

Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive degenerative disease of motor neurons. The inherited form of the disease, familial ALS, represents 5–10% of the total cases, and the best documented of these are due to lesions in SOD1, the gene encoding copper–zinc superoxide dismutase (CuZnSOD). The mechanism by which mutations in SOD1 cause familial ALS is currently unknown. Two hypotheses have dominated recent discussion of the toxicity of ALS mutant CuZnSOD proteins: the oligomerization hypothesis and the oxidative damage hypothesis. The oligomerization hypothesis maintains that mutant CuZnSOD proteins are, or become, misfolded and consequently oligomerize into increasingly high-molecular-weight species that ultimately lead to the death of motor neurons. The oxidative damage hypothesis maintains that ALS mutant CuZnSOD proteins catalyze oxidative reactions that damage substrates critical for viability of the affected cells. This perspective reviews some of the properties of both wild-type and mutant CuZnSOD proteins, suggests how these properties may be relevant to these two hypotheses, and proposes that these two hypotheses are not necessarily mutually exclusive.
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by the relentless degeneration of both upper and lower motor neurons (1, 2). Similar to other common neurodegenerative diseases, e.g., Alzheimer's disease, Parkinson's disease, Huntington's disease, and transmissible spongiform encephalopathies, ALS is characterized by mid-to-late-life onset, selective neuronal death, and the formation of protein deposits in affected neuronal tissues (3). Most cases (90–95%) of ALS are sporadic, i.e., occurring in individuals with no family history of ALS. The remaining cases are familial ALS (FALS), and the best documented of these are due to inherited mutations in SOD1, the gene encoding copper–zinc superoxide dismutase (CuZnSOD) (4, 5).
CuZnSOD, the product of the SOD1 gene, is a major antioxidant enzyme located predominantly in the cytosol, nucleus, and mitochondrial intermembrane space of eukaryotic cells and in the periplasmic space of bacteria (6–8). The eukaryotic enzyme is a 32-kDa homodimer with a highly conserved amino acid sequence, and it contains one copper and one zinc-binding site, as well as a disulfide bond in each of its two subunits. It catalyzes the disproportionation of superoxide, yielding hydrogen peroxide and dioxygen, thus reducing steady-state levels of superoxide in its surroundings.
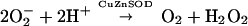 |
Superoxide, the substrate for CuZnSOD, is a potent, but chemically selective, reactive oxygen species produced in all aerobic cells. Sometimes it functions as a signaling agent (9), but it can also cause oxidative damage, particularly at excessive concentrations or when it reaches the wrong cellular locations. One of the best documented examples of superoxide-mediated damage occurs when it attacks and destroys labile iron–sulfur cluster cofactors of aconitase and other similar iron-containing proteins (10, 11).
Over 100 distinct SOD1 mutations (12) have been identified in FALS patients (most of them are listed at http://alsod.org). Most of the individual mutations result in substitution of one single amino acid by another; such substitutions have been identified at over one-third of the 153 amino acid residues of the wild-type CuZnSOD protein. In addition to the individual amino acid substitutions, there are also a smaller group of mutations resulting in amino acid deletions and truncations.
SOD1-associated FALS has been demonstrated to occur by a gain-of-function mechanism. Transgenic mice expressing the human ALS-mutant SOD1 transgene in addition to their normal mouse wild-type SOD1 gene develop a motor neuron disease similar to human ALS, whereas transgenic mice expressing the human wild-type SOD1 transgene in addition to their normal mouse wild-type SOD1 gene do not (13). In both cases, the transgenic mice have elevated levels of SOD activity relative to the nontransgenics, indicating that the disease is not caused by a lowering of SOD activity, but that the presence of the mutant CuZnSOD polypeptide itself is toxic to motor neurons (13–16).
Despite the apparent toxicity of superoxide, SOD1 knockout mice have a surprisingly mild phenotype. They develop relatively normally but display subtle motor symptoms by 6 months of age (17), as well as impaired fertility (18). They do not develop ALS, an observation that adds further support to the theory that ALS mutant CuZnSOD proteins are inherently toxic. Because the mitochondria are the main cellular source of superoxide, the mild phenotype of the SOD1 knockout mice could come from the fact that the gene encoding the mitochondrial manganese superoxide dismutase (SOD2) remains intact in these animals.
Oligomerization Versus Oxidative Damage
Two hypotheses (19, 20) have dominated recent discussion of the toxicity of ALS mutant CuZnSOD proteins: the oligomerization hypothesis and the oxidative damage hypothesis. The oligomerization hypothesis maintains (i) that mutant CuZnSOD proteins are or become misfolded and consequently oligomerize into increasingly high-molecular-weight species that ultimately aggregate and end up in proteinaceous inclusions and (ii) that the oligomerized or aggregated proteins are, at some stage in their formation, selectively toxic to motor neurons. A variation of the oligomerization hypothesis is that mutant CuZnSOD aggregates bind to other essential proteins, such as heat shock proteins (21) or mitochondrial electron transport proteins (see below), making them unavailable to perform their normal functions in the cell.
