

Abstract
The tumor suppressor p53 is the most commonly mutated gene in human cancers. Active p53 is able to stimulate the transcription of a variety of genes including the pro-apoptotic gene bax, as well as p21, a cell cycle regulator. In this study we produced novel zinc finger transcription factors that would selectively increase the expression of bax, but not of other p53 targets. Reporter gene assays in p53-negative Saos-2 cells showed that the novel zinc finger proteins stimulated transcription driven by a minimal bax promoter, but not that driven by a minimal p21 promoter. Moreover, electromobility shift assays demonstrated that the novel transcription factors could bind the bax promoter sequence with high affinity and selectivity. Expression of a five zinc finger protein (5ZFAV) in COS-7 cells resulted in an increase in Bax protein levels, indicating that this novel transcription factor could act on endogenous gene expression. Expression of 5ZFAV also drastically reduced Saos-2 cell survival; this effect could be reversed by the general caspase inhibitor B-D-FMK. These data suggest that 5ZFAV is able to induce apoptosis through increased Bax expression. Further, while expression of 5ZFAV in p53-deficient Saos-2 cells reduced cell survival, there was little effect on U-2 OS cells which have wild-type p53. Thus the selective induction of the pro-apoptotic bax gene may be a valuable adjunct to cancer chemotherapy by diminishing survival of p53-deficient tumor cells.
INTRODUCTION
The tumor suppressor p53 plays a key role in the cellular response to stress signals such as DNA damage, activated oncogenes, hypoxia or nucleotide depletion (1,2). Normal cells contain low levels of unstable and latent p53 wild-type protein. Upon activation and stabilization by post-translational modifications, p53 can function as a sequence-specific transcription factor. Activated p53 is able to regulate a number of target genes, including p21 (waf1/cip1) and bax (3). Increased transcription of the p21 gene leads to inactivation of cyclin-dependent kinases, which then results in G1 cell cycle arrest (4), while p53-mediated expression of the Bcl-2 family member Bax leads to apoptosis (5).
It has been reported that activation and/or overexpression of Bax results in its translocation to the outer mitochondrial membrane (6), where Bax induces the release of cytochrome c (7). Cytochrome c forms a complex with Apaf-1 and procaspase 9, which leads to the activation of the downstream caspase cascade, eventually resulting in apoptosis (5,8–10). The pro-apoptotic effects of Bax can be inhibited by the anti-apoptotic Bcl-2 family members (Bcl-2 and Bcl-XL), and it is commonly assumed that the ratio of pro- to anti-apoptotic Bcl-2 family member proteins determines the fate of the cell (11). Thus, influencing the ratio of pro- to anti-apoptotic Bcl-2 family member proteins by selective transcription of the bax gene should drive cells into apoptosis. This possibility identifies the bax gene as an interesting therapeutic target.
The p53 gene product is lost or inactivated by mutation in over 50% of cancers (12). This tends to make cancer cells refractory to chemotherapeutic agents, since p53 negative cells fail to induce bax, as well as other p53-regulated pro-apoptotic genes (13,14). These observations underlie recent approaches using transfection of bax with the goal of driving cancer cells into programmed cell death (15,16). As an alternative approach, in the studies described below, we use novel designed trancription factors to selectively regulate the expression of bax.
The design of artificial transcription factors to control the expression of targeted genes represents a rapidly growing area of research. Transcription factors are generally of modular structure and contain a DNA binding domain as well as a trans-regulating domain. The Cys2-His2 type of zinc finger (Zif) provides a particularly valuable tool for creating novel transcription factors with modified DNA binding specificities, since Zifs can function as semi-autonomous modules (17). Several groups have used phage display strategies to select novel zinc fingers (18–21), while our laboratory has identified zinc fingers with novel DNA binding abilities by using a yeast one hybrid approach (22). This strategy was used to develop zinc finger peptides that bind selectively to the MDR1 promoter, leading to the design of a novel repressor able to inhibit the transcription of the chromosomal MDR1 gene and modulate tumor cell drug resistance (23,24).
In the present study we designed novel transcription factors to selectively regulate expression of the bax gene. We used DNA binding domains comprised of three or five Zifs, which were based on the zinc fingers from the mouse transcription factor Zif268 (25). The Zifs were coupled to the VP16 transactivation domain (26). The novel transcription factors were able to increase transcription of a reporter gene driven by a truncated artificial bax promoter, but not a reporter gene driven by a truncated p21 promoter. Transiently transfected COS-7 cells carrying a novel five zinc finger transcription factor showed an increased Bax expression. Transfection of this factor also resulted in a drastic reduction of survival of p53-deficient Saos-2 cells; this effect could be reversed by the addition of caspase inhibitors. These data suggest that the designed zinc finger proteins are capable of driving Saos-2 cells into apoptosis through selective up-regulation of bax expression.
