for producing the pheromones are acetyl COA desaturases, which are encoded by a medium-sized gene family (133, 134). This gene family is also known to be subject to birth-and-death evolution.
BIRTH-AND-DEATH EVOLUTION WITH STRONG PURIFYING SELECTION
Histone Genes
As noted above, when genetic variation of multigene families was studied by restriction enzyme analysis, many gene families that are required to produce a large quantity of gene products were assumed to be subject to concerted evolution. One example is the histone gene family. In this gene family, even authors who studied nucleotide sequences maintained that histone gene families are subject to concerted evolution (82). This view arose from their preconception about gene conversion, as well as the fact that the number of sequences studied was small.
By the 1990s, however, a substantial number of sequences of histone genes had been accumulated from various species of animals, plants, fungi, and protists. Rooney et al. (136) and Piontkivska et al. (127) conducted an extensive statistical analysis of these data to examine whether the histone gene families are subject to concerted evolution or birth-and-death evolution. They reasoned that if concerted evolution is the main factor, both the number of synonymous differences per synonymous site (pS) and the number of nonsynonymous differences per nonsynonymous site (pN) must be virtually 0 for any pair of genes because gene conversion affects both synonymous and nonsynonymous sites in the same way. By contrast, if protein similarity is caused by purifying selection but every member gene evolves independently, pS is expected to be greater than pN because in this case synonymous substitutions accumulate continuously whereas nonsynonymous substitutions do not.
When this approach was applied to histone H3 and H4 genes from diverse groups of organisms, pS was clearly higher than pN in almost all cases (Table 3) (59a, 127, 136). Similar results were also obtained from an extensive study of the histone H1 gene family (26). These results therefore clearly show that the histone gene families are subject to strong purifying selection but all member genes evolve according to a birth-and-death process. Of course, occasionally there were cases in which pS and pN were both 0 or close to 0. For such a case,> there is a possibility that gene conversion has occurred. It can also be explained by recent gene duplication. Since the number of cases in which pS > pN were overwhelmingly large, Rooney et al. and Piontkivska et al. concluded that histone genes are generally subject to birth-and-death evolution with strong purifying selection.
Table 3.
The pS values (below diagonal) and pN values (above diagonal) ( × 100) observed between representative histones H3 sequences from human and mouse
| Human 1 | Human 4 | Human 16 | Mouse 2 | Mouse 8 | Mouse 10 | |
|---|---|---|---|---|---|---|
| Human 1 | — | 4.0 | 0.0 | 0.3 | 0.7 | 0.7 |
| Human 4 | 53.6 | — | 4.0 | 0.3 | 0.7 | 1.4 |
| Human 16 | 54.3 | 49.1 | — | 0.3 | 0.7 | 1.0 |
| Mouse 2 | 50.0 | 40.1 | 46.2 | — | 4.0 | 7.0 |
| Mouse 8 | 53.6 | 39.2 | 47.0 | 10.4 | — | 0.0 |
| Mouse 10 | 57.0 | 40.9 | 48.7 | 19.2 | 16.7 | — |
Ubiquitin Genes
Ubiquitin is a small protein consisting of 76 amino acids that plays a major role in both cellular processes and protein degradation in eukaryotes. It is one of the most conserved proteins, and 72 of the 76 amino acids appear to be invariant in animals, plants, and fungi (38). Ubiquitins are encoded by a small- to medium-size gene family, which comprises monomeric and polymeric genes. Monomeric genes consist of 228 nucleotides (76 codons) with an additional sequence that encodes a ribosomal protein (Figure 3b). By contrast, polymeric genes known as polyubiquitins (poly-u) are composed of tandem repeats of a 76-codon gene with no spacer sequence between them. The number of ubiquitin units in a poly-u locus, the number of poly-u loci, and the number of monomeric genes per genome vary considerably among eukaryotic species. Yet all ubiquitin genes encode the same amino acid sequence in each species.
These properties of the ubiquitin genes clearly indicate the importance of purifying selection. However, this multigene family was thought to be subject to concerted evolution until recently (88, 100, 139, 153, 159). This view rested partly on the preconception by many authors that homogenization of member genes of a family is caused by concerted evolution and partly on the fact that they generally worked with the genes from a few closely related species.
This view changed after 2000 when Nei et al. (99) conducted an extensive statistical analysis of all available data. The results of this study clearly indicated that it is purifying selection rather than concerted evolution that homogenizes protein sequences. In most species, nonsynonymous nucleotide differences among the member genes was 0, whereas the synonymous differences were virtually saturated (see Table 4). Of course, some pairs of member genes showed small nucleotide differences apparently because of recent gene duplication.
Table 4.
The pS values ( × 100) for intraspecific and interspecific comparisons of ubiquitin poly-u B loci from humans (H1, H2, and H3) and mice (M1, M2, M3, and M4). All the pN values were 0. From Nei et al. (99)
| Gene | H1 | H2 | H3 | M1 | M2 | M3 |
|---|---|---|---|---|---|---|
| H1 | ||||||
| H2 | 12 | |||||
| H3 | 19 | 7 | ||||
| M1 | 28 | 24 | 25 | |||
| M2 | 30 | 24 | 25 | 7 | ||
| M3 | 31 | 25 | 26 | 4 | 9 | |
| M4 | 27 | 22 | 24 | 15 | 7 | 16 |
MIXED PROCESS OF CONCERTED AND BIRTH-AND-DEATH EVOLUTION
As noted above, the pair of inverted repeat genes hsp70Aa and hsp70Ab for heat shock proteins in Drosophila has virtually identical nucleotide sequences and therefore it probably represents one of the best cases of gene conversion. However, this does not mean that all hsp70 genes for this highly conserved protein gene family are subject to concerted evolution. Figure 4b shows a phylogenetic tree for hsp70 genes from D. melanogaster, two species of nematodes (Caenorhabditis elegans and C. briggsae), and yeast. The sequence identity of hsp70Aa and hsp70Ab is clear from this tree, but other Drosophila hsp70 genes do not necessarily show a pattern of concerted evolution, though heat-inducible genes (marked *) are generally closely related. For example, hsp70-2, hsp70-3, and hsp70-4 are quite different from hsp70Aa or hsp70Ab.
In C. elegans, the protein sequences of hsp70-7 and hsp70-8, a pair of inverted repeats, are identical, but other hsp70 genes are not necessarily closely related. Similarly, in C. briggsae, genes hsp70-7 and hsp70-14 are closely related to each other but other genes are not necessarily so. Actually, there are many C. briggsae genes that pair with C. elegans genes. These pairs of genes diverged before the separation of the two species, and therefore they have evolved independently for a long time (50 ∼ 100 million years). Furthermore, yeast genes show no indication of concerted evolution. We can therefore conclude that the heat shock gene families have been subject to both concerted and birth-and-death evolution.
Essentially the same situation was observed with another highly conserved family of amylase genes in Drosophila (177). Some species (e.g., D. orena, D. lini, and D. kikkawai) of Drosophila have a pair of identical genes, but most other species do not. A phylogenetic analysis of 49 active genes by Zhang et al. (177) showed that some gene lineages were lost from the genome while some other genes were duplicated. These observations suggest that this gene family is subject to a mixture of concerted and birth-and-death evolution.
Historically, a well-known gene family that is apparently subject to a mixed process of concerted and birth-and-death evolution is the α-like globin gene family. The human genome contains 4 functional genes (α, α2, θ1, and ζ) and 3 pseudogenes (ψa1, ψα2, and ψζ). The α1 and α2 genes are virtually identical within hominoids (178), but other genes are considerably differentiated. In practice, the evolution of the globin families is much more complicated than previously thought (A.P. Rooney, unpublished).
ORIGINS OF NEW GENETIC SYSTEMS GENERATED BY BIRTH-AND-DEATH EVOLUTION
Although single-locus genes can be the major determinants of some phenotypic characters such as color vision and the oxygen-transporting function of hemoglobins, most genetic systems or phenotypic characters are controlled by the interaction of multigene families. Here genetic systems mean any functional units of biological organization such as the olfaction (odor recognition) and adaptive immune system in vertebrates, flower development in plants, meiosis, and mitosis. Evolution of these genetic systems is obviously very complicated, but it appears that the only way to understand it is to know the evolution of each component multigene family and their interaction with other gene families using the simplest possible organisms. Note that if multigene families evolve following the model of birth-and-death processes, groups of new functional genes often evolve, whereas concerted evolution does not allow the functional differentiation of genes because all member genes are supposed to evolve as a unit in concert.
