

Abstract
Understanding binding properties at protein–protein interfaces has been limited to structural and mutational analyses of natural binding partners or small peptides identified by phage display. Here, we present a high-resolution analysis of a nonpeptidyl small molecule, previously discovered by medicinal chemistry [Tilley, J. W., et al. (1997) J. Am. Chem. Soc. 119, 7589–7590], which binds to the cytokine IL-2. The small molecule binds to the same site that binds the IL-2 α receptor and buries into a groove not seen in the free structure of IL-2. Comparison of the bound and several free structures shows this site to be composed of two subsites: one is rigid, and the other is highly adaptive. Thermodynamic data suggest the energy barriers between these conformations are low. The subsites were dissected by using a site-directed screening method called tethering, in which small fragments were captured by disulfide interchange with cysteines introduced into IL-2 around these subsites. X-ray structures with the tethered fragments show that the subsite-binding interactions are similar to those observed with the original small molecule. Moreover, the adaptive subsite tethered many more compounds than did the rigid one. Thus, the adaptive nature of a protein–protein interface provides sites for small molecules to bind and underscores the challenge of applying structure-based design strategies that cannot accurately predict a dynamic protein surface.
Identifying small molecules that modulate protein–protein interactions continues to be a major challenge for drug discovery (for recent reviews, see refs. 1 and 2). Although traditional targets such as G-coupled receptors and enzymes have defined clefts that bind small molecules, protein–protein interfaces are large (typically 1,300–3,000 Å2), and their surfaces are relatively flat (for recent reviews, see refs. 3 and 4). Moreover, drug discovery is often facilitated by the knowledge of natural small molecule effectors or substrates, but such starting points rarely exist for protein–protein interfaces.
One of the best characterized examples of a small organic compound that can bind at a protein–protein interface is Compound 1 (Scheme S1), which binds to IL-2. This compound was discovered empirically from a medicinal chemistry effort designed to target the IL-2 α receptor (IL-2Rα) (5). Compound 1 binds to IL-2 with moderate affinity near the IL-2Rα-binding site and blocks receptor binding.
Scheme 1.

Structure of Compound 1.
To better understand how Compound 1 binds to IL-2 and to further probe the potential of this site for drug discovery, we solved high-resolution x-ray structures of IL-2 both free and in complex with Compound 1. We further dissected the site by using a drug discovery tool called tethering (6) to discover small organic fragments that can specifically interrogate these subsites. This combination of approaches revealed that a portion of the receptor-binding surface of IL-2 is highly adaptive, and that the most adaptive region is prone to bind or tether small molecules. Adaptive regions hold promise for drug discovery but pose challenges for rational design methods that do not consider flexibility of the protein.
Methods
Cloning and Expression of IL-2 and Mutants.
Full-length IL-2 cDNA (7, 8) was isolated by PCR from plasmid pTCGF-11 (American Type Culture Collection). The cDNA was then cloned into pRSET (Invitrogen) between the NdeI and XhoI restriction endonuclease sites and fully sequenced. Wild-type IL-2 and cysteine mutants, prepared by single-stranded mutagenesis (9), were expressed in Escherichia coli [BL21 (DE3) pLysS; Invitrogen] as inclusion bodies and refolded from 8 M guanidine hydrochloride. For a 1.5-liter culture, inclusion bodies were resuspended in 45 ml of guanidine, and the soluble material was then added to a buffer containing 1.1 M guanidine, 110 mM Tris (pH 8), 6.5 mM cysteamine, and 0.65 mM cystamine. This solution was allowed to equilibrate at room temperature for 5 h and was dialyzed into a buffer of 10 mM ammonium acetate, pH 6/25 mM sodium chloride. The soluble material was purified by chromatography on an S-Sepharose column by using a sodium chloride gradient (25 mM to 1 M NaCl) in a buffer of 25 mM ammonium acetate.
Cloning and Expression of IL-2 Receptors.
The secretion signal and extracellular portion of human IL-2Rα (expressed residues Glu-1 to Pro-179) (10) were isolated by PCR from pMLSV N1/N4-S (American Type Culture Collection) and cloned into a pFastBac1(GIBCO/BRL) vector that had been altered to contain a C-terminal rTEV protease cleavage site and a 6× His tag. The protein was expressed in insect cells via recombinant baculovirus infection and purified on a Ni-NTA superflow column (Qiagen, Chatsworth, CA).
