

Abstract
Gene-targeted mice deficient in the evolutionarily conserved uracil–DNA glycosylase encoded by the UNG gene surprisingly lack the mutator phenotype characteristic of bacterial and yeast ung– mutants. A complementary uracil–DNA glycosylase activity detected in ung–/– murine cells and tissues may be responsible for the repair of deaminated cytosine residues in vivo. Here, specific neutralizing antibodies were used to identify the SMUG1 enzyme as the major uracil–DNA glycosylase in UNG-deficient mice. SMUG1 is present at similar levels in cell nuclei of non-proliferating and proliferating tissues, indicating a replication- independent role in DNA repair. The SMUG1 enzyme is found in vertebrates and insects, whereas it is absent in nematodes, plants and fungi. We propose a model in which SMUG1 has evolved in higher eukaryotes as an anti-mutator distinct from the UNG enzyme, the latter being largely localized to replication foci in mammalian cells to counteract de novo dUMP incorporation into DNA.
Keywords: DNA repair/DNA glycosylases/UNG-deficient cells
Introduction
Hydrolytic deamination of cytosine residues in DNA generates the aberrant base uracil ∼100–500 times per human cell per day, as estimated from in vitro measurements of deamination rates (Frederico et al., 1990; Lindahl, 1993; Shen et al., 1994). If unrepaired, the resulting pre-mutagenic U:G mispairs would give rise to CG→TA transition mutations upon DNA replication. In addition, transcriptional bypass of U:G mispairs by RNA polymerase may result in the expression of altered proteins (Viswanathan et al., 1999). To protect the cell from these deleterious consequences, all organisms studied to date express uracil–DNA glycosylases, which excise uracil residues and initiate the base excision repair (BER) pathway (Lindahl and Barnes, 2000).
The most highly conserved family of uracil–DNA glycosylases is typified by the Escherichia coli Ung enzyme (Lindahl, 1974; Scharer and Jiricny, 2001). Members of the ubiquitous UNG family are present in most species analysed and are even encoded by some viruses (reviewed by Krokan et al., 1997), although UNG orthologues are notably absent from the genomes of Drosophila melanogaster and the Archaea (Aravind and Koonin, 2000). UNG family members are the principal repair enzymes responsible for the removal of pre-mutagenic uracil from U:G mispairs in E.coli (Duncan and Miller, 1980; Duncan and Weiss, 1982) and Saccharomyces cerevisiae (Impellizzeri et al., 1991), as ung–/– mutants in these organisms show a significantly increased spontaneous mutation frequency, mainly as a result of an increase in CG→TA transitions. Based on the assumption that the UNG enzymes were general anti-mutators, we chose to make an ung–/– knockout mouse model. Surprisingly, UNG-deficient mice showed only a marginal increase in mutation frequency in a lacI transgene, indicating that UNG is not the major enzyme removing pre-mutagenic uracil from DNA in mammals (Nilsen et al., 2000).
As well as resulting from hydrolytic deamination of cytosine, uracil can also occur in DNA through misincorporation of dUMP opposite A (adenine) residues during DNA replication (Brynolf et al., 1978; Tye et al., 1978). This has been considered relatively innocuous as U:A pairs have unchanged coding properties, and up to 20% of genomic thymine can be replaced with uracil with no obvious detrimental effect in E.coli dut– ung– mutants defective in both dUTPase and uracil–DNA glycosylase (Tye et al., 1978; Warner et al., 1981). In mammalian cells, two alternatively spliced forms of the UNG enzyme are sorted to the nuclei (UNG2) or to the mitochondria (UNG1) (Nilsen et al., 1997). The UNG2 isoform interacts with replication factor A (RPA) (Nagelhus et al., 1997) and proliferating cell nuclear antigen (PCNA), and is localized to replication foci during S phase (Otterlei et al., 1999). Moreover, dUMP incorporated instead of TMP persists in isolated ung–/– nuclei, consistent with a predominant in vivo role for UNG2 in removing uracil from newly synthesized DNA and resulting in a significantly increased steady-state level of uracil in the genome of UNG-deficient mice (Nilsen et al., 2000).
Biochemical analysis of cell and tissue extracts from UNG-deficient mice showed that a significant uracil–DNA glycosylase activity remained (Nilsen et al., 2000). The absence of a mutator phenotype in UNG-deficient mice makes it a reasonable assumption that this activity limits mutagenesis resulting from cytosine deamination. It was, therefore, of interest to identify this cryptic uracil–DNA glycosylase. In a parallel development, a previously unrecognized uracil–DNA glycosylase was identified by an in vitro expression cloning strategy screening for enzymes that would bind to synthetic DNA glycosylase inhibitors (Haushalter et al., 1999). The biochemical properties of this enzyme, denoted SMUG1, seemed similar to the activity revealed in UNG-deficient mice (Nilsen et al., 2000). Here, we identify and characterize SMUG1 as the major uracil–DNA glycosylase in UNG-deficient murine cells and tissues. We propose that SMUG1 has evolved in higher organisms to prevent accumulation of mutations resulting from deamination of cytosine residues in DNA.
Results
The prevalent uracil–DNA glycosylase activity in ung–/– cell extracts is inhibited by SMUG1 antibodies
Mice deficient in the UNG uracil–DNA glycosylase show little, if any, increase in spontaneous mutation frequency, and this lack of a mutator phenotype has been attributed to a complementary uracil–DNA glycosylase activity in ung–/– mice (Nilsen et al., 2000). In order to characterize this distinct uracil-excising activity further, we measured total uracil–DNA glycosylase activity in crude nuclear extracts of ung–/– mouse embryo fibroblast (MEF) cell lines using a double-stranded DNA substrate containing [3H]dUMP residues in U:A pairs (Figure 1). In agreement with earlier results, homozygous disruption of the UNG gene substantially, but not entirely, reduced the uracil–DNA glycosylase activity (Figure 1, white bars). Similarly, the majority of uracil-excising activity was ablated in UNG+/+ extracts by addition of Ugi, the structure-specific UNG inhibitor from PBS2 bacteriophage (Figure 1, hatched bar). Addition of Ugi to ung–/– extracts gave no further reduction in activity (Figure 1, hatched bar), clearly demonstrating that there is no residual UNG activity in the ung–/– mice and thus, that the remaining uracil–DNA glycosylase activity is a result of other gene products.

