

Abstract
The bioinorganic chemistry of iron is central to life processes. Organisms must recruit iron from their environment, control iron storage and trafficking within cells, assemble the complex, iron-containing redox cofactors of metalloproteins, and manage a myriad of biochemical transformations by those enzymes. The coordination chemistry and the variable oxidation states of iron provide the essential mechanistic machinery of this metabolism. Our current understanding of several aspects of the chemistry of iron in biology are discussed with an emphasis on the oxygen activation and transfer reactions mediated by heme and nonheme iron proteins and the interactions of amphiphilic iron siderophores with lipid membranes.
That an iron-containing enzyme mediated the activation and transfer of molecular oxygen into its substrate was first demonstrated by Hayaishi et al. (1) in the 1950s. It was shown, in some of the first mechanistically informative oxygen isotopic measurements, that both of the inserted oxygen atoms in the conversion of catechol to cis-muconic acid derived from O2 and not water. These findings challenged the then-firmly held view that oxygen in biomolecules was derived exclusively from water via hydration processes. The biosynthesis of cholesterol and its precursor, lanosterol, from the hydrocarbon squalene were also shown to derive their oxygen functionality from molecular oxygen (2). Here, a single oxygen atom derived from molecular oxygen while the other was transformed to water. Later, the prostaglandins were shown to derive from the incorporation of two molecules of oxygen to form, initially, an alkyl hydroperoxide-endoperoxide. Thus, what appeared at first to be an obscure process of bacteria and fungi became recognized as a major theme of aerobic metabolism in higher plants and animals. The subsequent search for “active oxygen species” and efforts to elucidate and understand the molecular mechanisms of oxygen activation and transfer have been richly rewarding. The roles of iron in these wonderfully varied processes have been a major force in the development of both bioinorganic chemistry and chemical catalysis over the past three decades. Novel and unusual iron redox chemistry has appeared as our understanding of biological iron acquisition, transport and storage, and enzymatic oxidation strategies has developed. The goal of this perspective is to discuss several aspects of the current state of bioinorganic chemistry relating to iron and our understanding of the chemical pathways and mechanisms by which iron–oxygen systems function.
Oxygen Activation by Heme Proteins
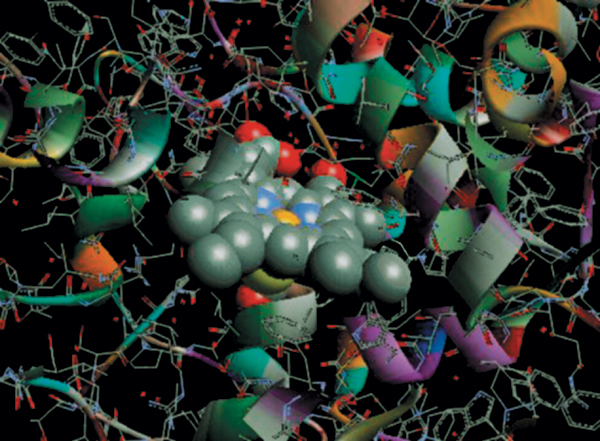
The heme-containing metalloenzymes cytochrome P450 (3), chloroperoxidase (CPO, refs. 4 and 5), NO synthase (NOS, ref. 6), and their relatives catalyze a host of crucial biological oxidation reactions. Highly specific P450s are involved in the selective oxygenations of steroid and prostaglandin biosynthesis. Myeloperoxidase, which is a CPO, is an integral part of the immune response, and NOS is the source of the highly regulated signal transducer NO. Certain fungal CPOs and bacterial P450s have been genetically engineered for large-scale biotransformations (7–10). The active sites of these three protein families, known in detail from a number of x-ray crystal structures (4, 11–13), are remarkably similar. All three have an iron-protoporphyrin IX center coordinated to a cysteine thiolate. All of them are oxidoreductases that activate molecular oxygen (O2), in the cases of P450 and NOS, or hydrogen peroxide in the case of CPO, at the iron center and incorporate one of the oxygen atoms into a wide variety of biological substrates, with concurrent transformation of the other oxygen atom to H2O. All three are proposed to initiate their chemistry through the oxidation of a resting iron(III) state (1) to a reactive oxoiron(IV)–porphyrin cation radical intermediate (2) (Fig. 1). A depiction of the CPO active site derived from the crystal structure of this protein from Calderiomyces fumago is shown in Fig. 5, which is published as supporting information on the PNAS web site, www.pnas.org. The structure, biochemistry, molecular biology, and the chemistry of cytochrome P450 and related model systems have been extensively reviewed (14–18).
Figure 1.

Iron(III) protoporphyrin IX with a cysteinate as the axial ligand (1), which is typical of cytochrome P450, CPO, and NOS enzymes. The active oxygen species of these proteins and related heme enzymes is an oxoiron(IV)porphyrin cation radical (2), often called compound I.
Our understanding of the mechanism of action of these heme proteins comes from the direct observation of intermediates in the catalytic cycle through a variety of spectroscopic techniques, the use of diagnostic substrates with mechanistically revealing rearrangements during oxidation, and the parallel development of the chemistry of synthetic metalloporphyrins. The principal features of the consensus mechanism of cytochrome P450 (19) are as outlined in Scheme S1: binding of substrate to the enzyme, sometimes accompanied by a spin-state change of the iron, to afford an enzyme-substrate adduct 3; reduction of the ferric cytochrome P450 by an associated reductase with an NADPH-derived electron to the ferrous cytochrome P450 4; binding of molecular oxygen to the ferrous heme to produce a ferrous cytochrome P450-dioxygen complex 5, similar to the situation in oxymyoglobin; a second one-electron reduction and protonation to arrive at the Fe(III)-hydroperoxy complex 6; protonation and heterolytic cleavage of the O—O bond in 6 with concurrent production of a water molecule to form a reactive iron-oxo intermediate 7; and, finally, oxygen atom transfer from this iron-oxo complex 7 to the bound substrate to form the oxygenated product complex 8. Product dissociation completes the cycle.