Previously, we noted that some MHC class Ib genes have functions different from those of Ia genes. For example, gene HFE in humans (Figure 4a) evolved independently of other class I genes and has acquired a new function. It now has the ability to form complexes with the receptor for iron-binding transferrins and thus regulate the uptake of dietary iron by cells of the intestine (27). A mutation of this gene is known to cause the genetic disease hemochromatosis. This is one example that was generated by a birth-and-death process. Figure 4b shows that heat shock protein genes are expressed in three different locations of a cell. These are also products of gene duplication and acquisition of new functions. Similarly, we have seen that olfactory genes diverged extensively from fish to mammals. In the following section, a few examples are considered in some details.
Adaptive Immune System
One of the best-studied cases of acquisition of new gene functions is the evolution of adaptive immune system in jawed vertebrates. In the adaptive immune system, lifelong immunity is maintained for certain groups of parasites (viruses, bacteria, fungi, and others) once the host is attacked by them (e.g., the immunity to smallpox viruses). However, the jawless vertebrates and other nonvertebrate animals do not have this system, though most animals have the so-called innate immune system, which defends the host from parasites but does not retain memory of past attacks (63).
How did the adaptive immune system evolve? This is still a mystery and is currently under active investigation (14, 64, 157). However, it is well known that this system works through the interaction of many different multigene families such as the MHC, immunoglobulin, and T-cell receptor gene families. Most of these multigene families are evolutionarily related and are apparently products of long-term birth-and-death evolution. Therefore, it seems that continuous operation of birth-and-death evolution has generated a new genetic system. Klein & Nikolaidis (64) argued that the adaptive immune system evolved by assembling elements that have evolved primarily to serve other functions and incorporated existing molecular cascades, resulting in the appearance of new organs and new types of cells.
Animal Development
Homeobox genes are member genes of an important supergene family that control animal and plant development. They encode transcription factors and can be divided into two groups; Typical and Atypical groups. Typical homeobox genes contain a homeobox of 60 codons, whereas Atypical group genes have a homeobox with either a few more or less codons (13). The Typical group includes several dozen gene families. A well-known example is the HOX gene family that plays a key role in animal body patterning. This family has also undergone birth-and-death evolution (1). Another example is the PAX6 gene family that controls the eye development (31). The Atypical group includes about seven gene families, five of which are called the TALE group. The TALE group genes are characterized by three extra codons between the helix 1 and helix 2 regions. These gene families were all concerned with some aspects of development in eukaryotes and were derived from a common ancestor that existed before the separation of animals, plants, and fungi. In other words, these diverse gene families were generated by successive gene duplication and differentiation over a long period of time. Interestingly, occasional loss of paralogous genes also appears to contribute to the differentiation of phenotypic characters (J. Nam & M. Nei, unpublished).
Olfactory System in Vertebrates
Another example of the origin of new genetic system by birth-and-death evolution is the evolution of the olfactory system in vertebrates. We have seen that fish and mammals have quite different OR gene families, which have apparently adapted to receive different odorants that are available. Furthermore, class II OR genes are a relatively new invention in vertebrate evolution, and this gene family contained an enormously large number of genes. Yet, many of the genes are pseudogenes. It is now known that the class II OR genes can be divided into many subfamilies, each specialized to receive a given group of orders such as good smell or bad smell (77, 140). Because this system falls within the realm of neuroscience (175), it is outside the scope of this review.
Flower Development in Plants
Flowers of angiosperms (flowering plants) are composed of sepals, petals, stamens, pistils, etc., and differ from poorly developed flower-like organs in gynosperms (seed plants). The development of flower organs are controlled by transcription factor genes called MADS-box genes. There are several classes of MADS-box genes that are essential for flower development (76, 155, 163). In a phylogenetic analysis of MADS-box genes, Nam et al. (93) predicted that MADS-box genes controlling flower development (floral MADS-box genes) originated about 650 mya. Tanabe et al. (154) identified floral MADS-box like genes in three species of green algae, which are believed to have originated about 700 mya. If we note that the oldest fossil records of angiosperms and gymnosperms are about 150 and 300 million years old, respectively, it appears that the ancestral genes of floral MADS-box existed a long time ago before the flowering system evolved. Tanabe et al. speculate that this group of genes originally controlled the development of haploid and diploid stages of green algae. MADS-box genes are ancient genes and are known to exist in plants, animals, and fungi. In animals they control muscle development. In the process of evolution of gymnosperms and angiosperms, however, different MADS-box genes appear to have evolved to form flowers (93).
CONCLUSIONS
In this paper we have discussed the controversy over the models of concerted and birth-and-death evolution. The model of concerted evolution was originally thought to apply to gene families that are responsible for producing a large quantity of the same gene products, as in the case of rRNA genes. However, the production of a large quantity of the same gene products can also be achieved by strong purifying selection without concerted evolution. In fact, the histone and ubiquitin genes use this strategy, and the underlying DNA evolution occurs as a birth-and-death process. It is therefore important to distinguish between purifying selection and concerted evolution in producing homogeneous gene products. In the past, a number of authors have assumed that gene products are homogenized only by concerted evolution.
There are some exceptions to the above statement. Relatively small gene families with strong purifying selection such as the heat shock protein and amylase gene families undergo a mixed process of concerted and birth-and-death evolution. In these families, a pair of inverted gene sequences appears to be particularly susceptible to gene conversion or gene conversion-like events that homogenize the pair of genes. It is possible that gene conversion occurs more easily for inverted gene pairs. However, other member genes are apparently subject to birth-and-death evolution.
The gene families that produce a variety of gene products are usually subject to birth-and-death evolution. This is quite reasonable, because this model of evolution promotes genetic variation. There are reports indicating that the extent of genetic variability is enhanced by gene conversion or unequal crossover. However, these processes are primarily for homogenization of member genes, and the interpretation of data supporting this idea should be reexamined. This does not mean that gene conversion or unequal crossover never occurs in these genes. Actually, there is some evidence for the occurrence of gene conversion or domain shuffling in MHC genes. However, the contribution of these events to the diversification of multigene families in long-term evolution seems to be minor. The controversy over the concerted and birth-and-death evolution has occurred partly because of misunderstandings and misconceptions. Since each author usually works with one gene family from a limited number of species, the results cannot be blindly extended to other genes or other organisms. It is important to examine each gene family carefully and derive objective conclusions.
We have indicated that the model of birth-and-death evolution would give a reasonable explanation of generation of new gene families but the model of concerted evolution cannot. However, how different subgroups of a gene family acquire new functions is not yet well understood. Since the generation of new gene clusters occurs largely by chance, the initial stage of evolution of multigene families could be fortuitous. However, the newly generated duplicate genes or gene families may evolve to interact with other existing gene families and promote the adaptation of organisms to new environments. In the future it will be important to examine the relative contribution of positive selection and random genetic drift in the evolution of multigene families.
We have also indicated that many genetic systems or important phenotypic characters are controlled by the interaction of a number of multigene families and therefore we may be able to understand the evolution of new genetic systems or new phenotypic characters by studying the evolution of component multigene families. We may then be able to study the interaction between different gene families. This will be one of the most important problems in evolutionary biology in the future.
In the 1960s little was known about the multigene families. Studying the rates of gene duplication and formation of pseudogenes, Nei (95) stated “there may be a great deal of duplicate genes and also nonsense DNA in today's vertebrates.” He also stated that “higher organisms including man have ample scope to evolve into various directions.” The first prediction is now confirmed, but the validity of the second prediction remains to be seen.
SUMMARY POINTS.
The controversy over the models of concerted and birth-and-death evolution of multigene families was reviewed with some historical background.
The model of concerted evolution is capable of explaining the evolutionary pattern of highly conserved ribosomal or other RNA genes that are essential for cell metabolism. However, the high conservation of the coding regions of these genes is primarily caused by purifying selection rather than by concerted evolution.
Until recently, the high conservation of some proteins such as histones was considered to be due to concerted evolution. Recent studies indicate that the conservation of these proteins is primarily caused by purifying selection, and their DNA sequences evolve following the model of birth-and-death evolution.
Some genes, such as heat shock protein and Drosophila amylase genes, appear to be subject to a mixed process of concerted and birth-and-death evolution.
Most gene families, particularly those producing variable gene products, are subject to birth-and-death evolution.
Although some investigators maintain that MHC polymorphism is enhanced by gene conversion, phylogenetic analysis of polymorphic alleles shows that the effect of this factor is minor in comparison to mutation and overdominant selection.
The model of birth-and-death evolution gives a reasonable explanation of the generation of new gene families, whereas the model of concerted evolution does not.
