

Abstract
We have reconstructed the group II intron from Pylaiella littoralis (PL) into a hydrolytic ribozyme, comprising domains 1–3 (D123) connected in cis plus domain 5 (D5) supplied in trans that efficiently cleaves spliced exon substrates. Using a novel gel-based fluorescence assay and nuclear magnetic resonance (NMR) spectroscopy, we monitored the direct binding of D5 to D123, characterized the kinetics of the spliced exon hydrolysis reaction (which is mechanistically analogous to the reverse of the second catalytic step of splicing), and identified the binding surface of D123 on D5. This PL ribozyme acts as an RNA endonuclease even at low monovalent (100 mM KCl) and divalent ion concentrations (1–10 mM MgCl2). This is in contrast to other group II intron ribozyme systems that require high levels of salt, making NMR analysis problematic. D5 binds tightly to D123 with a K d of 650 ± 250 nM, a K m of ∼300 nM, and a K cat of 0.02 min−1 under single turnover conditions. Within the ∼160-kDa D123–D5 binary complex, site-specific binding to D123 leads to dramatic chemical shift perturbation of residues localized to the tetraloop and internal bulge within D5, suggesting a structural switch model for D5-assisted splicing. This minimal ribozyme thus recapitulates the essential features of the reverse of the second catalytic step and represents a well-behaved system for ongoing high-resolution structural work to complement folding and catalytic functional studies.
Keywords: fluorescence gel shift assay, NMR chemical shift perturbation analysis, P. littoralis ribosomal RNA intron, RNA structure and catalysis
INTRODUCTION
Splicing, the removal of intron RNA from coding sequences, is essential for the expression of most eukaryotic genes, not only because accurate splicing is needed to avoid mutations that lead to translation of aberrant mRNAs, but also because alternative splicing is a major source for creating protein diversity in metazoan organisms (for recent reviews, see Black 2003; Nilsen 2003). Understanding the structural basis for the molecular mechanism of splicing is therefore of great importance in our understanding of biology and modern medicine (Sullenger and Gilboa 2002; Jones et al. 2005).
At the heart of splicing in eukaryotes is the very simple two-step transesterification reaction that is undertaken by a huge molecular machine, the spliceosome. The spliceosome is composed of >100 proteins and five small nuclear RNAs (snRNAs) (U1/U11, U2/U12, U4/U4atac, U5, and U6/U6atac) (Burge et al. 1999; Nilsen 2003). Remarkably, the chemical pathway by which the very complex spliceosome catalyzes the excision of nuclear introns is similar to the one used by group II introns. Yet group II introns can perform this same reaction without the help of any protein factors (Peebles et al. 1986; van der Veen et al. 1986). It is now well established that domains 1, 2, and 3 (D123) and domain 5 (D5) are essential for the optimal catalytic activity of group II introns (Qin and Pyle 1998; Bonen and Vogel 2001). D5 can functionally replace its structural analog, the U6/U6atac snRNA stem–loop, in spliceosomal splicing (Shukla and Padgett 2002). Group II ribozymes share striking structural and mechanistic similarities to the spliceosome, which makes them an attractive model for studying the basic biochemistry of splicing. Both D5 and U6 have been the focus of intense structural studies because they are the most conserved structures in each enzymatic system and each forms an integral part of their respective catalytic center (Zhang and Doudna 2002; Sashital et al. 2004; Sigel et al. 2004; Seetharaman et al. 2006). The D5 and U6 structures solved to date provide some insight into the catalytic mechanism, but these structures do not fully explain the function of three prominent and universally conserved features of D5/U6 known to be involved in binding and splicing, namely, a catalytic AGC triad, a tetraloop, and an internal bulge. Because D5 in isolation does not necessarily reflect the native catalytic conformation, examining the structure of D5 in the context of D123 and substrates (i.e., a tripartite system) becomes critical for advancing our knowledge of the atomic basis for catalysis.
Group II introns, like the spliceosome, use a two-step mechanism of concerted phosphodiester bond cleavage–ligation reactions to splice out introns from precursor RNAs. Both reaction steps in the two systems contain identical reactants and products (Fig. 1A; Konarska et al. 1985; Maschhoff and Padgett 1993; Moore and Sharp 1993; Padgett et al. 1994). In the first step, the 2′-hydroxyl (OH) of the branch site adenosine attacks the 5′-splice site guanosine, breaks the phosphodiester bond, and simultaneously forms a 2′–5′ phosphodiester linkage between this 5′-terminal nucleotide of the intron and the branch adenosine. In the second step, the newly liberated 3′-OH of the 5′-exon attacks the 3′-splice junction, breaks that phosphodiester bond, and forms a new bond between the 5′- and 3′-exons. Both steps of the splicing reaction proceed with a phosphorothioate in the pro-Sp but not pro-Rp configuration, and both steps proceed with stereochemical inversion (Fig. 1A; Padgett et al. 1994). By the principle of microscopic reversibility, the reverse of both reactions proceeds by pro-Rp but not pro-Sp configuration. This fact is exploited in the Spliced Exon Reopening (SER) reaction, which involves the cleavage of the ligated 5′- and 3′-exon product (Jarrell et al. 1988; Podar et al. 1995; Gordon et al. 2004). Intriguingly, for group II introns an alternative pathway for the first step exists that bypasses the requirement for a branchpoint adenosine provided by domain 6 (branchpoint pathway), and instead uses water or hydroxide ions (hydrolytic pathway). This hydrolytic pathway is obligatory for certain group II intron families that lack branchpoint structural elements (van der Veen et al. 1987; Michel et al. 1989; Bonen and Vogel 2001; Li-Pook-Than and Bonen 2006), and is similar to RNAse P phosphodiester cleavage using water or hydroxide as the nucleophile (Smith and Pace 1993). Additionally, both the branch-point and hydrolytic pathways occur in parallel in vivo and in vitro, and both pathways are followed by a successful second step of splicing (Schmidt et al. 1990; Daniels et al. 1996; Chu et al. 1998; Podar et al. 1998). Thus the hydrolytic and branching pathways likely occur in the same catalytic pocket. Similarly, both hydrolytic- and branch-mediated attacks at the 5′-splice site occur with an Sp phosphorothioate preference that leads to a successful second step of splicing (Podar et al. 1995). The hydrolytic pathway is thus likely to reflect a central mechanism in group II intron self-splicing and a useful reaction for its analysis. Research on group II intron self-splicing has used various constructs that omit domain 6 (Podar et al. 1995, 1998). These studies have primarily involved the use of multipartite assays that divide the intron into separate ribozyme and substrate portions of intron aI5γ derived from yeast mitochondria, which is able to undergo both the branchpoint and hydrolytic splicing pathways (Chanfreau and Jacquier 1994; Griffin et al. 1995; Michels and Pyle 1995; Peebles et al. 1995; Podar et al. 1995; Bar-Shalom and Moore 2000; Gordon et al. 2000, 2004).
FIGURE 1.

(A) Parallels between the splicing mechanisms of the spliceosome and group II introns. Group II intron and mRNA precursor splicing using branch and hydrolytic pathways are depicted, together with the diastereomeric preference (Rp or Sp) for each step of the splicing reaction. (B) The full “six-wheel” domain structure of group II introns. (C) A schematic of the tripartite model system. (D–F) Details of the secondary structures of the components of the new tripartite model system developed to probe structure and function. (D) D123 and D5 secondary structures. Major tertiary interactions between domains 1, 2, 3, and 5 are depicted. (E) Schematic of 3D structure of D5-PL with same color coding as the D5 secondary structure. Major elements of the tetraloop, bulge, and catalytic triad are shown. (F) E1E2 substrate sequence. The substrate cleavage site is indicated by “X”.