Scheme 1.

Consensus catalytic cycle for oxygen activation and transfer by cytochrome P450.
There were a number of important realizations in the course of elucidating this mechanism. That hydrogen peroxide, alkyl hydroperoxides, periodate, and iodosylbenzene were also functional with cytochrome P450 suggested that the chemistry of “oxygen activation” was the two-electron reduction of molecular oxygen to hydrogen peroxide and that, in analogy to the peroxidases, the active oxygen species was a ferryl (or oxene) complex Fe⩵O, formally iron(V). It was shown that a synthetic oxoiron(IV)porphyrin cation radical species could be formed at low temperature by the oxidation of an iron(III) precursor with peroxyacids (9 → 10) (20). Intermediate 10 did have the requisite reactivity to transfer an oxygen atom to hydrocarbon substrates. It is this oxygen atom transfer from the oxygen donor to form the Fe⩵O intermediate 7 and the subsequent oxygen transfer to form the substrate complex 8 that has been termed oxygen rebound. Such an iron-oxo species (compound I) has been observed for the CPO of C. fumago (21), but the active species of cytochrome P450 has remained elusive. Very recently, it has been shown that an intermediate with the spectral properties similar to those of CPO compound I and the model iron porphyrin systems is formed upon the oxidation of Cyp119, a thermostable cytochrome P450, with a peroxyacid, analogous to the model systems (22). Consistent with the high reactivity expected for P450 compound I, this intermediate decayed with a rate constant of 29 s−1 at 4°C. Interestingly, similar experiments with P450cam, the camphor oxidizing enzyme from Pseudomonas putida, resulted in an iron(IV)-protein tyrosine radical species, presumably via a one-electron oxidation of Tyr-96, which is only 9.4 Å from the iron center (21).
There has been much discussion in the field about the oxygen transfer process 6 → 7 → 8. Specifically, the hydroperoxy iron(III) complex 6 has also been suggested to effect substrate oxygenations based on observed changes in product ratios and loss of hydrogen peroxide (uncoupling) upon P450 active site mutations (23). An important recent advance has been the development of cryo-spectroscopic studies by Hoffman and colleagues (24) that have allowed the step-wise interrogation of intermediates depicted in Scheme S1. Thus, the injection of an electron into complex 5 via γ-radiation, followed by thermal annealing of the sample has produced EPR and electron nuclear double resonance evidence for the formation of first a hydrogen-bonded iron-peroxo species and then the iron–hydroperoxo complex 6. Although no ferryl intermediate 7 was observed, the product alcohol was found to be formed with its oxygen atom coordinated to the iron center and with the substrate-derived proton attached to the product alcohol as depicted in structure 8 (Scheme S1). This arrangement has important mechanistic implications because, if a ferryl species (7) is the immediate precursor of the product complex 8, then coordination of the product oxygen would be a necessary consequence. By contrast, if the hydroperoxo species 6 were the source of the electrophilic oxygen, then water would be coordinated to iron rather than the product alcohol. A product complex such as 8 could also be the source of cationic rearrangement products that are sometimes observed during P450 oxygenations.
Significant recent advances in computational approaches to the study of biological catalysis, and the applications of these techniques to the cytochrome P450 mechanism have also been illuminating. Thus, Shaik and colleagues (25) have presented the results of a density functional theory analysis of the reactivity of hydroperoxy iron(III) complexes such as 6. The protonation and heterolytic O—O bond cleavage of 6 to afford a ferryl species analogous to 7 was found to proceed with almost no energetic barrier, in accord with earlier experimental results for the oxidation of an iron(III) porphyrin 9 to an oxoiron(IV)porphyrin cation radical species 10 with a peroxyacid (26). Further, the oxygen transfer from 7 to ethylene to form an epoxide proceeded with only a low barrier. It was concluded that the density functional theory (DFT) calculations exclude a hydroperoxyiron(III) intermediate such as 6 as a reactive, electrophilic oxidant. Several modes of oxygen transfer from the hydroperoxide intermediate encountered exceedingly high barriers for reaction. The lowest energy of these was an interaction of the substrate ethylene with the proximal, iron-bound oxygen of the Fe(III)-OOH ensemble .
Nucleophilic reactions of a hydroperoxy iron(III) intermediate 6, as have been suggested by Akhtar et al. (27), Cole and Robinson (28), and Vaz et al. (29) for the deformylation reactions characteristic of the P450 aromatase, do seem to be suggested by the significant basicity of the distal, hydroxylic oxygen found in the calculations for the Fe(III)–OOH group. This mode of reactivity is highly analogous to the reactions of enzymes such as cyclohexanone monooxygenase that proceed through a flavin 4a-hydroperoxide (30). Here, only electron-deficient olefins react to afford epoxides even thought the flavin hydroperoxide is 2 × 105 times more reactive than a simple alkyl hydroperoxide (31).