MATERIALS AND METHODS
Plasmids
All protein-expressing plasmids used in this study are based on the mammalian expression vector pcDNA3.1(–)/Myc-HisA (Stratagene, La Jolla, CA). As previously described (23), we cloned the nuclear localization sequence (NLS) from the TAT protein [amino acids 37–60 (27)] into XbaI–EcoRI restricted pcDNA3.1. Using the restriction enzymes EcoRI–KpnI we added the zinc finger sequences in frame downstream of the NLS. In the final step we cloned the VP16 transactivation domain [amino acids 411–490 (26)] into a KpnI–HindIII restriction site C-terminal to the zinc finger coding regions (Fig. 1A). The resulting plasmids were named pcZFAV, pcZF123V, which code for 3-Zif proteins, and pc5ZFAV, which codes for a 5-Zif protein; all expressed proteins are C-terminal Myc and His tagged. The plasmid pCMV-p53 was purchased from Clontech (Palo Alto, CA), and a Bax expressing plasmid was purchased from Upstate Biotechnology (Lake Placid, NY). Reporter plasmids are based on the pFR-Luc plasmid (Stragene, La Jolla, CA). The five yeast Gal4 binding sites upstream of the luciferase gene were excised with HindIII/KpnI and replaced by a multiple cloning site (MCS) containing BglII, PstI, AgeI, PvuII, HindIII, a TATATAA-box and KpnI. Inserting the MCS into pFR-Luc resulted in the loss of the original HindIII site. Either two bax [two times –489 to –449: TGGGCTCACAAG TTAGAGACAAGCCTGGGCGTGGGCTATAT; (28)] or two p21 [two times –2300 to –2331: CAGGAACATGT CCCAACATGTTGAGCTCTGGC; (29)] promoter regions, both containing consensus p53 binding sites (underlined), were cloned into the BglII/HindIII sites of the MCS. The resulting vectors were named pFR*-2bax and pFR*-2p21. The MCS, as well as the promoter regions, were obtained by annealing two primers coding for these sequences. Primers were obtained from the Nucleic Acids Core Facility (Lineberger Comprehensive Cancer Center, UNC).
Figure 1.

(A) Structure of the novel three (ZFAV) and five (5ZFAV) zinc finger transcription factors binding to their cognate sites in the bax promoter. ZF1 and ZF2 are wild-type zinc fingers of the transcription factor Zif268. ZF1* and ZF3* are zinc fingers selected by screening combinatorial libraries (22). VP16 is a trans-activation domain and NLS is a nuclear localization sequence. The consensus p53 binding site is shown in the box and the zinc finger binding sites are shown in bold. (B) Protein expression. Saos-2 nuclear cell extracts were immunoblotted with anti-Myc (9E10) antibody. The black arrows show the positions of the zinc finger proteins; the gray arrowhead marks non-specific bands seen in controls.
Cells
Escherichia coli DH5α competent cells were obtained from Life Technologies (Gaithersburg, MD). The p53-deficient human osteosarcoma Saos-2 cell line, the wild-type p53-containing human osteosarcoma U-2 OS cell line, and the monkey kidney COS-7 cell line were obtained from the Lineberger Comprehensive Cancer Center (University of North Carolina). Saos-2 and U-2 OS cells were cultured in McCoy’s 5A medium with l-glutamine and COS-7 cells were cultured in Dulbecco’s Modified Eagle’s Medium. Both media were obtained from Life Technologies (Geithersburg, MD) and, if not otherwise described, were used supplemented with 10% fetal bovine serum (FBS).
Transient transfection
Saos-2 or U-2 OS cells were transiently transfected using LipofectAMINE™ reagent (Life Technologies, Geithersburg, MD) and COS-7 cells were transiently transfected using SuperFect™ reagent (Qiagen Inc., Valencia, CA), both according to the manufacturer’s recommendation.
CD4 enrichment
Saos-2 and U-2 OS cells were plated in 100 mm dishes (1.6 × 106 cells/dish) and transiently co-transfected with a CD4-expressing plasmid (4 µg) and 2 µg [0.255 ng/mm2 plate area] plasmids coding for 5ZFAV or Bax or the empty vector. The cells were harvested 15 h after transfection and selected for transfected cells with magnetic anti-CD4 Dynabeads M-450 (Dynal ASA, Oslo, Norway) according to the manufacturer’s recommendation. Transfected cells were lysed in modified RIPA buffer and used in western blot experiments.
Western blotting
Transfected cells were lysed in modified RIPA buffer (24). In some cases nuclear extracts were obtained with NE-PER™ nuclear and cytoplasmic extraction reagents (PIERCE, Rockford, IL) according to the manufacturer’s recommendation. Lysates and nuclear extracts were resolved by SDS–PAGE and proteins detected by western blotting as described (24). Expressed zinc finger proteins were detected by using a mouse monoclonal anti c-myc antibody 9E10 (BABCO, Richmond, CA) at a dilution of 1:2000. The secondary antibody was a peroxidase-conjugated goat anti mouse IgG antibody (Calbiochem, San Diego, CA) at a dilution of 1:5000. Bax proteins were detected using a rabbit polyclonal anti bax antibody (Upstate Biotechnology, Lake Placid, NY) at a dilution of 1:700. Actin was detected by using a rabbit anti actin antibody (Sigma-Aldrich Co., St Louis, MO), at a dilution of 1:7000. The secondary antibody was a peroxidase-conjugated goat anti rabbit IgG antibody (Calbiochem, San Diego, CA) at a dilution of 1:5000. Signals were detected by enhanced chemiluminescence (ECL kit, Amersham, Arlington Heights, IL) using X-ray films (Denville Scientific Inc., Metuchen, NJ). Densitometry data were collected with the Fluor-S™ MultiImager (Bio-Rad, Philadelphia, PA) and analyzed with Quantity One software (Bio-Rad, Philadelphia, PA).