While this system has yielded valuable insights into the function and architecture of a group II intron ribozyme, it has a major drawback in that it requires very high salt conditions (100 mM MgCl2, 500 mM (NH4)2SO4, or 0.5–1.0 KCl) to self-splice efficiently and fold correctly (Swisher et al. 2001; Su et al. 2003, 2005). Such high salt concentrations present a problem for high-resolution structural techniques such as NMR. They introduce high dielectric constant and electrical conductivity, which, in turn, severely compromises the NMR probe performance and leads to unavoidable radiofrequency-induced heating detrimental to both the sample and the NMR probe (Kelly et al. 2002; Stringer et al. 2005). In the past, this has limited the investigation of the structure of group II introns under functional conditions by NMR.
In contrast to aI5γ, the group II intron from the Pylaiella littoralis (PL) mitochondria large ribosomal RNA has been shown to retain the ability to self-splice using the branchpoint pathway under a broader range of ionic conditions including near physiological magnesium ion concentrations of 0.5–50 mM, monovalent salt concentrations of 0.1–1 M KCl or (NH4)2SO4, and a range of temperatures of 25°–45°C (Fontaine et al. 1995, 1997; Costa et al. 1997). However, extensive analysis in this system has only been carried out on the lariat form of the PL ribozyme (Costa et al. 1997, 1998, 2000; Costa and Michel 1999). The lariat form can undergo additional reactions due to reversibility of the transesterification reaction, further complicating the analysis. Using the tripartite system allows us to selectively label each component and monitor them exclusively in a binary or ternary complex within the rest of the group II intron context using powerful biophysical tools such as NMR and fluorescence spectroscopy to characterize the reverse of the second catalytic step, a chemical reaction relatively unexplored even in the better-characterized aI5γ system.
For all these reasons, we have developed a tripartite system for the PL group II intron comprising structural domains 1, 2, and 3 (D123, 493 nt), domain 5 (D5, 36 nt), and a fragment of 5′- and 3′-exon sequences reflecting the product of exon ligation (E1E2, 22 nt) (Fig. 1). Using this tripartite system and a novel gel-based fluorescence assay, we show that the PL ribozyme, indeed, acts as a highly specific RNA endonuclease under a wide range of salt conditions. In addition, we show that D5 binds D123 with a K d of 300–800 nM within a binary complex. Furthermore, structural analysis by NMR chemical shift perturbation reveals detailed D123-binding sites within D5 that suggest that the bulge and the tetraloop are two regions of D5 that undergo the most significant rearrangement on binding, with important implications for catalysis.
RESULTS
Development of a new tripartite model system for the PL group II intron ribozyme
Previous work on group II intron ribozyme activity used a model system derived from the yeast aI5γ intron under conditions of elevated levels of monovalent salt (0.5–1.0 M KCl) and divalent metal ion (100 mM Mg2+) that were inimical for optimal NMR performance: The high dielectric and conductivity of the resulting buffer can significantly reduce the sensitivity of the NMR experiments and/or cause sample aggregation. To address this issue, we have reconstructed a three-part model system derived from the brown algae P. littoralis (PL) large mitochondrial ribosomal RNA intron based on its ability to fold into a homogeneous population without the requirement for high salt (Fig. 1; Costa et al. 1997, 2000). This model system comprising D123 and D5 as a two-piece ribozyme can cleave small oligonucleotides containing sequences derived from the 5′-exon (E1) and the 3′-exon (E2) that mimic the product of exon ligation.
Direct nonradioactive observation of D5:D123 and E1E2:D123 complexes
We have adapted a fast and direct multicolor fluorescence-based gel shift assay to monitor RNA–RNA biomolecular interactions because one can use differential labeling (two or more fluorophores) that allows orthogonal detection and independent verification of binding affinity. Detection is orthogonal in that signals from one labeled component (say, in the red frequency regime) do not interfere with signals from other labeled components (say, in the blue frequency regime) so that we can simultaneously monitor both catalytic and binding events within this ribozyme and account for all RNAs and their interactions present in the reaction mixture at a given time. Similarly, equilibrium measurements such as fluorescence anisotropy can be made on the same samples to validate the nonequilibrium gel-based measurements.
For the three-piece ribozyme system, we therefore constructed an Alexa Fluor 647-labeled spliced exon 22-mer substrate (A647-E1E2) comprising the last 16 nt of the 5′-exon (E1) and the first 6 nt of the 3′-exon (E2) (A647-C6 linker-5′-GACUGUUUAUUAAAAA•CACAGG-3′; the cleavage site is indicated by •). We also made an Alexa Fluor 488-labeled 36-nt D5 RNA construct (A488-D5). This fluorophore pair was chosen because, compared to alternative dyes with similar spectral properties, the conjugates of these dyes have been shown to be brighter, more resistant to photobleaching, and exhibit little or no absorption abnormalities usually indicative of dye–dye interactions (Panchuk-Voloshina et al. 1999; Berlier et al. 2003). Because most of the work on group II introns using the yeast aI5γ intron were carried out under high salt conditions, all the work reported herein was similarly done under high salt conditions (1 M KCl and 50 mM MgCl2) to enable easy comparison to these earlier studies. The NMR titration data were carried out under low salt conditions that still support cleavage (100 mM KCl and 5 mM MgCl2).
D5 binds with high affinity to D123
Using these constructs, we next established how tightly D5 is capable of binding to D123 in vitro. Titrating increasing concentrations of D123 (0–6 μM) into A488-D5 (2 μM) resulted in a direct shift in D5 mobility, suggesting formation of an A488-D5:D123 binary complex (Fig. 2A,B). Extrapolating from the linear region of the binding isotherm to the point of intersection with a horizontal line indicates saturation at a 1:1 D123:D5 ratio, with an apparent equilibrium dissociation constant (K d app) of 300 ± 100 nM. Reactions repeated with 50 nM A488-D5 yielded similar K d app (data not shown). While saturation occurred at a D123 concentration of ∼2 μM, there is significant free D5 in the EMSA due to dissociation of the D5–D123 complex during the electrophoresis (see, e.g., Hoopes et al. 1992; Gumbs et al. 2003). Given that a gel-shift assay is a nonequilibrium measurement, we also performed equilibrium fluorescence anisotropy experiments by adding increasing amounts of D123 to free A488-D5 (2 μM). The average fluorescence emission of the fluorescently labeled A488-D5 was monitored from 510 to 540 nm after exciting at 494 nm. We observed a steady increase in A488-D5 anisotropy that saturated at an anisotropy of ∼0.12. This saturation is indicative of formation of a 1:1 A488-D5:D123 binary complex (Fig. 2C,D; Lakowicz 1999). For an Alexa 488 dye with a lifetime of 4.1 nsec, the ratio of the free and bound anisotropy of ∼2 is consistent with an ∼160-kDa complex (see Materials and Methods for calculation). Fitting of the data from the anisotropy measurements in Figure 2C, and D, to Equation 1 (see Materials and Methods) gave a K d app of 700 ± 200 nM (Fig. 2C,D), a value close to that reported by the gel mobility shift method. This suggests that the gel mobility shift method measures the equilibrium value.
FIGURE 2.

Monitoring D5–D123 binding using fluorescence spectroscopy. (A) Fluorescence gel shift of D5 binding to D123 using native PAGE analysis. The fraction of D5 bound by D123 is calculated as [(Ii − I 0)/(I B − I 0)], where Ii is the measured intensity of D5 during the titration, I 0 is the intensity of D5 in the absence D123, and I B is the intensity of D5 bound to D123. (B) Fit of data in A to binding isotherm. (C) Representative fluorescence anisotropy scan of D5 binding to D123. (D) Fit of the fluorescence anisotropy data in C to the binding isotherm discussed in Materials and Methods. The reactions were made on a 2 μM A488-D5 sample.
A fluorescence analysis of group II ribozyme's catalytic activity
Having shown direct interaction of PL D5 with D123, we next tested for direct biomolecular interaction of D123 with its substrate in the presence and absence of D5 to initiate a cleavage reaction. The ability of D123 and D5 to cleave A647-E1E2 was monitored by incubating 25 nM A647-E1E2 with 1 μM A488-D5 and 4 μM unlabeled D123 (Fig. 3). Cleavage of the A647-E1E2 substrate occurs (green bands) within 2 h in 10 mM MgCl2 and 100 mM KCl (Fig. 3, lane 6) at 42°C. No cleavage was observed in the absence of either magnesium (Fig. 3, lanes 4,11) or D5 (A488-labeled or unlabeled) (Fig. 3, lanes 2,9). These results suggest that PL does, indeed, function as a ribonuclease under a broad range of salt conditions.