The hydroxylation of a C—H bond does seem to require the full formation of a reactive ferryl intermediate as in 11. This applies both for the reductive activation of dioxygen and the very revealing cases of alkyl hydroperoxide isomerization catalyzed by P450 in which the alkyl group (R) occupies the substrate-binding cavity (32, 33). For P450s in which the proton relay system has been disrupted by active-site mutations, one would expect that particularly reactive substrates could interact with the proximal oxygen earlier in this reaction profile as shown in 12. Although similar atomic trajectories and electronic charge redistributions are followed in each case, the former (11) is analogous to the SN1 reaction in organic chemistry, generating a discreet ferryl intermediate, whereas the latter (12) is SN2-like, requiring assistance from the electron-rich substrate. Indeed, in a recent report by Jin et al. (34), mutation of the conserved active site threonine-252 to alanine in P450CAM was shown to disable camphor hydroxylation while maintaining some reactivity for more reactive olefinic substrates. Similarly, two reactive intermediates, as suggested by Volz et al. (35) for the reactions of a thioether substrate, and also for model porphyrin systems described by Nam et al. (36), could reasonably derive from a mechanistic spectrum of this type. An important precedent for this behavior is seen in the reactions of peroxyacids with model Fe(III) porphyrins. Thus, Machii, Watanabe, and Morishima (37, 38) have shown that the iron-coordinated peroxyacid 9 reacted with olefins at the iron-coordinated oxygen atom in nonpolar solvents to give epoxides but would not react with saturated hydrocarbons. By contrast, the same oxoiron(IV)porphyrin cation radical, 10, was formed with a variety of peroxyacids in more polar media. This effect is also seen in model compounds with a thiolate ligand to iron (39). The protein-derived hydrogen bonds to the axial thiolate ligand to iron in P450cam have been shown to affect the O—O bond cleavage (40).
Mechanisms and Molecular Trajectories for Hydroxylation by Cytochrome P450
Among all of the varied reactions mediated by cytochrome P450 none has captured the imagination of chemists more than the hydroxylation of saturated carbon centers. Metal-oxo reagents such as chromates and permanganate can perform reactions of this type but are notoriously nonselective and must be used under forcing conditions. The selective hydroxylation of hydrocarbons remains one of the grand challenges for the chemical catalysis community. How can a protein create an iron intermediate reactive enough to hydroxylate even as inert a substrate as cyclohexane and not oxidize the relatively fragile protein superstructure? What is the electronic structure of that intermediate and what are the molecular pathways for oxygen insertion into a C—H bond? Without clear answers to these questions, the chemical catalysis performed by these metalloenzymes will remain an enigma and our attempts to draw conclusions will be without physical meaning. Without knowledge of the mechanism we learn nothing of predictive value that could be applied to other systems such as the rational design of enzyme inhibitors or the development of enzymatically inspired catalysts.
Presented in Scheme S2 is the range of mechanisms that have been considered as likely candidates for the cytochrome P450-catalyzed hydroxylation of hydrocarbons and those of model iron, manganese, and ruthenium porphyrins. For P450 and model iron and manganese systems, very large hydrogen/deuterium isotope effects have been observed. Thus, a linear, homolytic transition state, as in intermediate H, best fits the available data. Indeed, extensive similarities to the hydrogen abstraction observed by cytochrome P450 and a t-butoxy radical have been presented by Manchester et al. (41) in support of this view. A nonconcerted pathway for C—H bond cleavage is strongly supported by the observations of a variety of molecular rearrangements that are known to accompany P450-mediated hydroxylation. Initially it was clear that the kinds of rearrangements observed were consistent with the formation of a caged substrate radical at the heme active site. The intermediate radical could be trapped in a subsequent step. Both P450 enzymes and model systems showed a nonstereospecificity for the hydrogen removal step from norbornane or camphor substrates. Such a process was counterindicative of a cationic pathway to explain the observed rearrangements. The results rule out freely diffusing radicals, but a short-lived substrate radical would explain the observed results.
Scheme 2.

Pathways for oxygen atom transfer from the active ferryl species 7 of heme-thiolate enzymes such as cytochrome P450 to form the product alcohol coordinated to the ferric, resting form of the protein (8).
It was shown by Ortiz de Montellano and Stearns (42) that bicyclo[2.1.0]pentane was oxidized by rat liver microsomes to a 7:1 mixture of endo-2-hydroxybicyclo[2.1.0]pentane and 3-cyclopenten-1-ol, consistent with a radical ring-opening reaction. Applications of the “radical clock” method by Atkinson and Ingold (43) and Newcomb and Toy (44) began to measure the lifetime of the suspected radical cage intermediate. The rate constant for the rearrangement of bicyclo[2.1.0]pent-2-yl radical to 3-cyclopenten-1-yl radical was determined to be 2.4 × 109 s−1 at room temperature by using laser flash photolysis techniques (45). Thus, a rate constant of kOH = 1.7 × 1010 M−1⋅s−1 was estimated for the rebound process. Radical clocks with very fast rearrangement times were shown to produce less rearrangement than slower clocks in the P450-mediated hydroxylations, however. The results led Newcomb et al. (46) to question whether a radical pathway existed because the apparent lifetimes revealed by these probes were in the range of 100 fs, too short to represent a bona fide intermediate. Thus, either there was something wrong with the clocks or there were unrecognized subtleties in radical rebound mechanism. Several suggestions have been considered to resolve this dilemma and the question is still an area of active experiment and debate. As shown in Scheme S2, the transition state for hydrogen abstraction will position the active oxygen only a few tenths of an Å farther from the hydroxylated carbon atom than the transition state for the ultimate C—O bond formation. Thus, the extent of radical rearrangement might be expected to depend critically on the tightness of the radical cage and the ensemble of steric and electronic forces experienced by the incipient radical within the cage. Even the molecular makeup of the active site will depend on how the substrate fills the site, leaving room for movement of amino acid side chains in the vicinity of the substrate or allowing additional water molecules into the active-site area. The extent of rearrangement detected by a particular probe may simply reflect a facile molecular trajectory from the hydrogen abstraction transition state to the hydroxylation transition state in this variable environment. For substrates with a very strong C—H bond and a small steric size, both effects would push the reaction coordinate toward a tighter radical cage. Indeed, it has been shown that the effective lifetime of a radical intermediate can even be affected by the stereochemistry of the hydrogen abstraction event (19). Here, the pro-R hydrogen of ethyl benzene was hydroxylated with nearly complete retention of configuration at carbon whereas the pro-S hydrogen underwent significant racemization.