Fig. 1. Uracil–DNA glycosylase activity in nuclear MEF extracts. Nuclear extracts from UNG+/+ and ung–/– MEF cell lines were incubated with a [3H]dUMP-containing double-stranded DNA substrate and release of acid soluble [3H]uracil was determined. Extracts were pre-incubated without additions (white bars), with Ugi (hatched bars), SMUG1 antibodies (black bars), or Ugi and SMUG1 antibodies together (dotted bars). Error bars show the SEM from three experiments.
SMUG1 has biochemical properties similar to the uracil–DNA glycosylase activity revealed in ung–/– mice (Nilsen et al., 2000). Polyclonal antibodies were raised against recombinant human SMUG1 (hSMUG1) in order to determine the contribution of the SMUG1 enzyme to total uracil–DNA glycosylase activity in ung–/– mice. Two different rabbit antisera did not detect murine SMUG1 (mSMUG1) by immunoblotting of nuclear extracts, indicating that mSMUG1 is a low abundance protein (data not shown). However, both antisera proved to be efficient neutralizing antibodies and gave similar results in all subsequent experiments. SMUG1 antibodies could be added to UNG+/+ extracts without diminishing total activity, which is largely a result of UNG in this assay system (Figure 1, black bar). In the presence of both Ugi and SMUG1 antibodies, uracil excision was synergistically inhibited in UNG+/+ extracts (Figure 1, dotted bar). In ung–/– extracts, pre-incubation with SMUG1 antibodies inhibited the majority of the remaining uracil–DNA glycosylase activity (Figure 1, black bar) and addition of Ugi produced no further reduction (Figure 1, dotted bar). More than 99% inhibition of recombinant hSMUG1 was achieved under the same assay conditions (data not shown). These data confirm the absence of UNG-encoded uracil–DNA glycosylase in the ung–/– MEF cell line and indicate that the remaining uracil–DNA glycosylase activity is predominantly a result of the SMUG1 gene product.
SMUG1 is a major uracil–DNA glycosylase activity on U:G mispairs
To mimic the activity on rare pre-mutagenic uracil residues, we measured uracil–DNA glycosylase activity in murine nuclear extracts using a double-stranded oligonucleotide substrate containing a single, centrally placed uracil residue paired opposite guanine. By using a low substrate concentration, this assay was designed to select for enzymes that are able to locate rare cytosine deamination products in the genome. There was efficient uracil release by ung–/– extracts (Figure 2A, lane 2) under these conditions. Significantly, the presence of the SMUG1 antibodies abolished this uracil–DNA glycosylase activity (Figure 2A, lane 4); no inhibition of the reaction was observed when control IgG was incubated with the extracts (Figure 2A, lane 3). As expected, addition of Ugi to the ung–/– extract did not detectably inhibit uracil release (Figure 2A, lane 5), and addition of Ugi to ung–/– extracts pre-treated with SMUG1 antibodies did not inhibit uracil release further (Figure 2A, lane 6). These results show that SMUG1 accounts for the uracil–DNA glycosylase in ung–/– MEF cells detected by this assay procedure.

Fig. 2. Uracil release from a double-stranded oligonucleotide substrate. Nuclear extracts from ung–/– (A) or UNG+/+ (B) MEF cell lines were incubated with a U:G-containing double-stranded oligonucleotide substrate with the uracil residue centrally placed in the 5′-32P-labelled strand (19mer; lane 1). Uracil release was determined following chemical cleavage of the abasic sites and resolution of the 9mer radio-labelled product by denaturing PAGE. Extracts were pre-incubated without additions (lane 2), with control IgG (lane 3), SMUG1 antibodies (lane 4), Ugi (lane 5) or SMUG1 antibodies and Ugi together (lane 6), as indicated. Release of uracil by purified recombinant human SMUG1 protein (C) or purified human UNG (UNGΔ84) protein (D) was similarly monitored.
Extending these experiments to UNG+/+ MEF extracts, pre-incubation of UNG+/+ extract with SMUG1 antibodies inhibited most, but not all, uracil release (Figure 2B, compare lanes 2 and 4). This demonstrates that the mSMUG1 enzyme is highly active in UNG+/+ extracts when using a low substrate concentration of U:G mispairs. In contrast, only modest inhibition of total activity in UNG+/+ extracts was observed after pre-incubation with Ugi (Figure 2B, lane 5), indicating weak UNG activity under these conditions. By combining the SMUG1 antibodies and Ugi, uracil release by the extract was almost obliterated (Figure 2B, lane 6). This indicates that SMUG1 and UNG are the two major uracil–DNA glycosylases in these extracts. As expected, the SMUG1 antibodies inhibit recombinant hSMUG1 protein (Figure 2C, lane 4), whereas Ugi does not (Figure 2C, lane 5). Conversely, SMUG1 antibodies do not inhibit human UNG (Figure 2D, lane 4), whereas Ugi inhibits the enzyme efficiently (Figure 2D, lane 5).
The two enzyme assays employed (Figures 1 and 2) gave different estimates of the relative amounts of UNG and SMUG1 activity in nuclear extracts. This is because of the different kinetic properties of the two enzymes (Slupphaug et al., 1995; Haushalter et al., 1999). When substrate is present in excess, UNG appears to be the dominant uracil–DNA glycosylase (Figure 1). However, under limiting substrate conditions (Figure 2A and B), SMUG1 is observed to be a major uracil–DNA glycosylase in both wild-type and UNG-deficient MEF extracts. Because of its high turnover number (Slupphaug et al., 1995), UNG would out-compete SMUG1 in processing large numbers of dUMP residues in DNA, as in Figure 1. On the other hand, the ∼100-fold lower Km of SMUG1 (Haushalter et al., 1999) would favour this enzyme over UNG when competing for limiting amounts of substrate uracil, such as under the conditions of the experiments shown in Figure 2.