FIGURE 3.

Tripartite reaction for the reverse of second chemical step using multicolored fluorescence-based trans-assay. The sequence of the synthetic oligonucleotide used is A647-C6 linker–5′-GACUGUUUAUUAAAAA•CACAGG-3′. The cleavage site is indicated by •. Denaturing PAGE analysis of the exon reopening reaction is shown. The PL ribozyme can cleave substrate under low salt conditions (cf. lanes 3 and 6). The reactions were carried out in 10 mM MgCl2. Tagging D5 with the fluorescent probe does not impair its catalytic function (cf. lanes 3 and 10).
Single turnover kinetic parameters for the SER reaction
Next we investigated the catalytic efficiency of this tripartite system. To determine K cat and K m for this PL system, we monitored the D5 concentration-dependent cleavage of E1E2. To date, K cat and K m have not been determined for the SER reaction under conditions of the reverse of the second catalytic step for any group II intron, and no hydrolytic pathway studies exist for the brown alga PL intron. The D5 concentration (0.1–1.0 μM) dependent kinetics of cleavage of 12.5 nM E1E2 substrate in the presence of 2 μM D123 is shown in Figure 4. Complete cleavage of the A647-E1E2 substrate occurs within 2 h. Fitting of this cleavage data as a function of time to a single exponential equation shows a steady increase in reaction kinetics (K obs) with increasing D5 concentration, with K obs saturating at ∼600 nM D5 (Fig. 4B). Under these saturating conditions, the apparent K obs may represent the catalytic rate of this enzymatic reaction (K obs = K cat) and/or conformational rearrangements preceding chemistry. Fitting the data in Figure 4B to Equation 1 (see Materials and Methods), we obtained a K m of ∼300 nM for D5–D123 binding, in good agreement with the K d we measured directly. This gives a second-order rate constant (K cat/K m D5) of 7.3 × 104 M−1 min−1.
FIGURE 4.

Single turnover kinetics of SER reaction catalyzed by D5. (A) Monoexponential fit of the time course for the formation of cleaved product in the presence of excess D123 (2 μM) and increasing concentration of D5 (0.1–1.0 μM). The fluorescent intensity in bands S and P (see Fig. 5A) was used to calculate the fraction of cleaved product. (B) Michaelis-Menten binding curve for data in A. K m is the [D5] at half-maximum rate and K cat is the maximal rate at D5 saturation. Please see Materials and Methods for details of extracting the kinetic constants from the experimental data.
The E1E2 cleavage rate shows log/linear pH dependence
The initial chemistry of the SER reaction involves the nucleophilic attack of a water molecule at the 5′/3′-exon junction. If the reaction is rate-limited by chemistry, we would expect a linear relationship between the pH of the reaction and the log of the observed reaction rate (Herschlag and Cech 1990). The slope of such a plot will indicate the number of protons involved in the rate-limiting step. We, therefore, investigated the effects of varying pH on the rate of catalysis using MES (pH 5.0–6.9), MOPS (pH 6.5–7.3), and HEPES (pH 6.5–8.2) buffers as described previously (Podar et al. 1995). No dependence on buffer type was observed at a constant pH 6.8. A representative time course is shown in Figure 5A. To evaluate K obs, we used two or three parameter fits and calculated the residuals based on a protocol by Fushman et al. (1999). As shown in Figure 5B, an increase in pH from 5.0 to 8.0 leads to an overall steady increase in the reaction rate. This indicates that we are, indeed, monitoring the pH-dependent chemistry or conformational change of the nucleophilic reaction. A plot of the data shows a log/linear pH dependence on the reaction rate with slope close to 1 (0.93) from pH 5.4 to 7.5, consistent with a chemical rate-limiting step with abstraction of a single proton (Fig. 5C).
FIGURE 5.

Rates of trans-splicing (in the SER reaction) as a function of pH. (A) A time course of the SER cleavage reaction at pH 6.8. The fluorescent intensity in bands S (substrate) and P (cleaved product) was used to calculate the fraction of cleaved product. (B) Monoexponential fit of the time course for the formation of cleaved product in the presence of excess D123 (2 μM) and increasing concentration of D5 (0.1–1.0 μM). (C) Log pH dependence of SER reaction rate.
Mapping the D123-binding surface on fully labeled D5 by NMR
Results from the fluorescence gel shifts and anisotropy measurements show that D5 binds quite tightly to D123 in the presence of Mg2+. Together with D123, D5 is able to cleave a 3′-exon–5′-exon substrate with a reaction rate likely limited by chemistry. Yet, until now, no direct structural evidence for the interaction between D5 and the rest of the intronic domains has been given. We, therefore, monitored the chemical shift changes of proton and carbon sites for every nucleotide in D5 (∼0.2 mM) in the presence of increasing amounts of D123 (0–0.22 mM) using resolution-optimized NMR spectroscopy experiments (Dayie 2005). Under identical experimental conditions, any observed differences in chemical shifts between unbound and D123-bound D5-PL would indicate a change in the local chemical environment of each nucleotide because of direct or indirect D5–D123 interactions. These experiments would enable us to pinpoint regions of D5 that interact directly with D123.
To this end, we probed the differential change in the resonances of a magnesium-bound 15N–13C-labeled D5 in the presence of unlabeled D123 by titrating increasing amounts of D123 (pre-folded in 5 mM Mg2+) into a 5 mM Mg2+-bound D5 and monitored the chemical shifts of the ribose H1′–C1′ and base H2–C2, H5–C5, H6–C6, and H8–C8 resonances. D5 (36 nt) is small enough to give a high-quality heteronuclear single quantum coherence (HSQC) spectrum without any interfering signals from the unlabeled D123. When D5 binds to the larger ∼160-kDa D123 (493 nt), the transverse relaxation rate (1/T 2) of the bound D5 within the complex increases significantly, and this leads to an overall broadening of all the NMR resonances at 25°C beyond detection (Fig. 6A,B). This is consistent with the formation of a large-molecular-weight D5–D123 complex (Riek et al. 2000; Wuthrich 2003; D'Silva et al. 2005). Raising the temperature to 45°C results in a decrease in line widths and sharpening of the D5 NMR resonances due to the decrease in the solvent viscosity (Fig. 6C,D). Because a stable complex forms at 45°C, all subsequent experiments on free D5 and D5–D123 complex were carried out at this temperature. In the ribose H1′–C1′ region, the titration resulted in large selective shifts of A24, A25, and G26 in the bulge region and A16, A17, and A18 in the tetraloop (Fig. 7A). Of note, A2 and G3 are not responsive to D123. In the base H8–C8 region, once again the triad A2 and G3 chemical shift resonances are not perturbed (Fig. 7B). However, G26 in the bulge and U14, G15, and A18 within the tetraloop are significantly shifted and/or broadened, suggesting that they are likely involved in the binding of D5 to D123 (Fig. 7B). Similarly in the base H6–C6 region, the resonances of U20 and C21 in the upper helix shift and/or broaden significantly (Fig. 7B). The most dramatic chemical shift perturbations occur within the base H2–C2 region. The internal bulge nucleotide A24 and the tetraloop nucleotides A16, A17, and A18 as well as the upper helix nucleotide A13 all undergo very large chemical shift changes (Fig. 7C). The resonance of A25 broadens beyond detection, whereas the lower helix nucleotides A2 and A28 remain unperturbed. We find the same trend in the base H5–C5 region. Here the resonances of the internal bulge C11 and U20 undergo large shifts and significant broadening, and those of C4 and C5 in the lower helix also undergo significant change (Fig. 7D). As a negative control, we titrated D5 into a 42-nt tetraloop receptor from domain 1 (Supplemental Fig. 1). This construct contains both the ζ–ζ′ and κ–κ′ elements. Previous work has shown that the isolated complex of a GAAA tetraloop/tetraloop receptor is only moderately stable with an apparent K d value of 0.4 mM (Qin et al. 2001). Consistent with this, we see no significant change in the chemical environment of the tetraloop nucleotides and no line broadening (Supplemental Fig. 1). This contrasts with the dramatic perturbation in the presence of D123 (Fig. 7B,C). This negative control also suggests that the perturbations of D5 bulge/tetraloop resonances are unlikely due to pre-melting at high temperature.