For a reaction that involves a paramagnetic iron-oxo intermediate and proceeds to produce paramagnetic radical intermediates, it is likely that spin-orbit coupling effects and the spin states of reacting intermediates may offer another significant consideration (47). Schwarz et al. (48) first suggested that the unusually slow reaction of FeO+ with hydrogen in the gas phase was caused by spin conservation effects that were imposed on these intermolecular encounters. Detailed density functional theory calculations on this simplest iron-oxo electrophile showed that there was a spin-state crossover during the H—H bond cleavage step to form a species H-Fe-OH+, and another spin crossover leading to the product Fe(OH2)+. Thus, the lowest energy pathway for reaction involved crossing from an initial high-spin, sextet state for the oxidant FeO+ to a low-spin quartet state near the transition state for H—H bond cleavage. Although such effects are common for first-row elements as, for example, with singlet and triplet carbenes, “spin forbidenness” has usually been discounted for reactions involving transition metals. However, the successful application of DFT calculations to explain the unusual behavior of FeO+ suggests that these effects may be significant in the area of oxidative catalysis.
Shaik and colleagues (49) have applied these considerations to examine interactions of a prototype substrate, methane, with a ferryl intermediate similar to 7 to probe this chemistry of P450. The results, as shown in Fig. 2, are very revealing. The ferryl intermediate was shown to have two nearly isoenergetic electron configurations, doublet and quartet, depending on whether the unpaired electron in the porphyrin cation radical is ferromagnetically or antiferromagnetically coupled to the triplet ferryl center. Indeed, both situations are known in enzymatic compounds I and model systems. The calculations indicate that the transition state for C—H bond cleavage does look like the extended arrangement H in Scheme S2. Here, however, the molecular trajectories for the high-spin and low-spin reaction coordinates diverge. For the high-spin pathway, there was a discernable intermediate caged radical state with the carbon center interacting weakly with the iron-hydroxide. A significant energy barrier was found for collapse of this high-spin intermediate to product via formation of a C—O bond. By contrast, the low-spin trajectory could proceed to products without encountering this barrier. This two-state hypothesis could provide a way out of the mechanistic dilemma presented by the radical clock results because the apparent timing of the clocks would depend on the relative importance of the high- and low-spin pathways.
Figure 2.

Energy level diagram and reaction coordinate for the hydroxylation of methane by a ferryl-porphyrin cation radical (7). This figure is adapted from ref. 47.
Is it possible to support this hypothesis by experiment? There is suggestive data in hand in the comparative behavior of P450 and a ruthenium porphyrin model system. Thus, evidence for short-lived substrate radicals has been presented recently for the oxidation of the mechanistically diagnostic probe molecule norcarane by cytochrome P450 (50). An alternate interpretation of similar data, involving unusual behavior of the probe molecule at the active site, has also been presented (51). In all known cases of reactions involving a radical intermediate, this norcarane probe produces a product derived from the 3-cyclohexenylmethyl radical, as the major rearrangement product. The rate constant for the radical rearrangement of the 2-norcaranyl radical has been found to be 2 × 108 s−1. By contrast, for reactions proceeding through discreet carbocations, rearrangement leads instead to 3-cycloheptenol as the major rearrangement product. The extent of observed rearrangement with a panel of P450 enzymes leads to a radical lifetime in the picosecond to nanosecond regime, certainly long enough to be considered an intermediate. A consistent timing was found for several similar probes. Smaller amounts of cation-derived products were also observed and attributed to a competing electron transfer oxidation of the incipient radical, a well precedented process. By contrast, the hydroxylation of norcarane with a ruthenium porphyrin catalyst that proceeds through a reactive oxoruthenium(V) porphyrin intermediate, afforded no detectable rearrangement. Density functional theory calculations on the ruthenium mediated hydroxylation show that the low-spin reaction trajectory is preferred throughout, in accord with general expectations for the behavior of second row transition metals (52). Thus, the data for the iron and ruthenium porphyrin systems is in accord with the predictions of theory that a radical rebound process is viable for iron, which has an accessible high-spin state but not for ruthenium that is always low-spin.
Other, more exotic factors such as nonstochastic behavior (53) and tunneling effects (54) could also be involved in causing the mistiming of events during C—H bond hydroxylation. Indeed, a carbene ring-expansion reaction was very recently found to have a large quantum tunneling effect that significantly affected the observed rate (55). High-level calculations indicated that a thermal, over-the-barrier, process and quantum tunneling of carbon were still competitive even at room temperature. Applied to C—H hydroxylation by a reactive oxidant, this situation could give the appearance of multiple oxidants and non-Arrhenius behavior. Thus, for a step-wise reaction via the caged radical intermediate in Scheme S2, a spectrum of apparent lifetimes, perhaps dependent on vibrational state, might be observed for rebound through transition state R to intermediate 8 (Scheme S2).
The nonheme diiron hydroxylases, such as methane monooxygenase (56) and AlkB, the ω-hydroxylase from P. putida, have also yielded to similar structural, spectroscopic, and mechanistic probes. Interestingly, there are striking similarities between the consensus mechanism for the heme and nonheme iron proteins. For methane monooxygenase (MMO) the resting enzyme has both iron centers in the ferric state. Reduction and binding of oxygen again produces a peroxo intermediate, which is oxidized to a reactive species, compound Q, that has been characterized as a bis-μ-oxo-iron(IV) intermediate. Both AlkB (57) and MMO (51, 58) have been interrogated recently with the diagnostic probe norcarane and both have shown the radical rearrangement product, hydroxymethylcyclohexene. For the histidine-rich hydroxylase AlkB, the results were particularly striking because fully 15% of the product was indicative of the radical rearrangement pathway. A significant aspect of this work was that it was performed on whole cells and clones into which the AlkB genes had been introduced. With MMO, it was possible to show that it was the reactive intermediate Q that was interacting with the substrate probe. Thus, mechanistically informative biochemistry can be obtained from this type of biological screen.