The secretion signal and extracellular portion of human IL-2 β receptor (IL-2Rβ) (residues Ala-1 to Thr-214) (11) were isolated by PCR from a Genestorm clone (Invitrogen) and cloned into a pFastBac1 (GIBCO/BRL) vector that had been altered to contain a C-terminal rTEV protease cleavage site and a 6× His tag. The protein was expressed in insect cells via recombinant baculovirus infection and purified on a Ni-NTA Superflow column (Qiagen).
X-Ray Crystallography.
Crystals were grown by standard hanging-drop vapor diffusion methods. In the case of the noncovalent complexes, a 1.1 molar excess of ligand was included. Before data collection, crystals were transferred to a reservoir solution supplemented with 20% glycerol. Diffraction data were collected at −180°C by using either an R-AXIS IV (Rigaku, Tokyo) detector mounted on an RU-3R generator and processed with d*trek (l = 1.54 Å) (12) or at beam line 7-1 (Stanford Synchrotron Research Laboratory, Stanford, CA) on a MAR345 (Mar Research, Hamburg, Germany) detector and processed with mosflm (λ = 1.08 Å) (13). The structures were determined by molecular replacement by using amore and refined with REFMAC5 (13). The protein models were adjusted by using o (14) and ligand models constructed in INSIGHT II (Accelrys, Waltham, MA). Solvent molecules were placed automatically by using arp_warp (no solvent was included for the 2.4-Å Native II model) (13) and refinement continued until no interpretable features remained in Fo − Fc difference maps.
Coordinates.
Coordinates have been deposited in the Protein Data Bank [PDB ID codes 1M47 (Native I), 1M4C (Native II), 1M48 (Compound 1 complex), 1M4A (Y31C tether), and 1M4B (K43C tether)].
Surface Plasmon Resonance (SPR).
IL-2 mutants were flowed over IL-2Rα or IL-2Rβ immobilized on a dextran-coated surface. By using standard amine coupling to a CM5 chip (Biacore, Uppsala), 4,000 relative units (RU) (≈160 fmol/mm2) of IL-2Rα and 3,000 RU (≈115 fmol/mm2) of IL-2Rβ were immobilized. Kd values were determined by using eight 2-fold serial dilutions of IL-2. Serial dilutions contained IL-2 in 100 ml of running buffer (PBS, 0.05% azide, 1% DMSO). The Kd value was determined by plotting RU at the plateau of the binding curve versus the compound concentration and fitting the binding curve using nonlinear regression analysis (KaleidaGraph, Synergy Software, Reading, PA). The stoichiometry of binding was found to be 1:1 IL-2/receptor for both receptors.
For van't Hoff analysis, the Kd of Compound 1 binding to IL-2 as a function of temperature was determined by SPR (15). IL-2 was immobilized to the SPR surface as described above. The enthalpy (ΔH) and entropy (ΔS) for the Compound 1/IL-2 binding interaction were calculated from the temperature-dependence of Kd measured by SPR from 5 to 40°C.
Data were analyzed by a van't Hoff plot, ln Kd vs. 1/T, which gives the enthalpy from the slope (ΔH/R) and the entropy from the Y-intercept (ΔS/R), according to the equation
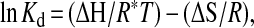 |
where R is the gas constant and T is the temperature in Kelvin.
Syntheses of Tethered Fragments.
Each tethered fragment contained a different small organic compound (Mr < 200) that was derivatized with a disulfide linker. The tethered fragment library of ≈7,000 compounds was prepared from a diverse set of commercially available amines, carboxylic acids, aldehydes, and ketones (6). The diversity element was coupled to a disulfide linker of variable length through either an amide or an oxime bond. Linkers were derived from cystamine, hydroxyethyl disulfide, dithiodiglycolic acid, or longer analogs. Monoprotection of the linker followed by coupling and subsequent deprotection provided the required monophores as a heterodisulfide with the diversity element on one side and a solubilizing group (aminoethane thiol) on the other. The library was divided into ≈700 stock pools, each containing 10 compounds at a final concentration of 10 mM each compound in DMSO solution. Compound pools were assembled such that each member differed by at least 4 atomic mass units, to allow them to be clearly resolved if tethered to IL-2.
Tethering Experiments.