Electromobility shift assays
Proteins were expressed using the TNT® quick coupled transcription/ translation system from Promega (Madison,WI). Double-stranded (ds) DNA sequences were produced by annealing complementary oligonucleotides containing zinc finger (bold) and p53 (underlined) binding sequences [5′bax TGGGCTCACAAGTTAGAGACAAGCCTGGGCGTGG GCTATATTGCTA; 3′bax TAGCAATATAGCCCACGC CC(AGGCTTGTCTCTAACTTGAGCCCA); 5′p21(p53BS) CTAGGAACATGTCCCAACATGTTGAGCTCTGGCA; 3′p21(p53BS) TGCCAGAGCTCAACATGTTGG(GACAT GTTCCTAG); 5′Zif268BS AGGAGGTGAAGCGTGGG CGTAGTGAGCT GAT; 3′Zif268BS ATCAGCTCACTA CGCCCA(GCTTCACCTCCT)]. For each oligonucleotide pair we designed a short version (without the nucleotides in the brackets) and a long version (with the nucleotides in the brackets) of the 3′ primer. Radiolabeled ds oligonucleotides were obtained from complexes using the short 3′-oligonucleotides by fill-in reactions with [α-32P]dCTP and Klenow Enzyme (Roche Diagnostics, Indianapolis, IN). Annealing of the long versions of the synthetic oligonucleotides resulted in unlabeled ds oligonucleotides. All oligonucleotides were purchased from the Nucleic Acids Core Facility (Lineberger Comprehensive Cancer Center, UNC). Proteins (5 µl samples) from the TNT® Quick reaction were pre-incubated at room temperature with the non-specific competitor poly dI-dC (1 µg/reaction) and PMSF (1 mM). After 10 min labeled probe (with or without unlabeled specific competitor) was added. The reactions were incubated for 20 min in binding buffer containing 50 mM Tris pH 7.6, 50% glycerol, 5 mM DTT and 2.5 mM EDTA. The samples were loaded on a 5% polyacrylamide gel (50% glycerol) and run at 150 V in 1× TGE buffer (Tris, glycine, EDTA) at room temperature for 2 h. The gel was dried and exposed to X-ray film.
Dual luciferase reporter assay
Saos-2 cells were plated in 12 well plates (1.3 × 105 cells/well) and 24 h later were transiently co-transfected with 0.33 µg (0.25 ng/mm2 plate area) protein expressing plasmids (pcZFAV, pc5ZFAV or pCMV-p53) or with pcDNA3.1, and with 1 µg reporter plasmid (pFR*-2bax or pFR*-2p21) and 0.1 µg renilla luciferase plasmid pRL-TK (Promega, Madison, WI) for 4 h. Cells were washed and incubated in 1% FBS-containing medium. Luciferase activities were determined using the dual-luciferase™ reporter assay system (Promega, Madison, WI) 18–20 h after transfection. Renilla luciferase activities were used as an internal control to normalize obtained activities. Measurements were performed on a Monolight 2010 instrument (Analytical Luminescence Laboratory, San Diego, CA).
Cells survival assays
Saos-2 or U-2 OS cells were plated in 60 mm dishes (8 × 105 cells/dish) and 24 h later were transiently co-transfected with a β-galactosidase-expressing plasmid (2 µg) and a protein-expressing plasmid [1 µg (0.35 ng/mm2 plate area) of pcZFAV, pc5ZFAV, pCMV-p53 or a Bax-expressing plasmid]. After transfection, cells were incubated in 1% FBS-containing medium. Caspase inhibitor [B-D-FMK (Enzyme Systems Products, Livermore, CA)] was used at a concentration of 40 µM and added to the medium 0 and 12 h after transfection. At 24 h post-transfection cells were washed, fixed and stained for β-galactosidase activity. Blue (β-galactosidase positive) cells were counted using a gridded 60 mm dish and a tissue culture microscope. The number of blue cells that could be observed in transfections with β-galactosidase plus ‘empty’ vector controls was taken as 100% and was used for normalization.
Enzymatic cell survival assay
Saos-2 or U-2 OS cells were plated in 12 well plates (7 × 104 cells/well) and 24 h later were transiently co-transfected with 0.08 µg plasmids (0.06 ng/mm2 plate area) coding for the various proteins indicated or the ‘empty’ vector and with a β-galactosidase plasmid (0.5 µg). After transfection, cells were washed and incubated in FBS-containing medium. β-Galactosidase activities were determined 96 h after transfection using the β-galactosidase Enzyme Assay System (Promega, Madison, WI). Measurements were performed on a Universal Microplate Reader ELX800 (Bio-Tek Instruments, Winooski, VT) at 405 nm. The average absorbance with β-galactosidase plasmid and pcDNA3.1 empty vector was set as 100% for normalization.