Iron in Supramolecular Assemblies
The self-assembled and highly ordered nature of phospholipid membranes is a central structural and functional motif in biology. Many of the iron-containing oxygenases, such as cytochrome P450s, mitochondrial NOS (59–67), and AlkB, are membrane bound. Synthetic phospholipid vesicles have presented the opportunity to construct novel supramolecular assemblies and elucidate the membrane-binding properties of biomolecules. For example, a synthetic multiheme assembly has been described that recruited cytochrome c to the outer surface of phospholipid vesicles (Fig. 3, ref. 68). The structure of the construct has been probed by observing electron transfer between cytochrome c and the redox centers embedded within a phospholipid bilayer at approximately known distances.
Figure 3.

Representation of a synthetic electron-transfer assembly in a vesicle bilayer, showing a membrane-spanning porphyrin, an amphiphilic zinc porphyrin recruiter, and an associated cytochrome c.
A very interesting class of iron-binding siderophores has amphiphilic properties, having a polar head group containing the iron binding site and one or two hydrophobic side chains reminiscent of a phospholipid. The first of these to be discovered were the exochelins and mycobactins of Mycobacter tuberculosis (69), rhizobactin 1021 from a terrestrial, nitrogen-fixing symbiont (70, 71), and acinetoferrin (72) from the pathogens Acinetobacter haemoliticus and Acinetobacter baumanii. The amphiphilic marinobactins and acquachelins have been discovered more recently in marine bacteria, (73), indicating that such structures are widely distributed in nature. Current interest in the iron-uptake strategies of pathogenic organisms stems from their increasing antibiotic resistance and the rising numbers of difficult-to-treat infections in humans (74, 75). Our interest in iron and membrane dynamics has led us to investigate how the amphiphilic nature of these compounds may be advantageous to the organisms.
The molecular architecture of Fe-mycobactin J, Fe-rhizobactin 1021, and Fe-marinobactin E, as depicted in Fig. 4, can be seen to be remarkably similar. Fig. 4 also depicts the dynamic interactions of marinobactin E and its iron complex with phospholipid vesicles that have recently been elucidated (76). Rates of iron chelation could be measured by stopped-flow spectroscopy by observing the UV absorption of the iron complex. The observed chelation rate decreased as the lipid concentration increased, indicating that nearly all of the siderophore molecules were associated with the vesicles and that the iron acquisition by free marinobactin (k1 in Fig. 4) is insignificant in comparison to the membrane-bound process k2. Iron acquisition by a membrane-associated siderophore would be one way to mitigate losses of the siderophore molecules caused by diffusion.
Figure 4.

Molecular models of the iron complexes of marinobactin E, mycobactin J, and rhizobactin 1021 showing the similarities in their amphiphilic structures. NMR data showing the line-broadening effect of Fe-marinobactin E on the choline methyl proton resonances. Proposed molecular mechanism of iron acquisition by marinobactin E (cf. ref. 76).
Because the iron(III) complex of marinobactin is paramagnetic, it was expected that binding of Fe-marinobactin to lipid membranes could be detected by NMR line-broadening techniques. The choline methyl groups of the inner and outer membrane leaflets of small unilamellar vesicles are spectroscopically distinguishable by proton NMR because of the membrane curvature. As shown in Fig. 4 (traces A and B), the addition of Fe-marinobactin E to a lipid vesicle suspension caused a dramatic broadening of the 1H-choline resonance corresponding to the outer membrane leaflet, whereas the inner membrane choline resonance was relatively unaffected, indicating rapid binding of the siderophore to the lipid phase.
Mixed vesicle experiments were designed to see whether Fe-marinobactin molecules could reversibly dissociate from the lipid. Here, Fe-marinobactin E was first bound to a suspension of small unilamellar vesicles composed of deuterated choline N-methyls. The NMR 1H-resonance of the choline N-methyl of proteo-dimyristoylphosphatidylcholine, but without the iron(III)-siderophore, was monitored and used as an indicator of the redistribution of the Fe-ME molecules. Broadening of the outer membrane leaflet choline resonance within 5 min indicated a fast redistribution of Fe-marinobactin E from one membrane population to the other with a dissociation rate, k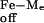 , of 4.4 × 10−3 s−1 (Fig. 4).
, of 4.4 × 10−3 s−1 (Fig. 4).
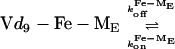 |
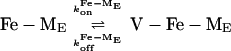 |
The lipid partition coefficient of Fe-marinobactin E was determined by this technique to be 2.3 × 102 M−1, ≈50 times smaller than that of the iron-free siderophore.
The interaction of the marinobactins and other amphiphilic siderophores with phospholipid vesicles is of interest for elucidating the molecular mechanisms involved in iron sequestration mediated by these unusual amphiphilic siderophores. The membrane-binding properties of marinobactin E are very similar to those of the lipopeptide, surfactin, a detergent-like peptide of similar overall constitution. As can be seen in Fig. 4, apo-marinobactin E has an extended molecular shape that folds into a more compact, spherical head group arrangement upon iron binding. Further, two of the iron-binding ligands are immediately adjacent to the point of attachment of the hydrophobic fatty acid appendage. This arrangement is a persistent feature in the small, but growing, family of amphiphilic siderophores from all sources. It is tempting to suggest a functional relationship wherein the membrane association is affected by iron binding to facilitate the receptor-mediated uptake process (77). The strong membrane affinity of marinobactin E would facilitate its retention by the bacterial cell or colony and protect against diffusive loss whereas the weaker binding of the iron complex would facilitate diffusive sharing of the iron resources among the cells in the colony (76).
