

Abstract
The recognition of specific signals encoded within the 3′-untranslated region of the newly transcribed mRNA triggers the assembly of a multiprotein machine that modifies its 3′-end. Hrp1 recognises one of such signals, the so-called polyadenylation enhancement element (PEE), promoting the recruitment of other polyadenylation factors in yeast. The molecular bases of this interaction are revealed here by the solution structure of a complex between Hrp1 and an oligonucleotide mimicking the PEE. Six consecutive bases (AUAUAU) are specifically recognised by two RNA-binding domains arranged in tandem. Both protein and RNA undergo significant conformational changes upon complex formation with a concomitant large surface burial of RNA bases. Key aspects of RNA specificity can be explained by the presence of intermolecular aromatic–aromatic contacts and hydrogen bonds. Altogether, the Hrp1–PEE structure represents one of the first steps towards understanding of the assembly of the cleavage and polyadenylation machinery at the atomic level.
Keywords: cleavage and polyadenylation, Hrp1, nuclear magnetic resonance, RNA-binding proteins, RNA processing
Introduction
The mechanics of 3′-end formation require a complex collection of trans-acting protein factors, 3′-untranslated region (3′-UTR) cis-acting RNA sequences and sometimes even gene-specific polyadenylation factors (Zhao et al, 1999; Shatkin and Manley, 2000; Proudfoot, 2004). The highly complex machinery ultimately responsible for the endonucleolytic cleavage and polyadenylation of the mRNA is assembled in a transcription coordinated manner, before the poly(A) site itself is transcribed. The same enzyme that synthesizes the nascent mRNA transcript (RNA pol II) is also crucial to coordinate all mRNA processing events: 5′-end capping, splicing and 3′-end formation. In particular, its carboxy terminal domain (CTD) is a vital docking platform that binds many mRNA processing factors. RNA pol II CTD is composed of a variable number of heptapeptide repeats (from 26 in yeast to 52 in mammals) with a consensus sequence YSPTSPS. The phosphorylation status of the RNA pol II CTD changes dynamically during transcription initiation, elongation and termination, determining at every stage which processing factors are recruited. In mammals, at least two cleavage and polyadenylation factors bind to the CTD during transcription initiation (Dantonel et al, 1997; McCracken et al, 1997). These factors are the cleavage stimulation factor (CstF) and the cleavage and polyadenylation specific factor (CPSF), both multicomponent protein complexes that interact mutually during the final assembly of the 3′-end processing machinery. Upon transcription of the pre-mRNA 3′-UTR, CstF-64, one of the three components of CstF recognises the G/U-rich region downstream of the poly(A) site, in coordination with CPSF-160 which specifically binds to the poly(A) signal itself (AAAUAA). RNA15 (Minvielle-Sebastia et al, 1994) and Yhh1p (Stumpf and Domdey, 1996) are the yeast homologues of these two mammalian factors and also interact with RNA cis-acting elements. However, the internal structure of the yeast 3′-UTR seems to be more complex (Guo and Sherman, 1996) and includes at least a third cis-acting element known as polyadenylation enhancement element (PEE), which is specifically recognised by yet another trans-acting factor Hrp1 (Kessler et al, 1997). This yeast-specific polyadenylation factor also plays important roles in mRNA export (Xu and Henry, 2004), mRNA surveillance and nonsense mediated decay (Gonzalez et al, 2000).
PEEs are mainly composed of several UA repeats, upstream of the RNA15 and Yhh1p recognition sites. Hrp1 central region contains two RNP-type RNA-binding domains (RBDs) arranged in tandem and by itself accounts for the RNA recognition activity. In many cases, the presence of two or more consecutive RBDs is necessary to increase the affinity constants to nM range (Maris et al, 2005). Although a very recent nuclear magnetic resonance (NMR) work has demonstrated that a single RBD can reach this range of affinities without the help of a second RBD or any extra structural element (Auweter et al, 2006). Some crystal structures have recently illustrated the mechanism of single-strand RNA recognition by RDB pairs. However, Hrp1 binds an RNA sequence (UA repeats) significantly different from poly(A) (in the PAPB complex (Deo et al, 1999)), or U-rich sequences (in the sex-lethal (Handa et al, 1999) and HuD (Wang and Tanaka Hall, 2001) complexes). Thus, investigation of the Hrp1–PEE complex increases our knowledge about how RBD pairs achieve their RNA specificity.
This work presents the solution structure of the RNA-binding region of Saccharomyces cerevisiae polyadenylation factor Hrp1 in complex with an RNA sequence G(UA)4 that mimics the PEE. The structure shows that only six bases (AU)3 form specific contacts to the protein. It presents novel features in comparison with previous examples of single-strand RNA recognition by two consecutive RBDs. The intermolecular interface is dominated by interactions between aromatic residues and RNA bases, with hydrophilic residues providing base specificity through a hydrogen bond network. Residues at the interface are mainly contributed by the canonical motifs in the β-sheet region, although loops and the interdomain linker also play a crucial role in Hrp1–RNA recognition. In particular, a conserved residue (Trp168) stacks on the second adenine (Ade4) and forms crucial base-specific hydrogen bonds with it. The importance of Trp168 has been tested here by in vitro competition experiments against two different mutants. Some of the interfacial residues have been previously identified in vivo as causing temperature-sensitive phenotypes (Kessler et al, 1997). The current structure explains the molecular basis of these phenotypes and can be used for rational design of new mutants with impaired function.
Results and discussion
Hrp1 recognises a short single-stranded RNA
The yeast Hrp1 is a key protein factor for mRNA processing and export. The protein is organised into a mutidomain structure showing as prominent features two consecutive RNP-type RBDs in the middle region (residues 156–321) and an RG-rich domain at the C-terminus. The NMR spectra of the protein fragment containing the RNA recognition domain (residues 156–321) (Figure 1) are characteristic of a well-folded polypeptide, they show disperse signals in the 1D proton and 2D 1H-15N HSQC spectra and proton amide T2 relaxation times (1D spin-echo experiment) in agreement with a monomeric state (data not shown).
Figure 1.
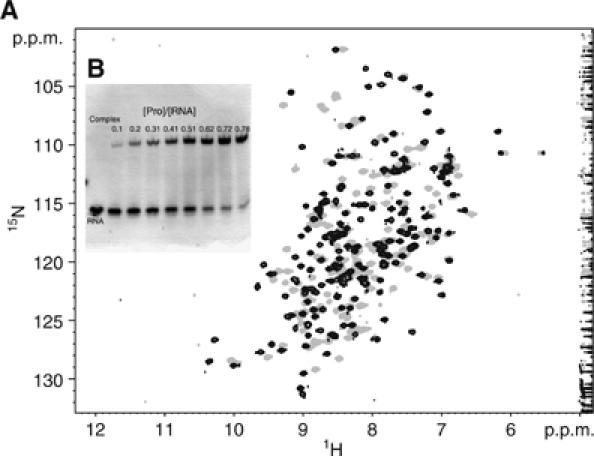
(A) Chemical shift perturbation of the protein 15N-HSQC spectrum upon complex formation with a G(UA)4. The position of most of the backbone and side-chain amide signals change between the free (grey) and RNA-bound (black) states. Both sets of signals exhibit slow exchange kinetics indicating that a well-defined high affinity complex is formed. (B) Protein–RNA titrations were monitored by electromobility assays in native gels (10% polyacrylamide). Saturation was reached at 1:1 stoichiometry for this short RNA sequence G(UA)4. Protein–RNA complex (upper band) and free RNA duplex (lower band) migrate as distinct and separate bands owing to the high affinity of the complex.