Single cysteine mutants of IL-2 at a concentration of 20 μM in 100 mM Hepes buffer, pH 7.4, containing 2 mM 2-mercaptoethanol (βME), were combined with 2 mM total disulfides (200 μM each of 10 different compounds) at a final DMSO concentration of 2%. After incubation at room temperature for 2–8 h, a portion of the reaction was analyzed by liquid chromatography/MS. Chromatography was conducted on a Phenomenex (Belmont, CA) Jupiter C5 column (50 × 2.0 mm) with a gradient from 5% acetonitrile in water containing 0.05% trifluoroacetic acid to 95% acetonitrile over 3 min on an HP 1100 HPLC with a Gilson 215 autosampler and Finnigan LCQ mass spectrometer. Spectra were deconvoluted by using the Finnigan biomass program. Hits were identified by the corresponding increase in mass of the protein conjugate and were confirmed by reanalysis at 4 mM βME; selected fragments with >20% conjugation were included in subsequent analysis. The amount of time required for selection to come to equilibrium was determined by examination of the rate of labeling with several different pools; once reactions had reached equilibrium, the analysis was consistent for up to 24 h.
Results
Crystallographic Characterization of IL-2 and IL-2–Ligand Complexes.
The x-ray structure of the complex between Compound 1 and IL-2 was solved to a resolution of 1.95 Å (Table 1 and Fig. 1). Compound 1 can be broken into two components: a hydrophilic fragment containing a piperidyl guanidine and a hydrophobic fragment containing a biaryl alkyne. The piperidyl guanidine forms a bidentate salt bridge with the side-chain carboxylate of E62 (2.8 Å) and makes a hydrogen bond to the backbone carbonyl oxygen of K43 (2.8 Å) (Fig. 1b). A similar hydrogen-bonding arrangement is observed for amidine- or guanidine-containing inhibitors bound in the P1 pocket of trypsin-like serine proteases (16). The piperidine ring also makes a hydrophobic contact with the ring of F42. On the hydrophobic side of the binding site, the biaryl alkyne occupies a narrow channel created by the hydrophobic side chains of M39, R38, F42, L72, and K76. Additionally, the carbonyl oxygen of the amide bond connecting the two components forms a hydrogen bond with the amide nitrogen of K43 (2.6 Å). The binding of Compound 1 to IL-2 buries ≈450 Å2 of the ligand surface area, and ≈40% of the Compound 1 surface remains solvent-exposed (calculated by using diffarea, ref. 13).
Table 1.
Crystallographic data
| Data set | Native I | Native II | Compound 1 complex | Y31C/tether | K43C/tether |
|---|---|---|---|---|---|
| Space group | P21212 | P21 | P212121 | P21 | C2 |
| Cell constants | a = 49.47 Å | a = 32.18 Å | a = 50.47 Å | a = 31.15 Å | a = 98.49 Å |
| b = 84.78 Å | b = 48.60 Å | b = 58.02 Å | b = 47.59 Å | b = 35.10 Å | |
| c = 31.71 Å | c = 79.20 Å | c = 93.08 Å | c = 42.63 Å | c = 36.64 Å | |
| β = 97.78° | β = 104.84° | β = 97.52° | |||
| Molecules/a.u., # | 1 | 2 | 2 | 1 | 1 |
| NCS restraints | — | tight | none | — | — |
| Resolution, Å | 10–1.95 | 10–2.40 | 10–1.95 | 10–2.18 | 10–2.15 |
| Wavelength | 1.08 | 1.54 | 1.54 | 1.54 | 1.54 |
| Unique reflections | 9,522 | 8,581 | 20,214 | 5,840 | 6,926 |
| Completeness,* % | 98.5 (91.9) | 94.8 (58.1) | 98.1 (94.3) | 91.7 (67.6) | 97.5 (78.0) |
| Rsym(I),† % | 6.1 (39.1) | 6.3 (25.9) | 5.9 (35.7) | 9.9 (24.6) | 6.7 (31.2) |
| I/σ | 13.2 (3.3) | 14.0 (4.1) | 11.4 (3.1) | 16.1 (3.2) | 12.7 (3.2) |
| Rcryst,‡ % | 22.0 | 28.6 | 19.6 | 26.9 | 24.2 |
| Rfree,§ % | 24.5 (399) | 33.3 (428) | 26.7 (974) | 32.2 (561) | 28.2 (320) |
| Average B factors, Å2 | |||||
| Protein | 24.8 | 44.0 | 32.2 | 43.6 | 36.7 |
| Solvent | 24.3 | — | 44.1 | 41.5 | 34.6 |
| Ligand | — | — | 23.8 | 44.2 | 23.9 |
a.u., asymmetric unit.
Values in parentheses are for the highest-resolution bin.
Rsym(I) = ∑hkl |Ihkl − 〈 Ihkl〉|/∑hklIhkl, where Ihkl is the intensity of reflection hkl.