Specificity of SMUG1 antibodies
The thymine–DNA glycosylases TDG (Hardeland et al., 2000) and MBD4 (Hendrich et al., 1999) exhibit some uracil–DNA glycosylase activity. A double-stranded 64mer oligonucleotide substrate with a single uracil residue in a UpG context (Figure 3A, lane 1), a preferred substrate for these enzymes, was used to assess the specificity of the neutralizing antibodies raised against hSMUG1 (lanes 2 and 3). As expected from the high degree of sequence conservation between mammalian SMUG1 orthologues (see below, Figure 7), the antibodies also efficiently inhibited the activity of mSMUG produced by coupled in vitro transcription–translation of a mSMUG1 cDNA clone (lanes 4 and 5). The antibodies did not detectably inhibit recombinant mTDG (lanes 6 and 7) or recombinant mMBD4 (lanes 8 and 9). This was expected as the enzymes, despite having retained a common glycosylase fold, share <10% amino acid sequence homology (Aravind and Koonin, 2000). Two different rabbit antisera were tested with identical results; both proved to be efficient and specific neutralizing antibodies of hSMUG1 and mSMUG1. In addition to the enzyme activity assays, immunoblotting experiments showed that mSMUG1 (Figure 3B, lane 1) was specifically recognized by the antibodies, as was the purified recombinant hSMUG1, which had been employed as antigen (lane 3).
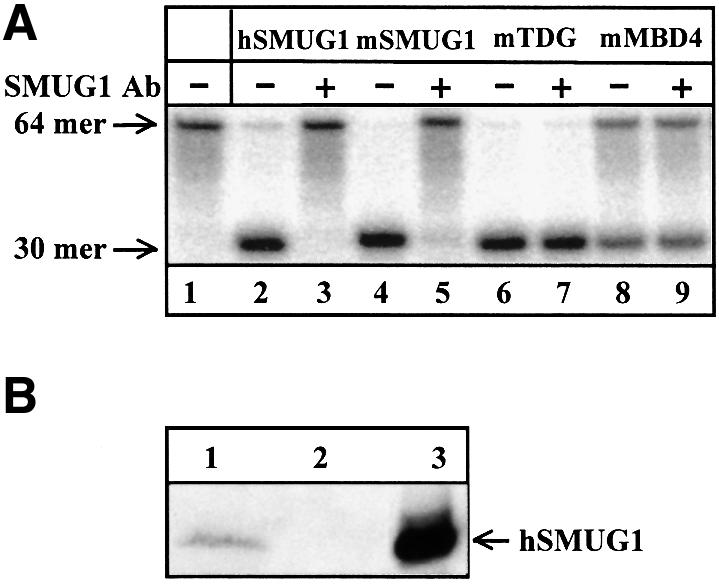
Fig. 3. Specificity of SMUG1 antibodies. The specificity of the antibodies raised against recombinant hSMUG1 was investigated. (A) Uracil release from a UpG-containing, double-stranded 64mer oligonucleotide substrate (lane 1) was determined as in Figure 2. Uracil release was measured directly (–) or after pre-incubation with SMUG1 antibodies (+) using recombinant hSMUG1 (lanes 2 and 3), mSMUG1 (lanes 4 and 5), recombinant mTDG (lanes 6 and 7), and recombinant mMBD4 (lanes 8 and 9). (B) Western blot of coupled in vitro transcription–translation reaction mixtures. The SMUG1 antibodies specifically recognized mSMUG1 (lane 1) and there was no cross-reaction with reticulocyte lysate proteins when mSMUG1 cDNA was excluded from the reaction mixture (lane 2). Recombinant hSMUG1 (lane 3) is shown as a reference.

Fig. 7. Alignment of putative SMUG1 orthologues. Complete cDNA sequences (Hs, Xl, Dm, Mm) and ESTs (Dr, Sp, Ag, Bm) were included in the alignment. Related sequences were not detected in plants, nematodes, or yeast and other fungi. Residues shaded magenta are proposed to be important for substrate recognition and catalysis (Haushalter et al., 1999; Aravind and Koonin, 2000). Conserved residues are highlighted yellow, and conservative substitutions are shaded grey. Gaps and missing residues are denoted by dashes and unassignable amino acid residues from the ESTs are denoted by the letter X. Species and DDBJ/EMBL/Genbank accession Nos are as follows: Hs, Homo sapiens (human), NP_055126.1; Mm, Mus musculus (mouse), BF467856; Xl, Xenopus laevis (African clawed toad), AAD17300.1; Dr, Danio rerio (zebrafish), compilation of AW419619, AI878196 and AW134258; Sp, Strongylocentrotus purpuratus (purple sea urchin), AF122749; Dm, Drosophila melanogaster (fruit fly), AAF55400; Ag, Anopheles gambiae (malaria mosquito), AJ282661; Bm, Bombyx mori (silk moth), AU004467.
Substrate specificity of uracil–DNA glycosylase in ung–/– MEF extracts
In order to characterize further uracil–DNA glycosylase activity in ung–/– MEF cells, nuclear extracts were fractionated by Mono S ion-exchange chromatography. Enzyme activity was resolved into two major peaks (Figure 4A): fractions 6–9 contained the bulk of the activity in the ung–/– extracts, but a smaller peak was also found in fraction 18. Fractions 6–9 were shown to contain the SMUG1 protein, as the activity of these fractions could be selectively inhibited by SMUG1 antibodies. As expected from the known properties of SMUG1 (Haushalter et al., 1999), these fractions could also excise uracil from U:A pairs, as well as from U:G mispairs, but were inactive on T:G containing double-stranded oligonucleotide substrates (Figure 4B). Furthermore, recombinant hSMUG1 protein elutes from a Mono S column at a salt concentration corresponding to fraction 8 in Figure 4A. The remaining active fraction 18 was not affected by the presence of SMUG1 antibodies, did not excise uracil from U:A pairs, but could excise thymine from substrates containing T:G mismatches (Figure 4B). The biochemical properties of the minor activity in fraction 18, which was only detected after the purification and concentration achieved by Mono S chromatography, seemed similar to the published properties of the thymine–DNA glycosylases TDG and MBD4 (Table I). However, changing the sequence of the double-stranded oligonucleotide substrate to place the uracil or thymine in a UpG or TpG context rather than UpA or TpA did not significantly affect the activity of fraction 18 (data not shown). As MBD4 has a distinct domain for binding deaminated CpG sequences and is much more active on uracil or thymine residues in a UpG or TpG context (Hendrich et al., 1999), we tentatively identify the minor activity observed here as TDG (Hardeland et al., 2000). Thus, ion-exchange chromatography experiments confirmed that the major uracil–DNA glycosylase activity in ung–/– extracts is due to SMUG1, while a minor activity with properties similar to TDG was also observed.