The oxidative damage hypothesis maintains that ALS mutant CuZnSOD proteins catalyze reactions with hydrogen peroxide or peroxynitrite that damage exogenous substrates critical for viability of the affected cells. A variation of the oxidation hypothesis is that mutant CuZnSOD proteins promote their own oxidative damage by similar mechanisms and that the oxidatively damaged mutant CuZnSOD molecules somehow cause the cells to die. The oxidative damage hypothesis would seem to require that copper (or some other redox active metal ion) be bound to the mutant CuZnSOD protein to promote the oxidation reaction.
Inclusions, Aggresomes, and Oligomers
Proteinaceous inclusions rich in mutant CuZnSOD protein, ubiquitin, and neurofilament proteins have been found in tissues from ALS patients (22–24) and ALS-SOD transgenic mice (25, 26). There is no direct evidence that their presence damages the cells in which they are formed (16, 27), and they appear to be formed relatively late in the disease in the ALS transgenic mice (28). In fact, there is good reason to believe that they are the ultimate product of a type of cellular defense mechanism that occurs generally in cells when the burden of misfolded or damaged proteins exceeds the capacity of the protein degradation machinery to eliminate them (29). In mammalian cells, this mechanism has been shown to involve transport of smaller protein aggregates along the microtubules to the microtubule organizing center, where they are incorporated into larger structures termed aggresomes. These cytoplasmic entities are structures in which aggregated, multiubiquitinated misfolded proteins are sequestered, where they await degradation by the proteasome or packaging into larger inclusion bodies (30, 31).
Although the larger, visible proteinaceous inclusions may not themselves be toxic, they are a sign that something is very wrong in the cell with respect to the stability or solubility of the mutant CuZnSOD proteins. High-molecular-weight protein complexes, rich in mutant CuZnSOD (32), that have features characteristic of aggresome formation (33) have been found in the ALS transgenic mice. These complexes of oligomerized mutant CuZnSOD are much smaller than the visible inclusions, and their formation occurs much earlier. Their presence suggests the possibility that soluble oligomers of mutant CuZnSOD are formed into aggresomes when their concentration exceeds the capacity of the proteasome to degrade them. These soluble oligomers may be the most toxic form of mutant CuZnSOD. Thus SOD1-associated ALS may be similar to other neurodegenerative diseases such as Alzheimer's disease, in which soluble protein oligomers increasingly are implicated as the pathogenic species, rather than the more visible fibrils or insoluble inclusions (29, 34, 35).
Both inclusion and aggresome formation appear to be part of a dynamic process, a finding of potential importance in designing therapeutic approaches to ALS and other neurodegenerative diseases (29). Inhibition of the proteasome accelerates formation of aggresomes (30), but decreasing the cellular burden of aggregating protein by turning off expression can cause visible cellular inclusions to disappear rapidly via a proteasomal pathway (36, 37). These findings suggest that the soluble oligomers are in equilibrium with the higher molecular weight complexes and aggresomes.
Mutant CuZnSOD proteins are degraded by the ubiquitin–proteasome pathway (38–42), and a progressive decline in proteasome function is observed in cell culture models in which mutant CuZnSODs are expressed (38). Involvement of the proteasome in the mechanism of mutant CuZnSOD-associated ALS could explain the late onset of the disease because proteasome function is known to decline with age (43).
Mitochondrial Dysfunction
Morphological mitochondrial abnormalities are some of the earliest signs of pathology in the ALS transgenic mice (44, 45), and the onset of disease is accompanied by a massive increase in the number of degenerating mitochondria (46). Inhibition of components of the mitochondrial electron transfer chain has been demonstrated in both a cell culture (43) and murine (44) models of the disease. Particularly striking in the latter case is the observation that the mitochondrial enzyme activities were progressively decreased in spinal cord, particularly in the ventral horn, but not at all decreased in the cerebellum and dorsal horn (47).
CuZnSOD protein has been shown to be abundant and enzymatically active in the mitochondrial intermembrane space in yeast (7), rat liver (8), and the ALS transgenic mice (48, 49). Inhibition of enzymes in the inner mitochondrial membrane in the ALS transgenic mice may be due to the presence of the mutant CuZnSOD in the intermembrane space, but the mechanism is unknown. Curiously, aggregated human CuZnSOD was not found in the intermembrane space but was found instead to be associated with the external side of the outer mitochondrial membrane mitochondrial membrane (48).
State of Metallation of Toxic Mutant CuZnSOD Proteins: Zinc
It has been proposed that the zinc-binding sites of the ALS mutant CuZnSOD proteins are compromised and that the proteins therefore have low affinities for zinc ions while retaining their high affinities for copper ions (50, 51). Our studies of ALS mutant CuZnSODs expressed in either yeast or baculovirus expression systems appear to contradict these findings, at least for the large number of mutants that we have termed “wild-type-like” (see below and refs. 52 and 53).