Previous in vitro selection experiments identified several RNA sequences binding with nM affinity to the protein (Valentini et al, 1999). Nearly all of these contain the six nucleotide sequence 5′-UAUAUA-3′, the consensus sequence for the PEE in yeast. Experiments using synthetic GAL7 3′-UTR showed binding of Hrp1 to the UA repeats, which is required for the efficient positioning of other RNA-binding proteins (e.g. RNA15) of the yeast cleavage/polyadenylation apparatus (Gross and Moore, 2001). The data suggest that Hrp1 binding is an early step in the assembly of the cleavage/polyadenylation supramolecular machine. Although there are structures illustrating how tandem RBDs recognize single-stranded RNA (Deo et al, 1999; Ding et al, 1999; Handa et al, 1999; Wang and Tanaka Hall, 2001), those data are insufficient to explain Hrp1 specificity. Hrp1 binds to RNA sequences that differ in both length (6mer, as opposed to 8 or 10 mer) and content from those bound by PABP, sex lethal or HuD proteins.
Hrp1 RNA-binding affinity has been explored using NMR titration experiments and polyacrylamide electromobility shift assays. Both techniques are quantitative and allow us to determine the stoichiometry of the complex. Furthermore, NMR is a sensitive technique to investigate binding events across a wide range of dissociation constants (from mM to nM) and provides residue-specific information. Several RNA constructs were synthesized containing between 4 and 8 UA repeats, mimicking the consensus sequence of the PEE. All RNAs cause extensive effects on the 15N-HSQC protein NMR spectra upon titration (Figure 1). Binding occurs in the slow exchange regime, an indirect indication of high binding affinity, and causes displacements on many of the protein signals relative to the free state. Changes of the protein spectra are the same regardless of the number of UA repeats (data not shown), showing that the all complexes share an identical binding mode. However, the protein–RNA ratios necessary to achieve RNA saturation are different, the data indicate a 1:1 stoichiometry for the shorter sequences (UA)4 and (UA)5 but an apparent 2:1 stoichiometry for the longer sequences (UA)6, (UA)7 and (UA)8. Gel electromobility assays also support 1:1 ratios for the shorter sequences (Figure 1), whereas the longer sequences show the presence of extra bands at high protein/RNA ratios. These bands are likely to arise from high molecular weight complexes containing more than one copy of Hrp1 per RNA.
The RNA constructs are auto-complementary, and hence can form duplexes in solution that are characterized by the presence of the typical proton imino signals of Watson–Crick base pairs between 12 and 14 p.p.m. The complex formation can be readily monitored by following the disappearance of these signals. Consistent with the gel data, longer RNAs require as much as two equivalents of protein for complete melting of the duplex, suggesting the presence of at least two binding sites.
In summary, the data demonstrate that the shortest repeat (UA)4 is both necessary and sufficient to bind to Hrp1 and that increasing the number of repeats does not affect the recognition mode of Hrp1. Many yeast mRNAs contain several consecutive copies of the PEE optimal consensus sequence within their 3′-UTRs (Guo and Sherman, 1995). It is interesting to hypothesize what would be the impact of several overlapping binding sites for the mechanism of polyadenylation complex assembly. It is possible that large PEE could accommodate more than one Hrp1 (as seems to occur for some of RNA probes tested here) or it perhaps Hrp1 can associate and dissociate at different binding sites, exchanging among them until the whole machinery is finally locked in position.
Conformational changes upon RNA binding
The two RBD domains of Hrp1 behave as independent rigid bodies in the free form. This can be demonstrated by the absence of interdomain nuclear overhausen enhancement (NOE) cross-peaks and the random coil chemical shift values observed for residues of the linker segment. The analysis of the NOE spectroscopy (NOESY) spectra of the free protein also confirms the βαββαβ fold for each of the two RBDs.
RNA titration causes dramatic changes in the 15N-HSQC spectrum of Hrp1 (156–321) (Figure 1), which are due both to conformational rearrangement of the protein and to the presence of many RNA–protein contacts. Comparison of 13Cα and 13Cβ chemical shifts between free and bound protein states reveals significant changes at the secondary structure level, mainly within the linker region, which folds up into a helical structure upon RNA binding (data not shown).
13C chemical shifts contain useful structural information (Wishart and Sykes, 1994); however, for the Hrp1–PEE structure calculation the great majority of conformational restraints used in this work came from NOE data (Table I) obtained from several 2D and 3D NOESY-type experiments under different solvent conditions. A large number of protein–protein and protein–RNA distance restraints were derived from these spectra (Table I) and used to calculate the structure of the complex between Hrp1 (156–321) and a RNA G(UA)4 construct mimicking the PEE.
Table 1.
Structure calculation statistics for the 25 accepted structures of the Hrp1–PEE complex
| Experimental restrains | |
| NOE-derived distance restrains | |
| Intraresidue (protein) | 1032 |
| Sequential (protein) | 591 |
| Medium range (protein) (2⩽∣i−j∣⩽4) | 363 |
| Long range (∣i−j∣>4) | 1094 |
| Intra-RNA | 49 |
| Intermolecular | 117 |
| Strong (1.8–2.8 Å) | 348 |
| Medium (1.8–3.3 Å) | 1075 |
| Weak (1.8–4.5 Å) | 833 |
| Very weak (1.8–5.5 Å) | 990 |
| Hydrogen bonds | 45 |
| RNA 2′endo sugar puckering | 8 |
| Structure statistics | |
| Mean XPLOR energy term (kcal mol-1±s.d.) | |
| E(total) | −4993±25 |
| E(van der Waals) | 151±13 |
| E(distance restraints) | 132±15 |
| mean: 4933.39s.d.: 48.4344 n: 25 | |
| R.m.s. deviations form ideal geometry used within CNS | |
| Bond lengths (Å) | 0.0027±0.0001 |
| Bond angles (deg) | 0.50±0.01 |
| Improper angles (deg) | 0.42±0.02 |
| Average atomic r.m.s.d. from the average structure (±s.d.) | |
| Protein | |
| N, Cα, C′ atoms (160–318) 0.92±0.21 Å | All heavy 1.34±0.17 Å |
| RBD 1+RNA | |
| N, Cα, C′ atoms (160–234) and O5′,C5′,C4′,C3′ O3′ P (Ade4–Ura7) 0.62±0.14 Å | All heavy 1.00±0.14 Å |
| RBD 2+RNA | |
| N, Cα, C′ atoms (242–318) and O5′,C5′,C4′,C3′ O3′ P (Ade2–Ura3) 0.68±0.10 Å | All heavy 1.19±0.11 Å |
| Ramachandran statistics | |
| Most favourable and allowed regions | 93.9% |
| Generously allowed regions | 3.0% |
| Disallowed regions | 3.1% |
The structure of the complex contains the two RBDs, each with their typical βαββαβ architecture, and arranged in tandem. The first RBD extends from residues Ser158 to Ala233 (Figure 2) and is built up by a central antiparallel four-stranded β-sheet with two long α-helices running across one face. The second RBD has the same topology and roughly the same size, extending from residues Lys244 to Ala318. The structure reveals that, in contrast to the free protein, in the complex the linker (Ile234–Gly243) connecting the two domains is partially structured forming a short two-turn α-helix (Arg236–Lys241). This helix is abundant in charged residues that probably help to stabilize it through salt-bridge interactions (Arg236–Asp240 and Asp237–Lys241).
Figure 2.