Most genetic systems such as the adaptive immune system in vertebrates are controlled by the interaction of many multigene families each of which undergoes a birth-and-death process. Therefore, the evolution of new genetic systems or phenotype characters is also caused by a large-scale birth-and-death evolution.
FUTURE DIRECTIONS.
The controversy over concerted and birth-and-death evolution is partly due to the fact that the molecular mechanism of gene conversion is not well understood except in fungi and bacteria and it is not clear how gene conversion occurs between distantly located member genes as in the case of MHC loci. It is therefore important to clarify the molecular mechanism of gene conversion in vertebrates.
Evolutionary relationships and functional differentiation of multigene families should be studied at the statistical and biochemical levels.
Evolution of new genetic systems or phenotypic characters should be studied by consideration of the interaction of multigene families.
ACKNOWLEDGMENT
We would like to thank Dan Graur, Li Hao, Jan Klein, Eddie Holmes, Jongmin Nam, Yoshihito Niimura, Naruya Saitou, Yoshiyuki Suzuki, and Jianzhi Zhang for their comments on an earlier version of the manuscript. This was supported by NIH grant GM020293-32 to M.N.
Footnotes
The U.S. Government has the right to retain a nonexclusive, royalty-free license in and to any copyright covering this paper.
LITERATURE CITED
- 1.Amores A, Suzuki T, Yan Y-L, Pomeroy J, Singer A, et al. Developmental roles of pufferfish Hox clusters and genome evolution in ray-fin fish. Genome Res. 2004;14:1–10. doi: 10.1101/gr.1717804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andersson L, Gustafsson K, Jonsson AK, Rask L. Concerted evolution in a segment of the first domain exon of polymorphic MHC class II beta loci. Immunogenetics. 1991;33:235–42. doi: 10.1007/BF00230500. [DOI] [PubMed] [Google Scholar]
- 3.Anjard C, Loomis WF. Evolutionary analyses of ABC transporters of Dictyostelium discoideum. Eukaryot. Cell. 2002;1:643–52. doi: 10.1128/EC.1.4.643-652.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Annilo T, Dean M. Degeneration of an ATP-binding cassette transporter gene, ABCC13, in different mammalian lineages. Genomics. 2004;84:34–46. doi: 10.1016/j.ygeno.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 5.Arden B, Klein J. Biochemical comparison of major histocompatibility complex molecules from different subspecies of Mus musculus: evidence for trans-specific evolution of alleles. Proc. Natl. Acad. Sci. USA. 1982;79:2342–46. doi: 10.1073/pnas.79.7.2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belich MP, Madrigal JA, Hildebrand WH, Zemmour J, Williams RC, et al. Unusual HLA-B alleles in two tribes of Brazilian Indians. Nature. 1992;357:326–29. doi: 10.1038/357326a0. [DOI] [PubMed] [Google Scholar]
- 7.Bettencourt BR, Feder ME. Rapid concerted evolution via gene conversion at the Drosophila hsp70 genes. J. Mol. Evol. 2002;54:569–86. doi: 10.1007/s00239-001-0044-7. [DOI] [PubMed] [Google Scholar]
- 8.Birky CWJ, Skavaril RV. Maintenance of genetic homogeneity in systems with multiple genomes. Genet. Res. 1976;27:249–65. doi: 10.1017/s001667230001644x. [DOI] [PubMed] [Google Scholar]
- 9.Brennan J, Lemieux S, Freeman JD, Mager DL, Takei F. Heterogeneity among Ly-49C natural killer (NK) cells: characterization of highly related receptors with differing functions and expression patterns. J. Exp. Med. 1996;184:2085–90. doi: 10.1084/jem.184.6.2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown DD, Sugimoto K. The structure and evolution of ribosomal and 5S DNAs in Xenopus laevis and Xenopus mulleri 505. Cold Spring Harbor Symp. Quant. Biol. 1974;38:501–8. doi: 10.1101/sqb.1974.038.01.054. [DOI] [PubMed] [Google Scholar]
- 11.Brown DD, Wensink PC, Jordan E. Xenopus laevis and Xenopus mulleri: the evolution of tandem genes. J. Mol. Biol. 1972;63:57–73. doi: 10.1016/0022-2836(72)90521-9. [DOI] [PubMed] [Google Scholar]
- 12.Brunsberg U, Edfors-Lilja I, Andersson L, Gustafsson K. Structure and organization of pig MHC class II DRB genes: evidence for genetic exchange between loci. Immunogenetics. 1996;44:1–8. doi: 10.1007/BF02602651. [DOI] [PubMed] [Google Scholar]
- 13.Burglin TR. Analysis of TALE superclass homeobox genes (MEIS, PBC, KNOX, Iroquois, TGIF) reveals a novel domain conserved between plants and animals. Nucleic Acids Res. 1997;25:4173–80. doi: 10.1093/nar/25.21.4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cannon JP, Haire RH, Litman GW. Identification of diversified genes that contain immunoglobulin-like variable regions in a protochordate. Nat. Immunol. 2002;3:1200–7. doi: 10.1038/ni849. [DOI] [PubMed] [Google Scholar]
- 14a.Carranza S, Giribet G, Ribera C, Baguna J, Riutort M. Evidence that two types of 18S rRNA coexist in the genome of Dugesia (Schmidtea) mediterranea (Platyhelminthes, Turbellaria, Tricladida) Mol. Biol. Evol. 1996;13:824–32. doi: 10.1093/oxfordjournals.molbev.a025643. [DOI] [PubMed] [Google Scholar]
- 14b.Carritt B, Kemp TJ, Poulter M. Evolution of the human RH (rhesus) blood group genes: a 50 year old prediction (partially) fulfilled. Hum. Mol. Genet. 1997;6:843–50. doi: 10.1093/hmg/6.6.843. [DOI] [PubMed] [Google Scholar]
- 15.Caudy AA, Pikaard CS. Xenopus ribosomal RNA gene intergenic spacer elements conferring transcriptional enhancement and nucleolar dominance-like competition in oocytes. J. Biol. Chem. 2002;277:31577–84. doi: 10.1074/jbc.M202737200. [DOI] [PubMed] [Google Scholar]
- 16.Childs G, Nocente-McGrath C, Lieber T, Holt C, Knowles JA. Sea urchin (Lytechinus pictus) late-stage histone H3 and H4 genes: characterization and mapping of a clustered but nontandemly linked multigene family. Cell. 1982;31:383–93. doi: 10.1016/0092-8674(82)90132-5. [DOI] [PubMed] [Google Scholar]
- 17.Coen E, Strachan T, Dover G. Dynamics of concerted evolution of ribosomal DNA and histone gene families in the melanogaster species subgroup of Drosophila. J. Mol. Biol. 1982;158:17–35. doi: 10.1016/0022-2836(82)90448-x. [DOI] [PubMed] [Google Scholar]
- 18.Coen ES, Thoday JM, Dover G. Rate of turnover of structural variants in the rDNA gene family of Drosophila melanogaster. Nature. 1982;18:564–68. doi: 10.1038/295564a0. [DOI] [PubMed] [Google Scholar]
- 19.Cohen LH, Newrock KM, Zweidler A. Stage-specific switches in histone synthesis during embryogenesis of the sea urchin. Science. 1975;190:994–97. doi: 10.1126/science.1237932. [DOI] [PubMed] [Google Scholar]
- 20.Cohn RH, Kedes LH. Nonallelic histone gene clusters of individual sea urchins (Lytechinus pictus): mapping of homologies in coding and spacer DNA. Cell. 1979;18:855–64. doi: 10.1016/0092-8674(79)90137-5. [DOI] [PubMed] [Google Scholar]
- 21.Cohn RH, Kedes LH. Nonallelic histone gene clusters of individual sea urchins (Lytechinus pictus): polarity and gene organization. Cell. 1979;18:843–53. doi: 10.1016/0092-8674(79)90136-3. [DOI] [PubMed] [Google Scholar]
- 22.Conte C, Ebeling M, Marcuz A, Nef P, Andres-Barquin J. Evolutionary relationships of the Tas2r receptor gene families in mouse and human. Physiol. Genomics. 2003;14:73–82. doi: 10.1152/physiolgenomics.00060.2003. [DOI] [PubMed] [Google Scholar]
- 23.Dean M, Rhetzky A, Allikmets R. The human ATP-Binding Cassette (ABC) transporter superfamily. Genome Res. 2001;11:1156–66. doi: 10.1101/gr.184901. [DOI] [PubMed] [Google Scholar]