We find that the ALS mutant CuZnSOD proteins that we have studied tend to fall into two groups. We have termed the first of these groups wild-type-like because the mutant proteins are isolated from their expression systems with copper and zinc levels very similar to those found for the wild-type protein expressed under the same conditions, i.e., high in zinc but with variable levels of copper (52). These wild-type-like mutants, when expressed in sod1− yeast (yeast lacking their own gene for yeast CuZnSOD) rescue the oxygen-sensitive phenotype of the sod1− strains just as well as expression of the human wild-type CuZnSOD protein itself in these strains (54, 55). The SOD activities and the spectroscopic characteristics of this class of ALS mutant CuZnSOD proteins are virtually identical to those of wild-type CuZnSOD (52, 53). We find no evidence that they exist in zinc-deficient forms in these eukaryotic expression systems, in which they are biologically metallated.
We term the second class of ALS mutant CuZnSOD proteins “metal-binding-region” mutants. These include mutations in the metal-binding ligands themselves or in the electrostatic and zinc loop elements that are intimately associated with metal binding. Fig. 1 shows the positions of these mutations. The side chain of Asp-124 directly links the electrostatic and zinc loop elements and is particularly important in that it contributes to the stabilization of both the copper and zinc-binding sites by forming hydrogen bonds simultaneously to the nonliganding imidazole nitrogen atoms of copper ligand His-46 and zinc ligand His-71. Because of this delicate interplay between the electrostatic and zinc loop elements, it is perhaps not too surprising that these mutants are isolated from the expression systems with very low zinc but also with very low copper. This class of ALS mutant CuZnSOD proteins is very likely to exist in vivo in zinc- and copper-deficient forms. We therefore think it very unlikely that members of this class of mutants, with the possible exception of H48Q, catalyze oxidative damage reactions. Because the members of the metal-binding-region mutant class demonstrate such different characteristics relative to the wild-type-like mutant class, understanding how they initiate motor neuron death could provide significant insight into ALS etiology across the board (see below).
Figure 1.

Structure of a monomer of wild-type-like pathogenic SOD1 mutant G37R [PDB ID code 1AZV (69)]. The positions of the G37R and A4V mutations are indicated with red spheres. The copper and zinc ions are represented by blue and magenta spheres, respectively. The wild-type-like mutations (most of which are not shown) are found scattered throughout the β-barrel of the protein, shown in gold. The electrostatic loop is cyan and the zinc loop and metal-binding ligands are shown in black. The positions of the metal-binding-region mutations are represented by yellow spheres. The position of Cys-111 is represented by a green sphere. Asp-124, a residue termed the “secondary bridge,” links the electrostatic and zinc loops and the copper- and zinc-binding sites. The second subunit of the G37R homodimer is generated by a rotation of 180° around the axis indicated.
His-48 is a copper-binding ligand, and yet the ALS mutant H48Q is unlike the other metal-binding-region mutants. It is the only mutant we have characterized to date that has high affinity for both copper and zinc and yet has very low SOD activity (52). Spectroscopic characterization of this mutant indicates that the copper site does not have the same geometry as that of wild-type CuZnSOD (52). This mutant also appears to have a redox chemistry quite different from other copper-binding mutants (56).
State of Metallation of Toxic Mutant CuZnSOD Proteins: Copper
Copper chelators have been shown to slow the course of the disease in both the mouse and cell-culture models of SOD-associated ALS (57–60). Nevertheless, it has been proposed by Wang et al. (61) that the toxicity of the ALS mutant CuZnSOD proteins does not require the presence of copper bound at the copper site of the mutant protein. They generated transgenic mice with a double mutation combining two known ALS mutations in the metal-binding region, H48Q and H46R, and showed that they developed the disease. We absolutely agree that those ALS mutant proteins with inherently compromised metal-binding abilities, such as the class of metal-binding-region mutants described above, may cause ALS without any involvement of copper. The H48Q/H46R double mutant adds another example to this class of mutant ALS proteins, but it does not represent them all. In our opinion, there remains strong evidence implicating copper in the mechanism of toxicity of the mice carrying wild-type-like mutant CuZnSOD (see below).
Another relevant experiment is the demonstration that ALS transgenic mice lacking CCS, the copper chaperone for SOD, have significantly less EDTA-resistant copper-loaded mutant CuZnSOD protein and yet show the same progression of disease as the ALS mice with CCS (62). However, it is difficult to interpret these findings until the state of metallation of the mutant CuZnSOD by both copper and zinc in the various tissues is confirmed. If the mutant CuZnSOD proteins are not properly metallated with zinc in the CCS knockout mice, it is likely that EDTA will remove any copper.