FIGURE 6.

Effect of temperature on the formation of D5–D123 complex. (A) NMR HSQC spectral evidence for the formation of a D5–D123 complex in the ribose region. At 25°C, addition of ∼0.22 mM unlabeled D123 to ∼0.2 mM 15N–13C-labeled D5-PL leads to disappearance of most of the D5 resonances. (B) NMR HSQC spectral evidence for the formation of a D5–D123 complex in the base region. At 25°C, addition of ∼0.22 mM unlabeled D123 to ∼0.2 mM 15N–13C-labeled D5-PL leads to disappearance of most of the D5 resonances. (C) NMR HSQC spectrum in the ribose region collected at 45°C leads to recovery of peaks. All the experiments were done on equimolar amounts of sample of ∼0.2 mM each and under identical conditions as in A. (D) NMR HSQC spectrum in the base region collected at 45°C leads to recovery of peaks. All the experiments were done on equimolar amounts of sample of ∼0.2 mM each and under identical conditions as in B.
FIGURE 7.

NMR spectra, at 45°C, showing the chemical shift perturbations of D5 nucleotides on binding D123 for resonances of the (A) ribose H1′–C1′, (B) H6–C6 and H8–C8, (C) base H2–C2, (D) cytosine base H5–C5, and (E) uracil base H5–C5. Arrows depict residues with most chemical shift perturbation with increasing D123 concentration (0 and ∼0.22 mM), indicative of likely binding sites. Note how A2 and G3 resonances are barely perturbed. For C and D, we depict three titration points to indicate the direction of the perturbations ([D123] mM: 0, black; 0.1, cyan; 0.22, magenta).
Overall, our data suggest that D5 resonances primarily from the tetraloop and internal bulge regions undergo the greatest chemical shift perturbation or extensive line broadening on binding to D123. The phylogenetically conserved A2 and G3 triad nucleotides strongly implicated in catalysis undergo very little change in chemical shift. Our data strongly implicate the D5 bulge and tetraloop regions rather than the AGC triad functional groups in binding to D123.
DISCUSSION
Why another tripartite ribozyme system?
Until now the only group II intron extensively characterized biochemically for both branching and hydrolytic reactions is aI5γ from yeast mitochondria (Griffin et al. 1995; Michels and Pyle 1995; Podar et al. 1995; Bar-Shalom and Moore 2000; Gordon et al. 2000, 2004). The discovery that the PL group II intron (Fontaine et al. 1995, 1997; Costa et al. 1997) retained the ability to self-splice at unusually low magnesium ion concentrations (0.5–10 mM) and moderate concentrations of monovalent salts (100 mM KCl or (NH4)2SO4) suggested that transforming the PL group II intron into a tripartite hydrolytic ribozyme would be valuable and desirable both as an independent model system from a different group II intron ribozyme and as a molecular probe to further our understanding of the mechanism of splicing at the structural level. Extensive footprinting and kinetic analyses have been carried out on the lariat form of the PL intron in debranching reactions using 5′-exons or ligated exons. These studies showed that the reaction is extremely fast, requiring low pH to slow the reaction to be measurable (Costa et al. 1997, 1998, 2000; Costa and Michel 1999). In addition, the lariat form can undergo additional reactions due to reversibility of the transesterification reaction, further complicating the analysis. The modular nature of the hydrolytic tripartite system circumvents these problems and allows us to investigate, comparatively, the principles of RNA splicing, folding, and structure.
Direct binding, kinetic parameters, and comparison to other ribozymes
Group II introns catalyze the splicing reaction in the absence of branch formation by hydrolysis of the 5′-splice junction in step 1 (Jarrell et al. 1988; Franzen et al. 1993; Pyle and Green 1994; Peebles et al. 1995; Podar et al. 1998; Gordon and Piccirilli 2001), and hydrolysis of the phosphodiester bond joining two spliced exons in the SER reaction (Jarrell et al. 1988; Podar et al. 1995; Gordon et al. 2004), thereby acting as true ribozymes. The SER reaction, shown earlier to be mechanistically analogous to the reverse of the second catalytic step by hydrolysis (Podar et al. 1995), has been partially characterized for aI5γ but not for PL (Podar et al. 1995; Gordon et al. 2004). Here we show, using a newly developed fluorescence-based RNA–RNA gel shift assay, that (1) D5-PL binds fairly tightly (300–800 nM) to D123 in the presence of magnesium ions with an affinity similar to that measured for aI5γ (400–870 nM); (2) this tripartite system obeys simple Michaelis-Menten kinetics with a chemical rate-limiting step; and, importantly, (3) the favorable salt requirements have enabled us, for the first time, to initiate high-resolution structural work on this class of ribozymes using NMR.
Agreement between binding constants from direct binding and K m from single turnover kinetics can provide valuable insight into the rate-limiting step in a reaction mechanism. Under single turnover conditions, D5 displayed saturation kinetics with a K m of ∼300 nM (Fig. 4), suggesting that a step following substrate binding is rate-limiting. The on rate must therefore be fast relative to chemistry. Otherwise slow association and fast chemistry would result in complete reaction of the intermediate D123•substrate complex as soon as it is formed. In that case, K obs would not plateau, as we observe, at the [D5] just above K d (Fig. 4B). For Michaelis-Menten reaction kinetics, the on and off rates for substrate are fast relative to the rate of chemistry (Fersht 2000), and because D5 binding displays this classic square-hyperbolic dependence, it is possible that the derived K cat could represent a step upstream of binding and/or the rate of the chemical step (K chem). Further evidence that K cat represents K chem is provided by the log-linear pH dependence of K cat under single turnover conditions in the pH range 5.4–7.5. Similar observations that K cat is log-linear with pH for D5-catalyzed hydrolysis of exD123 or ligation of intron–3′-exon (intron-E2) and the 5′-exon (E1) led to the hypothesis that the rate of the reaction was limited by chemistry in the pH range of 5–7 in aI5γ (Pyle and Green 1994; Podar et al. 1998; Gordon et al. 2004). Log linearity is not an ironclad proof of chemistry. One could undertake an extensive study using an array of nucleoside analogs for the 2′-hydroxyl group at the cleavage site of a SER substrate, as reported recently by Piccirilli and colleagues (Gordon et al. 2004). This is, however, beyond the scope of the present study and will be the focus of future investigations. Nonetheless, collectively, these previous reports on aI5γ and our data for PL are consistent with a model in which the RNA subunits form a binary complex that cleaves short RNA oligonucleotides that mimic the spliced exons E1E2. In particular, the PL ribozyme construct described above provides a 107-fold rate enhancement over the uncatalyzed rate (Herschlag and Cech 1990) for the hydrolytic cleavage of the phosphodiester linkage at the boundary between exon 1 and exon 2, consistent with similar rate enhancements for the hydrolytic cleavage at the 5′-splice site (Michels and Pyle 1995). The substantial agreement of PL's K m to those determined previously for two- and three-part systems for aI5γ argue that our new tripartite PL system is an attractive alternative model especially for NMR structural characterization. Akin to chemical footprinting studies for the analysis of tertiary contacts between D5 and D123, NMR chemical shift perturbation analysis potentially provides exquisite site-specific information for D5 atomic positions that directly contact D123 and/or respond to induced structural changes.