RESULTS
Construction and expression of novel Zif transcription factors
In examining the bax promoter we detected consensus binding sites for zinc fingers one (ZF1) and two (ZF2) of the native Zif268 transcription factor in close proximity to the p53 consensus binding site; the promoters of other p53 responsive genes, such as p21, lack such sites. In addition, previous screening in yeast for Zifs with novel DNA-recognition capabilities (23) yielded two novel Zifs termed ZF3* and ZF1* that could recognize additional sites overlapping or adjacent to the bax p53 consensus site; these novel zinc fingers were based on Zif268, but had altered residues at critical DNA binding positions. Using ZF3*, which binds the nucleotide triplet ‘GCC’, as well as the native ZF1 and ZF2 modules, we constructed a three zinc finger protein termed ZFAV. This novel transcription factor is designed to bind the DNA sequence ‘GCCTGGGCG’, which overlaps with the bax promoter p53 consensus binding site (Fig. 1A). To seek stronger binding to the bax promoter, we added two extra zinc fingers N-terminal to the ZFAV DNA-binding modules; these were native ZF2, and ZF1* which was selected to bind the ‘GCT’ nucleotide triplet. This resulted in a five zinc finger transcription factor that we termed 5ZFAV that was designed to bind the DNA sequence ‘GCCTGGGCGTGGGCT’ which overlaps the p53 binding site. Both ZFAV and 5ZFAV contained a NLS and the mammalian trans-activator domain of VP16 (Fig. 1A). As a control we constructed a similar transcription factor carrying wild-type zinc fingers one (ZF1), two (ZF2) and three (ZF3) of Zif268; the resulting plasmid was named pcZF123V.
The plasmids coding for the various novel transcription factors were transiently transfected into p53 deficient Saos-2 cells and expression as well as nuclear localization was detected by western blot analysis. Immunoblotting showed a strong expression of the three zinc finger (ZFAV, ZF123V) and five zinc finger (5ZFAV) proteins in the nucleus of Saos-2 cells (Fig. 1B).
Selective activation of a minimal Bax promoter by the novel transcription factors
To test the effects of the novel zinc finger proteins on the p53 binding site containing bax or p21 promoters we performed Dual Luciferase assays (Promega). Human p53 deficient Saos-2 cells were cotransfected with plasmids coding for the transcription factors and with the reporter plasmids pFR*-2bax or pFR*-2p21. The novel zinc finger proteins ZFAV (2) and 5ZFAV (3) were able to increase expression of the luciferase gene under the control of the truncated bax promoter 9- to 10-fold (Fig. 2). In comparison, these proteins had no effect on luciferase gene expression regulated by the truncated p21 promoter. Both ZFAV (2) and 5ZFAV (3) displayed only modest activation of the bax reporter as compared with the effect of native p53 (4); however, the effect of the novel factors was very selective in favoring bax over p21, whereas p53 (4) activated both reporters (Fig. 2).
Figure 2.

Transcriptional activities of ZFAV and 5ZFAV in Saos-2 cells. Luciferase activities were measured 24 h after transfection and normalized against renilla luciferase activities. Luciferase activities obtained with the reporter vector pFR*-2p21 are in black bars and activities obtained with the reporter vector pFR*-2bax are in gray bars. Normalized luciferase activities obtained with cell lysate from cells transfected with the empty vector pcDNA3.1 were set as one. The transfected plasmids were (1) pcDNA, (2) ZFAV, (3) 5ZFAV, (4) p53.
ZFAV and 5ZFAV bind to bax promoter sequences
We examined the ability of the novel transcription factors to bind to sequences in the bax promoter using electromobility shift assays (EMSAs) (Fig. 3). We expressed ZFAV, 5ZFAV and ZF123V using an in vitro transcription/translation system. ZF123V, carrying the wild-type zinc fingers of the transcription factor Zif268, was used as a control, since the binding affinities are well known (30). ZFAV and 5ZFAV bound to the bax ds oligonucleotide (Fig. 3A, lanes 10 and 15), and the binding could be competed by the addition of increasing amounts of unlabeled bax oligonucleotide (lanes 11–13 and 16–18). ZF123V did not bind the bax sequence (lane 20), but rather bound the consensus Zif268 binding sequence (lane 1) and this binding could also be competed by increasing amounts of unlabeled oligonucleotide (lanes 2–4). The EMSAs also indicated the selective binding of ZFAV and 5ZFAV to the bax but not to the p21 promoter sequences, since the binding to the bax sequence could be competed by unlabeled bax but not p21 competitor oligonucleotides (Fig. 3B). The selectivity of 5ZFAV for its targeted sequence was also confirmed by the fact that bax competitor oligonucleotides blocked binding of 5ZFAV to the bax sequence while zif268BS competitor oligonucleotides had only a slight effect (Fig. 3C).
Figure 3.