It should also be noted that human CuZnSOD has a cysteine, Cys-111 (Fig. 1), that has been shown to be capable of binding copper ions in vitro in H46R CuZnSOD (63). Copper ions bound at this location to mutant CuZnSOD proteins could conceivably catalyze oxidative damage reactions in vivo, but it would not have been detected in the studies described above because it would not survive treatment with EDTA.
Are the Zinc-Binding Sites Defective in ALS Mutant CuZnSODs?
All of the metal-binding-region mutant CuZnSOD proteins that we have studied, except for H48Q, fail to be properly metallated with zinc in the yeast or insect cell expression systems, but the wild-type-like CuZnSOD mutants are properly metallated by zinc (52). We conclude that the affinity of the zinc site for zinc is not inherently compromised in these latter mutants, so long as the protein can achieve the correct conformation in the zinc site. Wild-type-like mutant CuZnSOD proteins appear to be properly metallated by zinc and copper in the two eukaryotic expression systems studied (52), and there is no evidence for altered conformations of the protein when this class of mutants is biologically metallated.
By contrast, in vitro metallation studies of isolated wild-type-like mutant CuZnSOD apoproteins, prepared by removal of the metal ions at low pH, suggest that the mutant apoproteins, under these conditions, unlike the wild type, readily convert to a misfolded form that will bind metal ions at the zinc site, but in an altered geometry. This is particularly evident from the visible absorption spectral titrations of the mutant apoproteins with either Co2+ or Cu2+, which show relatively high-affinity binding of those metal ions to the zinc sites, but in altered geometries based on the spectral characteristics of that metal ion (53, 64–66). The misfolding of the zinc site is probably responsible also for the loss of in vitro metal ion-binding specificity of many of the mutant proteins (53).
The altered conformation of the zinc site apparently leaves the copper site relatively unaffected. This conclusion can be seen most clearly in the case of the wild-type-like mutants G93A, A4V, and L38V, in which the apoproteins have been remetallated with Cu2+ ions in both their copper and zinc sites. These four-copper derivatives have SOD activities identical to wild type biologically metallated with copper and zinc, indicating that the copper ions in the copper sites are fully SOD active. It is highly likely that their modes of copper binding are identical to that of copper in the wild-type protein. By contrast, the visible spectral characteristics of these four-copper derivatives are quite different from each other and from the four-copper derivative of the wild-type protein (53). We conclude that the change in the spectrum is due to distinct differences in the geometries of the copper bound to the zinc sites of the different mutants.
Structures of Wild-Type and ALS Mutant CuZnSODs
Fig. 1 shows that each subunit of SOD1 possesses an eight-stranded Greek key β-barrel fold. However, not all of the strands forming the β-barrel are continuously hydrogen bonded to each other as is observed in “true” β-barrel proteins such as triose phosphate isomerase. For the purposes of this discussion, it is perhaps more useful to think of the SOD1 fold as a β-sandwich, where β-strands 5 and 6 are considered edge strands (Fig. 2). Edge strands are potentially dangerous for proteins because they provide a hydrogen-bonding surface that naturally accommodates interaction with other edge strands, which in turn can lead to the formation of higher-order structures such as those found in amyloid fibrils. As β-sheet-containing proteins evolve, they remain under constant selective pressure to protect their edge strands from such nonproductive associative interactions by using a wide variety of strategies (reviewed in ref. 67). As shown in Fig. 2, wild-type SOD1 makes its edge strands unavailable for self-association by sterically blocking such interactions with its well ordered electrostatic and zinc loop elements.
Figure 2.

Structure of a monomer of wild-type-like pathogenic SOD1 mutant G37R, rotated 180° relative to the view shown in Fig. 1. The electrostatic loop is shown in cyan and the zinc loop is shown in black. β-Strands 5 and 6, the edge strands of the SOD1 β-sandwich (see text), are shown in red. The zinc loop projects out of the plane of the paper toward the viewer such that when two SOD1 proteins encounter each other in solution, they are sterically hindered from participating in protein–protein interactions at the edge strands. This edge strand protection could be lost if the zinc loop loses its wild-type conformation or becomes mobile because of mutation, metal deficiency, or both. The second subunit of the G37R homodimer is generated by a 180° rotation around the axis indicated.
Given the assumption that ALS SOD1 oligomerization or aggregation is mediated through nonnative SOD1–SOD1 protein–protein interactions, it is difficult to envision how members of the wild-type-like class, when fully metallated, might be prone to participate in such nonproductive interactions. In this context, the crystal structures of wild-type-like pathogenic SOD1 mutants G37R and A4V have been determined (68, 69). Not surprisingly, both proteins are metal-loaded, and their overall structures, including the conformations of the electrostatic and zinc loops, are nearly indistinguishable from the wild-type protein (Fig. 1). Taken alone, these structures shed little insight into how mutant SOD1 oligomerization or aggregation might occur.