Rcryst = ∑hkl | |Fobs| − |Fcalc| |/|Fobs|, where Fobs and Fcalc are the observed and calculated structure factors, respectively, for the data used in refinement.
Rfree = ∑hkl | |Fobs| − |Fcalc| |/|Fobs|, where Fobs and Fcalc are the observed and calculated structure factors, respectively, for data omitted from refinement (number of included reflections in parentheses).
Figure 1.

(a) Compound 1/IL-2 complex determined by x-ray crystallography (Table 1). Compound 1 is shown in green sticks, IL-2 in white ribbon. The B–C loop is not defined by the electron density and is shown schematically in white spheres. (b) Interaction of Compound 1 (green sticks) with IL-2 (white sticks) taken from x-ray coordinates. Key contact side chains are labeled; H-bonds and distances are shown by yellow dotted lines. All molecular graphic images were produced with PYMOL (W. L. DeLano, San Carlos, CA).
Overall, both the hydrophilic and hydrophobic components of Compound 1 show good complementarity to the surface of IL-2 (Fig. 2a). Previous mutational work (17–19) has identified residues important for binding of IL-2 to the IL-2 receptor (a “hot spot”). Even though Compound 1 was not designed to bind IL-2, it does bind directly over the region of IL-2 that is functionally critical for binding to IL-2Rα (Fig. 2a).
Figure 2.

Adaptivity of the IL-2-binding surface. (a) Structure of the complex of IL-2 and Compound 1, shown as a surface representation of IL-2 (white and orange) and stick representation of Compound 1 (green). The residues that comprise the IL-2Rα-binding hot spot are shown in orange. Residues contacting the molecule are labeled. (b–d) Surface representation of three different unliganded structures (21) (b) and Native I and II (c and d; Table 1). Compound 1 is overlaid to emphasize the extent of rearrangement that occurs in both of the unliganded structures and in the liganded form. Movie 1 (RIGIMOL, W. L. DeLano) highlights the dynamic nature of this site across this series of structures.
Comparison of the bound structure (Fig. 2a) and the unliganded structure of IL-2 (Fig. 2b) solved by McKay and coworkers (20) reveals significant structural changes on binding. To further probe the conformational variability on the surface of unliganded IL-2, we solved the structure in two additional crystal forms (Native I and II; Table 1, Fig. 2 c and d). These structures provide two additional independent views of free IL-2 in distinct crystalline environments. From these data, it is apparent that the surface of IL-2 where the small molecule and receptor binds is dynamic and capable of adopting multiple conformations on the surface of IL-2. Moreover, the surface is capable of providing a hydrophobic channel to bind Compound 1 that is not apparent in free structures of IL-2.
The ligand-binding site on IL-2 can be dissected into two distinct subsites that correspond to the two fragments that compose Compound 1. The binding site for the hydrophilic piperidyl guanidine fragment is relatively fixed and includes the center of helix B and the A′–B loop (Fig. 1a). By contrast, the binding site for the hydrophobic biaryl alkyne fragment is highly mobile. This mobile region comprises the C-terminal end of helix B, the loop connecting helices A and A′, and the loop connecting helix B to helix C.
The free structures of IL-2 show that large changes are observed in the loop connecting helices A and A′. Additionally, the backbone structure of a mutant of IL-2 (F42A¶) has been solved by NMR spectroscopy (21). Although the side chains were not well-defined, the NMR structure showed that the loops that connect the four helices are mobile. The two structures presented here (Fig. 2 c and d) and the NMR structure (21) show this loop is in a closed conformation that packs the side chains of residues 30–32 within the interior of the protein. By contrast, the earlier x-ray structure (20) features an open conformation where these side chains point away from the protein. In this structure, Y31 is tightly packed against a neighboring molecule in the unit cell, making close contacts with H16 and D20. It is also notable that, in the open loop conformation, the side chain of N33 faces the interior of the protein, occupying the same site that Y31 occupies in the closed loop conformation (Fig. 6, which is published as supporting information on the PNAS web site, www.pnas.org). These variations suggest that the loop structure is not disordered but can toggle between at least two stable conformations.
In addition to loop flexibility, important contact residues for Compound 1 (K35, R38, M39, and F42) display multiple conformations in the unliganded structures (Fig. 2 b–d). F42 has a χ1 angle of –60° in the unliganded structures that shifts to 180° when it packs against the piperidyl ring of Compound 1. This new conformation for F42 exposes a hydrophobic channel, blocked in the unliganded structures, which is then occupied by the biaryl alkyne. In the bound structure, the side chains of K76, L72, and R38 pack against both sides of the terminal aromatic ring of the ligand, burying Compound 1 in the newly created hydrophobic pocket. Thus, the flexibility of the receptor-binding site on IL-2 creates a binding groove for the small molecule. This can be further viewed in Movie 1, which is published as supporting information on the PNAS web site.