Fig. 4. Fractionation of ung–/– MEF nuclear extract by Mono S chromatography. Nuclear extract was prepared from ung–/– MEFs (108 cells) and the extract loaded onto a Mono S column. A linear NaCl gradient (50–500 mM) was applied and fractions collected (2–18), followed by elution in 1 M NaCl. (A) An aliquot of each fraction was assayed using the 19mer U:G-containing oligonucleotide substrate (as in Figure 2); uracil release (%) in each fraction is shown as open bars. (B) Nuclear extract (NE) and selected fractions were assayed for activity on oligonucleotide substrates containing U:G, U:A or T:G base pairs. The 9mer product is shown (see Figure 2 legend). Activity on the U:G substrate in fractions pre-incubated with SMUG1 antibodies is indicated by an asterisk.
Table I. Properties of mammalian enzymes with uracil-excising activity.
| Gene product | Molecular mass (kDa) | Substrate specificity |
Inhibition by Ugi | Inhibition by SMUG1 Ab | |||
|---|---|---|---|---|---|---|---|
| U:G | U:A | T:G | U | ||||
| UNG | 34 | yes | yes | no | yes | yes | no |
| TDG | 46 | yes | no | yes | no | no | no |
| MBD4 | 63 | yes | no | yes | no | no | no |
| SMUG1 | 30 | yes | yes | no | yes | no | yes |
| Activity in ung–/– cells | 30–40 | yes | yes | no | yes | no | yes |
Kinetic properties of the SMUG1 enzyme
The efficient action of SMUG1 on double-stranded DNA reported here contrasts with the earlier characterization of this enzyme as a single-strand selective uracil–DNA glycosylase (Haushalter et al., 1999). Subsequently, we have shown that under single-turnover conditions (limiting substrate and excess enzyme), SMUG1 is able to process efficiently double-stranded substrates with U paired opposite A or G (Figures 2 and 4). When substrate is in excess, the slow turnover of SMUG1 on double-stranded DNA might be attributed to end-product inhibition by abasic sites, a common property of diverse uracil–DNA glycosylases. Prompted by recent reports that these enzymes can be potently stimulated by AP-endonuclease (APE1) (Waters et al., 1999; Mol et al., 2000; Hill et al., 2001; Vidal et al., 2001; Yang et al., 2001), we investigated the effect of APE1 on recombinant hSMUG1 activity.
As shown in Figure 5, addition of APE1 under multiple-turnover conditions accelerated the processing by hSMUG1 of both single-stranded and double-stranded oligonucleotides containing uracil. The activity of hSMUG1 on double-stranded DNA was especially strongly stimulated by APE1, to a level well above the activity of SMUG1 on single-stranded DNA. Addition of excess EDTA to impair the catalytic activity but not substrate binding of APE1 did not diminish the stimulation of hSMUG1 turnover on double-stranded DNA, indicating that the strong binding of APE1 to its substrate DNA served to displace the DNA glycosylase, in agreement with similar observations by Vidal et al. (2001).

Fig. 5. Stimulation of SMUG1 activity by APE1. Oligonucleotides containing uracil, as a single-stranded substrate (squares) or paired opposite G as a double-stranded substrate (triangles), were incubated with SMUG1 (open symbols) or SMUG1 plus APE1 (closed symbols). Following NaOH treatment to cleave AP sites and resolution of the product band by denaturing polyacrylamide gel electrophoresis, uracil release was quantitated by phosphoimager analysis.
Given the ability of APE1 to promote the activity of several DNA glycosylases, now including SMUG1, it has been debated whether APE1 physically interacts with any of the DNA glycosylases (Waters et al., 1999; Parker et al., 2001; Hill et al., 2001). To test this possibility, co-immunoprecipitation experiments were performed with epitope-tagged hSMUG1 overproduced by transient transfection in HeLa cells. No protein from the HeLa cell lysate was clearly retained by antibody-bound hSMUG1 (data not shown). These experiments provide no support for a model of direct protein–protein interaction between SMUG1 and the abundant APE1, but do not rule out the possibility that APE1 recognizes a composite surface of SMUG1 bound to DNA.
SMUG1 activity is not correlated with cell proliferation
It was observed previously that the uracil–DNA glycosyl ase activity in ung–/– cell extracts was high in kidney but low in thymus and spleen when measured with a multiple-turnover substrate (Nilsen et al., 2000). This prompted us to measure uracil–DNA glycosylase activity with the U:G containing double-stranded oligonucleotide substrate in nuclear extracts from organs taken from 10-week-old ung–/– mice (Figure 6). By pre-incubating extracts with SMUG1 antibodies (Figure 6, lanes labelled +), the uracil-excising activity was dramatically reduced in extracts from all organs. In some organs, such as heart and thymus, the uracil–DNA glycosylase activity appeared to be completely abolished by the SMUG1 antibodies. Most importantly, there was no apparent correlation between SMUG1 activity and cell renewal rate in these tissues. Thus, there is no clear difference in SMUG1 activity between non-replicative tissue, such as heart and kidney, and proliferating tissue, such as spleen and thymus. While replication factors, including the UNG protein (Aprelikova and Tomilin, 1982; Haug et al., 1998), show much higher activity in proliferating tissue, the tissue distribution of SMUG1 activity resembles that found for constitutively expressed repair proteins such as DNA polymerase β (Zmudzka et al., 1988; Hirose et al., 1989).