Direct probing of the D123 binding surface on D5-PL by NMR
Even though the D5–D123 complex has a high molecular mass (∼160 kDa) for NMR studies, the observed spectral complexity is limited to only the labeled D5 segment of the complex, and, remarkably, the 1H–13C NMR data at 45°C are of such excellent quality that we could follow most of the D5 resonances during the titration with unlabeled D123. This is the largest RNA–RNA binding interaction characterized by NMR. This is made possible by a tight binding interaction, a well-behaved complex, new NMR experiments designed for resolving the usually crowded ribose H1′–C1′ spectral region, and the ability to work at high temperatures. Previous RNA or RNA complexes investigated were ∼40 kDa or less (Lukavsky et al. 2003; D'Souza et al. 2004; Davis et al. 2005).
ζ–ζ′ tetraloop–receptor interactions
Starting from 5 mM Mg2+-bound D5, binding of D123 prefolded in 5 mM Mg2+ leads to dramatic (>0.2 ppm) and selective perturbation of resonances of the minor groove H2–C2 resonances and H1′–C1′ resonances of A16, A17, and A18 in the tetraloop (Figs. 7A–C, 8). These dramatic chemical shifts observed for the minor groove H2–C2 atoms of the second, third, and fourth tetraloop nucleotides as well as the surrounding nucleotides (U14 and G19) are not only consistent with previous studies (Cate et al. 1996a,b; Pley et al. 1994; Costa and Michel 1995; Abramovitz et al. 1996; Golden et al. 1998, 2005; Boudvillain et al. 2000; Juneau et al. 2001; Adams et al. 2004; Davis et al. 2005) but also indicate they can serve as key indicators of the tetraloop–tetraloop receptor interactions in solution. Our observation that the minor groove H2–C2 atoms of the second and fourth tetraloop nucleotides undergo dramatic chemical shifts parallels those reported recently (Davis et al. 2005), only in our case, the shifts are more dramatic in the carbon dimension than in the proton dimension.
FIGURE 8.

Combined proton (1H) and carbon (13C) chemical shift perturbations {(Δδ) = [(Δ1H)2 + (Δ13Cχ)2]1/2} between free D5-PL and D123-bound D5-PL as a function of nucleotide sequence in (A) the ribose H1′–C1′ and aromatic (B) H2–C2 and H8–C8 and (C) H5–C5 and H6–C6 regions. Nucleotides that disappear at the titration end point (∼0.22 mM D123) are not shown. The horizontal line in each panel represents the calculated average chemical shift perturbation. χ = 0.18 and 0.24 for the base and the ribose carbons, respectively. The 3′-AGC label refers to nucleotides GUU that base-pair with the AGC triad nucleotides in the 5′-end of D5.
ε–ε′ and Ψ2 internal bulge and J2/3 interactions
Selective and dramatic perturbations (>0.2 ppm) also occur for the minor groove H2–C2 and H1′–C1′ resonances of A24 and A25, and the H1′–C1′ resonance of G26 in the internal bulge region. These results are consistent with previous work that has implicated bulge nucleotides 24 and 25 mostly in catalysis (Chanfreau and Jacquier 1994; Abramovitz et al. 1996; Schmidt et al. 1996; Boudvillain and Pyle 1998; Gordon and Piccirilli 2001) and their likely proximity to a highly conserved nucleotide in the junction of domains 1 and 2 (J2/3) and nucleotides flanking the ε–ε′ substructure (de Lencastre et al. 2005). Based on chemical shift arguments alone, the dramatic change in chemical environment of the base H2–C2 resonances of A24 and A25 suggests these residues are likely forming extrahelical stacking interactions with other intronic elements. For this to happen, D5 would have to undergo significant conformational change within the bulge. Thus, our NMR perturbation analysis provides a facile handle on identifying key tertiary RNA–RNA interactions within PL.
κ–κ′ and λ–λ′ helical stem–receptor interactions
Significantly, C4 and C11 within D5 also undergo chemical shift perturbation in the base H5–C5 region (Figs. 7D, 8). These nucleotides or those in close proximity to them (U9, G10, C23) (Figs. 7, 8) are postulated to form a network of base triple λ–λ′ interactions (Boudvillain et al. 2000). Our results therefore provide the first direct structural evidence for these previous biochemical observations.
Absence of triad A2 and G3 interactions
Finally, what is perhaps most surprising is the lack of responsiveness of the triad nucleotides A2 and G3 to D123 binding. This result is at first puzzling given that extensive studies have shown that nucleotides and individual atoms along the AGC triad are critical for the chemical steps of splicing (Chanfreau and Jacquier 1994; Boulanger et al. 1995; Peebles et al. 1995; Abramovitz et al. 1996; Konforti et al. 1998; Boudvillain et al. 2000). More than any other nucleotide, substitution of G3 with A, C, or U, replacement by an Rp phosphorothioate, or modification of any of the major groove functional groups (O6 or N7) completely abrogated D5 activity, suggesting that G3 is the single most important residue within the group II active site. Yet, paradoxically, the G3 residue is unperturbed by D123 (Figs. 7, 8). Our results argue against G3 participating in D123 binding in the ground state. This may not be unexpected for a catalytic residue.
Previous biochemical work and our present NMR titration and fluorescence gel shift data strongly suggest that, in the absence of substrate, D5 binds to D123 primarily through the tetraloop–tetraloop receptor ζ– ζ′, base triple λ– λ′, and κ–κ′ interactions without engaging the triad G3 nucleotide.
A molecular handclasp-like interaction between D5 and D123 suggests a structural switch model
To quantitatively define residues at the binding interface, we used all residues whose chemical shift perturbations are greater than the average (>0.1 ppm) and/or selectively broadened beyond detection (Figs. 7, 8) and whose base and ribose functional groups are solvent-exposed in the free D5 structure. The nucleotides that satisfy both criteria (C4, G12, U14, G15, A16, A17, A18, G19, A24, A25, and G26) are likely at the D5–D123 binding site. These residues are defined as “active residues” (Dominguez et al. 2003). The neighboring residues that are also solvent-exposed, defined as “passive residues,” are G3, C5, G8, C23, and C27. We projected the “active” binding site residues in the tetraloop and lower helix onto the 3D structure of D5 (Seetharaman et al. 2006) and the corresponding nucleotides identified in the tetraloop receptor structural element represented as a computer model (Fig. 9A; Costa et al. 2000). Strikingly, two surfaces are revealed in both D5 and D1 that allow each structural element to dock into the other, reminiscent of a handshake. However, the free D5 structure would have to undergo a conformational change to fit snugly into the D1 binding site and/or vice versa. This suggests the need for conformational rearrangement in D5 for catalysis.
FIGURE 9.

(A) Yin-Yang of the D5–D1 binding interface showing D5 docking surface in the D1 tetraloop receptor, and the D1 tetraloop receptor docking site on D5-PL. (B) Cartoon of a structural switch model of the group II intron catalysis. The bulge likely undergoes significant change on binding D123.
Thus our working model is that D5 likely undergoes a conformational change on interacting with other intronic components. The evidence on which this is based is as follows: (1) Ribozymes derived from PL that lacked D5 were shown to be defective in binding their 5′-exon substrate by ∼100-fold (Costa and Michel 1999). (2) In the presence of the 5′-exon and intron D1, G3 within the catalytic triad of D5 becomes more reactive to hydroxyl radicals, indicating likely conformational rearrangement in the presence of substrate (Costa et al. 2000). (3) This triad G3 participates in catalysis using normally inaccessible major groove functional groups (N7 or O6), hinting at a likely conformational change (Konforti et al. 1998). (4) From NMR relaxation data, the bulge functional groups (A24 and A25) are very dynamic, suggesting deformability of the D5 structure on timescales that might be relevant for catalysis (N.V. Eldho and K.T. Dayie, unpubl.). (5) From NMR titration data, the bulge functional groups A24, A25, and G26 undergo substantial chemical shift perturbation on binding D123, suggesting a stacking interaction with other intronic elements (this study). These data collectively point to a central hypothesis: that binding of D123 would likely trigger a conformational change in the D5 bulge region that may be required to correctly juxtapose different regions of the intron to form a competent catalytic structure. We call this the structural switch model (Fig. 9B). Although the information gathered here is not sufficient to derive a high-resolution 3D model of D5 bound to D123, future NMR studies will address this and other questions to further illuminate the structural basis for group II intron catalytic function.