Thermodynamic Analysis of Compound 1 Binding to IL-2.
To better understand the enthalpic and entropic parameters for binding Compound 1, we measured the van't Hoff enthalpy of interaction by monitoring the effect of temperature on the dissociation constant (Kd) (Fig. 3). The binding of the compound is enthalpically driven (ΔH = −8.9 ± 0.35 kcal/mol) and entropically disfavored by a small amount (ΔS = −6.8 ± 0.56 cal/mol K), with a ΔG = −6.9 ± 0.06 kcal/mol at 298 K (Kd = 8.2 μM). Because the compound has few rotatable bonds, this cost in conformational entropy must come from the protein and/or solvent. Two water molecules are also displaced from the E62-binding pocket; these will have a small effect on ΔCp and how entropy changes are partitioned (22). The structural and thermodynamic data are consistent with the view that unbound IL-2 adopts many conformations in solution, and that ligand binding restricts the number of conformations at a modest entropic cost (+2 kcal/mol at 298 K).
Figure 3.

van't Hoff analysis of Compound 1 binding to IL-2. Kd values were determined by SPR at four temperatures (see Methods). (ΔH = −8.9 ± 0.35 kcal/mol, ΔS =−6.8 ± 0.56 cal/mol K.) Data are an average of three measurements; the standard deviations for each temperature are shown on the graph.
Discovery of Fragments in the Compound 1-Binding Site Through Tethering.
To further dissect the binding site, we screened for novel small molecule ligands by using a site-specific fragment-discovery method called tethering. The tethering method identifies fragments (Mr typically <200) that are selected through disulfide exchange on the basis of their noncovalent association with the protein (6, 23). A library of small drug-like fragments containing a common disulfide is allowed to reach equilibrium with a set of single-cysteine mutants of the target protein (Scheme S2). Disulfide exchange reactions are performed under reducing conditions, such that the stabilization of a specific disulfide bond requires favorable noncovalent interactions between the fragment and the targeted binding site. Selected compounds are then identified by MS. This method was previously applied to two enzymes, thymidylate synthase (6) and caspase-3 (23), where tethered fragments were identified and rapidly optimized by structure-based and combinatorial methods to yield noncovalent submicromolar inhibitors. Structural analysis showed that the tethered and free fragments bound in virtually the same fashion, indicating that the disulfide tether does not distort the nature of binding.
Scheme 2.

Tethering method for fragment assembly.
The site-specific nature of tethering is well suited for identifying subsites and the chemical properties of selected fragments. These fragments bind weakly (typically with Kd values in the low millimolar range) as noncovalent ligands, and, without the disulfide bond, they are difficult to detect. To apply tethering, we produced 11 individual cysteine mutants around the perimeter of the binding site for Compound 1 (Fig. 4a). None of the cysteine substitutions caused a substantial change in the ability of IL-2 to bind to IL-2Rβ (Table 2), suggesting they did not significantly perturb the conformation of the protein. Some of the mutants (Y45C, L72C, and K43C) did interfere with IL-2Rα binding, as anticipated from previous mutagenesis studies that define the IL-2 hot spot (17–19).
Figure 4.

Results from tethering experiments at cysteine variants surrounding the IL-2 hot spot. (a) The cysteine mutations located near the flexible region (shown in red; N30C, Y31C, N33C, L72C, and N77) selected >20 tethered fragments, whereas those made around the more ordered region of the protein (shown in blue; R38C, F42C, K43C, and Y45C) selected fewer than seven fragments. (b) The number of tether hits (z axis) versus the cysteine mutant position (y axis) and each of seven different tether compound libraries, each containing ≈1,000 compounds (x axis). The chemical structures represented in this graph are published as Table 3 in supporting information on the PNAS web site.
Table 2.