The amphiphilic marine aquachelins have recently been shown by Butler and colleagues (78) to be photoreactive. The iron-hydroxamate chromophore absorbs visible light and the resulting ligand-to-metal charge-transfer chemistry causes an internal reduction of the iron(III) to iron(II) and oxidation of the organic portion of the siderophore. Significantly, this photochemical redox transformation causes a cleavage of the molecule such that the hydrophobic side chain and two of the iron-binding ligands are lost. This could have important implications for how iron is recycled in a marine environment that is critically short of iron near the surface.
Supplementary Material
Figure.

Structures 9–12.
Acknowledgments
Special thanks are due to all of the members of my research group and my collaborators who participated in these projects and contributed so many ideas, inspirations, and insights. Their names are indicated in the referenced papers. Research described here from our laboratory was supported by the National Institutes of Health (National Institute of General Medical Sciences) and the National Science Foundation. I also thank the National Science Foundation and the U.S. Department of Energy for their support of the Environmental Molecular Science Institute (the Center for Environmental Bioinorganic Chemistry) at Princeton University.
References
- 1.Hayaishi O, Katagiri M, Rothberg S. J Am Chem Soc. 1955;77:5450–5451. [Google Scholar]
- 2.Tchen T T, Block K. J Am Chem Soc. 1956;78:1516–1517. [Google Scholar]
- 3.Ortiz de Montellano P R, De Voss J J. Nat Prod Rep. 2002;19:477–493. doi: 10.1039/b101297p. [DOI] [PubMed] [Google Scholar]
- 4.Sundaramoorty M, Terner J, Poulos T L. Structure (London) 1995;3:1367–1377. doi: 10.1016/s0969-2126(01)00274-x. [DOI] [PubMed] [Google Scholar]
- 5.Manoj K M, Hager L P. Biochim Biophys Acta. 2001;1547:408–417. doi: 10.1016/s0167-4838(01)00210-2. [DOI] [PubMed] [Google Scholar]
- 6.Groves J T, Wang C C-Y. Curr Opin Chem Biol. 2000;4:687–695. doi: 10.1016/s1367-5931(00)00146-0. [DOI] [PubMed] [Google Scholar]
- 7.Santhanam L, Dordick J S. Biocatal Biotransform. 2002;20:265–274. [Google Scholar]
- 8.Rantwijk F, Sheldon R A. Curr Opin Biotechnol. 2000;11:554–564. doi: 10.1016/s0958-1669(00)00143-9. [DOI] [PubMed] [Google Scholar]
- 9.Van Beilen J B, Li Z. Curr Opin Biotechnol. 2002;13:338–344. doi: 10.1016/s0958-1669(02)00334-8. [DOI] [PubMed] [Google Scholar]
- 10.Glieder A, Farinas E T, Arnold F H. Nat Biotechnol. 2002;20:1135–1139. doi: 10.1038/nbt744. [DOI] [PubMed] [Google Scholar]
- 11.Crane B R, Arvai A S, Ghosh S, Getzoff E D, Stuehr D J, Tainer J A. Biochemistry. 2000;39:4608–4621. doi: 10.1021/bi992409a. [DOI] [PubMed] [Google Scholar]
- 12.Raman C S, Li H Y, Martasek P, Southan G, Masters B S S, Poulos T L. Biochemistry. 2001;40:13448–13455. doi: 10.1021/bi010957u. [DOI] [PubMed] [Google Scholar]
- 13.Li H Y, Raman C S, Martasek P, Masters B S S, Poulos T L. Biochemistry. 2001;40:5399–5406. doi: 10.1021/bi002658v. [DOI] [PubMed] [Google Scholar]
- 14.Fujii H. Coord Chem Rev. 2002;226:51–60. [Google Scholar]
- 15.Woggon W-D, Wagenknecht H-A, Claude C. J Inorg Biochem. 2001;83:289–300. doi: 10.1016/s0162-0134(00)00175-6. [DOI] [PubMed] [Google Scholar]
- 16.Ortiz de Montellano P R E. Cytochrome P-450: Structure, Mechanism, and Biochemistry. 2nd Ed. New York: Plenum; 1995. [Google Scholar]
- 17.Guengerich F P. Chem Res Toxicol. 2001;14:611–650. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- 18.Meunier B, Bernadou J. Top Catalysis. 2002;21:47–54. [Google Scholar]
- 19.McLain J, Lee J, Groves J T. In: Biomimetic Oxidations. Meunier B, editor. London: Imperial College Press; 1999. pp. 91–170. [Google Scholar]
- 20.Groves J T, Haushalter R C, Nakamura M, Nemo T E, Evans B J. J Am Chem Soc. 1981;102:2884–2886. [Google Scholar]
- 21.Schunemann V, Jung C, Terner J, Trautwein A X, Weiss R. J Inorg Biochem. 2002;91:586–596. doi: 10.1016/s0162-0134(02)00476-2. [DOI] [PubMed] [Google Scholar]