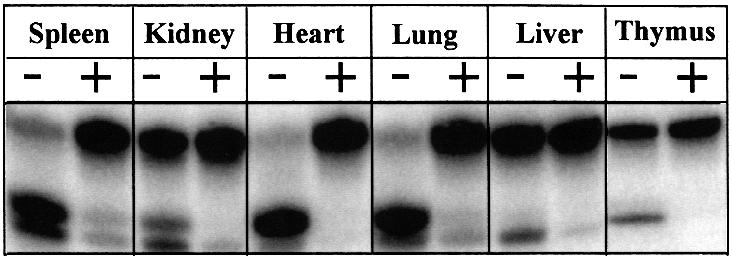
Fig. 6. Tissue distribution of SMUG1 activity. Nuclear extracts were prepared of different organs from ung–/– mice. Uracil-excising activity was measured using the 19mer U:G containing double-stranded oligonucleotide substrate (as in Figure 2). Uracil release was measured directly (–) or after pre-incubating the extracts with SMUG1 antibodies (+).
Evolution of SMUG1
In considering in vivo roles of SMUG1, it is relevant to estimate when, during the course of evolution, SMUG1 arose. While SMUG1 has been predicted to retain the common core fold of the uracil–DNA glycosylase superfamily (Haushalter et al., 1999; Aravind and Koonin, 2000), the primary structure of SMUG1 has diverged almost completely from other superfamily members. BLAST searching of the non-redundant DDBJ/EMBL/GenBank database retrieved a sequence corresponding to a putative SMUG1 protein coding sequence from D.melanogaster, in addition to the previously characterized Xenopus laevis and Homo sapiens SMUG1 (Haushalter et al., 1999). Further BLAST searching of the NCBI expressed sequence tag (EST) database yielded sequences with high similarity (e <0.1) to SMUG1 in cDNAs isolated from Mus musculus, Danio rerio, Strongylocentrotus purpuratus, Anopheles gambiae and Bombyx mori (Figure 7). The translated sequences of the putative SMUG1 orthologues were aligned using Clustal_W (Thompson et al., 1994) and were shown to retain the key amino acid residues believed to be important for substrate binding and catalysis (Haushalter et al., 1999; Aravind and Koonin, 2000). The apparent absence of SMUG1 from Caenorhabditis elegans, Arabidopsis thaliana, Schizosaccharomyces pombe, S.cerevisiae, and the completely sequenced members of the Eubacteria and Archaea suggests that the SMUG1 gene arose relatively recently, apparently some time shortly after the last common ancestor of nematodes and man, ∼550 million years ago.
Discussion
Compared with microbial genomes, the much larger and more slowly replicating mammalian genome would be expected to be considerably more susceptible to endogenously produced DNA damage and thus, offer a bigger target for mutagenesis. However, E.coli and S.cerevisiae ung– mutants deficient in uracil–DNA glycosylase exhibit a distinct mutator phenotype because of their inability to repair spontaneously deaminated cytosine in their DNA, whereas gene-targeted knockout mice lacking the homologous enzyme do not. These initially puzzling results can now be explained. The uracil–DNA glycosylase detected in murine ung–/– cells (Nilsen et al., 2000) has been identified here as the SMUG1 enzyme (Haushalter et al., 1999), which is not present in E.coli or yeast. Neutralizing antibodies against SMUG1 did not inhibit other uracil–DNA glycosylases (summarized in Table I). However, they did strongly suppress the enzyme activity in extracts from ung–/– cells (Figure 2), showing that SMUG1 accounts for almost all measurable activity against uracil in DNA in ung–/– cells.
In this regard, the evolution of another uracil–DNA glycosylase with a separate physiological role most likely occurred through duplication of the UNG gene ∼550 million years ago, followed by genetic drift of the primary sequence (only 8% homology remaining), but with retention of the general folding pattern of the protein (Haushalter et al., 1999; Aravind and Koonin, 2000). This general strategy is common during evolution to more complex organisms; for example, mammalian cells have several related cyclin-dependent kinases that act consecutively and precisely at defined stages of the cell cycle, replacing a single yeast enzyme of this type. The mammalian UNG enzyme has retained only one of its two key roles in yeast, i.e. the removal of uracil from newly incorporated dUMP opposite A, when dUTP is used as a precursor during DNA replication (Nilsen et al., 2000). The mammalian UNG protein appears to have improved properties in this regard, as it binds to PCNA, which helps sequester it to sites of DNA replication in proliferating cells (Otterlei et al., 1999). In contrast, the consensus PCNA binding motif QxxL/I/MxxF/HF/Y (Warbrick, 1998) is not present in SMUG1. Therefore, SMUG1 is not likely to be able to serve as a backup for UNG at replication forks, and instead has a different function.
The relative amounts of UNG and SMUG1 activity in wild-type murine nuclear extracts appear similar (Figure 2). In previously published work on mammalian systems, the assay conditions were optimized to measure UNG activity (Figure 1), which would make it easy to overlook SMUG1 because of the different kinetic properties of the two enzymes. The UNG enzyme has a higher turnover number (kcat = 4.6 s–1) than other DNA glycosylases, consistent with a role at the replication fork (Otterlei et al., 1999). SMUG1 has a much more modest turnover (kcat = 0.0014 s–1), so UNG dominates under assay conditions of substrate excess (Haushalter et al., 1999). On the other hand, SMUG1 has a low Km of 0.035 µM, whereas UNG has a high Km of 4.5 µM. Consequently, SMUG1 has a clear kinetic advantage at low substrate concentrations (Figure 2). This could be a highly relevant property for a DNA repair function that needs to detect rare deaminated cytosine residues in the genome in an efficient way. It remains to be seen whether the minor uracil–DNA glycosylase activities of the TDG and MBD4 enzymes can serve as general backup enzymes in this respect or whether these functions might have more specialized roles in the cell, for example in specific sequence contexts or at specific stages of the cell cycle.
In the only previous study on SMUG1, the enzyme was considered to show a preference for a single-stranded substrate (Haushalter et al., 1999). This remains correct when the enzyme is assayed in isolation, but under the physiologically more relevant conditions of measuring activity in the presence of APE1, product inhibition by abasic sites in the double-stranded substrate is prevented (Figure 5), and SMUG1 functions efficiently on a double-stranded substrate.
In conclusion, a putative role for SMUG1 as an anti-mutator has been revealed in UNG-deficient mice. SMUG1 has many expected properties of a DNA repair enzyme, such as being present in similar amounts in non-replicating and replicating tissues, and efficiency in detection of low levels of DNA damage. Genetic and biochemical studies with SMUG1 knockout mice should serve to clarify further the strategies employed by mammalian cells to counteract the pre-mutagenic effects of cytosine deamination in DNA.