- 24.Dormoy A, Reviron DV, Froelich N, Weiller PJ, Mercier PJ, Tongio MM. Birth of a new allele in a sibling: cis or trans gene conversion during meiosis? Immunogenetics. 1997;46:520–23. doi: 10.1007/s002510050314. [DOI] [PubMed] [Google Scholar]
- 25.Dulac C, Axel R. A novel family of genes encoding putative pheromone receptors in mammals. Cell. 1995;83:195–206. doi: 10.1016/0092-8674(95)90161-2. [DOI] [PubMed] [Google Scholar]
- 26.Eirin-Lopez JM, Gonzalez-Tizon AM, Martinez A, Mendez J. Birth-and-death evolution with strong purifying selection in the histone H1 multigene family and the origin of orphon H1 genes. Mol. Biol. Evol. 2004;21:1992–2003. doi: 10.1093/molbev/msh213. [DOI] [PubMed] [Google Scholar]
- 27.Feder JN, Penny DM, Irrinki A, Lee VK, Lebron JA, et al. The hemochromatosis gene product complexes with the transferrin receptor and lowers its affinity for ligand binding. Proc. Natl. Acad. Sci. USA. 1998;95:1472–77. doi: 10.1073/pnas.95.4.1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Figueroa F, Gunther E, Klein J. MHC polymorphism pre-dating speciation. Nature. 1988;335:265–67. doi: 10.1038/335265a0. [DOI] [PubMed] [Google Scholar]
- 29.Freitag J, Ludwig G, Andreini I, Rossler P, Breer H. Olfactory receptors in aquatic and terrestrial vertebrates. J. Comp. Physiol. A. 1998;183:635–50. doi: 10.1007/s003590050287. [DOI] [PubMed] [Google Scholar]
- 30.Fry BG, Wuster W, Kini RM, Brusic V, Khan S, et al. Molecular evolution and phylogeny of elapid snake venom three-finger toxins. J. Mol. Evol. 2003;57:110–29. doi: 10.1007/s00239-003-2461-2. [DOI] [PubMed] [Google Scholar]
- 31.Gehring W. Master Control Genes in Development and Evolution: The Homeobox Story. Yale Univ. Press; New Haven/London: 1998. [Google Scholar]
- 32.Glusman G, Yanai I, Rubin I, Lancet D. The complete human olfactory subgenome. Genome Res. 2001;11:685–702. doi: 10.1101/gr.171001. [DOI] [PubMed] [Google Scholar]
- 33.Go Y, Satta Y, Kawamoto Y, Rakotoarisoa G, Randrianjafy A, et al. Frequent segmental sequence exchanges and rapid gene duplication characterize the MHC class I genes in lemurs. Immunogenetics. 2003;55:450–61. doi: 10.1007/s00251-003-0613-6. [DOI] [PubMed] [Google Scholar]
- 34.Godfrey PA, Malnic B, Buck LB. The mouse olfactory receptor gene family. Proc. Natl. Acad. Sci. USA. 2004;101:2156–61. doi: 10.1073/pnas.0308051100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gonzalez IL, Sylvester JE. Human rDNA: evolutionary patterns within the genes and tandem arrays derived from multiple chromosomes. Genomics. 2001;736:255–63. doi: 10.1006/geno.2001.6540. [DOI] [PubMed] [Google Scholar]
- 36.Gorski J, Mach B. Polymorphism of human Ia antigens: gene conversion between two DR beta loci results in a new HLA-D/DR specificity. Nature. 1986;322:67–70. doi: 10.1038/322067a0. [DOI] [PubMed] [Google Scholar]
- 37.Gotesson A, Marshall JS, Jones DA, Hardham AR. Characterization and evolutionary analysis of a large polygalacturonase gene family in the oomycete plant pathogen Phytophthora cinnamomi. Mol. Plant-Microbe Interact. 2002;15:907–21. doi: 10.1094/MPMI.2002.15.9.907. [DOI] [PubMed] [Google Scholar]
- 38.Graham RW, Jones D, Candido EP. UbiA, the major polyubiquitin locus in Caenorhabditis elegans, has unusual structural features and is constitutively expressed. Mol. Cell Biol. 1989;9:268–77. doi: 10.1128/mcb.9.1.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grus WE, Shi P, Zhang Y-P, Zhang J. Dramatic variation of the vomeronasal pheromone receptor gene repertoire among five orders of placental and marsupial mammals. Proc. Natl. Acad. Sci. USA. 2005;102:5767–72. doi: 10.1073/pnas.0501589102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gu X, Nei M. Locus specificity of polymorphic alleles and evolution by a birth-and-death process in mammalian MHC genes. Mol. Biol. Evol. 1999;16:147–56. doi: 10.1093/oxfordjournals.molbev.a026097. [DOI] [PubMed] [Google Scholar]
- 41.Hanneman WH, Schimenti KJ, Schimenti JC. Molecular analysis of gene conversion in spermatids from transgenic mice. Gene. 1997;200:185–92. doi: 10.1016/s0378-1119(97)00416-2. [DOI] [PubMed] [Google Scholar]
- 42.Hao L, Nei M. Genomic organization and evolutionary analysis of Ly49 genes encoding the rodent natural killer cell receptors: rapid evolution by repeated gene duplication. Immunogenetics. 2004;56:343–54. doi: 10.1007/s00251-004-0703-0. [DOI] [PubMed] [Google Scholar]
- 43.Hao L, Nei M. Rapid expansion of killer cell immunoglobulin-like receptor genes in primates and their coevolution with MHC Class I genes. Gene. 2005;347:149–59. doi: 10.1016/j.gene.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 44.Hentschel CC, Birnstiel ML. The organization and expression of histone gene families. Cell. 1981;25:301–13. doi: 10.1016/0092-8674(81)90048-9. [DOI] [PubMed] [Google Scholar]
- 45.Hogstrand K, Bohme J. A determination of the frequency of gene conversion in unmanipulated mouse sperm. Proc. Natl. Acad. Sci. USA. 1994;91:9921–25. doi: 10.1073/pnas.91.21.9921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hogstrand K, Bohme J. Gene conversion of major histocompatibility complex genes is associated with CpG-rich regions. Immunogenetics. 1999;49:446–55. doi: 10.1007/s002510050518. [DOI] [PubMed] [Google Scholar]
- 47.Holt CA, Childs G. A new family of tandem repetitive early histone genes in the sea urchin Lytechinus pictus: evidence for concerted evolution within tandem arrays. Nucleic Acids Res. 1984;12:6455–71. doi: 10.1093/nar/12.16.6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hood L, Campbell JH, Elgin SCR. The organization, expression, and evolution of antibody genes and other multigene families. Annu. Rev. Genet. 1975;9:305–53. doi: 10.1146/annurev.ge.09.120175.001513. [DOI] [PubMed] [Google Scholar]
- 49.Hughes AL. Birth-and-death evolution of protein-coding regions and concerted evolution of non-coding regions in the multi-component genomes of nanoviruses. Mol. Phylogenet. Evol. 2004;30:287–94. doi: 10.1016/s1055-7903(03)00189-1. [DOI] [PubMed] [Google Scholar]
- 50.Hughes AL, Nei M. Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature. 1988;335:167–70. doi: 10.1038/335167a0. [DOI] [PubMed] [Google Scholar]
- 51.Hughes AL, Nei M. Evolution of the major histocompatibility complex: independent origin of nonclassical class I genes in different groups of mammals. Mol. Biol. Evol. 1989;6:559–79. doi: 10.1093/oxfordjournals.molbev.a040573. [DOI] [PubMed] [Google Scholar]
- 52.Hughes AL, Nei M. Nucleotide substitution at major histocompatibility complex II loci: evidence for overdominant selection. Proc. Natl. Acad. Sci. USA. 1989;86:958–62. doi: 10.1073/pnas.86.3.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hughes AL, Nei M. Evolutionary relationships of class II major-histocompatibility-complex genes in mammals. Mol. Biol. Evol. 1990;7:491–514. doi: 10.1093/oxfordjournals.molbev.a040622. [DOI] [PubMed] [Google Scholar]
- 54.Ingram VM. Gene evolution and the haemoglobins. Nature. 1961;189:704–8. doi: 10.1038/189704a0. [DOI] [PubMed] [Google Scholar]
- 55.Jeffreys A. DNA sequence variants in the Gγ-, Aγ-, δ- and β-globin genes of man. Cell. 1979;18:1–10. doi: 10.1016/0092-8674(79)90348-9. [DOI] [PubMed] [Google Scholar]