On the other hand, it is not too difficult to envision how members of the metal-binding-region mutant class of ALS SOD proteins might be prone to oligomerization or aggregation. Because metal binding plays an enormous role in stabilizing the conformations of the electrostatic and zinc loops, these metal-deficient proteins likely have significantly increased conformational mobility of these loop elements relative to the wild-type enzyme, permitting nonnative SOD1–SOD1 protein–protein interactions at or near the deprotected edge strands. Because SOD1 is a homodimer, such interactions can occur at either end of the molecule and propagate in two directions. Preliminary structural data gathered on several metal-deficient representatives of the metal-binding-region class of mutant SOD1 proteins reveal that the electrostatic and zinc loops do indeed become disordered, which in turn permits a gain-of-interaction between SOD1 molecules that leads to their incorporation into higher-order assemblies.
Oxidative Damage to CuZnSOD Occurs Predominantly in the Metal-Binding Region
There is considerable evidence indicating that elevated oxidative stress and elevated oxidative damage are present in the tissues of the ALS transgenic mice (reviewed in ref. 19). In the G93A mouse, one of the most heavily oxidized proteins identified was the mutant CuZnSOD itself (70).
Most oxidative damage to proteins in vivo is believed to occur by a site-specific, metal-mediated mechanism in which hydrogen peroxide reacts with a redox-active metal ion to generate a hydroxyl radical, ⋅OH, which immediately oxidizes an amino acid residue in close proximity to the redox metal ion-binding site (71). The reaction of hydrogen peroxide with CuZnSOD is an excellent example of such site-specific oxidative damage, which occurs in the metal-binding region and results in enzyme inactivation (72–74). Specific residues that have been reported to be oxidized by hydrogen peroxide are His-46, His-48, Pro-62, His-63, and His-120 (human numbering; refs. 75 and 76). In recent studies, we have discovered that the presence of bicarbonate ion significantly enhances the rate of inactivation of CuZnSOD by hydrogen peroxide. We therefore suggest here that the wild-type-like mutant CuZnSOD proteins may be oxidized in vivo by hydrogen peroxide via a bicarbonate-mediated, site-specific oxidation mechanism and that the product would be mutant CuZnSOD that has been specifically oxidized in the metal-binding region; we predict that such oxidatively damaged mutant CuZnSOD will have compromised metal-binding ability and, consequently, properties similar to those of the metal-binding-region mutants.
Conclusions
Oligomerization of ALS mutant CuZnSOD proteins appears to be important in explaining the toxicity of these proteins. The metal-binding-region mutant CuZnSOD proteins are poorly metallated in vivo with either copper or zinc and appear to be primed for aggregation with no further modification. By contrast, the wild-type-like mutants are metallated well in vivo and appear to be very similar to the wild-type protein in solubility. Site-specific oxidative damage to the metal-binding regions of these mutant proteins by reaction with hydrogen peroxide, a chemistry known to be enhanced in the wild-type-like mutant class, would compromise the ability of these mutants to bind metal ions and may convert them to forms that are much more prone to aggregation. In this way, the wild-type-like and the metal-binding-region classes of pathogenic SOD1 mutants may become fused into a single class of molecules that can oligomerize and wreak havoc in the cell.
Acknowledgments
We are grateful to Alex Taylor for help with the figures. This work was supported by National Institutes of Health Grants GM28222 (to J.S.V.) and NS39112 (to P.J.H.), the Robert A. Welch Foundation (P.J.H.), and the Amyotrophic Lateral Sclerosis Association (J.S.V. and P.J.H.).