Effect of cysteine mutations on the binding of IL-2 to its receptors
| Mutant* | IC50 (IL-2Rα), nM | Fold reduction vs. WT IL-2 | IC50 (IL-2Rβ), nM | Fold reduction vs. WT IL-2 |
|---|---|---|---|---|
| WT | 30 | — | 1,200 | — |
| M23C | 30 | 1 | 350 | 0.3 |
| N30C | 4 | 0.13 | 1,200 | 1 |
| Y31C | 5 | 0.16 | 700 | 0.6 |
| N33C | 45 | 1.5 | 300 | 0.3 |
| K43C | 100 | 3.3 | 1,000 | 0.8 |
| Y45C | >300 | >10 | 1,200 | 1 |
| L72C | >300 | >10 | 800 | 0.7 |
M23C is near the IL-2Rβ-binding site; the rest of the mutations are near the IL-2Rα-binding site. The other five mutants were not tested but were expected to be properly folded based on data from alanine mutations at these sites (C. Thanos and J.A.W., unpublished results). All measurements were made by SPR, as described in Methods.
Each cysteine variant was screened against a 7,000-compound tethering library (6) in pools of 10 compounds each. These were grouped into seven bins containing ≈1,000 compounds each (S1–S7, Fig. 4b), based on the general chemical features of the fragment pools. In general, “hits” were rare events (frequency of 0.1–1%) over the entire set of cysteine mutants. A plot of the number of hits produced from each of the seven compound bins for the cysteine positions shows that each position produces a different set of hits from each of the compound bins (Fig. 4b). That hits were rare and no two cysteine variants captured the same set of tethered fragments reinforces the specificity and selectivity of the tethering method. Some cysteine variants produced a large number of hits [N30C (0.8%), Y31C (1.1%), N33C (1.3%), L72C (0.2%), and N77C (0.4%)], whereas others produced few, if any [R38C (0 hits), F42C (0 hits), K43C (0 hits), and Y45C (0.08%)]. The cysteine variants that produced the largest number of hits cluster in the region that was found to be most adaptive (Fig. 4a).
Many of the strong hits in the adaptive region contained multiple aromatic or fused ring structures similar in character to the biaryl alkyne portion of Compound 1. To further characterize how one of these strong hits bound to the most hit-rich tethering site (Y31C), we chose an indole glyoxylate fragment and solved the x-ray structure of the tethered complex (Fig. 5a). This site encompasses part of the IL-2Rα-binding site and includes residues K35, R38, L72, and A73. Interestingly, the indole ring of the fragment overlays the terminal aromatic ring of Compound 1, consistent with the tethering results that this adaptive site has a strong propensity for binding aromatic groups.
Figure 5.

Tethering hits (in yellow) in the hydrophobic and hydrophilic subsites of IL-2 in comparison with Compound 1 (overlaid in green). The chemical structure of the fragment is shown above the structure. (a) X-ray structure of IL-2 with the indole glyoxylate fragment tethered at Y31C (Y31C/tether, Table 1). (b) X-ray structure of one of the guanidine tethering hits at K43C (K43C/tether). Movie 2, which is published as supporting information on the PNAS web site, highlights the different protein conformations.
Comparison of the structures of IL-2 in Figs. 2a and 5a reveals an interesting difference. In the tethered indole glyoxylate structure, F42 is found in a conformation similar to that observed in the unliganded structures (Fig. 2 b–d), where it closes off the right-hand portion of the hydrophobic channel used to bind Compound 1 (Fig. 5a). Thus, the aromatic subsite can exist independently. The hydrophilic site remains relatively fixed even when unoccupied as seen in all of the structures.
We then applied tethering to probe the nonadaptive, guanidine-binding region of IL-2. Accordingly, we prepared a series of tethering fragments consisting of guanidine-containing dipeptides with different spacers and found several strong hits when tethering from the K43C mutant. These hits showed a very strict linker length dependence, indicating that a highly specific interaction was needed for the fragment to be selected. The x-ray structure of the tethered complex with one of the strongest hits (Fig. 5b) showed that the tethered fragment formed some of the same polar interactions as seen when Compound 1 bound to IL-2. In particular, the guanidine fragment forms the same bidentate salt bridge with the side chain of E62 and makes a hydrogen bond to the backbone carbonyl of K43. The additional cyclohexyl portion of the tethered guanidine fragment makes new hydrophobic interactions not seen in Compound 1. Moreover, the hydrophobic channel is in a closed conformation, suggesting that the formation of the channel is independent of binding at the guanidine site. Thus, tethering identifies fragments with similar functional groups to those seen in Compound 1 in both the hydrophobic and hydrophilic subsites. The guanidine-binding portion of IL-2 is relatively static and is more restrictive in selecting fragments, whereas the hydrophobic binding region is adaptive and able to bind a larger number of structurally related fragments.