- 56.Jeffreys AJ, May CA. Intense and highly localized gene conversion activity in human meiotic crossover hot spots. Nat. Genet. 2004;36:151–56. doi: 10.1038/ng1287. [DOI] [PubMed] [Google Scholar]
- 57.Kedes LH. Histone genes and histone messengers. Annu. Rev. Biochem. 1979;48:837–70. doi: 10.1146/annurev.bi.48.070179.004201. [DOI] [PubMed] [Google Scholar]
- 58.Kelley J, Walter L, Trowsdale J. Comparative genomics of major histocompatibility complexes. Immunogenetics. 2005;56:683–95. doi: 10.1007/s00251-004-0717-7. [DOI] [PubMed] [Google Scholar]
- 58a.Kellogg EA, Appels R. Intraspecific and interspecific variation in 5S RNA genes are decoupled in diploid wheat relatives. Genetics. 1995;140:325–43. doi: 10.1093/genetics/140.1.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Khakoo SI, Rajalingam R, Shum BP, Weidenbach K, Flodin L, et al. Rapid evolution of NK cell receptor systems demonstrated by comparison of chimpanzees and humans. Immunity. 2000;12:687–98. doi: 10.1016/s1074-7613(00)80219-8. [DOI] [PubMed] [Google Scholar]
- 59a.Kim HN, Yamazaki T. Nonconcerted evolution of histone 3 genes in a liverwort, Conocephalum conicum. Genes Genet. Syst. 2004;79:331–44. doi: 10.1266/ggs.79.331. [DOI] [PubMed] [Google Scholar]
- 60.Kitano T, Saitou N. Evolution of Rh blood group genes have experienced gene conversions and positive selection. J. Mol. Evol. 1999;49:615–26. doi: 10.1007/pl00006583. [DOI] [PubMed] [Google Scholar]
- 61. Deleted in proof. [Google Scholar]
- 62.Klein J, Figueroa F. Evolution of the major histocompatibility complex. Crit. Rev. Immunol. 1986;6:295–386. [PubMed] [Google Scholar]
- 63.Klein J, Horejsi V. Immunology. 2nd ed. Blackwell Sci.; London: 1997. [Google Scholar]
- 64.Klein J, Nikolaidis N. The descent of the antibody-based immune system by gradual evolution. Proc. Natl. Acad. Sci. USA. 2005;102:169–74. doi: 10.1073/pnas.0408480102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klein J, Ono H, Klein D, O'hUigin C. The accordion model of MHC evolution. Prog. Immunol. 1993;8:137–43. [Google Scholar]
- 65a.Kriener K, O'hUigin C, Klein J. Conversion or convergence? Introns of primate DRB genes tell the true story. In: Kasahara M, editor. Major Histocompatibility Complex; Evolution, Structure, and Function. Springer-Verlag; Tokyo: 2000. pp. 354–76. [Google Scholar]
- 66.Lawlor DA, Ward FE, Ennis PD, Jackson AP, Parham P. HLA-A and B polymorphisms predate the divergence of humans and chimpanzees. Nature. 1988;335:268–71. doi: 10.1038/335268a0. [DOI] [PubMed] [Google Scholar]
- 67.Leh-Louis V, Wirth B, Despons L, Wain-Hobson S, Potier S, Souciet JL. Differential evolution of the Saccharomyces cerevisiae DUP240 paralogs and implication of recombination in phylogeny. Nucleic Acids Res. 2004;32:2069–78. doi: 10.1093/nar/gkh529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leigh Brown AJ, Ish-Horowicz D. Evolution of the 87A and 87C heat-shock loci in Drosophila. Nature. 1981;290:677–82. doi: 10.1038/290677a0. [DOI] [PubMed] [Google Scholar]
- 68a.Li J, Gutell RR, Damberger SH, Wirtz RA, Kissinger JC, et al. Regulation and trafficking of three distinct 18 S ribosomal RNAs during development of the malaria parasite. J. Mol. Biol. 1997;269:203–13. doi: 10.1006/jmbi.1997.1038. [DOI] [PubMed] [Google Scholar]
- 69.Liao D. Gene conversion drives within genic sequences: converted evolution of ribosomal RNA genes in bacteria and archaea. J. Mol. Evol. 2000;51:305–17. doi: 10.1007/s002390010093. [DOI] [PubMed] [Google Scholar]
- 70.Liao D, Pavelitz T, Weiner AM. Characterization of a novel class of interspersed LTR elements in primate genomes: structure, genomic distribution, and evolution. J. Mol. Evol. 1998;46:649–60. doi: 10.1007/pl00006345. [DOI] [PubMed] [Google Scholar]
- 71.Lifton RP, Goldberg ML, Karp RW, Hogness DS. The organization of the histone genes in Drosophila melanogaster; functional and evolutionary implications. Cold Spring Harbor Symp. Quant. Biol. 1977;2:1047–51. doi: 10.1101/sqb.1978.042.01.105. [DOI] [PubMed] [Google Scholar]
- 72.Liu W, Rooney AP, Xue B, Roelofs WL. Desaturases from the spotted fireworm moth (Choristoneura parallela) shed light on the evolutionary origins of novel moth sex pheromone desaturases. Gene. 2004;342:303–11. doi: 10.1016/j.gene.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 73.Loh DY, Baltimore D. Sexual preference of apparent gene conversion events in MHC genes of mice. Nature. 1984;309:639–40. doi: 10.1038/309639a0. [DOI] [PubMed] [Google Scholar]
- 74.Lopez de Castro JA, Strominger JL, Strong DM, Orr HT. Structure of cross-reactive human histocompatibility antigens HLA-A28 and HLA-A2: possible implications for the generation of HLA polymorphism. Proc. Natl. Acad. Sci. USA. 1982;79:3813–17. doi: 10.1073/pnas.79.12.3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lynn DJ, Lloyd AT, Fares MA, O'Farrelly C. Evidence of positively selected sites in mammalian alpha-defensins. Mol. Biol. Evol. 2004;21:819–27. doi: 10.1093/molbev/msh084. [DOI] [PubMed] [Google Scholar]
- 76.Ma H, de Pamphilis CW. The ABCs of floral evolution. Cell. 2000;101:5–8. doi: 10.1016/S0092-8674(00)80618-2. [DOI] [PubMed] [Google Scholar]
- 77.Malnic B, Godfrey PA, Buck LB. The human olfactory receptor gene family. Proc. Natl. Acad. Sci. USA. 2004;101:2584–89. doi: 10.1073/pnas.0307882100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mandl B, Brandt WF, Superti-Furga G, Graninger PG, Birnstiel ML, Busslinger M. The five cleavage-stage (CS) histones of the sea urchin are encoded by a maternally expressed family of replacement histone genes: functional equivalence of the CS H1 and frog H1M(B4) proteins. Mol. Cell Biol. 1997;17:1189–200. doi: 10.1128/mcb.17.3.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Marcos CY, Fernandez-Vina MA, Lazaro AM, Nulf CJ, Raimondi EH, Stastny P. Novel HLA-B35 subtypes: putative gene conversion events with donor sequences from alleles common in native Americans (HLA-B*4002 or B*4801) Hum. Immunol. 1997;53:148–55. doi: 10.1016/S0198-8859(97)00035-9. [DOI] [PubMed] [Google Scholar]
- 80.Martinsohn JT, Sousa AB, Guethlein LA, Howard JC. The gene conversion hypothesis of MHC evolution: a review. Immunogenetics. 1999;50:168–200. doi: 10.1007/s002510050593. [DOI] [PubMed] [Google Scholar]
- 81.Matsuda F, Ishii K, Bouvagnet P, Kuma K, Hyashida H, et al. The complete nucleotide sequence of the human immunoglobulin heavy chain variable region locus. J. Exp. Med. 1998;188:2151–62. doi: 10.1084/jem.188.11.2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Matsuo Y, Yamazaki T. tRNA derived insertion element in histone gene repeating unit of Drosophila melanogaster. Nucleic Acids Res. 1989;17:225–38. doi: 10.1093/nar/17.1.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maxson R, Cohn RH, Kedes L, Mohun T. Expression and organization of histone genes. Annu. Rev. Genet. 1983;17:239–77. doi: 10.1146/annurev.ge.17.120183.001323. [DOI] [PubMed] [Google Scholar]
- 84.McAdam SN, Boyson JE, Liu X, Garber TL, Hughes AL, et al. A uniquely high level of recombination at the HLA-B locus. Proc. Natl. Acad. Sci. USA. 1994;91:5893–97. doi: 10.1073/pnas.91.13.5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McConnell TJ, Talbot WS, McIndoe RA, Wakeland EK. The origin of MHC class II gene polymorphism within the genus Mus. Nature. 1988;332:651–54. doi: 10.1038/332651a0. [DOI] [PubMed] [Google Scholar]