MATERIALS AND METHODS
D123 (493 nt) ribozyme sample preparation
RNA samples were synthesized by in vitro transcription with T7 RNA polymerase from plasmid using established protocols (Milligan and Uhlenbeck 1989). An N-terminal His-tag containing T7 RNA polymerase was overexpressed in Escherichia coli BL21 (DE3) and purified on a Ni-chelating Sepharose column (Pharmacia). An RNA containing intron domains 1–3 (D123) was transcribed from DNAs produced by PCR amplification from the original construct containing the complete domains 1–6 (D1 to D6) (Costa et al. 1997). D123 was amplified using an upstream primer containing the T7 RNA polymerase promoter sequence and the first 19 nt from the 5′-end of the D1 sequence and downstream primers containing 19 nt of the 3′-end of the intron domain 3. The D123 DNA fragment was amplified and digested, and the purified D123 fragment was ligated into an EcoRI- and BamHI-linearized pUC19 vector. Positive colonies, screened on the basis of ampicillin resistance, were checked by standard sequencing procedures for the presence of the correct pUC19-D123 construct. The plasmid DNA was digested with SmaI yielding linearized DNA ready for several T7 RNA polymerase-directed in vitro transcription reactions. The optimal transcription conditions, found by a systematic sparse matrix variation of Mg2+, NTP, and enzyme concentrations, were 10–15 mM total NTP and 10–25 mM Mg2+ in a transcription buffer of 40 mM Tris-HCl (pH 8.1), 1 mM spermidine, 5 mM dithiothreitol (DTT), 0.01% Triton X-100, 200 μg/mL SmaI-linearized plasmid, 0.5 μg/mL inorganic pyrophosphatase, and 0.0685 mg/mL T7 polymerase per 40 μL of transcription volume (Milligan and Uhlenbeck 1989). However, for our NMR experiments, these were scaled up to 400–500 mL of transcription reaction. After 3 h of incubation at 37°C, the transcription reaction was extracted with phenol/chloroform/isoamyl alcohol (25:24:1) to remove the T7 RNA polymerase, followed by ethanol precipitation. The crude RNA precipitate was purified using a 4% denaturing preparative gel at 65 W in a 1× TBE using 3 mm × 45 cm × 35 cm gels. The product band was detected by briefly UV-shadowing the gel, and excised and electro-eluted in an Elutrap electro-separation system (Schleicher and Schuell) at 200 V for a total of 5 h, with aliquots taken every 2 h until the gel slices were clear under the UV lamp. The purified RNA was precipitated with two to three volumes of absolute ethanol and 0.3 M sodium acetate. The RNA pellet was then resuspended in water and dialyzed in a Biodialyzer (Nestgroup) with a 500 MWCO membrane (Nestgroup) for 12–24 h each against 100 mM potassium phosphate buffer (pH 6.0), 0.5 M KCl, 10 mM EDTA, and then 1 and 0.1 mM EDTA, and finally against two changes of ddH2O water. After dialysis, the RNA was lyophilized. RNA concentration was then determined by sodium hydroxide hydrolysis for 2 h at 70°C, and UV absorbance was used to calculate the concentration based on the extinction coefficients of the individual nucleotides. The extinction coefficient was calculated as a sum contribution of each nucleotide in the RNA sequence.
Unlabeled D5 (36 nt) RNA sample preparation
The D5 RNA samples were synthesized by in vitro transcription with T7 RNA polymerase as described above for D123 (Milligan and Uhlenbeck 1989) using synthetic DNA templates (Operon Technologies, Inc.). The DNA promoter sequence, or top strand, has a C nucleotide at the −18 T7 promoter region (CTOP) (Baklanov et al. 1996) and has the sequence 5′-CTAATACGACTCACTATAG. The corresponding wild-type template strand, or bottom strand, used for the analysis is 5′-ggAACCGTGCTTGCGACTTTCATCGCACACGGCTCCTATAGTGAGTCGTATTAG-3′. Use of ribose C2′-methoxyl modification of the 5′ two nucleotides of the template strands substantially reduced the amount of N + 1 and higher add-on transcripts (Kao et al. 1999). The optimal transcription conditions for D5 were also found by a systematic sparse matrix variation of Mg2+, NTP, and T7 RNAP enzyme concentrations as described above using identical transcription buffer supplemented with 80 mg/mL PEG 8000 and 300 nM each DNA strand, and 0.0685 mg/mL T7 RNA polymerase (optimized amount) per 40 μL of transcription volume. The resulting RNAs were gel-purified as described above for D123 RNA, except 15% urea gel was used for the gel purification.
Labeling of D5 and E1E2 substrate (22 nt) RNAs with Alexa 488 and 647 fluorophores
For the fluorescence studies, Alexa 488 and 647 carboxylic acid, succinimidyl esters were purchased from Invitrogen/Molecular Probes Inc. and the D5 and E1E2 RNAs from Dharmacon Inc. For all fluorescence experiments, Alexa 488 or 647 were site-specifically incorporated at the 5′-end of either D5 or E1E2 RNA via a primary amine C-6 linker. The RNAs were labeled using 10–15 molar excess of dye to RNA in 0.1 M sodium tetraborate (pH 8.5) at room temperature for 12 h with gentle mixing. The labeled RNA was then purified over a G-25 Sephadex gel filtration column. D5 fractions were then pooled and ethanol-precipitated. The resulting RNAs were gel-purified using a 15% urea gel prior to electroelution. The pooled fractions were ethanol-precipitated and dissolved in 500 μL of RNA MOPS buffer (40 mM MOPS at pH 7.5, 100 mM KCl), and dialyzed extensively in buffers A (100 mM potassium phosphate at pH 6.0, 10 mM EDTA, 0.5 M KCl), B (100 mM potassium phosphate at pH 6.0, 1 mM EDTA, 0.1 M KCl), and C (100 mM potassium phosphate at pH 6.0, 0.1 mM EDTA, 0.1 M KCL) and several times in H2O before lyophilizing and dissolving in MOPS storage buffer (40 mM MOPS at pH 7.5, 100 mM KCl).
Direct measurement of D5:D123 binding by fluorescence gel-shift experiments
The fluorescence mobility shift assay for the determination of D5–D123 binding constant used unlabeled D123 and Alexa-488 D5 (A488-D5). Complex formation was monitored on a native 5% polyacrylamide gel (37.5:1, acrylamide:bis ratio). Various permutations of folding and preincubations were tried for folding each RNA component separately or together. For instance, each RNA stock solution in storage buffer was heated to either 90°C or 55°C for 1 min or 3 min, then allowed to cool to 42°C. A 2× salt solution (high salt: 2 M KCl, 100 mM MgCl2; low salt: 200 mM KCl, 20 mM MgCl2) was added to each RNA (12.5 nM A647-E1E2, 2 μM D5, and 4 μM D123) and allowed to fold for various times (0, 5, 10, 20, 30, 60, 120 min), and the RNAs were combined to initiate the reaction. At each specific time point, 8-μL aliquots were taken and added to 6 μL of stop buffer (50% glycerol and 100 mM EDTA), and loaded onto an 8 M urea 15% polyacrylamide gel running at 23 W for 2 h. We noticed a lag in initiation of the SER reaction with this protocol that went away for increased times (0 min, 10 min, 20 min, 1 h, 2 h, 3 h) of benchtop pre-folding at each MgCl2 concentration (0, 10, 50 mM) with the optimum at 2 h in the presence of 10–50 mM MgCl2. If only D123 was pre-folded as described above, the reaction kinetics were indistinguishable from pre-folding all three RNAs. These observations suggest that D123 pre-folding is the single most important factor that affects the rate of the reaction. Thus, prior to binding, all RNAs were heated at 90°C for 1.5 min, and cooled on the bench top for at least 10 min in RNA MOPS storage buffer to allow for proper folding. For the binding experiments, increasing concentrations of D123 (0–6 μM) were added to 0.05 or 2 μM A488-D5, in binding buffer containing specific concentrations of KCl (100 mM or 1 M) and MgCl2 (0–50 mM). Reaction mixtures were incubated for at least 2 h at 45°C prior to running on a 10 × 10 cm 5% native gel (37.5:1 acrylamide:bis ratio) at 200 V for 45 min. The A488-D5 complexes were then visualized using a fluorescence scanner (Bio-Rad Molecular Imager pro-plus Fx) equipped with 635- and 488-nm diode pumped solid-state external lasers.