NMR solution structure of the RNA-binding domain of Saccharomices cerevisiae Hr1p (residues 156–321) in complex with an RNA molecule ((UA)4) representing the PEE (PDB: 2CJK). (A) Backbone superposition of the 56 selected structures comprising the ensemble. The two RBD domains define a deep cleft that constitutes the binding site for the RNA molecule. The structure is colour-coded according to sequence, from yellow (N-terminus) to dark orange (C-terminus) for the protein and from green (3′-end) to blue (5′-end) for the RNA. (B) Cartoon representation showing the overall architecture of the complex (same colour code as in 2A). The C-terminal RBD domain recognizes bases 2 and 3, whereas the N-terminal domain recognizes bases 4–7. The canonical RNP motifs in the two β-sheets participate in the recognition of Ade2, Ura3, Ura5, Ade6 and Ura7. The remaining base, Ade4, is recognized by a stacking interaction with Trp168, outside the canonical RNP motif (see text and Figure 3 for details). All figures were generated with the program PyMol (DeLano, 2002). (C) Comparison between protein–RNA recognition modes of Hrp1 and other 2 × RBD protein–RNA complexes. Structure-based protein sequence alignment between Hrp1–PEE S. cerevisiae (PDB 2CJK, this work), hnRNP A1 Homo sapiens (PDB 2U1P; Ding et al, 1999), HuD-(c-fos) H. sapiens (PDB 1FXL; Wang and Tanaka Hall, 2001), sex lethal Drosophila melanogaster (PDB 1B7F; Handa et al, 1999) and PABP H. sapiens (PDB 1CVJ; Deo et al, 1999). Secondary structural elements have been depicted above the sequences and numbered according to the Hrp1 sequence. Conserved residues have been shaded in blue (the darkest the most conserved). Residues participating in aromatic stacking or van der Waals interactions have been marked by red hexagons and residues forming hydrogen bonds with the RNA molecule are marked by green pentagons. Hrp1 aromatic residues contacting the RNA in a nonparallel way have been indicated by orange hexagons and structurally ill-defined contacts are represented by open green pentagons. Finally, the graph bars below the sequence alignment show the backbone amide proton chemical shifts changes upon complex formation. The protons that are involved in base-specific contacts (see the text) have been highlighted in red.
The structure represents a further example of single strand RNA recognition with distinct features. The association of the two RBD domains creates a deep and highly positively charged cleft between them, and it is largely this which accommodates the RNA molecule, in a roughly v-shaped conformation (Figure 2A and B). The two antiparallel β-sheets are arranged almost parallel to each other, with all the RNA bases buried inside the cleft whereas the phosphate backbone appears largely exposed to solvent (Supplementary Figure 1). Inspection of the H1′–H2′ correlations on the 2D correlation spectroscopy (COSY) spectra showed that all the sugars are in 2′ endo conformation.
Electrostatic surface representation (Supplementary Figure 1) reveals that some of the RNA bases (e.g. Ade6 and Ura5, for which only phosphate backbone can be seen in the figure) are highly buried into protein binding pockets, whereas others (e.g. Ade2 and Ade4) are sandwiched between protein loops (Supplementary Figure 1B). The recognition mode is entirely dominated by protein–RNA contacts, with a remarkable absence of RNA base–base stacking as has been observed in similar complexes (Deo et al, 1999; Ding et al, 1999; Handa et al, 1999; Wang and Tanaka Hall, 2001) or RNA base pairs (Auweter et al, 2006). Absence of these intra-RNA interactions allows the RNA molecule to adopt a more extended conformation and perhaps explains why Hrp1 targets a shorter sequence than those recognised in structurally related complexes.
Structural origins of base and ribose specificity
The RNA-binding region of Hrp1 (156–321) specifically recognises six bases (5′-GUAUAUAUA-3′). The first two bases at the 5′-end of the RNA (Ade2 and Ura3) bind to the second RBD of the protein whereas the following four (Ade4, Ura5, Ade6 and Ura7) are recognised by the first RBD (Figure 2A and B) (for convenience 5′-G has been numbered as residue 0). As expected, most of the protein residues at the protein–RNA interface are contributed from the exposed faces of the antiparallel β-sheets of the two RBDs, particularly residues of the RNP motifs, which are highly conserved among other hnRNPs (Figure 2C). The linker residues also participate actively in the recognition, as do other side chains outside the canonical RNP. Previous biochemical studies have shown that Hrp1 has a specific preference for UA-rich sequences, with a consensus 5′-UAUAUA-3′. The present structure describes a complex in which only Ade2 to Ura7 participate in protein–RNA contacts (5′-AUAUAU-3′), which is one nucleotide frame-shifted from the consensus. No intermolecular NOEs were found for the 5′ uracil (Ura1) and 3′ adenine (Ade8) residues, indicating that they do not participate in the protein–RNA interface. The NMR structure of the complex presented here gives insights into the base specificity at each position of the RNA.
Adenine recognition
The three adenines at the interface (Ade2, Ade4 and Ade6) are deeply buried within hydrophobic pockets. Parts of these recognition sites are probably preformed in the free form (i.e. the aromatics in the RNP motifs of RDB1 and RBD2). However, Ade6 binding pocket (Figure 3C and E) is also constituted by residues of the linker region, which are likely to be unfolded in the free form. Two protein side chains, those of Phe162 and Ile234, sandwich the Ade6 ring, largely burying its surface. Similarly, Ade4 is highly buried in a well-defined pocket formed by the side chains of Trp168 and Lys226 (Figure 3C and D). The tryptophan stacking appears as a novel feature of this structure without parallel in other single-stranded RNA 2 × RBDs complexes. Interestingly, this tryptophan is conserved in other fungal Hrp1-like proteins (Supplementary Figure 1). Finally, yet another aromatic side chain, that of Phe246, forms the base of the Ade2 recognition pocket (Figure 3A and B). The C-terminal region of the protein closes the opposite site of the Ade2 ring (Supplementary Figure 1A), likely through a π-cation interaction with Arg321, as supported by weak NOE interactions between the Ade2 H2 proton and the Hδ protons of Arg321. Moreover, the positive character of this side chain seems conserved in other Hrp1-like proteins (Supplementary Figure 2).
Figure 3.
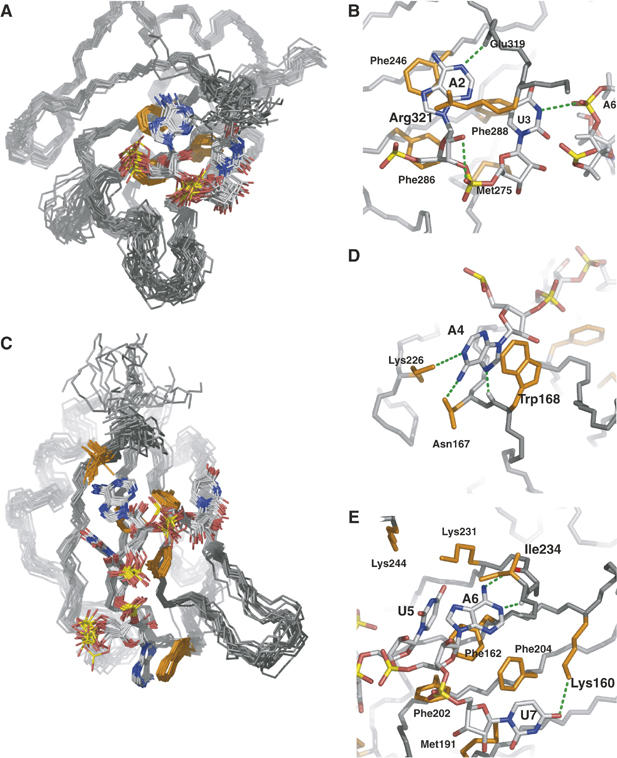
Detailed views of protein–RNA interface of the Hrp1–PEE complex. (A) The C-terminal RBD of Hrp1 recognizes Ade2 and Ura3. (B) Both bases form aromatic interactions with protein residues Phe246 and Phe288. The ribose of Ade2 form van der Waals contacts with Phe286 and Met245, with the last one also contacting the ribose of Ura3. The adenine is specified by a hydrogen bond between the position N1 and the Glu 319 backbone amide whereas the pyrimidine might be specified by a hydrogen bond contact (H3) to Ade6 phosphate group. The ensemble also shows a high population of a ribose-selective hydrogen bond between Ade2 OH2′ and the Ura3 phosphate oxygen. (C) The N-terminal RBD of Hrp1 recognizes four bases from Ade4 to Ura7. (D) One of the novel features of the Hrp1 RNA-binding mode is the well-defined aromatic stacking between Ade4 and Trp168. Adenine selectivity is accomplished by specific hydrogen bonds to positions N1 (Lys226 side chain), NH2(6) (Asn167 side chain) and N7 (Trp168 backbone amide). (E) Another two aromatic stacking interactions between Ade6 and Phe162 and Ura7 and Phe204 dominate the recognition of the 3′-end of the RNA. Ura5 forms van der Waals contacts with Phe162, with two lysine side chains (Lys244 and Lys231) likely involved in the recognition of the carbonyl groups at positions 2 and 4 of the base. Another lysine (Lys160) provides uracil specificity for the seventh base of the RNA by forming a hydrogen bond to the O2 position of this base. Adenine at position five is doubly specified by two hydrogen bonds involving its Watson-Crick face (exocycle NH2 and N1) and protein backbone groups (Ile234 carbonyl and Arg232 amide). Other protein residues (Met191 and Phe216) complete the interactions map by contacting ribose moieties of Ade6 and Ura7.