- 86.Mellor AL, Weiss EH, Ramachandran K, Flavell RA. A potential donor gene for the bm1 gene conversion event in the C57BL mouse. Nature. 1983;306:792–95. doi: 10.1038/306792a0. [DOI] [PubMed] [Google Scholar]
- 86a.Mercereau-Puijalon O, Barale JC, Bischoff E. Three multigene families in plasmodium parasites: facts and questions. Int. J. Parasitol. 2002;32:1323–44. doi: 10.1016/s0020-7519(02)00111-x. [DOI] [PubMed] [Google Scholar]
- 87.Michelmore RW, Meyers BC. Clusters of resistance genes in plants evolve by divergent selection and a birth-and-death process. Genome Res. 1998;8:1113–30. doi: 10.1101/gr.8.11.1113. [DOI] [PubMed] [Google Scholar]
- 88.Mita K, Ichimura S, Nenoi M. Essential factors determining codon usage in ubiquitin genes. J. Mol. Evol. 1991;33:216–25. doi: 10.1007/BF02100672. [DOI] [PubMed] [Google Scholar]
- 89.Morrison C, Vagnarelli P, Sonoda E, Takeda S, Earnshaw WC. Sister chromatid cohesion and genome stability in vertebrate cells. Biochem. Soc. Trans. 2003;31:263–65. doi: 10.1042/bst0310263. [DOI] [PubMed] [Google Scholar]
- 89a.Muir G, Fleming CC, Schlotterer C. Three divergent rDNA clusters predates species divergence in Quercus petraea (Matt.) Liebl. and Quercus robur L. Mol. Biol. Evol. 2001;18:112–19. doi: 10.1093/oxfordjournals.molbev.a003785. [DOI] [PubMed] [Google Scholar]
- 90.Nagylaki T. Evolution of multigene families under interchromosomal gene conversion. Proc. Natl. Acad. Sci. USA. 1984;81:3796–800. doi: 10.1073/pnas.81.12.3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nagylaki T. The evolution of multigene families under intrachromosomal gene conversion. Genetics. 1984;106:529–48. doi: 10.1093/genetics/106.3.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nagylaki T, Petes TD. Intrachromosomal gene conversion and the maintenance of sequence homogeneity among repeated genes. Genetics. 1982;100:315–37. doi: 10.1093/genetics/100.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nam J, de Pamphilis CW, Ma H, Nei M. Antiquity and evolution of the MADS-box gene family controlling flower development in plants. Mol. Biol. Evol. 2003;20:1435–47. doi: 10.1093/molbev/msg152. [DOI] [PubMed] [Google Scholar]
- 94.Nam J, Kim J, Lee S, An G, Ma H, Nei M. Type I MADS-box genes have experienced faster birth-and-death evolution than type II MADS-box genes in angiosperms. Proc. Natl. Acad. Sci. USA. 2004;101:1910–15. doi: 10.1073/pnas.0308430100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nei M. Gene duplication and nucleotide substitution in evolution. Nature. 1969;221:40–42. doi: 10.1038/221040a0. [DOI] [PubMed] [Google Scholar]
- 96.Nei M, Gu X, Sitnikova T. Evolution by the birth-and-death process in multigene families of the vertebrate immune system. Proc. Natl. Acad. Sci. USA. 1997;94:7799–806. doi: 10.1073/pnas.94.15.7799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nei M, Hughes AL. Polymorphism and evolution of the major histocompatibility complex loci in mammals. In: Selander R, Clark A, Whittam T, editors. Evolution at the Molecular Level. Sinauer; Sunderland, MA:: 1991. pp. 222–47. [Google Scholar]
- 98.Nei M, Hughes AL. Balanced polymorphism and evolution by the birth-and-death process in the MHC loci. In: Tsuji K, Aizawa M, Sasazuki T, editors. 11th Histocompatibility Workshop and Conference. Oxford Univ. Press; Oxford, UK: 1992. [Google Scholar]
- 99.Nei M, Rogozin IB, Piontkivska H. Purifying selection and birth-and-death evolution in the ubiquitin gene family. Proc. Natl. Acad. Sci. USA. 2000;97:10866–71. doi: 10.1073/pnas.97.20.10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nenoi M, Mita K, Ichimura S, Kawano A. Higher frequency of concerted evolutionary events in rodents than in man at the polyubiquitin gene VNTR locus. Genetics. 1998;148:867–76. doi: 10.1093/genetics/148.2.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Newrock KM, Alfageme CR, Nardi RV, Cohen LH. Histone changes during chromatin remodeling in embryogenesis. Cold Spring Harbor Symp. Quant. Biol. 1978;1:421–31. doi: 10.1101/sqb.1978.042.01.045. [DOI] [PubMed] [Google Scholar]
- 102.Newrock KM, Cohen LH, Hendricks MB, Donnelly RJ, Weinberg ES. Stage-specific mRNAs coding for subtypes of H2A and H2B histones in the sea urchin embryo. Cell. 1978;14:327–36. doi: 10.1016/0092-8674(78)90118-6. [DOI] [PubMed] [Google Scholar]
- 103.Niimura Y, Nei M. Evolution of olfactory receptor genes in the human genome. Proc. Natl. Acad. Sci. USA. 2003;100:12235–40. doi: 10.1073/pnas.1635157100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Niimura Y, Nei M. Comparative evolutionary analysis of olfactory receptor gene clusters between humans and mice. Gene. 2005;346:13–21. doi: 10.1016/j.gene.2004.09.025. [DOI] [PubMed] [Google Scholar]
- 105.Niimura Y, Nei M. Evolutionary changes of the number of olfactory receptor genes in the human and mouse lineages. Gene. 2005;346:23–28. doi: 10.1016/j.gene.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 106.Niimura Y, Nei M. Evolutionary dynamics of olfactory receptor genes in fishes and tetrapods. Proc. Natl. Acad. Sci. USA. 2005 doi: 10.1073/pnas.0501922102. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nikolaidis N, Klein J, Nei M. Origin and evolution of the Ig-like domains present in mammalian leukocyte receptors: insights from chicken, frog, and fish homologues. Immunogenetics. 2005;57:151–57. doi: 10.1007/s00251-004-0764-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nikolaidis N, Makalowska I, Chalkia D, Makalowski W, Klein J, Nei M. Origin and evolution of the chicken leukocyte receptor complex. Proc. Natl. Acad. Sci. USA. 2005;102:4057–62. doi: 10.1073/pnas.0501040102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nikolaidis N, Nei M. Concerted and nonconcerted evolution of the Hsp70 gene superfamily in two sibling species of nematodes. Mol. Biol. Evol. 2004;21:498–505. doi: 10.1093/molbev/msh041. [DOI] [PubMed] [Google Scholar]
- 110.Ohta T. Evolution and Variation of Multigene Families. Springer-Verlag; Berlin: 1980. [Google Scholar]
- 111.Ohta T. Allelic and nonallelic homology of a supergene family. Proc. Natl. Acad. Sci. USA. 1982;79:3251–54. doi: 10.1073/pnas.79.10.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ohta T. On the evolution of multigene families. Theor. Popul. Biol. 1983;23:216–40. doi: 10.1016/0040-5809(83)90015-1. [DOI] [PubMed] [Google Scholar]
- 113.Ohta T. A model of duplicative transposition and gene conversion for repetitive DNA families. Genetics. 1985;110:513–24. doi: 10.1093/genetics/110.3.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ohta T. Role of diversifying selection and gene conversion in evolution of major histocompatibility complex loci. Proc. Natl. Acad. Sci. USA. 1991;88:6716–20. doi: 10.1073/pnas.88.15.6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ohta T, Dover GA. The cohesive population genetics of molecular drive. Genetics. 1984;108:501–21. doi: 10.1093/genetics/108.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ota T, Nei M. Divergent evolution and evolution by the birth-and-death process in the immunoglobulin VH gene family. Mol. Biol. Evol. 1994;11:469–82. doi: 10.1093/oxfordjournals.molbev.a040127. [DOI] [PubMed] [Google Scholar]
- 117.Ota T, Nei M. Evolution of immunoglobulin VH pseudogenes in chickens. Mol. Biol. Evol. 1995;12:94–102. doi: 10.1093/oxfordjournals.molbev.a040194. [DOI] [PubMed] [Google Scholar]