References
- 1.Rowland L P, Shneider N A. N Engl J Med. 2001;344:1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- 2.Hand C K, Rouleau G A. Muscle Nerve. 2002;25:135–159. doi: 10.1002/mus.10001. [DOI] [PubMed] [Google Scholar]
- 3.Soto C. Nat Rev Neurosci. 2003;4:49–60. doi: 10.1038/nrn1007. [DOI] [PubMed] [Google Scholar]
- 4.Rosen D R, Siddique T, Patterson D, Figlewicz D A, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan J P, Deng H X, et al. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 5.Deng H X, Hentati A, Tainer J A, Iqbal Z, Cayabyab A, Hung W Y, Getzoff E D, Hu P, Herzfeldt B, Roos R P, et al. Science. 1993;261:1047–1051. doi: 10.1126/science.8351519. [DOI] [PubMed] [Google Scholar]
- 6.Lyons T J, Gralla E B, Valentine J S. Metal Ions Biol Syst. 1999;36:125–177. [PubMed] [Google Scholar]
- 7.Sturtz L A, Diekert K, Jensen L T, Lill R, Culotta V C. J Biol Chem. 2001;276:38084–38089. doi: 10.1074/jbc.M105296200. [DOI] [PubMed] [Google Scholar]
- 8.Okado-Matsumoto A, Fridovich I. J Biol Chem. 2001;276:38388–38393. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki Y J, Forman H J, Sevanian A. Free Radical Biol Med. 1997;22:269–285. doi: 10.1016/s0891-5849(96)00275-4. [DOI] [PubMed] [Google Scholar]
- 10.Valentine J S, Wertz D L, Lyons T J, Liou L L, Goto J J, Gralla E B. Curr Opin Chem Biol. 1998;2:253–262. doi: 10.1016/s1367-5931(98)80067-7. [DOI] [PubMed] [Google Scholar]
- 11.Liochev S I, Fridovich I. IUBMB Life. 1999;48:157–161. doi: 10.1080/713803492. [DOI] [PubMed] [Google Scholar]
- 12.Guegan C, Przedborski S. J Clin Invest. 2003;111:153–161. doi: 10.1172/JCI17610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gurney M E, Pu H, Chiu A Y, Dal Canto M C, Polchow C Y, Alexander D D, Caliendo J, Hentati A, Kwon Y W, Deng H X, et al. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 14.Gurney M E. J Neurol. 1997;244, Suppl. 2:S15–S20. doi: 10.1007/BF03160575. [DOI] [PubMed] [Google Scholar]
- 15.Gurney M E. BioEssays. 2000;22:297–304. doi: 10.1002/(SICI)1521-1878(200003)22:3<297::AID-BIES12>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 16.Shibata N. Neuropathology. 2001;21:82–92. doi: 10.1046/j.1440-1789.2001.00361.x. [DOI] [PubMed] [Google Scholar]
- 17.Shefner J M, Reaume A G, Flood D G, Scott R W, Kowall N W, Ferrante R J, Siwek D F, Upton-Rice M, Brown R H., Jr Neurology. 1999;53:1239–1246. doi: 10.1212/wnl.53.6.1239. [DOI] [PubMed] [Google Scholar]
- 18.Ho Y S, Gargano M, Cao J, Bronson R T, Heimler I, Hutz R J. J Biol Chem. 1998;273:7765–7769. doi: 10.1074/jbc.273.13.7765. [DOI] [PubMed] [Google Scholar]
- 19.Valentine J S. Free Radical Biol Med. 2002;33:1314–1320. doi: 10.1016/s0891-5849(02)01080-8. [DOI] [PubMed] [Google Scholar]
- 20.Cleveland D W, Rothstein J D. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- 21.Okado-Matsumoto A, Fridovich I. Proc Natl Acad Sci USA. 2002;99:9010–9014. doi: 10.1073/pnas.132260399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shibata N, Hirano A, Kobayashi M, Sasaki S, Kato T, Matsumoto S, Shiozawa Z, Komori T, Ikemoto A, Umahara T, et al. Neurosci Lett. 1994;179:149–152. doi: 10.1016/0304-3940(94)90956-3. [DOI] [PubMed] [Google Scholar]
- 23.Matsumoto S, Kusaka H, Ito H, Shibata N, Asayama T, Imai T. Clin Neuropathol. 1996;15:41–46. [PubMed] [Google Scholar]
- 24.Shibata N, Asayama K, Hirano A, Kobayashi M. Dev Neurosci. 1996;18:492–498. doi: 10.1159/000111445. [DOI] [PubMed] [Google Scholar]
- 25.Bruijn L I, Becher M W, Lee M K, Anderson K L, Jenkins N A, Copeland N G, Sisodia S S, Rothstein J D, Borchelt D R, Price D L, Cleveland D W. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 26.Bruijn L I, Houseweart M K, Kato S, Anderson K L, Anderson S D, Ohama E, Reaume A G, Scott R W, Cleveland D W. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- 27.Lee J P, Gerin C, Bindokas V P, Miller R, Ghadge G, Roos R P. J Neurochem. 2002;82:1229–1238. doi: 10.1046/j.1471-4159.2002.01056.x. [DOI] [PubMed] [Google Scholar]