Many of the residues participating in van der Waals interactions are also involved in hydrogen bonds to the bases that determine specificity. Three backbone amides (Glu319 HN, Trp168 HN and Ile234 HN) form specific hydrogen bonds to nitrogen atoms of the three adenine bases (Ade2 N1, Ade4 N7 and Ade6 N1) (Figure 3B, D and E). Consistent with their new role, these three protons suffer the largest downfield shifts observed upon complex formation (Figure 2C). Two other contacts to the exocyclic NH2(6) specify adenine at positions 4 and 6, with the protein side chain acting here as donor (Asn167 Oδ1–Ade4 NH2 and Arg232 O–Ade6 NH2). Ade4 shows a third base-specific contact (Lys226 NɛH3–Ade4 N1). For the first adenine, the structure only shows one base-specific contact (Glu319 NH–Ade2 N1), although it is likely that other residues at the protein C-terminus participate in its recognition (Figure 3B). However, none of the potential interactions show high occupancy owing to the lower precision of the protein structure towards the C-terminal.
Uracil recognition
The three uracil bases of the PEE contact residues of the canonical RNA-binding motifs in the β-sheet of each RBD domain. Uridines interact with the protein mainly through van der Waals contacts. However, the low surface accessibility of Ura3 and Ura5 contrasts with that of Ura7. For all three uridines, the protein–RNA contacts involve aromatic protein side chains: Ura3 and Ura5 interact with Phe288 and Phe162 in a nonplanar fashion (Figure 3C and E) whereas Ura7 form a planar stacking arrangement with the ring of Phe202 (Figure 3E). Discrimination against cytosine relies heavily on the interactions with position four of the heterocycle. The NɛH3 group of Lys160 forms a direct hydrogen bond to this position of Ura7 (Figure 3E). Although less evident, Lys244 and Lys231 are close enough to the O2 and O4 positions of the Ura5 to be involved in base discrimination. These two lysine residues are conserved in the sequence-related hnRNP A1, where they form interfacial hydrogen bonds (Figure 2C). Lys244 is changed to Asn in HuD, sex lethal and PABP complexes, but the interaction seems to be conserved (Figure 2C). RNA interacting residues are also found at positions equivalent to Lys160 in all the five complexes. Overall these comparisons reinforce the putative recognition role of Lys160, Lys244 and Lys231 in the Hrp1–PEE complex. The origin of discrimination in the case of Ura3 is less clear but many structures in the ensemble suggest that it might be mediated by recognition of the imino NH (3) position by the backbone phosphate of Ade6. However, this imino proton could not be assigned, perhaps because it overlaps with other protons of the protein or because it is in fast or intermediate exchange with solvent.
Ribose recognition
The structure ensemble provides indirect information concerning the role played by the ribose HO2′ groups in the complex, because separate signals are not observed for these protons. Ribose-specific hydrogen bonds like that the Ade2 ribose hydroxyl to Ura3 phosphate (Figure 3B) and another between Ade6 OH2′ and Ura7 O5′, appear highly populated in the ensemble and offer hints about the discrimination between RNA and DNA.
Comparison with similar structures
The RNP-type RBD is perhaps one of the most important protein domains involved in RNA recognition (Burd and Dreyfuss, 1994; Maris et al, 2005), playing principal roles in mRNA capping, splicing, 3′-end formation and transport. The intrinsic capacity of these protein domains to bind different RNAs seems to require arrangements of two or more consecutive RBDs to increase binding affinity and achieve selectivity. During the last 5 years, a number of crystal structures exhibiting recognition of single-strand RNA by tandem repeats of RBDs have been reported. These include the recognition of polyadenine by PABP (Deo et al, 1999) and uracil-rich sequences by sex-lethal (Handa et al, 1999) and HuD (Wang and Tanaka Hall, 2001) proteins. The structure of hnRNPA1 in complex with telomeric DNA (Ding et al, 1999) has a related architecture to these two complexes. NMR solution structures of nucleolin (Allain et al, 2000) and U1A (Varani et al, 2000) can also be classified within this group taking into account that their RNA molecules contain regular secondary structures elements together with the single-strand sections. More recently, several new NMR structures: the splicing factor Fox-1 RBD bound to UGCAUGU (Auweter et al, 2006) and complexes of individual PTB RBD domains (1–4) with different RNA molecules (Oberstrass et al, 2005) have increased substantially our knowledge of nucleic acid single-strand recognition. The structure of the Hrp1–PEE complex brings a new example of single-strand RNA recognition that shares similarities with previous structures but, interestingly, identifies new structural features also.
The two RBDs in the Hrp1–PEE complex have a nearly identical orientation as that observed in HuD and sex-lethal complexes (Figure 4), but is maintained by different interdomain contacts. In crystal structures, there are conserved hydrogen bonds between the two RBDs (Lys197 Nζ–Val238 O in sex-lethal and Lys111 Nζ–Ile52 O in HuD), between the linker and RBD2 (Arg202 Nη1–Asn260 Oδ in sex-lethal and Arg116 Nη1–Asp174Oδ1) and between the helix in the linker and the second helix of RBD2. In the Hrp1–PEE complex, RBD1 and RBD2 interact through single salt-bridge (Lys231 Nζ–Asp271 Oδ1) (Figure 5C). Remarkably, Lys231 is not sequence homologous to Lys197/Lys111 in sex-lethal/HuD complexes Figure 2C. The hydrogen bond between the linker's helix and helix 2 of RBD2 is also conserved in the Hrp1 complex (Figure 5C). The major differences with the HuD or sex-lethal complexes involve the region of the linker that participates actively in RNA recognition. The relative domain orientation is heavily determined by these protein–RNA and other protein–protein contacts (Figure 5C) between linker and RBD2 residues. In conclusion, it seems that the Hrp1–PEE structure contains features that resembles the HuD and sex-lethal complexes (i.e., interdomain orientation) but also other characteristics more similar to the PABP recognition mode (i.e., linker residues form van der Waals contacts with Ade6).
Figure 4.