Steady-state fluorescence anisotropy binding experiments
Steady-state equilibrium fluorescence anisotropy measurements were performed on an L-format SLM spectrofluorimeter equipped with a 450 W xenon arch lamp. Experiments were performed at 45°C, and slit widths were set to 4 nm overall. In the binding experiments of D123 to A488-D5, 0.05 μM or 2.0 μM A488-D5 was titrated with increasing concentrations of D123 (0, 0.042, 0.082, 0.162, 0.33, 0.496, 0.659, 0.817, 1.7, 3.26, 4.6, 5.38, 6.13 μM) in 40 mM MOPS (pH 7.5) containing 50 mM MgCl2 and 1.0 M KCl in total final volumes of 80 μL. Samples were excited at 494 nm, and average fluorescence emission was monitored between 510 and 540 nm. Prior to use, all RNAs were heated to 90°C in binding buffer for 1.5 min and cooled at room temperature for a minimum of 10 min. To a first approximation, assuming the D123–D5 complex tumbles isotropically, we can estimate the ratio of the rotational anisotropy of the free (R f) and bound (R b) D5 using R b/R f = {(θ1 + τ)/(θ2 + τ)}(θ2/θ1), where τ is the lifetime of the Alexa 488 dye (4.1 nsec), and the rotational correlation times (θ) for D5 and D123 are, respectively, θ1 and θ2. θ is related to the molecular weight (M), degree of hydration (h = ∼0.3 g H2O/g RNA), specific volume of the RNA (ν = 0.569 mL/g) (Voss and Gerstein 2005), solvent viscosity (η = 0.597 cP), temperature (45°C), and gas constant (R = 8.31 J K−1 mol−1); thus θ = ηM(h + ν)/RT. The experimental ratio is close to the calculated ratio of ∼2.
Multicolored fluorescence gel-shift of E1E2 substrate splicing
To test the activity of the D5 and D123 ribozyme, we adapted the spliced exon reopening (SER) reaction (Jarrell et al. 1988; Podar et al. 1995). However, instead of using radioactive 32P labeling, we used fluorescently labeled A647-E1E2 substrate and tested the ability of unlabeled D123 and fluorescently labeled D5 (A488-D5) or unlabeled D5 to cleave this substrate. Here specified concentrations of A647-E1E2 substrate were mixed with A488-D5 or unlabeled D5 and D123 in 40 mM MOPS binding buffer at indicated concentrations of KCl (100 mM–1 M) and MgCl2 (0–100 mM) in a final volume of 10 or 100 μL. Addition of either magnesium ions or A647-E1E2 was used to initiate the cleavage reaction, and the reaction was incubated for 2 h at 45°C. For kinetics studies, a 100-μL reaction mixture was set up and time points were taken at 0, 5, 15, 30, 45, 60, 75, 90, 120, 150, and 180 min. At these specific times, 8-μL aliquots of the reaction mixture were taken and quenched with 6-μL aliquots of stop buffer (100 mM EDTA in 10% glycerol) and placed in a dry ice ethanol bath. Samples were later run on a 15% denaturing gel at 23 W for 1.5–2 h. Immediately following electrophoresis, the gel was directly scanned, without any other workup, with a Bio-Rad pro-plus FX scanner using 488- and 635-nm external lasers, along with appropriate filter sets.
Data analysis
The apparent dissociation constant for D5 binding to D123 was determined assuming 1:1 bimolecular association using the equation:
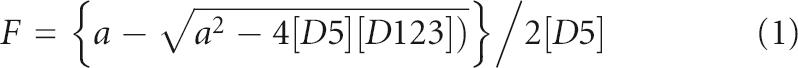 |
where a = ([D5] + [D123] + Kd).
The experimentally measured fraction bound (F) and total concentrations of D123 ([D123]) and D5 ([D5]) are used to extract the equilibrium dissociation constant (Kd). The fraction bound was computed from the ratio of the volumes of the free and the sum of the free and bound peak positions using the Quantity One software (Bio-Rad). Global background correction was applied using similar-sized rectangles for regions devoid of signal. The binding between wild-type PL D123 ribozyme and wild-type D5-PL was monitored by both FIGEMSA and equilibrium fluorescence anisotropy measurements. For both measurements, the normalized fraction bound was calculated and fitted using Equation 1.
Single turnover kinetics as a function of [D5]
Reactions were carried out at 42°C with saturating D123 (4 μM) and D5 (1 μM), 12.5 nM A647-E1E2 in 50 mM MgCl2, 40 mM MOPS-K (pH 7.5), and 1 M KCl. D5, D123, and substrate were heated separately at 90°C for 1 min and cooled to 42°C. D123 and substrate were combined and allowed to fold in MgCl2 for 20 min at 42°C. Reactions were initiated with addition of D5, and the reactions were quenched with 8 μL of stop solution (100 mM EDTA, 50% glycerol) added to 4-μL aliquots of the reaction mixture taken at specified times (0, 5, 15, 30, 45, 60, 75, 90, 120, 150, and 180 min). The RNAs were separated by denaturing PAGE (8 M urea, 15%, 19:1 acrylamide:bis), and the ratio of product and substrates at each time point was quantitated on a Molecular Imager Pro-Fx. The extent of the reaction was calculated from the fraction of product at each time point. Reaction rates were then obtained by fitting the extent of the reaction to a two- or three-parameter exponential equation. The apparent Michaelis-Menten curve was obtained by plotting the K obs as a function of [D5]. The maximal rate at saturating [D5] represents K cat, analogous to K chem, and the [D5] at half-maximal rate represents K M. The [D5] concentrations used were 0, 100, 250, 400, 500, 750, and 1000 nM. The kinetic constants are obtained from fitting of these data to a binding isotherm (Equation 1) as described previously (Pyle and Green 1994).
Single turnover kinetics as a function of [pH]
Reactions were carried out at 42°C and contained saturating D5 (1 μM) and D123 (4 μM each), 25.0 nM A647-E1E2, and 50 mM MgCl2; the 40 mM buffers used were MES-K (pH 5.0–6.9), MOPS-K (pH 6.5–7.3), and HEPES-K (pH 6.5–7.5). Trial reactions were carried out in different buffers at pH 6.8 to control for specific effects of buffers. None of the buffers used showed any specific effects. The pH was determined at the reaction temperature in the presence of reaction components except RNA. D5, D123, and substrate were heated separately in the presence of MgCl2 at 90°C for 1 min, cooled on the bench top to room temperature for 1–2 h. D123 and D5 were combined in the various buffers, and reactions were initiated with addition of substrate. The reactions were quenched with 4 μL of stop solution (100 mM EDTA, 50% glycerol) added to 8-μL aliquots of the reaction mixture taken at specified times and placed on a dry-ice bath. The RNAs were separated by denaturing PAGE, and the ratio of product and substrates at each time point was quantitated on a Molecular Imager Pro-Fx. RNAs were also heated to 90°C in the absence of MgCl2 and cooled to bench top for 10 min before combining all the RNAs, and MgCl2 was added last to initiate reaction at 42°C. This order of addition did not influence the derived rate constants.