- 28.Morrison B M, Morrison J H, Gordon J W. J Exp Zool. 1998;282:32–47. [PubMed] [Google Scholar]
- 29.Sherman M Y, Goldberg A L. Neuron. 2001;29:15–32. doi: 10.1016/s0896-6273(01)00177-5. [DOI] [PubMed] [Google Scholar]
- 30.Johnston J A, Ward C L, Kopito R R. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnston J A, Illing M E, Kopito R R. Cell Motil Cytoskeleton. 2002;53:26–38. doi: 10.1002/cm.10057. [DOI] [PubMed] [Google Scholar]
- 32.Wang J, Xu G, Borchelt D R. Neurobiol Dis. 2002;9:139–148. doi: 10.1006/nbdi.2001.0471. [DOI] [PubMed] [Google Scholar]
- 33.Johnston J A, Dalton M J, Gurney M E, Kopito R R. Proc Natl Acad Sci USA. 2000;97:12571–12576. doi: 10.1073/pnas.220417997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kirkitadze M D, Bitan G, Teplow D B. J Neurosci Res. 2002;69:567–577. doi: 10.1002/jnr.10328. [DOI] [PubMed] [Google Scholar]
- 35.Lashuel H A, Hartley D, Petre B M, Walz T, Lansbury P T., Jr Nature. 2002;418:291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- 36.Martin-Aparicio E, Yamamoto A, Hernandez F, Hen R, Avila J, Lucas J J. J Neurosci. 2001;21:8772–8781. doi: 10.1523/JNEUROSCI.21-22-08772.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamamoto A, Lucas J J, Hen R. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- 38.Urushitani M, Kurisu J, Tsukita K, Takahashi R. J Neurochem. 2002;83:1030–1042. doi: 10.1046/j.1471-4159.2002.01211.x. [DOI] [PubMed] [Google Scholar]
- 39.Niwa J, Ishigaki S, Hishikawa N, Yamamoto M, Doyu M, Murata S, Tanaka K, Taniguchi N, Sobue G. J Biol Chem. 2002;277:36793–36798. doi: 10.1074/jbc.M206559200. [DOI] [PubMed] [Google Scholar]
- 40.Allen S, Heath P R, Kirby J, Wharton S B, Cookson M R, Menzies F M, Banks R E, Shaw P J. J Biol Chem. 2003;278:6371–6383. doi: 10.1074/jbc.M209915200. [DOI] [PubMed] [Google Scholar]
- 41.Watanabe M, Dykes-Hoberg M, Culotta V C, Price D L, Wong P C, Rothstein J D. Neurobiol Dis. 2001;8:933–941. doi: 10.1006/nbdi.2001.0443. [DOI] [PubMed] [Google Scholar]
- 42.Hoffman E K, Wilcox H M, Scott R W, Siman R. J Neurol Sci. 1996;139:15–20. [PubMed] [Google Scholar]
- 43.Szweda P A, Friguet B, Szweda L I. Free Radical Biol Med. 2002;33:29–36. doi: 10.1016/s0891-5849(02)00837-7. [DOI] [PubMed] [Google Scholar]
- 44.Dal Canto M C, Gurney M E. Am J Pathol. 1994;145:1271–1279. [PMC free article] [PubMed] [Google Scholar]
- 45.Wong P C, Pardo C A, Borchelt D R, Lee M K, Copeland N G, Jenkins N A, Sisodia S S, Cleveland D W, Price D L. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- 46.Kong J, Xu Z. J Neurosci. 1998;18:3241–3250. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jung C, Higgins C M, Xu Z. J Neurochem. 2002;83:535–545. doi: 10.1046/j.1471-4159.2002.01112.x. [DOI] [PubMed] [Google Scholar]
- 48.Mattiazzi M, D'Aurelio M, Gajewski C D, Martushova K, Kiaei M, Beal M F, Manfredi G. J Biol Chem. 2002;277:29626–29633. doi: 10.1074/jbc.M203065200. [DOI] [PubMed] [Google Scholar]
- 49.Higgins C M, Jung C, Ding H, Xu Z. J Neurosci. 2002;22:RC215. doi: 10.1523/JNEUROSCI.22-06-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beckman J S, Estevez A G, Crow J P, Barbeito L. Trends Neurosci. 2001;24:S15–S20. doi: 10.1016/s0166-2236(00)01981-0. [DOI] [PubMed] [Google Scholar]
- 51.Rakhit R, Cunningham P, Furtos-Matei A, Dahan S, Qi X F, Crow J P, Cashman N R, Kondejewski L H, Chakrabartty A. J Biol Chem. 2002;277:47551–47556. doi: 10.1074/jbc.M207356200. [DOI] [PubMed] [Google Scholar]
- 52.Hayward L J, Rodriguez J A, Kim J W, Tiwari A, Goto J J, Cabelli D E, Valentine J S, Brown R H., Jr J Biol Chem. 2002;277:15923–15931. doi: 10.1074/jbc.M112087200. [DOI] [PubMed] [Google Scholar]
- 53.Goto J J, Zhu H, Sanchez R J, Nersissian A, Gralla E B, Valentine J S, Cabelli D E. J Biol Chem. 2000;275:1007–1014. doi: 10.1074/jbc.275.2.1007. [DOI] [PubMed] [Google Scholar]