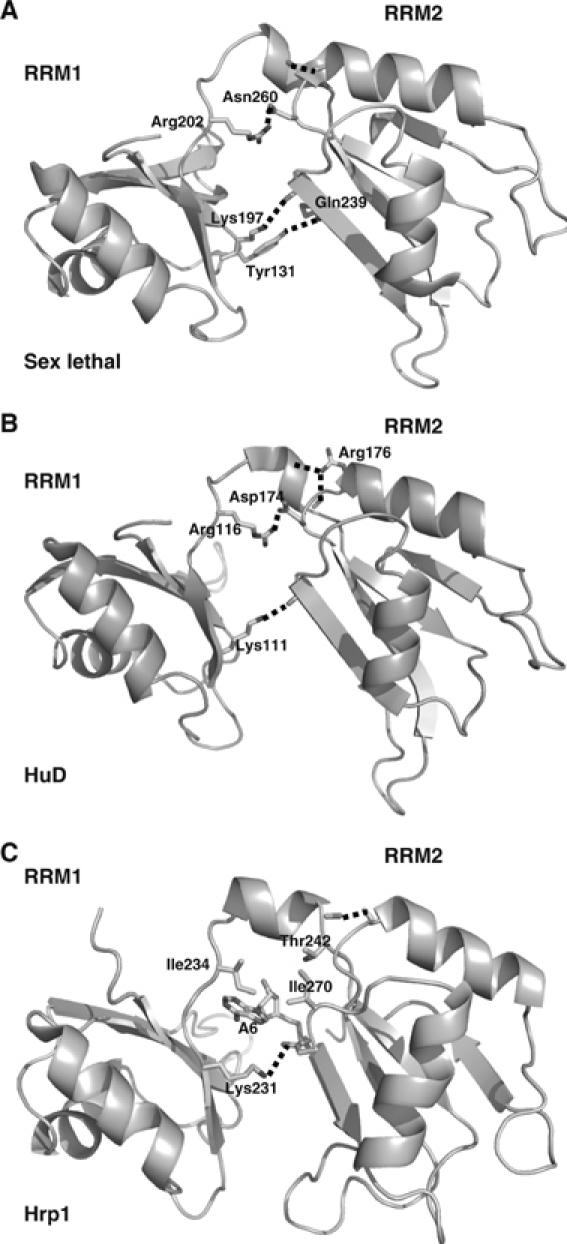
Structural comparison of the interdomain contacts between (A) sex lethal (PDB: 1B7F), (B) HuD-(c-fos) (PDB: 1FXL) and (C) Hrp1–PEE (PDB: 2CJK). Structures are represented facing the opposite side of the RNA-binding interface and the RNA molecules have been removed for clarity (with the exception of Ade6 in the Hrp1 structure). Residues involved in key interactions between RBD1, RBD2 and the linker between them have been represented in each case.
Figure 5.
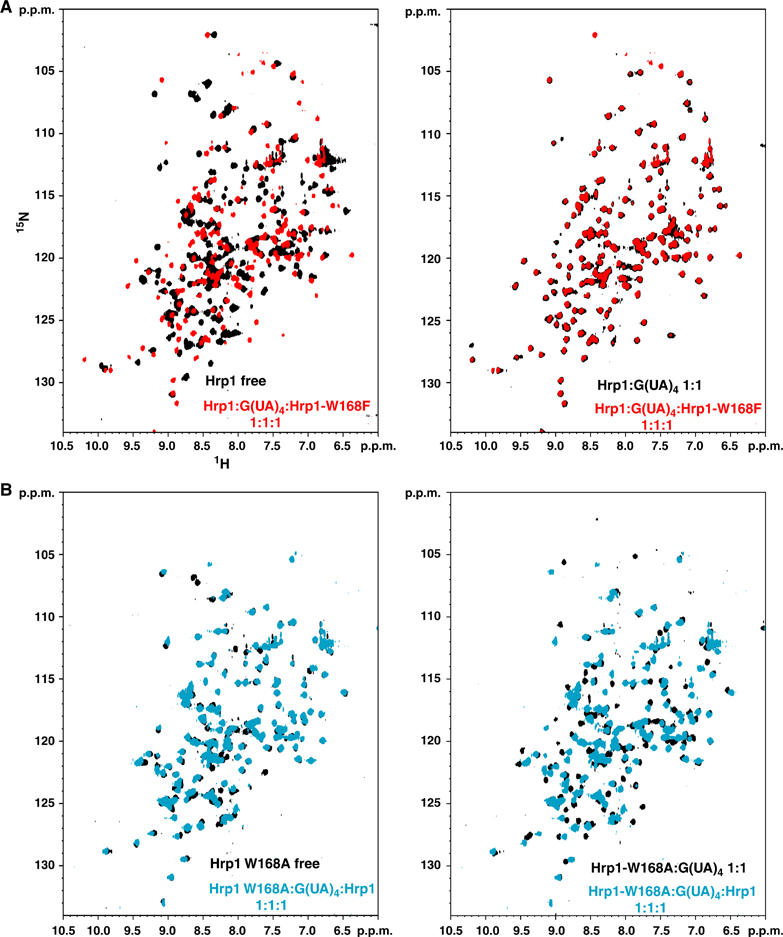
Results from the in vitro competition assays performed with Trp168 mutants. (A) Comparison of the 15N-HSQC spectrum (red) of the equimolecular ternary mixture: 15N-Hrp1 wild-type: G(UA)4: Hrp1 Trp168Phe, against the reference spectra (in black) of Hrp1 wild-type in its free form (left panel) or in 1:1 complex with G(UA)4 (right panel). (B) A similar comparison between the 15N-HSQC spectrum (cyan) of the equimolecular ternary mixture: Hrp1 wild-type: G(UA)4: 15N Hrp1 Trp168Ala, against the reference spectra (in black) of Hrp1–Trp168Ala in its free form (left panel) or in 1:1 complex with G(UA)4 (right panel).
Hrp1 binds single-strand RNA in the same direction (5′–3′) as do PABP, sex-lethal and HuD proteins, a feature seems now to be established as general in these kinds of interactions (Perez-Canadillas and Varani, 2001). An interesting difference is that Hrp1 covers a shorter RNA region that its structural partners. PABP, sex-lethal and HuD accumulate between 8 and 10 nucleotides at the binding interface, and always include at least one intra-RNA base–base stacking interaction, but, interestingly, this feature is absent in the Hrp1–PEE complex. In this regard, Ade5–Ade6 stacking in the PABP complex has some structural resemblance to the Ura6–Ade7 stacking in HuD complex or the Ura7–Ura8 stacking in sex-lethal. Structurally, these intra-RNA interactions seem to be replaced by a Trp168–Ade4 stacking interaction in the Hrp1 complex, which perhaps explains why the protein targets a much shorter and elongated RNA region in this case. In this sense, it is interesting to compare the differential role of loop 1 residues in Hrp1 and HuD or sex-lethal complexes. In these last two structures, a tyrosine residue (Tyr45/Tyr131) is making a stacking with the uracil base 3′ to the intra-RNA stacking mentioned above. Hrp1 does not contain an equivalent residue at this position (Figure 2C) and neither HuD nor sex-lethal have an aromatic residue equivalent to Trp168. In the Hrp1 complex, the Tyr–Ura and Ura–Ura (or Ura–Ade) stacking interaction seems to be replaced by the Trp168–Ade6 interaction, eliminating two bases from the protein–RNA interface. Recently, the structure of Fox-1–RNA complex has provided more evidences showing that loop 1 aromatic residues might play an important and perhaps more common role in RBD–RNA recognition (Auweter et al, 2006). Mutagenesis studies have demonstrated that the aromatic character of the Phe162 side chain is crucial to explain the unusually low dissociation constant of this complex. Structurally, Phe162 is surrounded by three RNA bases (two of them stacking on each side of the Phe ring) and represents a new recognition mode that differs from that in HuD (sex-lethal) and Hrp1 complexes. Previous studies have shown the important role that other regions outside the RNP motifs (i.e. C-terminal helices) play in modulation of RNA recognition by RBDs. The comparison among HuD, sex-lethal, Fox-1 and Hrp1 binding modes shows that loop 1 aromatic residues can be also strong determinants of RNA specificity.
Trp168 is crucial for binding affinity and specificity
The interaction between Trp168 and Ade4 constitutes the most prominent and novel feature of the Hrp1–PEE complex. The structural role of this residue seems to be crucial for the RNA-binding mode of Hrp1. Furthermore, it is totally conserved in several fungal Hrp1-like proteins (Supplementary Figure 2), but not in PABP, HuD and sex-lethal proteins that have Pro or Gln at equivalent positions (Figure 2C).