Materials and methods
Recombinant enzymes
SMUG1. The hSMUG1 coding sequence, PCR amplified from the corresponding cDNA (Haushalter et al., 1999), was cloned into pGEX-3X (Amersham Pharmacia Biotech) as a BamHI–EcoRI fragment in order to express the protein as a fusion protein containing an N-terminal glutathione S-transferase (GST) domain. The resulting expression construct (hSMUG1/pGEX-3X) was transformed into E.coli BL21-DE3 (Novagen) and expression was induced in mid-log phase by the addition of isopropyl-β-d-thiogalactopyranoside (IPTG) to 1 mM. The cells were induced for 4 h at 30°C and then harvested by centrifugation. The cell pellet was resuspended in buffer I [50 mM Tris–HCl, 1 mM EDTA, 1 mM dithiothreitol (DTT), 10% glycerol pH 8.0] plus 500 mM KCl, and a cocktail of protease inhibitors (Sigma). The cells were lysed in a French Press and the resulting lysate was clarified by centrifugation at 20 000 g for 30 min. To the clarified supernatant was added 4 ml (bed volume) of glutathione–Sepharose 4B (Amersham Pharmacia Biotech), pre-equilibrated in buffer I plus 500 mM KCl. The cell lysate and glutathione resin were allowed to mix for 30 min using a mechanical rotary platform and then transferred to a disposable gravity-flow column. The resin was washed with 4 column vol. of buffer I containing 500 mM KCl and 8 vol. of buffer II (buffer I with 100 mM KCl), and then eluted with 10 mM glutathione in buffer II. The eluate was dialysed into buffer III (buffer I with 50 mM KCl) and then treated with Factor Xa (New England Biolabs) at a concentration of 1 mg/ml overnight at 4°C. The reaction mixture was then concentrated by centrifugal dialysis (Centriprep-10; Amicon) and loaded onto a 1 ml Mono S FPLC column (Amersham Pharmacia Biotech), washed with buffer III, and then eluted with a 25 ml linear gradient of KCl (50–700 mM) in buffer I. Fractions containing hSMUG1 were collected, pooled and dialysed into buffer II before loading onto a Hi-Load 16/60 Superdex 75 prep grade gel filtration column (Amersham Pharmacia Biotech). After elution with buffer II and concentration by centrifugal dialysis, aliquots were snap-frozen and stored at –80°C.
mSMUG1. Mouse SMUG1 was produced by coupled in vitro transcription–translation of a murine cDNA clone (DDBJ/EMBL/GenBank accession No. BF467856) using the TNT® Coupled Reticulocyte Lysate system (Promega) according to the manufacturer’s recommendations. The presence of mSMUG1 protein in the reaction mixture was confirmed by standard immunoblotting of a 10% SDS–PAGE gel followed by detection using the enhanced chemiluminescence ECL Kit (Amersham Pharmacia Biotech).
hUNG. Recombinant human UNG protein lacking the N-terminal 84 amino acids (UNGΔ84), expressed and purified as described in Slupphaug et al., (1995), was a gift from Hans Krokan.
mTDG. Recombinant murine TDG (Hardeland et al., 2000), expressed in E.coli and purified free from bacterial uracil–DNA glycosylase, was a gift from Primo Schar and Josef Jiricny.
mMBD4. Recombinant, His-tagged murine MBD4 (Hendrich et al., 1999), expressed in Sf9 insect cells using the Baculovirus system and purified over Ni–NTA agarose, was a gift from Catherine Millar and Adrian Bird.
APE1. Recombinant human AP endonuclease 1, APE1 (also called HAP1), was over-expressed and purified according to a two-step chromatography protocol (B.Demple, unpublished data). The expression construct consists of APE1 fused in-frame with a His6 tag in the pET28b vector.
Nuclear extracts
MEF cell cultures were collected during exponential growth and harvested after trypsin treatment. Snap-frozen organs from ung–/– mice were partly thawed and finely minced with scissors. All following procedures were performed at 0°C. The cell pellet or organ suspensions were resuspended and lysed by incubation in 2× vol. of hypotonic buffer A [10 mM HEPES–KOH pH 7.7, 0.5 mM MgCl2, 10 mM KCl, 1 mM DTT, 0.2 mM phenylmethylsulfonyl fluroride (PMSF)]. After 15 min the nuclei were recovered by centrifugation at 2000 g for 10 min and extracted with 2× vol. of buffer B (20 mM HEPES–KOH pH 7.7, 0.5 mM MgCl2, 0.42 M NaCl, 0.2 mM EDTA, 1 mM DTT, 0.2 mM PMSF, 25% glycerol). After 20 min the preparations were centrifuged at 14 000 g for 10 min and the supernatants recovered and briefly dialysed against buffer C (25 mM HEPES–KOH pH 7.7, 50 mM KCl, 2 mM DTT). The extracts were recovered and quick-frozen in small aliquots.
Antibodies
Polyclonal antibodies were raised against purified recombinant hSMUG1 protein in two rabbits (Research Genetics). SMUG1 antibodies were obtained as a partly purified γ-globulin fraction, by the addition of ammonium sulfate to 50% saturation to serum diluted 2-fold with phosphate-buffered saline (PBS). The resulting precipitate was collected, dissolved in PBS and ammonium sulfate again added to 50% saturation. The final precipitate was collected, dissolved in PBS, dialysed extensively, and frozen in aliquots. Control IgG was prepared from pre-immune rabbit serum in the same way.
DNA substrates
A 19mer (5′-CATAAAGTGUAAAGCCTGG-3′) or a 64mer (5′-GCG ATTTTAATCACAATTCCACACATGACGUGAGCCGGAAGCATAAAGTGAAGTAGCATGACGG-3′) oligonucleotide containing a single, centrally placed uracil residue was 5′-32P-end-labelled with T4 polynucleotide kinase and annealed to a complementary strand containing a G residue opposite uracil to generate a U:G containing substrate, unless stated otherwise. Similarly, T:G substrates were synthesized containing a T rather than a U in the radiolabelled strand.
The double-stranded DNA substrate containing [3H]dUMP was a gift from H.Krokan and was prepared from calf thymus DNA nick-translated in the presence of [3H]dUTP (Krokan and Wittwer, 1981).