Binding of ZFAV and 5ZFAV to bax ds oligonucleotides. (A) The three zinc finger proteins ZFAV, ZF123V and the five zinc finger protein 5ZFAV were incubated with the indicated labeled dsDNA and the binding was competed by the addition of increasing amounts of unlabeled oligonucleotide competitor. (B) The binding of ZFAV and 5ZFAV to the bax promoter was competed by increasing amounts of bax or p21 unlabeled oligonucleotide and in (C) with increasing amounts of bax or zif268BS unlabeled oligonucleotide. The concentrations of radiolabeled probe in (A) were 1.7 pmol/ml for zif268BS and 2.1 pmol/ml for bax. The concentrations of unlabeled competitor ranged from 0–346 pmol/ml for zif268BS, and 0–416 pmol/ml for bax. In (B) the concentration of radiolabeled bax probe was 3.8 pmol/ml, while the ranges of unlabeled competitors were 0–1.54 nmol/ml for bax and 0–2.55 nmol/ml for p21. In (C), 0.4 pmol/ml labeled bax was used, while the unlabeled competitors ranged from 0–174 pmol/ml bax and 0–174 pmol/ml zif268BS.
5ZFAV induces Bax expression in COS-7 cells
Since Saos-2 cells have very low transfection and protein expression efficiency, we used COS-7 cells to test whether ZFAV or 5ZFAV could up-regulate expression of endogenous Bax. Constructs expressing ZFAV, 5ZFAV or Bax proteins were transiently transfected into the cells, and whole cell lysates were harvested after 24 h. The lysates were immunoblotted with an anti Bax antibody. These experiments showed an increase of Bax expression in cells carrying the five zinc finger protein 5ZFAV as compared with control cells tranfected with empty vector (Fig. 4). As a positive control we transfected COS-7 cells with various amounts of a Bax-expressing plasmid; this also led to increased Bax protein levels. Transfection of 5ZFAV did not affect levels of p21 protein in these experiments (not shown). Thus 5ZFAV selectively induced a moderate overexpression of Bax in COS-7 cells. In similar experiments in Saos-2 cells we were not able to detect increased Bax expression by western blotting of whole cell lysates (even with transfection of Bax vector) presumably because of poor transfection efficiency and low levels of protein production (data not shown).
Figure 4.
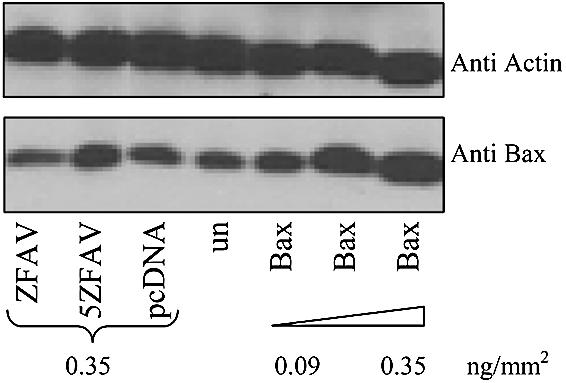
Bax expression in transiently transfected COS-7 cells. Whole cell lysates of cells transfected with pCMV-p53, pcZFAV, pc5ZFAV, pcDNA3.1 or a Bax expressing plasmid were immunoblotted with an anti-Bax or anti-actin antibody. un = untransfected cells.
5ZFAV causes caspase-dependent cell death
Since the novel transcription factor 5ZFAV could up-regulate Bax expression, we were interested in determining whether it could cause apoptosis. Here we used p53-deficient Saos-2 cells so that the effects of 5ZFAV could be examined independently of p53. Thus cells were transfected with a β-galactosidase plasmid as a marker for transfection, and co-transfected with vectors expressing ZFAV, 5ZFAV, Bax or p53, or with pcDNA3.1 ‘empty’ vector as a control. In these experiments (Fig. 5A) it was clear that Bax, p53 and 5ZFAV, but not ZFAV, could dramatically reduce cell survival. This effect was fully reversed by the general caspase inhibitor B-D-FMK for the cases of Bax and 5ZFAV, but only partially reversed for p53 (Fig. 5B). Thus the novel transcription factor 5ZFAV reduces cell survival by a caspase-dependent mechanism.
Figure 5.

Cell survival assay. (A) Co-transfected Saos-2 cells (β-galactosidase plasmid plus plasmids coding for the various proteins indicated) were fixed and stained for β-galactosidase activity. Blue cells per microscope field were counted. The average number of blue cells in samples transfected only with β-galactosidase plasmid and pcDNA3.1 empty vector was set as 100% for normalization. (B) Cell survival with a caspase inhibitor. Co-transfected cells treated or untreated with the caspase inhibitor B-D-FMK (200 nM) were fixed and stained for β-galactosidase. The samples were normalized as in (A). Transfection efficiency was ∼10% in these assays.
Effects of 5ZFAV are dependent on p53 status
The studies above suggest that the five Zif transcription factor can upregulate Bax levels and induce caspase-dependent cell death. However, it is clear that the effects of 5ZFAV are weaker than those of Bax itself or of p53. Thus it seemed important to test the effects of 5ZFAV in cell lines that possessed or lacked wild-type p53. As seen in Figure 6A, over-expression of Bax led to impaired cell viability in both p53-deficient Saos-2 cells and p53 replete U-2 OS cells. However, over-expression of 5ZFAV reduced the viability of Saos-2 cells but not U-2 OS cells. Thus the effects of 5ZFAV on cell survival seems to depend on the p53 status of the cell. To examine Bax expression levels we selected for transfected cells with CD4 Dynabeads. In agreement with the cell survival assay, pc5ZFAV transfected Saos2 cells, but not pc5ZFAV transfected U-2 OS cells showed a modest but constant increase in Bax expression compared with empty vector transfected cells (Fig. 6B and C).