The effect on binding affinity and specificity of this protein side chain has been studied by replacing Trp168 with Phe and Ala side chains. Both Trp168Phe and Trp168Phe mutants retain the ability to form 1:1 complexes with the G(UA)4 RNA probe. However, unlike for the wild-type protein, the mutants NMR signals in intermediate to fast exchange regime suggest that the binding affinity may be weaker than the wild-type protein. In order to test this hypothesis, in vitro competition experiments were performed. The ternary mixtures were prepared (see Materials and methods), with the amount of RNA being limiting (1:2) compared to the total protein (wild type and mutant). The mixtures contained the same amount of both proteins, and only one of them (wild type and mutant) was 15N labelled. With this experimental set-up, it is possible to analyse the effect of the competitor (unlabelled component) on the binding of the 15N labelled component by simply comparing the spectrum of the ternary mixture against reference spectra of the free and bound state of the labelled component The results (Figure 5 and Supplementary Figure 3) shows that both mutants have weaker affinity for the native-like RNA probe (G(AU)4) than wild-type protein. The 15N-HSQC spectra of the mixtures with the wild-type15N labelled protein overlap with the reference spectra of the Hrp1–PEE complex (Figure 5A, right panel and Supplementary Figure 3A, right panel). No Hrp1 wild-type signals corresponding to the free form (Figure 5A, left panel and Supplementary Figure 3A, left panel) have been found, indicating that the amount of wild-type protein competed out by the mutant was below the detection limit (<5%). Complementary experiments with ternary mixtures in which only the mutant was 15N labelled showed that most of the signals overlap with the reference 15N-HSQC spectrum mutant in the free form (Figure 5B, left panel and Supplementary Figure 3A, left panel). In this case, a small number of signals is missing and presumably in intermediate exchange with a minor proportion of RNA-bound mutant.
The 15N-HSQC spectrum is very sensitive to structural changes and therefore a powerful tool to compare wild-type and Trp168 mutant RNA-binding modes. The superposition of the 15N-HSQC spectrum of Hrp1–Trp168Phe complex with the spectrum of the Hrp1 wild-type complex shows that only very few protein signals overlap (Figure 6B), that contrast with the high degree of similarity between the spectra of the Hrp1–Trp168Ala and Hrp1–Trp168Phe complexes (Figure 6A). The comparison strongly suggests that both mutants share a similar RNA-binding mode, which is totally different from that of the wild-type protein. These results allow conclusion that mutation of the crucial Trp168 affects the affinity, binding mode and even perhaps base specificity for RNA.
Figure 6.
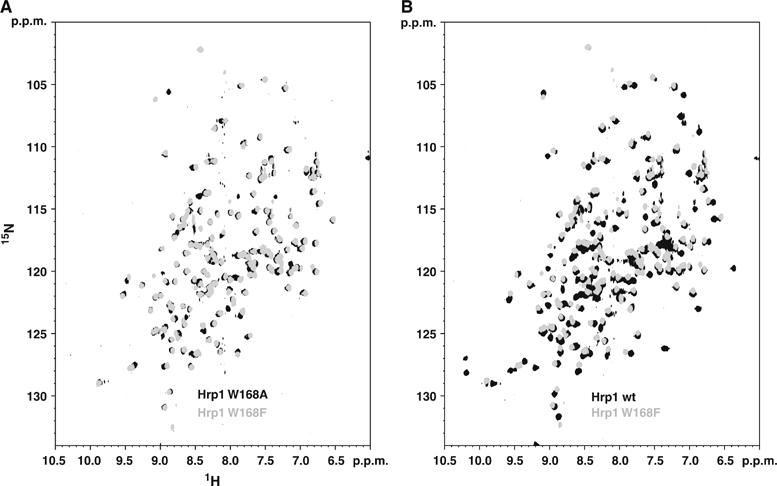
Comparison between 15N-HSQC spectra Hrp1 and Hrp1 Trp168 complexes. (A) The spectra of Hrp1 Trp168Ala (black) and Trp168Phe (grey) RNA complexes are highly superimposable, which is consistent with a similar binding mode. (B) In contrast, the comparison between the 15N-HSQC spectra of wild-type (black) and Trp168Phe (grey) mutant complexes show significant differences indicating that the binding mode differs between the wild-type and mutant proteins.
The recently published structure of the splicing factor Fox-1–RNA complex (Auweter et al, 2006) identifies a phenylalanine (Phe126) that is at equivalent position of Trp168 in Hrp1. The authors evaluated the impact of several mutations at this site on the dissociation constant of the complex, finding that the aromatic character of Phe126 is very important for affinity. This can be explained structurally as Phe126 makes two planar stacking interactions with two RNA bases and contacts a third one. The competition experiments presented here are very qualitative but allow to state that the binding affinity of the two Hrp1 Trp168 mutants is at least 10 times weaker compared with the wild-type one. In Fox-1 replacement of Phe126 by other aromatics has little impact on the affinity indicating that these mutants presumably share a similar binding mode than wild type. This does not seem to be the case in Hrp1, in which the two mutants seem to share a similar binding mode that is different to wild-type one (Figure 6A and B) and that perhaps the sequence requirements in Hrp1 are stricter than in Fox-1.
Structural basis for Hrp1 temperature-sensitive phenotypes
Previous genetic studies have identified at least eight mutations that are crucial for temperature viability of mutant-harboring yeast strains (Kessler et al, 1997). The high-resolution structure described here allows discussing about the possible structural causes underlying the mutant phenotypes. For instance, two of these mutations (Leu205Ser and Ile313Asn) are in the protein core, so that their phenotypes are presumably associated with protein instability at the nonpermissive temperature. Together with these structural mutants, there are others that could be classified as contact mutants, as they might interfere with the protein–RNA recognition (just like the Trp168 mutants presented here). One of this contact mutations could be Lys160Glu (in allele hrp1–1) as the NɛH3 group makes an intermolecular hydrogen bond to the O4 atom of Ura7. The double mutant alleles hrp1–7 (Met191Ser, Ala195Pro) and hrp1–4 (Tyr182Cys, Met191Gly) also both involve an interfacial residue (Met191, which is in van der Waals contact with the ribose of Ura7), but here it is unclear in either case whether the mutation at position 191 is necessarily the one responsible for the phenotype.
In summary, the structure of the Hrp1–PEE complex presented here is a powerful tool to rationalise the mechanism of functionally defective mutants and, what it is more important, to predict and design new mutations that interfere with the wild-type RNA-binding mode (i.e. Trp168 mutants). Knowing the structural basis of the interaction will now improve the design of future experiments and to have more control in the way that mutations interfere with organization of the polyadenylation machinery.
Materials and methods
Protein expression and purification
The DNA sequence corresponding to Hrp1 156–321 was amplified by polymerase chain reaction from the plasmid for full-length protein (courtesy of C Moore) and subcloned into a pET-15 vector (Novagen), which includes an N-terminal His6 tag followed by a thrombin cleavage that precedes the Hrp1 sequence. The protein was overexpressed in Escherichia coli BL21(DE3) cells grown in minimal medium (M9) containing 80 mg/l of ampicillin, vitamin and trace elements solutions. Glucose and ammonium chloride (13C and 15N labelled when required) were used as the carbon and nitrogen sources. Cells were gown at 37°C and induced using IPTG at OD 0.6 and harvested after 4 h at 37°C. Cell pellets were disrupted by mild sonication and ultracentrifuged and the soluble fraction, that contains the fusion protein of interest, purified by methal-chelate chromatography (Qiagen Ni-NTA column). The His6 tag was removed by thrombin digestion (SIGMA) and at the same time the sample was dialysed overnight against low imidazol buffer (150 mM NaCl, 25 mM phosphate pH 7.5, 10 mM β-mercaptoethanol). The sample was passed again through the nickel column, then subjected to size exclusion chromatography (Amersham G75 column), concentrated to around 1 mM and microdialysed against NMR buffer (25 mM phosphate (pH 6.0), 10 mM dithiothreitol (DTT)). No detectable impurities were found in the purified protein by polyacrylamide gel electrophoresis (PAGE) or mass spectroscopy.