Discussion
These studies provide a high-resolution analysis of a nonpeptidyl small molecule binding at a protein–protein interface. Given the empirical approach by which Compound 1 was discovered (5), it is notable that it binds the same region that mutations suggest is critical for binding the IL-2α receptor. This site is adaptive and can exist in a variety of conformations. A number of protein–protein binding sites have been shown to be highly adaptive (24–26), and phage display efforts have identified naïve peptides that can bind at these sites (24, 27, 28). Enzyme active sites have also revealed structural rearrangements, suggesting new possibilities for inhibitor design (29, 30). Surface adaptability provides multiple conformations and thus may facilitate binding of multiple partners of either natural or synthetic origin. Conformational flexibility comes with an entropic cost; indeed, thermodynamic analysis shows that binding of Compound 1 is entropically disfavored. However, the entropic cost is modest (+2 kcal/mol) compared with the strong enthalpic term (−8.9 kcal/mol). Thus, it appears that accessing the Compound 1-binding mode does not present a large thermodynamic barrier.
The binding site for Compound 1 site could be divided into two subsites. The subsite that binds the hydrophobic biaryl alkyne portion is adaptive, whereas that which binds the polar guanidine portion is very rigid. Tethering experiments at each of these subsites readily identified the ligand preferences and tolerances. Many fragments were found in the highly adaptive subsite. Structural characterization of one of these fragments (Fig. 5a) shows that it binds in the same pocket created by binding of the hydrophobic portion of Compound 1. In contrast, far fewer hits were found in the rigid and polar subsite. Selection of the guanidine fragment was highly sensitive to the length of the spacer and the structure of the fragment. This result is not unexpected, because the hydrophilic fragment must retain a number of polar interactions and shape complementarity to a relatively fixed region of protein structure.
The dynamic nature of the IL-2-binding surface suggests two points that are important for drug discovery. First, it would not have been possible to predict the Compound 1-binding site by analyzing the surfaces of the unliganded IL-2 structures (Fig. 2 b–d). Although there is tremendous interest in applying structure-based methods for drug discovery, methods that do not accurately account for the dynamics of protein–protein surfaces will have difficulty in providing a robust docking algorithm. Second, the dynamic surface of IL-2 is not “flat” but can provide grooves for binding small molecules. Although parts of the binding groove in IL-2 were visible in the unliganded structures of IL-2, the complete small-molecule-binding conformation was observed only in the presence of Compound 1. Tethering effectively probed these subsites and ligand tolerances. Such information can be useful for designing small molecules that can bind these targets. Although protein–protein interfaces are challenging drug targets, their adaptive character can provide binding sites for small molecules and thus opportunities for drug discovery.
Supplementary Material
Acknowledgments
Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory (SSRL), a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and by the National Institute of General Medical Sciences.
Abbreviations
- IL-2Rα
IL-2 α receptor
- IL-2Rβ
IL-2 β receptor
Footnotes
Data deposition: The coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID codes 1M47, 1M4C, 1M48, 1M4A, and 1M4B).
Mutants are designated by the single-letter code for the wild-type residue followed by its position in the mature coding sequence and the single-letter code for the mutant.
References
- 1.Cochran A G. Curr Opin Chem Biol. 2001;5:654–659. doi: 10.1016/s1367-5931(01)00262-9. [DOI] [PubMed] [Google Scholar]
- 2.Toogood P L. J Med Chem. 2002;45:1–16. doi: 10.1021/jm010468s. [DOI] [PubMed] [Google Scholar]