Figure 6.

Survival of Saos-2 versus U-2 OS cells. (A) Saos-2 or U-2 OS cells were plated in 12 well plates (1.3 × 105 cells/well) and 24 h later were transiently co-transfected with 0.08 µg plasmids (0.06 ng/mm2 plate area) coding for the various proteins indicated or the ‘empty’ vector and with a β-galactosidase plasmid (0.5 µg). Cells were washed and incubated in FBS-containing medium. β-Galactosidase activities were determined 96 h after transfection. The average absorbance with β-galactosidase plasmid and pcDNA3.1 empty vector was set as 100% for normalization. (B) Bax expression. Co-transfected Saos-2 and U-2 OS cells (CD4-expressing plasmid plus plasmids coding for the various proteins indicated) were CD4 enriched for transfected cells and their lysates were immunoblotted with anti-Bax or anti-actin antibody. (C) Densitometry data were used to determine the ratios of Bax to actin expression in cells carrying 5ZFAV, Bax or empty vector pcDNA3.1. The Bax to actin ratio of empty vector pcDNA3.1 transfected cells was set as one. Results represent the means and standard errors of three experiments.
DISCUSSION
When p53 is up-regulated in response to DNA damage or to other stresses, a complex cell regulatory network is entrained. P53 increases expression of pro-apoptotic genes such as Bax and Noxa (1,14), as well as cell cycle control genes such as p21 (29), and genes involved in DNA repair (31). Thus the ultimate outcome in terms of cell death versus cell cycle arrest depends on the interplay of multiple genes downstream of p53 (32). In some cases it may be desirable to selectively regulate one or more p53 target genes. In particular, the selective up-regulation of pro-apoptotic genes in cancer cells could result in enhanced effectiveness for chemotherapy agents. Thus in this study we have described novel zinc-finger transcription factors that selectively activate the bax gene, but not other p53 responsive genes, with p21 serving as a model.
The ZFAV and 5ZFAV proteins selectively activate a minimal Bax promoter, but not a minimal p21 promoter, although both share p53 consensus sites. Thus these novel Zif transcription factors are selective. In terms of efficacy, ZFAV and 5ZFAV are much weaker than native p53 in driving the Bax promoter. However, this is not unexpected since p53 binds to its consensus sites as a tetramer and thus brings four transactivating domains into play (33), while ZFAV and 5ZFAV are designed to bind as monomers. The selectivity of ZFAV and 5ZFAV was confirmed in EMSA where these proteins were shown to bind to oligonucleotides reflecting the bax promoter but not those reflecting the p21 promoter. Surprisingly, there was only a modest difference in the binding of 5ZFAV and ZFAV, despite the presence of two extra Zifs in the former (data not shown). Interestingly, although ZFAV and 5ZFAV behaved fairly similarly in in vitro binding assays and in reporter gene assays, 5ZFAV was much more effective in inducing Bax protein expression and in triggering cell death. The basis for this difference is not clear at this time; however, it is possible that 5ZFAV is more effective at accessing chromosomal sites than is ZFAV.
A cell survival assay showed that the expression of 5ZFAV in Saos-2 cells led to cell death. To test whether this was due to apoptosis or to a general toxic effect we blocked caspase activation by the addition of the general caspase inhibitor B-D-FMK. It is known that overexpression and/or activation of Bax leads to apoptosis via the activation of a caspase cascade, and that inhibition of caspase activation blocks Bax-mediated apoptosis (34). This was true in the present studies, since the addition of B-D-FMK led to a complete inhibition of apoptosis in Saos-2 cells ectopically overexpressing Bax. Since a similar caspase inhibitor effect was observed for 5ZFAV-induced cell death, this suggests that 5ZFAV causes a form of caspase-dependent apoptosis. However, we have not yet extensively characterized the cell death process in the 5ZFAV transfectants. Regulation of apoptosis is complex (35), but clearly enhanced Bax levels are likely to play a role in this process.
The development of bax-gene targeted novel transcription factors might be a useful tool for selectively killing p53-deficient tumor cells. These novel transcription factors may have an advantage over the reintroduction of wild-type p53 proteins, or functional active monomeric p53 proteins, into p53 mutated cancer cells, since mutated p53 proteins frequently bind to wild-type p53, thus causing dominant negative effects (36,37). Although Bax has a role in tumor cell death (15,38), the biology of Bax induced cell death is quite complicated (39). Simple transfection of Bax or other pro-apoptotic proteins would not seem to offer any selectivity between normal cells and p53-deficient tumor cells. However, 5ZFAV, the novel bax-promoter targeted transcription factor, seems to be more effective in p53-deficient Saos-2 cells than in U-2 OS cells that have wild-type p53. Since 5ZFAV is a relatively weak transactivator compared with p53, its bax-selective effects may be overcome by the presence of wild-type p53. This may lead to a balanced expression of pro- and anti-apoptotic genes in p53-replete cells. Thus 5ZFAV might be considered a prototype for novel transcription factors that act preferentially in p53 deficient tumor cells rather than normal cells having wild-type p53.