Hrp1 mutants (Trp168Phe and Trp168Ala) were made by standard site-directed mutagenesis using the appropriate DNA oligos and subcloned into a pET28 vector that contains an N-terminal thioredoxin tag, followed by a His6 and thrombin cleavage site prior the Hrp1 mutant sequences. Protein expression and purification were performed in a similar manner as for the wild-type protein.
RNA synthesis
RNA samples were made by in vitro transcription using home-made RNA T7 polymerase following a previously reported protocol (Price et al, 1998). Seven different RNA samples were produced containing a number of increasing repeats of the dinucleotide sequence (UA)n (n=4, 5, 6, 7, 8 and 10), which constitutes the binding site of Hrp1. The run-off transcription protocol results in an extra G at the 5′ end of all products.
Samples were purified by quantitative PAGE electrophoresis under denaturing conditions (gels contain 7 M Urea), isolation of the appropriate band, then electroelution for 4 h at 4°C (yield>90%). Samples were ethanol precipitated twice and dialysed four times against buffer (25 mM phosphate pH 8.0, 1 mM ethylene diaminetetraacetic acid) containing decreasing amounts of salt (from 1 M to no salt). The product was freeze-dried, resuspended in water and microdialysed against NMR buffer (see above).
Electromobility shifts assays
Complex formation and stoichiometry of RNA–Hrp1 was monitored by gel retardation experiments. Protein and RNA were mixed at different ratios and brought to a final concentration of 1 × TB buffer pH 7.4 and 10% glycerol. Mixtures were incubated for 10 min at room temperature, loaded into native gels (10% acrylamide) and run at low voltage at 4°C. The resulting gels were stained in 1% toloudine. NMR sample preparation of different protein–RNA complexes was also monitored by this method, resulting in very precise stoichiometries.
NMR spectroscopy
NMR spectra were acquired at 27°C on Bruker Avance 800, DMX600 and DRX500 spectrometers, the DRX500 being equipped with a triple-resonance (1H/15N/13C) cryoprobe. Sample concentrations of Hrp1–RNA complex were approximately 1 mM in NMR buffer (25 mM phosphate buffer, 10 mM deuterated DTT, pH 6.0), and optimum stoichiometry (1:1) was obtained by stepwise titration of protein into RNA, monitoring disappearance of 1H imino resonances at ∼13–14 ppm (arising from RNA duplex) and also using gel electromobility assays (see above). On one occasion, RNA was added to protein to observe how the protein 15N HSQC spectrum changed during the titration; this demonstrated that the system is in slow exchange on the chemical shift timescale. Protein backbone assignments (Cα, Cβ, C′, N, HN) were obtained using triple resonance experiments including HNCA, HN(CO)CA, HNCO and CBCA(CO)HN (Sattler et al, 1999), and side-chain assignments were completed using an HCCH-COSY experiment. An equivalent protocol was followed to assign protein signals in the RNA-bound spectrum. NOE-derived distance restraints were obtained from a series of 2D NOESY spectra (H2O or 2H2O) and 3D 15N and 13C edited NOESY spectra. All NOE-based experiments were recorded at 800 MHz using 80 ms mixing time (to minimise spin diffusion). Homonuclear 2D NOESY (i.e. Supplementary Figure 4), COSY and total correlation spectroscopy (TOCSY) (60 ms of DIPSI-3 mixing) experiments in 2H2O were used to assign resonances of G(UA)4 in the bound state. The H1′ and H2′ and uracil H5 and H6 were assigned from their COSY peaks. Connection between sugar and aromatic spin systems was made on the basis of the strong NOEs between H1′ and H2′ protons with adenine H8 and uracil H6. These sugar protons give sequential NOEs with the next base (adenine or uracil) allowing the sequential assignment. Owing to the repetitive nature of the RNA, complexes with sequences longer than four AU repeats (not in the G(UA)4 complex) show exchange peaks between consecutive Ade and Ura residues (i, i+2) in the TOCSY experiment that were very useful to confirm the sequential assignment of the RNA. Preliminary structures were calculated with the information of these primary assignments. This preliminary structural information allowed assigning the H2 resonances (downfield shifted in the spectra) of the adenine residues on the basis of their pattern of intermolecular NOEs. Sugar resonances H3′, H4′, H5′ and H5″ can be observed in the TOCSY experiment from the H1′, but are difficult to assign specifically. NOEs involving these ambiguous resonances were referenced to the C4′ atom and corrected by adding 2.0 Å, which corresponds to the distance between H3′, H5′ or H5″ and the C4′ atom.
Trp168 mutant's in vitro competition assays
Four different ternary mixture were prepared containing Hrp1 wild-type, Hrp1 mutant and G(UA)4 RNA in equimolecular amount (400 μM each). In all the cases, the amount of RNA is limiting and unable to saturate all the proteins (wild-type and mutant) present. Only one of the protein components (Hrp1 wild-type, Trp168Ala or Trp168Phe mutants) was 15N labelled whereas the other remained at natural abundance isotope composition, with the aim to follow the competition effect in only one of the protein components. The reference spectra comprise three 15N-HSQC of Hrp1 wild-type, Hrp1–Trp168Ala and Hrp1–Trp168Phe, and another three 15N-HSQC of these three proteins in complex with G(UA)4 (1-to-1 complexes). All the samples were prepared in a 3-mm NMR tube (150 μl) and 15N-HSQC spectra were recorded on a 600 MHz spectrometer equipped with a cryoprobe. The temperature and buffer conditions are identical to that of the experiments used for the structure calculation (see above). The 15N-HSQC spectra of the ternary mixtures (that shows only the signals of wild-type, Trp168Ala or Trp168Phe) were compared with the relevant 15N-HSQC reference spectra to determine the ability of unlabelled protein (wild-type or mutant) or mutant (unlabelled) to compete out the labelled protein (mutant or wild type).
Structure calculation
NOE-derived distance restraints were extracted from NOESY spectra at 80 ms mixing time and classified according to crosspeaks intensity into four different groups (Table I). The relative intensity boundaries for each class (as defined in the program ANSIG) were chosen so that the NH(i)-NH(i+1) correlations in known helical regions fell into the strong (1.8–2.8 Å) or medium categories (1.8–3.3 Å) and the HA(i)-NH(i+3) correlations into the medium (1.8–3.3 Å) or weak (1.8–4.5 Å) categories. Relative intensities weaker than these were classified as very weak (1.8–5.5 Å). Angular restraints were introduced during the structure calculation to restrict the ribose puckering to the 2′-endo conformation, as determined by the inspection of the H1′–H2′ correlations in a 2D COSY spectrum. A total of 50 structures were calculated with the program CNS (Brunger et al, 1998) using a standard protocol that starts from extended chains representing the protein and RNA molecules. The procedure began with 1000 high-temperature (50 000 K) torsion angle dynamics steps followed by 7000 torsion angle dynamics steps from 50 000 to 0 K, 1000 cartesian dynamics steps from 2000 to 0 K and a final 4000 energy minimization steps. The resulting structures were subjected to a simple refinement protocol with the program XPLOR-NIH (Schwieters et al, 2003), which includes Ramachandran (Kuszewski and Clore, 2000) and hydrogen bond (Lipsitz et al, 2002)-derived potentials. Hydrogen bond restraints were introduced for backbone amides groups that show some degree of protection in deuterium oxide solutions. The 50 individual structures were subjected to 3000 steps of cartesian dynamics from 1000 to 0 K followed by 1000 steps of energy minimization. The program CLUSTERPOSE (Diamond, 1995) was used to calculate the mean r.m.s.d. of ensembles to their mean structure (Supplementary Figure 5). The atomic coordinates of the Hrp1–PEE complex have been deposited on the PDB database (id: 2CJK).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Acknowledgments
I thank Drs Peter Lukavsky, David Neuhaus, Clara M Santiveri and Douglas V Laurents for their critical reading and suggestions during the preparation of the manuscript and additionally to Dr Neuhaus for the help with the program CLUSTERPOSE. This research would not have been possible without the infrastructure and support provided by members of Dr Lukavsky's, Dr Neuhaus's and Professor Manuel Rico's laboratories to which I am profoundly grateful. The original clone for the Hrp1 was kindly provided by Professor Claire Moore. This work has been financed by a individual Marie Curie Fellowship of the European Community human potential program (HPRMF-CT-2000-00722).