- 22.Kellner D G, Hung S-C, Weiss K E, Sligar S G. J Biol Chem. 2002;277:9641–9644. doi: 10.1074/jbc.C100745200. [DOI] [PubMed] [Google Scholar]
- 23.Vaz A D N, McGinnity D F, Coon M J. Proc Natl Acad Sci USA. 1998;95:3555–3560. doi: 10.1073/pnas.95.7.3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davydov R, Markis T M, Kofman V, Werst D E, Sligar S G, Hoffman B M. J Am Chem Soc. 2001;123:1413–1415. doi: 10.1021/ja003583l. [DOI] [PubMed] [Google Scholar]
- 25.Ogliaro F, de Visser S P, Cohen S, Sharma P K, Shaik S. J Am Chem Soc. 2002;124:2806–2817. doi: 10.1021/ja0171963. [DOI] [PubMed] [Google Scholar]
- 26.Groves J T, Watanabe Y. J Am Chem Soc. 1988;110:8443–8452. [Google Scholar]
- 27.Akhtar M, Corina D, Miller S, Shyadehi A Z, Wright J N. Biochemistry. 1994;33:4410–4418. doi: 10.1021/bi00180a039. [DOI] [PubMed] [Google Scholar]
- 28.Cole P A, Robinson C H. J Am Chem Soc. 1988;110:1284–1285. [Google Scholar]
- 29.Vaz A D N, Roberts E S, Coon M J. J Am Chem Soc. 1991;113:5886–5887. [Google Scholar]
- 30.Sheng D, Ballou D P, Massey V. Biochemistry. 2001;40:11156–11167. doi: 10.1021/bi011153h. [DOI] [PubMed] [Google Scholar]
- 31.Colonna S, Gaggero N, Carrea G, Ottolina G, Pasta P, Zambianchi F. Tetrahedron Lett. 2002;43:1797–1799. [Google Scholar]
- 32.Fish K M, Avaria G E, Groves J T. In: Microsomes and Drug Oxidations. Miners J, Birkett D J, Dew R, May B K, McManus M E, editors. New York: Taylor and Francis; 1988. pp. 176–183. [Google Scholar]
- 33.Kupfer R, Liu S Y, Allentoff A J, Thompson J A. Biochemistry. 2001;40:11490–11501. doi: 10.1021/bi010914d. [DOI] [PubMed] [Google Scholar]
- 34. Jin, S., Makris, T. M., Bryson, T. A., Sligar, S. G. & Dawson, J. H. (2003) J. Am. Chem. Soc., 10.1021/ja 029272n. [DOI] [PubMed]
- 35.Volz T J, Rock D A, Jones J P. J Am Chem Soc. 2002;124:9724–9725. doi: 10.1021/ja026699l. [DOI] [PubMed] [Google Scholar]
- 36.Nam W, Jin S W, Lim M H, Ryu J Y, Kim C. Inorg Chem. 2002;41:3647–3652. doi: 10.1021/ic011145p. [DOI] [PubMed] [Google Scholar]
- 37.Machii K, Watanabe Y, Morishima I. J Am Chem Soc. 1995;117:6691–6697. [Google Scholar]
- 38.Watanabe Y. J Biol Inorg Chem. 2001;6:846–856. doi: 10.1007/s007750100278. [DOI] [PubMed] [Google Scholar]
- 39.Suzuki N, Higuchi T, Nagano T. J Am Chem Soc. 2002;124:9622–9628. doi: 10.1021/ja0115013. [DOI] [PubMed] [Google Scholar]
- 40.Yoshioka S, Tosha T, Takahashi S, Ishimori K, Hori H, Morishima I. J Am Chem Soc. 2002;124:14571–14579. doi: 10.1021/ja0265409. [DOI] [PubMed] [Google Scholar]
- 41.Manchester J I, Dinnocenzo J P, Higgins L A, Jones J P. J Am Chem Soc. 1997;119:5069–5070. [Google Scholar]
- 42.Ortiz de Montellano P R, Stearns R A. J Am Chem Soc. 1987;109:3415–3420. [Google Scholar]
- 43.Atkinson J K, Ingold K U. Biochemistry. 1993;32:9209–9214. doi: 10.1021/bi00086a028. [DOI] [PubMed] [Google Scholar]
- 44.Newcomb M, Toy P H. Acc Chem Res. 2000;33:449–455. doi: 10.1021/ar960058b. [DOI] [PubMed] [Google Scholar]
- 45.Bowry V W, Ingold K U. J Am Chem Soc. 1991;113:5699–5707. [Google Scholar]
- 46.Newcomb M, Shen R, Choi S-Y, Toy P T, Hollenberg P F, Vaz A D N, Coon M J. J Am Chem Soc. 2000;122:2677–2686. [Google Scholar]
- 47.Shaik S, de Visser S P, Ogliaro F, Schwarz H, Schröder D. Curr Opin Chem Biol. 2002;6:556–567. doi: 10.1016/s1367-5931(02)00363-0. [DOI] [PubMed] [Google Scholar]
- 48.Schröder D, Fiedler A, Ryan M F, Schwarz H. J Phys Chem. 1994;98:68–70. [Google Scholar]
- 49.Schoneboom J C, Lin H, Reuter N, Thiel W, Cohen S, Ogliaro F, Shaik S. J Am Chem Soc. 2002;124:8142–8151. doi: 10.1021/ja026279w. [DOI] [PubMed] [Google Scholar]
- 50.Auclaire K, Hu Z, Little D M, Ortiz de Montellano P R, Groves J T. J Am Chem Soc. 2002;124:6020–6027. doi: 10.1021/ja025608h. [DOI] [PubMed] [Google Scholar]
- 51.Newcomb M, Shen R, Lu Y, Coon M J, Hollenberg P F, Kopp D A, Lippard S J. J Am Chem Soc. 2002;124:6879–6886. doi: 10.1021/ja017858o. [DOI] [PubMed] [Google Scholar]