- 118.Ota T, Sitnikova T, Nei M. Evolution of vertebrate immunoglobulin variable gene segments. Curr. Top. Microbiol. Immunol. 2000;248:221–45. doi: 10.1007/978-3-642-59674-2_10. [DOI] [PubMed] [Google Scholar]
- 119.Palmer D, Snyder LA, Blumenfeld M. Drosophila nucleosomes contain an unusual histone-like protein. Proc. Natl. Acad. Sci. USA. 1980;77:2671–75. doi: 10.1073/pnas.77.5.2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Paques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pardue ML, Kedes LH, Weinberg ES, Birnstiel ML. Localization of sequences coding for histone messenger RNA in the chromosomes of Drosophila melanogaster. Chromosoma. 1977;63:135–51. doi: 10.1007/BF00292726. [DOI] [PubMed] [Google Scholar]
- 122.Pavelitz T, Liao D, Weiner AM. Concerted evolution of the tandem array encoding primate U2 snRNA (the RNU2 locus) is accompanied by dramatic remodeling of the junctions with flanking chromosomal sequences. EMBO J. 1999;18:3783–92. doi: 10.1093/emboj/18.13.3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pavelitz T, Rusche L, Matera AG, Scharf JM, Weiner AM. Concerted evolution of the tandem array encoding primate U2 snRNA occurs in situ, without changing the cytological context of the RNU2 locus. EMBO J. 1995;14:169–77. doi: 10.1002/j.1460-2075.1995.tb06987.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pease LR, Horton RM, Pullen JK, Yun TJ. Unusual mutation clusters provide insight into class I gene conversion mechanisms. Mol. Cell Biol. 1993;13:4374–81. doi: 10.1128/mcb.13.7.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Petes TD. Unequal meiotic recombination within tandem arrays of yeast ribosomal DNA genes. Cell. 1980;19:765–74. doi: 10.1016/s0092-8674(80)80052-3. [DOI] [PubMed] [Google Scholar]
- 126.Piontkivska H, Nei M. Birth-and-death evolution in primate MHC class I genes: divergence time estimates. Mol. Biol. Evol. 2003;20:601–9. doi: 10.1093/molbev/msg064. [DOI] [PubMed] [Google Scholar]
- 127.Piontkivska H, Rooney AP, Nei M. Purifying selection and birth-and-death evolution in the histone H4 gene family. Mol. Biol. Evol. 2002;19:689–97. doi: 10.1093/oxfordjournals.molbev.a004127. [DOI] [PubMed] [Google Scholar]
- 128.Quesada H, Ramos-Onsins SE, Aguade M. Birth-and-death evolution of the Cecropin multigene family in Drosophila. J. Mol. Evol. 2005;60:1–11. doi: 10.1007/s00239-004-0053-4. [DOI] [PubMed] [Google Scholar]
- 129.Rajalingam R, Parham P, Abi-Rached L. Domain shuffling has been the main mechanism forming new hominoid killer cell Ig-like receptors. J. Immunol. 2004;172:356–69. doi: 10.4049/jimmunol.172.1.356. [DOI] [PubMed] [Google Scholar]
- 130.Roberts SB, Weisser KE, Childs G. Sequence comparisons of non-allelic late histone genes and their early stage counterparts. Evidence for gene conversion within the sea urchin late stage gene family. J. Mol. Biol. 1984;174:647–62. doi: 10.1016/0022-2836(84)90088-3. [DOI] [PubMed] [Google Scholar]
- 131.Robertson HM. Two large families of chemoreceptor genes in the nematodes Caenorhabditis elegans and Caenorhabditis briggsae reveal extensive gene duplication, diversification, movement, and intron loss. Genome Res. 1998;8:449–63. doi: 10.1101/gr.8.5.449. [DOI] [PubMed] [Google Scholar]
- 132.Robinett CC, O'Connor A, Dunaway M. The repeat organizer, a specialized insulator element within the intergenic spacer of the Xenopus rRNA genes. Mol. Cell Biol. 1997;17:2866–75. doi: 10.1128/mcb.17.5.2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Roelofs WL, Liu W, Hao G, Jiao H, Rooney AP, Linn CEJ. Evolution of moth sex pheromones via ancestral genes. Proc. Natl. Acad. Sci. USA. 2002;99:13621–26. doi: 10.1073/pnas.152445399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Roelofs WL, Rooney AP. Molecular genetics and evolution of pheromone biosynthesis in Lepidoptera. Proc. Natl. Acad. Sci. USA. 2003;100:9179–84. doi: 10.1073/pnas.1233767100a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134a.Roesner A, Fuchs C, Hankeln T, Burmester T. A globin gene of ancient evolutionary origin in lower vertebrates: evidence for two distinct globin families in animals. Mol. Biol. Evol. 2005;22:12–20. doi: 10.1093/molbev/msh258. [DOI] [PubMed] [Google Scholar]
- 135.Rooney AP. Mechanisms underlying the evolution and maintenance of functionally heterogeneous 18S rRNA genes in apicomplexans. Mol. Biol. Evol. 2004;21:1704–11. doi: 10.1093/molbev/msh178. [DOI] [PubMed] [Google Scholar]
- 136.Rooney AP, Piontkivska H, Nei M. Molecular evolution of the nontandemly repeated genes of the histone 3 multigene family. Mol. Biol. Evol. 2002;19:68–75. doi: 10.1093/oxfordjournals.molbev.a003983. [DOI] [PubMed] [Google Scholar]
- 137.Rooney AP, Ward TJ. Evolution of a large ribosomal RNA multigene family in filamentous fungi: birth and death of a concerted evolution paradigm. Proc. Natl. Acad. Sci. USA. 2005;102:5084–89. doi: 10.1073/pnas.0409689102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Schaschl H, Suchentrunk F, Hammer S, Goodman SJ. Recombination and the origin of sequence diversity in the DRB MHC class II locus in chamois (Rupicapra spp.) Immunogenetics. 2005;57:108–15. doi: 10.1007/s00251-005-0784-4. [DOI] [PubMed] [Google Scholar]
- 139.Sharp PM, Li W-H. The codon adaptation index a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15:1281–95. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Shepherd GM. The human sense of smell: Are we better than we think? PLOS Biol. 2004;2:572–75. doi: 10.1371/journal.pbio.0020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sitnikova T, Nei M. Evolution of immunoglobulin kappa chain variable region genes in vertebrates. Mol. Biol. Evol. 1998;15:50–60. doi: 10.1093/oxfordjournals.molbev.a025846. [DOI] [PubMed] [Google Scholar]
- 142.Sitnikova T, Su C. Coevolution of immunoglobulin heavy- and light-chain variable-region gene families. Mol. Biol. Evol. 1998;15:617–25. doi: 10.1093/oxfordjournals.molbev.a025965. [DOI] [PubMed] [Google Scholar]
- 143.Slightom JL, Blechl AE, Smithies O. Human fetal Gγ_ and Aγ_ globin genes: complete nucleotide sequences suggest that DNA can be exchanged between these duplicated genes. Cell. 1980;21:627–38. doi: 10.1016/0092-8674(80)90426-2. [DOI] [PubMed] [Google Scholar]
- 144.Smith GP. Unequal crossover and the evolution of multigene families. Cold Spring Harbor Symp. Quant. Biol. 1974;38:507–13. doi: 10.1101/sqb.1974.038.01.055. [DOI] [PubMed] [Google Scholar]
- 145.Smith GP. Evolution of repeated DNA sequences by unequal crossovers. Science. 1976;191:528–34. doi: 10.1126/science.1251186. [DOI] [PubMed] [Google Scholar]
- 146.Srivastava AK, Schlessinger D. Structure and organization of ribosomal DNA. Biochimie. 1991;73:631–38. doi: 10.1016/0300-9084(91)90042-y. [DOI] [PubMed] [Google Scholar]
- 147.Su C, Jakobsen IB, Gu X, Nei M. Diversity and evolution of T-cell receptor variable region genes in mammals and birds. Immunogenetics. 1999;50:301–8. doi: 10.1007/s002510050606. [DOI] [PubMed] [Google Scholar]
- 148.Su C, Nei M. Fifty-million-year old polymorphism at an immunoglobulin variable region gene locus in the rabbit evolutionary lineage. Proc. Natl. Acad. Sci. USA. 1999;96:9710–15. doi: 10.1073/pnas.96.17.9710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Su C, Nei M. Evolutionary dynamics of the T-cell receptor VB gene family as inferred from the human and mouse genomic sequences. Mol. Biol. Evol. 2001;18:503–13. doi: 10.1093/oxfordjournals.molbev.a003829. [DOI] [PubMed] [Google Scholar]