References
- Allain FH, Bouvet P, Dieckmann T, Feigon J (2000) Molecular basis of sequence-specific recognition of pre-ribosomal RNA by nucleolin. EMBO J 19: 6870–6881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auweter SD, Fasan R, Reymond L, Underwood JG, Black DL, Pitsch S, Allain FH (2006) Molecular basis of RNA recognition by the human alternative splicing factor Fox-1. EMBO J 25: 163–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54 (Part 5): 905–921 [DOI] [PubMed] [Google Scholar]
- Burd CG, Dreyfuss G (1994) Conserved structures and diversity of functions of RNA-binding proteins. Science 265: 615–621 [DOI] [PubMed] [Google Scholar]
- Dantonel JC, Murthy KG, Manley JL, Tora L (1997) Transcription factor TFIID recruits factor CPSF for formation of 3′ end of mRNA. Nature 389: 399–402 [DOI] [PubMed] [Google Scholar]
- DeLano WL (2002) The PyMOL Molecular Graphics System. San Carlos, CA, USA: DeLano Scientific LLC [Google Scholar]
- Deo RC, Bonanno JB, Sonenberg N, Burley SK (1999) Recognition of polyadenylate RNA by the poly(A)-binding protein. Cell 98: 835–845 [DOI] [PubMed] [Google Scholar]
- Diamond R (1995) Coordinate-based cluster analysis. Acta Crystallogr Sect D 51: 127–135 [DOI] [PubMed] [Google Scholar]
- Ding J, Hayashi MK, Zhang Y, Manche L, Krainer AR, Xu RM (1999) Crystal structure of the two-RRM domain of hnRNP A1 (UP1) complexed with single-stranded telomeric DNA. Genes Dev 13: 1102–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez CI, Ruiz-Echevarria MJ, Vasudevan S, Henry MF, Peltz SW (2000) The yeast hnRNP-like protein Hrp1/Nab4 marks a transcript for nonsense-mediated mRNA decay. Mol Cell 5: 489–499 [DOI] [PubMed] [Google Scholar]
- Gross S, Moore CL (2001) Rna15 interaction with the A-rich yeast polyadenylation signal is an essential step in mRNA 3′-end formation. Mol Cell Biol 21: 8045–8055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Sherman F (1995) 3′-end-forming signals of yeast mRNA. Mol Cell Biol 15: 5983–5990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Sherman F (1996) 3′-end-forming signals of yeast mRNA. Trends Biochem Sci 21: 477–481 [DOI] [PubMed] [Google Scholar]
- Handa N, Nureki O, Kurimoto K, Kim I, Sakamoto H, Shimura Y, Muto Y, Yokoyama S (1999) Structural basis for recognition of the tra mRNA precursor by the sex-lethal protein. Nature 398: 579–585 [DOI] [PubMed] [Google Scholar]
- Kessler MM, Henry MF, Shen E, Zhao J, Gross S, Silver PA, Moore CL (1997) Hrp1, a sequence-specific RNA-binding protein that shuttles between the nucleus and the cytoplasm, is required for mRNA 3′-end formation in yeast. Genes Dev 11: 2545–2556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuszewski J, Clore GM (2000) Sources of and solutions to problems in the refinement of protein NMR structures against torsion angle potentials of mean force. J Magn Reson 146: 249–254 [DOI] [PubMed] [Google Scholar]
- Lipsitz RS, Sharma Y, Brooks BR, Tjandra N (2002) Hydrogen bonding in high-resolution protein structures: a new method to assess NMR protein geometry. J Am Chem Soc 124: 10621–10626 [DOI] [PubMed] [Google Scholar]
- Maris C, Dominguez C, Allain FH (2005) The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J 272: 2118–2131 [DOI] [PubMed] [Google Scholar]
- McCracken S, Fong N, Yankulov K, Ballantyne S, Pan G, Greenblatt J, Patterson SD, Wickens M, Bentley DL (1997) The C-terminal domain of RNA polymerase II couples mRNA processing to transcription. Nature 385: 357–361 [DOI] [PubMed] [Google Scholar]
- Minvielle-Sebastia L, Preker PJ, Keller W (1994) RNA14 and RNA15 proteins as components of a yeast pre-mRNA 3′-end processing factor. Science 266: 1702–1705 [DOI] [PubMed] [Google Scholar]
- Oberstrass FC, Auweter SD, Erat M, Hargous Y, Henning A, Wenter P, Reymond L, Amir-Ahmady B, Pitsch S, Black DL, Allain FH (2005) Structure of PTB bound to RNA: specific binding and implications for splicing regulation. Science 309: 2054–2057 [DOI] [PubMed] [Google Scholar]
- Perez-Canadillas JM, Varani G (2001) Recent advances in RNA–protein recognition. Curr Opin Struct Biol 11: 53–58 [DOI] [PubMed] [Google Scholar]
- Price SR, Oubridge C, Varani G, Nagai K (1998) Preparation of RNA–protein complexes for X-ray crystallography and NMR. In RNA-Protein Interactions: A Practical Approach, Smith C (ed), Vol. 1, pp 30–68. Oxford, UK: Oxford University Press [Google Scholar]
- Proudfoot N (2004) New perspectives on connecting messenger RNA 3′ end formation to transcription. Curr Opin Cell Biol 16: 272–278 [DOI] [PubMed] [Google Scholar]
- Sattler M, Schleucher J, Griesinger C (1999) Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog NMR Spectrosc 34: 93–158 [Google Scholar]
- Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM (2003) The Xplor-NIH NMR molecular structure determination package. J Magn Reson 160: 65–73 [DOI] [PubMed] [Google Scholar]
- Shatkin AJ, Manley JL (2000) The ends of the affair: capping and polyadenylation. Nat Struct Biol 7: 838–842 [DOI] [PubMed] [Google Scholar]
- Stumpf G, Domdey H (1996) Dependence of yeast pre-mRNA 3′-end processing on CFT1: a sequence homolog of the mammalian AAUAAA binding factor. Science 274: 1517–1520 [DOI] [PubMed] [Google Scholar]
- Valentini SR, Weiss VH, Silver PA (1999) Arginine methylation and binding of Hrp1p to the efficiency element for mRNA 3′-end formation. RNA 5: 272–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varani L, Gunderson SI, Mattaj IW, Kay LE, Neuhaus D, Varani G (2000) The NMR structure of the 38 kDa U1A protein – PIE RNA complex reveals the basis of cooperativity in regulation of polyadenylation by human U1A protein. Nat Struct Biol 7: 329–335 [DOI] [PubMed] [Google Scholar]
- Wang X, Tanaka Hall TM (2001) Structural basis for recognition of AU-rich element RNA by the HuD protein. Nat Struct Biol 8: 141–145 [DOI] [PubMed] [Google Scholar]
- Wishart DS, Sykes BD (1994) Chemical shifts as a tool for structure determination. Methods Enzymol 239: 363–392 [DOI] [PubMed] [Google Scholar]
- Xu C, Henry MF (2004) Nuclear export of hnRNP Hrp1p and nuclear export of hnRNP Npl3p are linked and influenced by the methylation state of Npl3p. Mol Cell Biol 24: 10742–10756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Hyman L, Moore C (1999) Formation of mRNA 3′ ends in eukaryotes: mechanism, regulation, and interrelationships with other steps in mRNA synthesis. Microbiol Mol Biol Rev 63: 405–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5