Enzyme assays
Uracil release was routinely determined by incubating 0.17 pmol of double-stranded oligonucleotide substrate (giving a final concentration of 8.5 nM dUMP residues) at 37°C for 1 h in the presence of 5 µg of nuclear extract in reaction mixtures containing 20 mM Tris–HCl pH 8, 50 mM NaCl, 1 mM DTT, 1 mM EDTA, 1 ng of APE1 and 100 µg/ml bovine serum albumin (BSA). Extracts were pre-incubated for 10 min on ice with either control IgG (10 µg), SMUG1 antibodies (10 µg), 2 U of Ugi (New England Biolabs) or SMUG1 antibodies and Ugi together. Similarly, uracil release was measured using purified recombinant enzymes, hSMUG1 (50 ng), hUNG (1 ng), mMBD4 (100 ng), mTDG (50 ng) or 5 µl of the 50 µl reaction mixture from coupled in vitro transcription–translation of mSMUG1 cDNA. Reactions were stopped by adding 1 M piperidine and abasic (AP) sites cleaved by incubation at 90°C for 20 min. The samples were dried under vacuum, resuspended in 95% formamide/dyes, electrophoresed in 20% denaturing polyacrylamide gels and visualized on a phosphorimager.
Release of acid-soluble [3H]uracil from the [3H]dUMP containing double-stranded DNA substrate was measured as described previously (Nilsen et al., 2000). Briefly, 5 µg of nuclear extract were incubated with 5 µl of DNA substrate (35.4 ng/µl) in reactions as above for 30 min at 37°C. The specific activity of the substrate was 0.5 mCi/µmol, giving a final concentration of 1.8 µM [3H]dUMP in the assay mixture. After precipitation with 5% TCA and 50 µg of sonicated salmon sperm DNA as carrier, acid-soluble [3H]uracil released into the supernatant was determined by scintillation counting.
Multiple-turnover assays with hSMUG1 were performed by incubating oligonucleotide substrate (1 µM), either single-stranded or with U paired opposite G, with hSMUG1 (2 nM) and APE1 (0 or 30 nM) in a reaction mixture containing 25 mM HEPES–KOH pH 7.6, 50 mM KCl, 2 mM MgCl2, 0.2 mM EDTA, 2 mM DTT and 0.5 mg/ml BSA, at 37°C. At various time points, aliquots were removed from the reaction mixtures, treated with 1 M NaOH in formamide, incubated at 90°C for 10 min, and then analysed in 20% denaturing polyacrylamide gels as above.
Fractionation of ung–/– MEF extracts by Mono S column chromatography
Nuclear extract was dialysed into buffer D (50 mM Tris–HCl pH 7.5, 50 mM NaCl, 1 mM EDTA, 1 mM DTT, 10% glycerol). The protein was recovered and loaded onto a SMART Mono S PC 1.6/5 column (Amersham Pharmacia Biotech) pre-equilibrated with buffer D. Protein was eluted with a 1 ml linear gradient of NaCl (50–500 mM) in buffer D and collected in 50 µl fractions, followed by 1 M NaCl in buffer D. A 2 µl aliquot of each fraction was assayed using a 19mer double-stranded oligonucleotide substrate.
Acknowledgments
Acknowledgements
We thank Hans Krokan (Institute of Cancer Research and Molecular Biology, Norwegian University of Science and Technology, Trondheim, Norway) for providing a uracil-containing DNA substrate and for recombinant human UNGΔ84; Primo Schar and Josef Jiricny (Institute for Medical Radiobiology, Zurich, Switzerland) for recombinant murine TDG; Catherine Millar and Adrian Bird (Wellcome Trust Centre for Cell Biology, University of Edinburgh, UK) for recombinant murine MBD4; and Bruce Demple and Donny Wong (Harvard School of Public Health, Boston, MA) for providing the APE1 over-expression construct. H.N. was the recipient of a long-term EMBO fellowship and K.A.H. was supported by graduate fellowships from the National Science Foundation and the American Chemical Society Division of Organic Chemistry. This work was supported by grants from the Imperial Cancer Research Fund (T.L.) and the NIH (G.L.V., GM51330).
References
- Aprelikova O.N. and Tomilin,N.V. (1982) Activity of uracil–DNA glycosylase in different rat tissues and in regenerating rat liver. FEBS Lett., 137, 193–195. [DOI] [PubMed] [Google Scholar]
- Aravind L. and Koonin,E.V. (2000) The α/β fold uracil-DNA glycosylases: a common origin with diverse fates. Genome Biol., 1, 0007.1–0007.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brynolf K., Eliasson,R. and Reichard,P. (1978) Formation of Okazaki fragments in polyoma DNA synthesis caused by misincorporation of uracil. Cell, 13, 573–580. [DOI] [PubMed] [Google Scholar]
- Duncan B.K. and Miller,J.H. (1980) Mutagenic deamination of cytosine residues in DNA. Nature, 287, 560–561. [DOI] [PubMed] [Google Scholar]
- Duncan B.K. and Weiss,B. (1982) Specific mutator effects of ung (uracil–DNA glycosylase) mutations in Escherichia coli. J. Bacteriol., 151, 750–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederico L.A., Kunkel,T.A. and Shaw,B.R. (1990) A sensitive genetic assay for the detection of cytosine deamination: determination of rate constants and activation energy. Biochemistry, 29, 2532–2537. [DOI] [PubMed] [Google Scholar]
- Hardeland U., Bentele,M., Jiricny,J. and Schar,P. (2000) Separating substrate recognition from base hydrolysis in human thymine–DNA glycosylase by mutational analysis. J. Biol. Chem., 275, 33449–33456. [DOI] [PubMed] [Google Scholar]
- Haug T., Skorpen,F., Aas,P.A., Malm,V., Skjelbred,C. and Krokan,H.E. (1998) Regulation of expression of nuclear and mitochondrial forms of human uracil–DNA glycosylase. Nucleic Acids Res., 26, 1449–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haushalter K.A., Stukenberg,P.T., Kirschner,M.W. and Verdine,G.L. (1999) Identification of a new uracil-DNA glycosylase family by expression cloning using synthetic inhibitors. Curr. Biol., 9, 174–185. [DOI] [PubMed] [Google Scholar]
- Hendrich B., Hardeland,U., Ng,H.-H., Jiricny,J. and Bird,A. (1999) The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature, 401, 301–304. [DOI] [PubMed] [Google Scholar]