- 3.Lo Conte L, Chothia C, Janin J. J Mol Biol. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 4.Stites W E. Chem Rev. 1997;97:1233–1250. doi: 10.1021/cr960387h. [DOI] [PubMed] [Google Scholar]
- 5.Tilley J W, Chen L, Fry D C, Emerson S D, Powers G D, Biondi D, Varnell T, Trilles R, Guthrie R, Mennona F, et al. J Am Chem Soc. 1997;119:7589–7590. [Google Scholar]
- 6.Erlanson D A, Braisted A C, Raphael D R, Randal M, Stroud R M, Gordon E M, Wells J A. Proc Natl Acad Sci USA. 2000;97:9367–9372. doi: 10.1073/pnas.97.17.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taniguchi T, Matsui H, Fujita T, Takaoka C, Kashima N, Yoshimoto R, Hamuro J. Nature. 1983;302:305–310. doi: 10.1038/302305a0. [DOI] [PubMed] [Google Scholar]
- 8.Devos R, Plaetinck G, Cheroutre H, Simons G, Degrave W, Tavernier J, Remaut E, Fiers W. Nucleic Acids Res. 1983;11:4307–4323. doi: 10.1093/nar/11.13.4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kunkel T A. Proc Natl Acad Sci USA. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nikaido T, Shimizu A, Ishida N, Sabe H, Teshigawara K, Maeda M, Uchiyama T, Yodoi J, Honjo T. Nature. 1984;311:631–635. doi: 10.1038/311631a0. [DOI] [PubMed] [Google Scholar]
- 11.Taniguchi T, Hatakeyama M, Minamoto S, Kono T, Doi T, Tsudo M, Miyasaka M, Miyata T. Cold Spring Harbor Symp Quant Biol. 1989;54:689–694. doi: 10.1101/sqb.1989.054.01.081. [DOI] [PubMed] [Google Scholar]
- 12.Pflugrath J W. Acta Crystallogr D. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 13.CCP4. Acta Crystallogr D. 1994;55:1718–1725. [Google Scholar]
- 14.Jones T A, Zou J Y, Cowan S W, Kjeldgaard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 15.Deinum J, Gustavsson L, Gyzander E, Kullman-Magnusson M, Edstrom A, Karlsson R. Anal Biochem. 2002;300:152–162. doi: 10.1006/abio.2001.5454. [DOI] [PubMed] [Google Scholar]
- 16.Krieger M, Kay L M, Stroud R M. J Mol Biol. 1974;83:209–230. doi: 10.1016/0022-2836(74)90388-x. [DOI] [PubMed] [Google Scholar]
- 17.Sauve K, Nachman M, Spence C, Bailon P, Campbell E, Tsien W H, Kondas J A, Hakimi J, Ju G. Proc Natl Acad Sci USA. 1991;88:4636–4640. doi: 10.1073/pnas.88.11.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zurawski S M, Vega F, Jr, Doyle E L, Huyghe B, Flaherty K, McKay D B, Zurawski G. EMBO J. 1993;12:5113–5119. doi: 10.1002/j.1460-2075.1993.tb06206.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weigel U, Meyer M, Sebald W. Eur J Biochem. 1989;180:295–300. doi: 10.1111/j.1432-1033.1989.tb14647.x. [DOI] [PubMed] [Google Scholar]
- 20.McKay D B. Science. 1992;257:412–413. doi: 10.1126/science.257.5068.412. [DOI] [PubMed] [Google Scholar]
- 21.Mott H R, Baines B S, Hall R M, Cooke R M, Driscoll P C, Weir M P, Campbell I D. J Mol Biol. 1995;247:979–994. doi: 10.1006/jmbi.1994.0194. [DOI] [PubMed] [Google Scholar]
- 22.Murphy K P, Freire E, Paterson Y. Proteins. 1995;21:83–90. doi: 10.1002/prot.340210202. [DOI] [PubMed] [Google Scholar]
- 23. Erlanson, D. A., Lam, J. W., Wiesmann, C., Luong, T. N., Simmons, R. L., DeLano, W. L., Choong, I. C., Burdett, M. T., Flanagan, W. M., Lee, D., et al. (2003) Nat. Biotechnol., in press. [DOI] [PubMed]
- 24.DeLano W L, Ultsch M H, de Vos A M, Wells J A. Science. 2000;287:1279–1283. doi: 10.1126/science.287.5456.1279. [DOI] [PubMed] [Google Scholar]
- 25.Atwell S, Ultsch M, De Vos A M, Wells J A. Science. 1997;278:1125–1128. doi: 10.1126/science.278.5340.1125. [DOI] [PubMed] [Google Scholar]
- 26.Ma B, Shatsky M, Wolfson H J, Nussinov R. Protein Sci. 2002;11:184–197. doi: 10.1110/ps.21302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Livnah O, Stura E A, Johnson D L, Middleton S A, Mulcahy L S, Wrighton N C, Dower W J, Jolliffe L K, Wilson I A. Science. 1996;273:464–471. doi: 10.1126/science.273.5274.464. [DOI] [PubMed] [Google Scholar]
- 28.Pan B, Li B, Russell S J, Tom J Y, Cochran A G, Fairbrother W J. J Mol Biol. 2002;316:769–787. doi: 10.1006/jmbi.2001.5370. [DOI] [PubMed] [Google Scholar]
- 29.Rockwell A, Melden M, Copeland R A, Hardman K, Decicco C P, DeGrado W F. J Am Chem Soc. 1996;118:10337–10338. [Google Scholar]
- 30.Bursavich M G, Rich D H. J Med Chem. 2002;45:541–558. doi: 10.1021/jm010425b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.