- 54.Rabizadeh S, Gralla E B, Borchelt D R, Gwinn R, Valentine J S, Sisodia S, Wong P, Lee M, Hahn H, Bredesen D E. Proc Natl Acad Sci USA. 1995;92:3024–3028. doi: 10.1073/pnas.92.7.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roe J A, Wiedau-Pazos M, Moy V N, Goto J J, Gralla E B, Valentine J S. Free Radical Biol Med. 2002;32:169–174. doi: 10.1016/s0891-5849(01)00778-x. [DOI] [PubMed] [Google Scholar]
- 56.Liochev S I, Chen L L, Hallewell R A, Fridovich I. Arch Biochem Biophys. 1997;346:263–268. doi: 10.1006/abbi.1997.0298. [DOI] [PubMed] [Google Scholar]
- 57.Wiedau-Pazos M, Goto J J, Rabizadeh S, Gralla E B, Roe J A, Lee M K, Valentine J S, Bredesen D E. Science. 1996;271:515–518. doi: 10.1126/science.271.5248.515. [DOI] [PubMed] [Google Scholar]
- 58.Ghadge G D, Lee J P, Bindokas V P, Jordan J, Ma L, Miller R J, Roos R P. J Neurosci. 1997;17:8756–8766. doi: 10.1523/JNEUROSCI.17-22-08756.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hottinger A F, Fine E G, Gurney M E, Zurn A D, Aebischer P. Eur J Neurosci. 1997;9:1548–1551. doi: 10.1111/j.1460-9568.1997.tb01511.x. [DOI] [PubMed] [Google Scholar]
- 60.Azzouz M, Poindron P, Guettier S, Leclerc N, Andres C, Warter J M, Borg J. J Neurobiol. 2000;42:49–55. doi: 10.1002/(sici)1097-4695(200001)42:1<49::aid-neu5>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 61.Wang J, Xu G, Gonzales V, Coonfield M, Fromholt D, Copeland N G, Jenkins N A, Borchelt D R. Neurobiol Dis. 2002;10:128–138. doi: 10.1006/nbdi.2002.0498. [DOI] [PubMed] [Google Scholar]
- 62.Subramaniam J R, Lyons W E, Liu J, Bartnikas T B, Rothstein J, Price D L, Cleveland D W, Gitlin J D, Wong P C. Nat Neurosci. 2002;5:301–307. doi: 10.1038/nn823. [DOI] [PubMed] [Google Scholar]
- 63.Liu H, Zhu H, Eggers D K, Nersissian A M, Faull K F, Goto J J, Ai J, Sanders-Loehr J, Gralla E B, Valentine J S. Biochemistry. 2000;39:8125–8132. doi: 10.1021/bi000846f. [DOI] [PubMed] [Google Scholar]
- 64.Nishida C R, Gralla E B, Valentine J S. Proc Natl Acad Sci USA. 1994;91:9906–9910. doi: 10.1073/pnas.91.21.9906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lyons T J, Liu H, Goto J J, Nersissian A, Roe J A, Graden J A, Cafe C, Ellerby L M, Bredesen D E, Gralla E B, Valentine J S. Proc Natl Acad Sci USA. 1996;93:12240–4. doi: 10.1073/pnas.93.22.12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lyons T J, Nersissian A, Huang H, Yeom H, Nishida C R, Graden J A, Gralla E B, Valentine J S. J Biol Inorg Chem. 2000;5:189–203. doi: 10.1007/s007750050363. [DOI] [PubMed] [Google Scholar]
- 67.Richardson J S, Richardson D C. Proc Natl Acad Sci USA. 2002;99:2754–2759. doi: 10.1073/pnas.052706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cardoso R M, Thayer M M, DiDonato M, Lo T P, Bruns C K, Getzoff E D, Tainer J A. J Mol Biol. 2002;324:247–256. doi: 10.1016/s0022-2836(02)01090-2. [DOI] [PubMed] [Google Scholar]
- 69.Hart P J, Liu H, Pellegrini M, Nersissian A M, Gralla E B, Valentine J S, Eisenberg D. Protein Sci. 1998;7:545–555. doi: 10.1002/pro.5560070302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Andrus P K, Fleck T J, Gurney M E, Hall E D. J Neurochem. 1998;71:2041–2048. doi: 10.1046/j.1471-4159.1998.71052041.x. [DOI] [PubMed] [Google Scholar]
- 71.Stadtman E R, Berlett B S. Chem Res Toxicol. 1997;10:485–494. doi: 10.1021/tx960133r. [DOI] [PubMed] [Google Scholar]
- 72.Hodgson E K, Fridovich I. Biochemistry. 1975;14:5294–5299. doi: 10.1021/bi00695a010. [DOI] [PubMed] [Google Scholar]
- 73.Cabelli D E, Allen D, Bielski B H J, Holcman J. J Biol Chem. 1989;264:9967–9971. [PubMed] [Google Scholar]
- 74.Yim M B, Chock P B, Stadtman E R. Proc Natl Acad Sci USA. 1990;87:5006–5010. doi: 10.1073/pnas.87.13.5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kurahashi T, Miyazaki A, Suwan S, Isobe M. J Am Chem Soc. 2001;123:9268–9278. doi: 10.1021/ja015953r. [DOI] [PubMed] [Google Scholar]
- 76.Uchida K, Kawakishi S. J Biol Chem. 1994;269:2405–2410. [PubMed] [Google Scholar]