- Hill J.W., Hazra,T.K., Izumi,T. and Mitra,S. (2001) Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential co-ordination of the initial steps in base excision repair. Nucleic Acids Res., 29, 430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose F., Hotta,Y., Yamaguchi,M. and Matsukage,A. (1989) Difference in the expression level of DNA polymerase β among mouse tissues: high expression in the pachytene spermatocyte. Exp. Cell Res., 181, 169–180. [DOI] [PubMed] [Google Scholar]
- Impellizzeri K.J., Anderson,B. and Burgers,P.M. (1991) The spectrum of spontaneous mutations in a Saccharomyces cerevisiae uracil–DNA glycosylase mutant limits the function of this enzyme to cytosine deamination repair. J. Bacteriol., 173, 6807–6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krokan H. and Wittwer,C.U. (1981) Uracil–DNA glycosylase from HeLa cells: General properties, substrate specificity and effects of uracil analogues. Nucleic Acids Res., 9, 2599–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krokan H.E., Standal,R. and Slupphaug,G. (1997) DNA glycosylases in the base excision repair of DNA. Biochem. J., 325, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl T. (1974) An N-glycosidase from Escherichia coli that releases free uracil from DNA containing deaminated cytosine residues. Proc. Natl Acad. Sci. USA, 71, 3649–3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl T. (1993) Instability and decay of the primary structure of DNA. Nature, 362, 709–715. [DOI] [PubMed] [Google Scholar]
- Lindahl T. and Barnes,D.E. (2000) Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol., 65, 127–133. [DOI] [PubMed] [Google Scholar]
- Mol C.D., Izumi,T., Mitra,S. and Tainer,J.A. (2000) DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair co-ordination. Nature, 403, 451–455. [DOI] [PubMed] [Google Scholar]
- Nagelhus T.A. et al. (1997) A sequence in the N-terminal region of human uracil–DNA glycosylase with homology to XPA interacts with the C-terminal part of the 34-kDa subunit of replication protein A. J. Biol. Chem., 272, 6561–6566. [DOI] [PubMed] [Google Scholar]
- Nilsen H., Otterlei,M., Haug,T., Solum,K., Naglehus,T.A., Skorpen,F. and Krokan,H.E. (1997) Nuclear and mitochondrial uracil–DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res., 25, 750–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen H. et al. (2000) UNG-deficient mice reveal a primary role of the uracil–DNA glycosylase enzyme during DNA replication. Mol. Cell, 5, 1059–1065. [DOI] [PubMed] [Google Scholar]
- Otterlei M. et al. (1999) Post-replicative base excision repair in replication foci. EMBO J., 18, 3834–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker A., Gu,Y., Mahoney,W., Lee,S.H., Singh,K.K. and Lu,A.L. (2001) Human homolog of the MutY repair protein (hMYH) physically interacts with proteins involved in long-patch DNA base excision repair. J. Biol. Chem., 276, 5547–5555. [DOI] [PubMed] [Google Scholar]
- Scharer O.D. and Jiricny,J. (2001) Recent progress in the biology, chemistry and structural biology of DNA glycosylases. BioEssays, 23, 270–281. [DOI] [PubMed] [Google Scholar]
- Shen J.C., Rideout,W.M.,III and Jones,P.A. (1994) The rate of hydrolytic deamination of 5-methylcytosine in double-stranded DNA. Nucleic Acids Res., 22, 972–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slupphaug G., Eftedal,I., Kavli,B., Bharati,S., Helle,N.M., Haug,T., Levine,D.W. and Krokan,H.E. (1995) Properties of a recombinant human uracil–DNA glycosylase from the UNG gene and evidence that UNG encodes the major uracil–DNA glycosylase. Biochemistry, 34, 128–138. [DOI] [PubMed] [Google Scholar]
- Thompson J.D., Higgins,D.G. and Gibson,T.J. (1994) CLUSTAL_W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res., 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tye B-K., Chien,J., Lehman,I.R., Duncan,B.K. and Warner,H.R. (1978) Uracil incorporation: a source of pulse-labeled DNA fragments in the replication of the Escherichia coli chromosome. Proc. Natl Acad. Sci. USA, 75, 233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal A.E., Hickson,I.D., Boiteux,S. and Radicella,J.P. (2001) Mechanism of stimulation of the DNA glycosylase activity of hOGG1 by the major human AP endonuclease: bypass of the AP lyase activity step. Nucleic Acids Res., 29, 1285–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan A., You,H.J. and Doetsch,P.W. (1999) Phenotypic change caused by transcriptional bypass of uracil in non-dividing cells. Science, 284, 159–162. [DOI] [PubMed] [Google Scholar]
- Warbrick E. (1998) PCNA binding motif through a conserved motif. BioEssays, 20, 195–199. [DOI] [PubMed] [Google Scholar]
- Warner H.R., Duncan,B.K., Garrett,C. and Neuhard,J. (1981) Synthesis and metabolism of uracil-containing deoxyribonucleic acid in Escherichia coli. J. Bacteriol., 145, 687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters T.R., Gallinari,P., Jiricny,J. and Swann,P.F. (1999) Human thymine–DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. J. Biol. Chem., 274, 67–74. [DOI] [PubMed] [Google Scholar]
- Yang H., Clendenin,W.M., Wong,D., Demple,B., Slupska,M.M., Chiang,J.H. and Miller,J.H. (2001) Enhanced activity of adenine-DNA glycosylase (Myh) by apurinic/apyrimidinic endonuclease (Ape1) in mammalian base excision repair of an A/OG mismatch. Nucleic Acids Res., 29, 743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmudzka B.Z., Fornace,A., Collins,J. and Wilson,S.H. (1988) Characterization of DNA polymerase β mRNA: cell-cycle and growth response in cultured human cells. Nucleic Acids Res., 16, 9587–9596. [DOI] [PMC free article] [PubMed] [Google Scholar]