- 150.Su C, Nguyen VK, Nei M. Adaptive evolution of variable region genes encoding an unusual type of immunoglobulin in camelids. Mol. Biol. Evol. 2002;19:205–15. doi: 10.1093/oxfordjournals.molbev.a004073. [DOI] [PubMed] [Google Scholar]
- 151.Szostak JW, Wu R. Unequal crossing over in the ribosomal DNA of Saccharomyces cerevisiae. Nature. 1980;284:426–30. doi: 10.1038/284426a0. [DOI] [PubMed] [Google Scholar]
- 152.Takahashi K, Rooney AP, Nei M. Origins and divergence times of mammalian class II MHC gene clusters. J. Hered. 2000;19:198–204. doi: 10.1093/jhered/91.3.198. [DOI] [PubMed] [Google Scholar]
- 153.Tan Y, Bishoff ST, Riley MA. Ubiquitins revisited: further examples of within-and between-locus concerted evolution. Mol. Phylogenet. Evol. 1993;2:351–60. doi: 10.1006/mpev.1993.1035. [DOI] [PubMed] [Google Scholar]
- 154.Tanabe Y, Hasebe M, Sekimoto H, Nishiyama T, Kitani M, et al. Characterization of MADS-box genes in charophycean green algae and its implication for the evolution of MADS-box genes. Proc. Natl. Acad. Sci. USA. 2005;102:2436–41. doi: 10.1073/pnas.0409860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Theissen G. Development of floral organ identity: stories from the MADS house. Curr. Opin. Plant Biol. 2001;4:75–85. doi: 10.1016/s1369-5266(00)00139-4. [DOI] [PubMed] [Google Scholar]
- 156.Trowsdale J, Barten R, Haude A, Stewart CA, Beck S, Wilson MJ. The genomic context of natural killer receptor extended gene families. Immunol. Rev. 2001;181:20–38. doi: 10.1034/j.1600-065x.2001.1810102.x. [DOI] [PubMed] [Google Scholar]
- 157.van den Berg TK, Yoder JA, Litman GW. On the origins of adaptive immunity: innate immune receptors join the tale. Trends Immunol. 2004;25:11–16. doi: 10.1016/j.it.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 158.Verica JA, Chae L, Tong H, Ingmire P, He ZH. Tissue-specific and developmentally regulated expression of a cluster of tandemly arrayed cell wall-associated kinase-like kinase genes in Arabidopsis. Plant Physiol. 2003;133:1732–46. doi: 10.1104/pp.103.028530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Vrana PB, Wheeler WC. Molecular evolution and phylogenetic utility of the polyubiquitin locus in mammals and higher vertebrates. Mol. Phylogenet. Evol. 1996;6:259–69. doi: 10.1006/mpev.1996.0075. [DOI] [PubMed] [Google Scholar]
- 160.Wagner GP, Amemiya C, Ruddle F. Hox cluster duplications and the opportunity for evolutionary novelties. Proc. Natl. Acad. Sci. USA. 2003;100:14603–6. doi: 10.1073/pnas.2536656100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Walsh JB. Sequence-dependent gene conversion—can duplicated genes diverge fast enough to escape conversion. Genetics. 1987;117:543–57. doi: 10.1093/genetics/117.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Watkins DI, McAdam SN, Liu XM, Strang CR, Milford EL, et al. New recombinant HLA-B alleles in a tribe of South-American Amerindians indicate rapid evolution of MHC class-1 loci. Nature. 1992;357:329–33. doi: 10.1038/357329a0. [DOI] [PubMed] [Google Scholar]
- 162a.Wegnez M, Monier R, Denis H. Sequence heterogeneity of 5S rRNA in Xenopus laevis. FEBS Lett. 1972;25:13–20. doi: 10.1016/0014-5793(72)80443-5. [DOI] [PubMed] [Google Scholar]
- 163.Weigel D, Meyerowitz EM. The ABCs of floral homeotic genes. Cell. 1994;78:203–9. doi: 10.1016/0092-8674(94)90291-7. [DOI] [PubMed] [Google Scholar]
- 164.Weiss EH, Mellor AL, Golden L, Fahrner K, Simpson E, et al. The structure of a mutant H-2 gene suggests that the generation of polymorphism in H-2 genes may occur by gene conversion-like events. Nature. 1983;301:671–74. doi: 10.1038/301671a0. [DOI] [PubMed] [Google Scholar]
- 165.Whinnett A, Mundy NI. Isolation of novel olfactory receptor genes in marmosets (Callithrix): insights into pseudogene formation and evidence for functional degeneracy in non-human primates. Gene. 2003;304:87–96. doi: 10.1016/s0378-1119(02)01182-4. [DOI] [PubMed] [Google Scholar]
- 166.Wilhelm BT, Gagnier L, Mager DL. Sequence analysis of the Ly49 cluster in C57BL/6 mice: a rapidly evolving multigene family in the immune system. Genomics. 2002;80:646–61. doi: 10.1006/geno.2002.7004. [DOI] [PubMed] [Google Scholar]
- 167.Wu S, Saunders TL, Bach FH. Polymorphism of human Ia antigens generated by reciprocal intergenic exchange between two DR loci. Nature. 1986;324:676–79. doi: 10.1038/324676a0. [DOI] [PubMed] [Google Scholar]
- 168.Young JM, Friedman C, Williams EM, Ross JA, Tonnes-Priddy L, Trask BJ. Different evolutionary processes shaped the mouse and human olfactory receptor gene families. Hum. Mol. Genet. 2002;11:535–46. doi: 10.1093/hmg/11.5.535. [DOI] [PubMed] [Google Scholar]
- 169.Yun TJ, Melvold RW, Pease LR. A complex major histocompatibility complex D locus variant generated by an unusual recombination mechanism in mice. Proc. Natl. Acad. Sci. USA. 1997;94:1384–89. doi: 10.1073/pnas.94.4.1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zamb TJ, Petes TD. Analysis of the junction between ribosomal RNA genes and single-copy chromosomal sequences in the yeast Saccharomyces cerevisiae. Cell. 1982;28:355–64. doi: 10.1016/0092-8674(82)90353-1. [DOI] [PubMed] [Google Scholar]
- 171.Zamocky M. Phylogenetic relationships in class I of the superfamily of bacterial, fungal, and plant peroxidases. Eur. J. Biochem. 2004;271:3297–309. doi: 10.1111/j.1432-1033.2004.04262.x. [DOI] [PubMed] [Google Scholar]
- 172.Zangenberg G, Huang MM, Arnheim N, Erlich H. New HLA-DPB1 alleles generated by interallelic gene conversion detected by analysis of sperm. Nat. Genet. 1995;10:407–14. doi: 10.1038/ng0895-407. [DOI] [PubMed] [Google Scholar]
- 173.Zhang J, Dyer KD, Rosenberg HF. Evolution of the rodent eosinophil-associated RNase gene family by rapid gene sorting and positive selection. Proc. Natl. Acad. Sci. USA. 2000;97:4701–6. doi: 10.1073/pnas.080071397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zhang J, Rosenberg HF, Nei M. Positive Darwinian selection after gene duplication in primate ribonuclease genes. Proc. Natl. Acad. Sci. USA. 1998;95:3708–13. doi: 10.1073/pnas.95.7.3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhang X, Firestein S. The olfactory receptor gene superfamily of the mouse. Nat. Neurosci. 2002;5:124–33. doi: 10.1038/nn800. [DOI] [PubMed] [Google Scholar]
- 176.Zhang Z, Carriero N, Gerstein M. Comparative analysis of processed pseudogenes in the mouse and human genomes. Trends Genet. 2004;20:62–67. doi: 10.1016/j.tig.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 177.Zhang Z, Inomata N, Yamazaki T, Kishino H. Evolutionary history and mode of the amylase multigene family in Drosophila. J. Mol. Evol. 2003;57:702–9. doi: 10.1007/s00239-003-2521-7. [DOI] [PubMed] [Google Scholar]
- 178.Zimmer EA, Martin SL, Beverley SM, Kan YW, Wilson AC. Rapid duplication and loss of genes coding for the chains of hemoglobin. Proc. Natl. Acad. Sci. USA. 1980;77:2158–62. doi: 10.1073/pnas.77.4.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zozulya S, Echeverri F, Nguyen T. The human olfactory receptor repertoire. Genome Biol. 2001;2:RESEARCH0018. doi: 10.1186/gb-2001-2-6-research0018. [DOI] [PMC free article] [PubMed] [Google Scholar]