REFERENCES
- 1.Ko L.J. and Prives,C. (1996) p53: puzzle and paradigm. Genes Dev., 10, 1054–1072. [DOI] [PubMed] [Google Scholar]
- 2.Levine A.J. (1997) p53, the cellular gatekeeper for growth and division. Cell, 88, 323–331. [DOI] [PubMed] [Google Scholar]
- 3.Agarwal M.L., Taylor,W.R., Chernov,M.V., Chernova,O.B. and Stark,G.R. (1998) The p53 network. J. Biol. Chem., 273, 1–4. [DOI] [PubMed] [Google Scholar]
- 4.Sheikh M.S., Rochefort,H. and Garcia,M. (1995) Overexpression of p21WAF1/CIP1 induces growth arrest, giant cell formation and apoptosis in human breast carcinoma cell lines. Oncogene, 11, 1899–1905. [PubMed] [Google Scholar]
- 5.Finucane D.M., Bossy-Wetzel,E., Waterhouse,N.J., Cotter,T.G. and Green,D.R. (1999) Bax-induced caspase activation and apoptosis via cytochrome c release from mitochondria is inhibitable by Bcl-xL. J. Biol. Chem., 274, 2225–2233. [DOI] [PubMed] [Google Scholar]
- 6.Hsu Y.T., Wolter,K.G. and Youle,R.J. (1997) Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc. Natl Acad. Sci. USA, 94, 3668–3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jurgensmeier J.M., Xie,Z., Deveraux,Q., Ellerby,L., Bredesen,D. and Reed,J.C. (1998) Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl Acad. Sci. USA, 95, 4997–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li P., Nijhawan,D., Budihardjo,I., Srinivasula,S.M., Ahmad,M., Alnemri,E.S. and Wang,X. (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell, 91, 479–489. [DOI] [PubMed] [Google Scholar]
- 9.Kobayashi T., Sawa,H., Morikawa,J., Zhang,W. and Shiku,H. (2000) Bax induction activates apoptotic cascade via mitochondrial cytochrome c release and Bax overexpression enhances apoptosis induced by chemotherapeutic agents in DLD-1 colon cancer cells. Jpn J. Cancer Res., 91, 1264–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei M.C., Zong,W.X., Cheng,E.H., Lindsten,T., Panoutsakopoulou,V., Ross,A.J., Roth,K.A., MacGregor,G.R., Thompson,C.B. and Korsmeyer,S.J. (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science, 292, 727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mikhailov V., Mikhailova,M., Pulkrabek,D.J., Dong,Z., Venkatachalam,M.A. and Saikumar,P. (2001) Bcl-2 prevents Bax oligomerization in the mitochondrial outer membrane. J. Biol. Chem., 276, 18361–18374. [DOI] [PubMed] [Google Scholar]
- 12.Levine A.J., Momand,J. and Finlay,C.A. (1991) The p53 tumour suppressor gene. Nature, 351, 453–456. [DOI] [PubMed] [Google Scholar]
- 13.Nakano K. and Vousden,K.H. (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell, 7, 683–694. [DOI] [PubMed] [Google Scholar]
- 14.Oda E., Ohki,R., Murasawa,H., Nemoto,J., Shibue,T., Yamashita,T., Tokino,T., Taniguchi,T. and Tanaka,N. (2000) Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science, 288, 1053–1058. [DOI] [PubMed] [Google Scholar]
- 15.Li X., Marani,M., Yu,J., Nan,B., Roth,J.A., Kagawa,S., Fang,B., Denner,L. and Marcelli,M. (2001) Adenovirus-mediated Bax overexpression for the induction of therapeutic apoptosis in prostate cancer. Cancer Res., 61, 186–191. [PubMed] [Google Scholar]
- 16.Honda T., Gjertsen,B.T., Spurgers,K.B., Briones,F., Lee,S.J., Hobbs,M.L., Meyn,R.E., Roth,J.A., Logothetis,C. and McDonnell,T.J. (2001) Restoration of bax in prostate cancer suppresses tumor growth and augments therapeutic cell death induction. Anticancer Res., 21, 3141–3146. [PubMed] [Google Scholar]
- 17.Pabo C.O. and Sauer,R.T. (1992) Transcription factors: structural families and principles of DNA recognition. Annu. Rev. Biochem., 61, 1053–1095. [DOI] [PubMed] [Google Scholar]
- 18.Kim J.S. and Pabo,C.O. (1998) Getting a handhold on DNA: design of poly-zinc finger proteins with femtomolar dissociation constants. Proc. Natl Acad. Sci. USA, 95, 2812–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Isalan M., Klug,A. and Choo,Y. (2001) A rapid, generally applicable method to engineer zinc fingers illustrated by targeting the HIV-1 promoter. Nat. Biotechnol., 19, 656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dreier B., Beerli,R.R., Segal,D.J., Flippin,J.D. and Barbas,C.F.,III (2001) Development of zinc finger domains for recognition of the 5′-ANN-3′ family of DNA sequences and their use in the construction of artificial transcription factors. J. Biol. Chem., 276, 29466–29478. [DOI] [PubMed] [Google Scholar]