- 52.Ogliaro F, de Visser S P, Groves J T, Shaik S. Angew Chem Int Ed. 2001;40:2874–2878. doi: 10.1002/1521-3773(20010803)40:15<2874::AID-ANIE2874>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 53.Reyes M B, Carpenter B K. J Am Chem Soc. 1998;120:1641–1642. [Google Scholar]
- 54.McMahon R J. Science. 2003;299:833–834. doi: 10.1126/science.1080715. [DOI] [PubMed] [Google Scholar]
- 55.Zuev P S, Sheridan R S, Albu T V, Truhlar D G, Hrovat D A, Borden W T. Science. 2003;299:867–870. doi: 10.1126/science.1079294. [DOI] [PubMed] [Google Scholar]
- 56.Kopp D A, Lippard S J. Curr Opin Chem Biol. 2002;6:568–576. doi: 10.1016/s1367-5931(02)00366-6. [DOI] [PubMed] [Google Scholar]
- 57.Austin R N, Chang H-K, Zylstra G J, Groves J T. J Am Chem Soc. 2000;122:11747–11748. [Google Scholar]
- 58.Brazeau B J, Austin R N, Tarr C, Groves J T, Lipscomb J D. J Am Chem Soc. 2001;123:11831–11837. doi: 10.1021/ja016376+. [DOI] [PubMed] [Google Scholar]
- 59.Wei C C, Wang Z-Q, Meade A L, McDonald J F, Stuehr D J. J Inorg Biochem. 2002;91:618–624. doi: 10.1016/s0162-0134(02)00432-4. [DOI] [PubMed] [Google Scholar]
- 60.Wei C C, Wang Z Q, Wang Q, Meade A L, Hemann C, Hille R, Stuehr D J. J Biol Chem. 2001;276:315–319. doi: 10.1074/jbc.M008441200. [DOI] [PubMed] [Google Scholar]
- 61.Hurshman A R, Krebs C, Edmondson D E, Huynh B H, Marletta M A. Biochemistry. 1999;38:15689–15696. doi: 10.1021/bi992026c. [DOI] [PubMed] [Google Scholar]
- 62.Hurshman A R, Marletta M A. Biochemistry. 2002;41:3439–3456. doi: 10.1021/bi012002h. [DOI] [PubMed] [Google Scholar]
- 63.Rosen G M, Tsai P, Pou S. Chem Rev. 2002;102:1191–1199. doi: 10.1021/cr010187s. [DOI] [PubMed] [Google Scholar]
- 64.Blasko E, Glaser C B, Devlin J J, Xia W, Feldman R I, Polokoff M A, Phillips G B, Whitlow M, Auld D S, McMillan K, et al. J Biol Chem. 2002;277:295–302. doi: 10.1074/jbc.M105691200. [DOI] [PubMed] [Google Scholar]
- 65.Davydov R, Ledbetter-Rogers A, Martasek P, Larukhin M, Sono M, Dawson J H, Masters B S S, Hoffman B M. Biochemistry. 2002;41:10375–10381. doi: 10.1021/bi0260637. [DOI] [PubMed] [Google Scholar]
- 66.Huang H, Hah J M, Silverman R B. J Am Chem Soc. 2001;123:2674–2676. doi: 10.1021/ja005900u. [DOI] [PubMed] [Google Scholar]
- 67.Li H, Shimizu H, Flinspach M, Jamal J, Yang W, Xian M, Cai T, Wen E Z, Jia Q, Wang P G, Poulos T L. Biochemistry. 2002;41:13868–13875. doi: 10.1021/bi020417c. [DOI] [PubMed] [Google Scholar]
- 68.Lahiri J, Fate G D, Ungashe S B, Groves J T. J Am Chem Soc. 1996;118:2347–2358. [Google Scholar]
- 69.Snow G A. Bacteriol Rev. 1970;34:99–125. doi: 10.1128/br.34.2.99-125.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Persmark M, Pittman P, Buyer J S, Schwyn S, Gill P R, Neilands J B. J Am Chem Soc. 1993;115:3950–3956. [Google Scholar]
- 71.Lynch D, O'Brien J, Welch T, Clarke P, Cuiv P O, Crosa J H, O'Connell M. J Bacteriol. 2001;183:2576–2585. doi: 10.1128/JB.183.8.2576-2585.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Okujo N, Sahahibara Y, Yoshida T, Yamamoto S. Biometals. 1994;7:170–176. doi: 10.1007/BF00140488. [DOI] [PubMed] [Google Scholar]
- 73.Martinez J S, Zhang G P, Holt P D, Jung H-T, Carrano C J, Haygood M G, Butler A. Science. 2000;287:1245–1247. doi: 10.1126/science.287.5456.1245. [DOI] [PubMed] [Google Scholar]
- 74.Horwitz L D, Sherman N A, Kong Y N, Pike A W, Gobin J, Fennessey P V, Horwitz M A. Proc Natl Acad Sci USA. 1998;95:5263–5268. doi: 10.1073/pnas.95.9.5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guo H, Naser S A, Ghobrial G, Phanstiel O, IV. J Med Chem. 2002;45:2056–2063. doi: 10.1021/jm0104522. [DOI] [PubMed] [Google Scholar]
- 76.Xu G, Martinez J S, Groves J T, Butler A. J Am Chem Soc. 2002;124:13408–13415. doi: 10.1021/ja026768w. [DOI] [PubMed] [Google Scholar]
- 77.Clarke T E, Braun V, Winkelmann G, Tari L W, Vogel H J. J Biol Chem. 2002;277:13966–13972. doi: 10.1074/jbc.M109385200. [DOI] [PubMed] [Google Scholar]
- 78.Barbeau K, Rue E L, Bruland K W, Butler A. Nature. 2001;413:409–413. doi: 10.1038/35096545. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}