- 21.Beerli R.R., Dreier,B. and Barbas,C.F.,III (2000) Positive and negative regulation of endogenous genes by designed transcription factors. Proc. Natl Acad. Sci. USA, 97, 1495–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng X., Boyer,J.L. and Juliano,R.L. (1997) Selection of peptides that functionally replace a zinc finger in the Sp1 transcription factor by using a yeast combinatorial library. Proc. Natl Acad. Sci. USA, 94, 14120–14125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bartsevich V.V. and Juliano,R.L. (2000) Regulation of the MDR1 gene by transcriptional repressors selected using peptide combinatorial libraries. Mol. Pharmacol., 58, 1–10. [DOI] [PubMed] [Google Scholar]
- 24.Xu D., Fisher,M., Ye,D. and Juliano,R. (2002) Selective inhibition of P-glycoprotein expression in multi-drug resistant tumor cells by a designed transcriptional repressor. J. Pharmacol. Exp. Ther., 302, 963–971. [DOI] [PubMed] [Google Scholar]
- 25.Christy B.A., Lau,L.F. and Nathans,D. (1988) A gene activated in mouse 3T3 cells by serum growth factors encodes a protein with ‘zinc finger’ sequences. Proc. Natl Acad. Sci. USA, 85, 7857–7861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Triezenberg S.J., Kingsbury,R.C. and McKnight,S.L. (1988) Functional dissection of VP16, the trans-activator of herpes simplex virus immediate early gene expression. Genes Dev., 2, 718–729. [DOI] [PubMed] [Google Scholar]
- 27.Ruben S., Perkins,A., Purcell,R., Joung,K., Sia,R., Burghoff,R., Haseltine,W.A. and Rosen,C.A. (1989) Structural and functional characterization of human immunodeficiency virus tat protein. J. Virol., 63, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyashita T. and Reed,J.C. (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell, 80, 293–299. [DOI] [PubMed] [Google Scholar]
- 29.el-Deiry W.S., Tokino,T., Waldman,T., Oliner,J.D., Velculescu,V.E., Burrell,M., Hill,D.E., Healy,E., Rees,J.L., Hamilton,S.R. et al. (1995) Topological control of p21WAF1/CIP1 expression in normal and neoplastic tissues. Cancer Res., 55, 2910–2919. [PubMed] [Google Scholar]
- 30.Elrod-Erickson M. and Pabo,C.O. (1999) Binding studies with mutants of Zif268. Contribution of individual side chains to binding affinity and specificity in the Zif268 zinc finger-DNA complex. J. Biol. Chem., 274, 19281–19285. [DOI] [PubMed] [Google Scholar]
- 31.Seo Y.R., Fishel,M.L., Amundson,S., Kelley,M.R. and Smith,M.L. (2002) Implication of p53 in base excision DNA repair: in vivo evidence. Oncogene, 21, 731–737. [DOI] [PubMed] [Google Scholar]
- 32.el-Deiry W.S. (1998) Regulation of p53 downstream genes. Semin. Cancer Biol., 8, 345–357. [DOI] [PubMed] [Google Scholar]
- 33.Woods D.B. and Vousden,K.H. (2001) Regulation of p53 function. Exp. Cell Res., 264, 56–66. [DOI] [PubMed] [Google Scholar]
- 34.Slee E.A., Harte,M.T., Kluck,R.M., Wolf,B.B., Casiano,C.A., Newmeyer,D.D., Wang,H.G., Reed,J.C., Nicholson,D.W., Alnemri,E.S., Green,D.R. and Martin,S.J. (1999) Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8 and -10 in a caspase-9-dependent manner. J. Cell Biol., 144, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bortner C.D. and Cidlowski,J.A. (2002) Cellular mechanisms for the repression of apoptosis. Annu. Rev. Pharmacol. Toxicol., 42, 259–281. [DOI] [PubMed] [Google Scholar]
- 36.Sigal A. and Rotter,V. (2000) Oncogenic mutations of the p53 tumor suppressor: the demons of the guardian of the genome. Cancer Res., 60, 6788–6793. [PubMed] [Google Scholar]
- 37.Joers A., Kristjuhan,A., Kadaja,L. and Maimets,T. (1998) Tumour associated mutants of p53 can inhibit transcriptional activity of p53 without heterooligomerization. Oncogene, 17, 2351–2358. [DOI] [PubMed] [Google Scholar]
- 38.Yin C., Knudson,C.M., Korsmeyer,S.J. and Van Dyke,T. (1997) Bax suppresses tumorigenesis and stimulates apoptosis in vivo. Nature, 385, 637–640. [DOI] [PubMed] [Google Scholar]
- 39.Nechushtan A., Smith,C.L., Lamensdorf,I., Yoon,S.H., and Youle,R.J. (2001) Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J. Cell Biol., 153, 1265–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]