D123 NMR binding studies
Following extensive dialysis in water, D5 RNA at a concentration of 0.2 mM was lyophilized and exchanged twice in 99.99% D2O and dissolved in NMR buffer (40 mM MOPS buffer, 100 mM KCl at pH 6.5, 5 mM MgCl2, 99.99% D2O, trace sodium azide, and DSS). NMR experiments were performed on fully labeled D5 in the absence and presence of 5 mM MgCl2. Similarly, D123 was exchanged twice in 99.99% D2O and resuspended in NMR buffer. The optimal folding conditions found for the cleavage reaction were also used for the NMR experiments. The D5–D123 RNA complex was incubated for 2 h at 45°C prior to transferring to a Shigemi NMR tube. NMR experiments were run on a four-channel UNITYINOVA 500 spectrometer equipped with an actively shielded Z-axis gradient triple resonance probe. Two-dimensional 1H–13C HSQC/TROSY experiments selective for C1′–H1′, aromatic C2–H2, C5–H5, C6–H8, and C8–H8 regions (Brutscher et al. 2001; Dayie 2005) were acquired on each RNA sample. Titrations were performed to final total D123 concentrations of 0–0.22 mM. The D5 peaks within the D123–D5 complex are extremely broadened at 25°C with hardly any signal above the noise level. Performing the experiments at 45°C alleviated this problem because of decrease in line width of the resonances, consistent with the decrease in solvent viscosity predicted by Stokes's Law. Therefore all the D123 titrations were done at 45°C. To rule out nonspecific electrostatic binding and/or pre-melting at 45°C, the titration was repeated with a 42-nt tetraloop receptor sequence derived from the PL ribozyme. For the tetraloop receptor (5′-GUGAGCCUAAAGCUUCGCCUAUGGCUAGCGAAAGCGAAUACC-3′)–D5 interaction, we used 0.1 mM D5 and varying concentrations of the receptor (0, 0.05, 0.15 mM). Once again, these RNAs were identically folded as described above. All the NMR experiments described above were processed by NMRPipe (Delaglio et al. 1995) and analyzed by SPARKY (T.D. Goddard and D.G. Kneller, University of California, San Francisco). The combined ribose and base chemical shift perturbations were calculated as Δδ = [(ΔδH)2 + (ΔδC · χ)2]1/2, where ΔδH and ΔδC are the chemical shift changes (for 1H and 13C, respectively) observed in D5 on adding D123, and the scaling factor χ reflects the relative chemical shifts for both protons and carbons in each region (χ=0.42 and 0.26, respectively for the ribose and aromatic regions) (see, e.g., Seetharaman et al. 2006).
SUPPLEMENTAL DATA
Supplemental material is available from the corresponding author (please send an e-mail to dayiek@ccf.org message containing the keyword “D123_D5”).
ACKNOWLEDGMENTS
We thank F. Michel for the PL.LSU/2 intron constructs, X. Wang for making the D123 DNA constructs, and N. Eldho for assisting with NMR experiments and making the tetraloop receptor RNA. We are grateful to E. Christian for critical reading of the manuscript. NMR studies were carried out at the Cleveland Center for Structural Biology with support from the Cleveland Foundation, Ohio State Hayes Investment Trust Fund, and additional equipment funding from the NIH Shared Instrumentation Program of the National Center for Research Resources. This investigation was supported by the Cleveland Clinic and U.S. NSF grant MCB-0316783 (to K.T.D.) and a structural supplement to NIH grant GM55105 (to R.A.P.).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.137006.
REFERENCES
- Abramovitz D.L., Friedman R.A., Pyle A.M. Catalytic role of 2′-hydroxyl groups within a group II intron active site. Science. 1996;271:1410–1413. doi: 10.1126/science.271.5254.1410. [DOI] [PubMed] [Google Scholar]
- Adams P.L., Stahley M.R., Gill M.L., Kosek A.B., Wang J., Strobel S.A. Crystal structure of a group I intron splicing intermediate. RNA. 2004;10:1867–1887. doi: 10.1261/rna.7140504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baklanov M.M., Golikova L.N., Malygin E.G. Effect of DNA transcription of nucleotide sequences upstream to T7 promoter. Nucleic Acids Res. 1996;24:3659–3660. doi: 10.1093/nar/24.18.3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Shalom A., Moore M.J. Tri-partite assay for studying exon ligation by the aI5γ group II intron. Biochemistry. 2000;39:10207–10218. doi: 10.1021/bi000808w. [DOI] [PubMed] [Google Scholar]
- Berlier J.E., Rothe A., Buller G., Bradford J., Gray D.R., Filanoski B.J., Telford W.G., Yue S., Liu J., Cheung C.Y., et al. Quantitative comparison of long-wavelength alexa fluor dyes to cy dyes: Fluorescence of the dyes and their bioconjugates. J. Histochem. Cytochem. 2003;51:1699–1712. doi: 10.1177/002215540305101214. [DOI] [PubMed] [Google Scholar]
- Black D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- Bonen L., Vogel L. The ins and outs of group II introns. Trends Genet. 2001;17:322–331. doi: 10.1016/s0168-9525(01)02324-1. [DOI] [PubMed] [Google Scholar]
- Boudvillain M., Pyle A.M. Defining functional groups, core structural features and inter-domain tertiary contacts essential for group II intron self-splicing: A NAIM analysis. EMBO J. 1998;17:7091–7104. doi: 10.1093/emboj/17.23.7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudvillain M., de Lencastre A., Pyle A.M. A tertiary interaction that links active-site domains to the 5′ splice site of a group II intron. Nature. 2000;406:315–318. doi: 10.1038/35018589. [DOI] [PubMed] [Google Scholar]
- Boulanger S.C., Belcher S.M., Schmidt U., Dib-Hajj S.D., Schmidt T., Perlman P.S. Studies of point mutants define three essential paired nucleotides in the domain 5 substructure of a group II intron. Mol. Cell. Biol. 1995;15:4479–4488. doi: 10.1128/mcb.15.8.4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brutscher B., Boisbouvier J., Kupce E., Tisne C., Dardel F., Marion D., Simorre J.P. Base-type-selective high-resolution 13C edited NOESY for sequential assignment of large RNAs. J. Biomol. NMR. 2001;19:141–151. doi: 10.1023/a:1008340210079. [DOI] [PubMed] [Google Scholar]
- Burge C.B., Tuschl T., Sharp P.A. Splicing of precursors to mRNAs by the spliceosome. In: Gesteland R.F., et al., editors. The RNA world. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1999. pp. 525–560. [Google Scholar]
- Cate J.H., Gooding A.R., Podell E., Zhou K., Golden B.L., Kundrot C.E., Cech T.R., Doudna J.A. Crystal structure of a group I ribozyme domain: Principles of RNA packing. Science. 1996a;273:1678–1685. doi: 10.1126/science.273.5282.1678. [DOI] [PubMed] [Google Scholar]
- Cate J.H., Gooding A.R., Podell E., Zhou K., Golden B.L., Szewczak A.A., Kundrot C.E., Cech T.R., Doudna J.A. RNA tertiary structure mediation by adenosine platforms. Science. 1994b;273:1696–1699. doi: 10.1126/science.273.5282.1696. [DOI] [PubMed] [Google Scholar]
- Chanfreau G., Jacquier A. A catalytic site component common to both splicing steps of a group II intron. Science. 1994;266:1383–1387. doi: 10.1126/science.7973729. [DOI] [PubMed] [Google Scholar]
- Chu V.T., Liu Q., Podar M., Perlman P.S., Pyle A.M. More than one way to splice an RNA: Branching without a bulge and splicing without branching in group II introns. RNA. 1998;4:1186–1202. doi: 10.1017/s1355838298980724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa M., Michel F. Frequent use of the same tertiary motif by self-folding RNAs. EMBO J. 1995;14:1276–1285. doi: 10.1002/j.1460-2075.1995.tb07111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa M., Michel F. Tight binding of the 5′ exon to domain I of a group II self-splicing intron requires completion of the intron active site. EMBO J. 1999;18:1025–1037. doi: 10.1093/emboj/18.4.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa M., Fontaine J.M., Loiseaux-de Goer S., Michel F. A group II self-splicing intron from the brown alga Pylaiella littoralis is active at unusually low magnesium concentrations and forms populations of molecules with a uniform conformation. J. Mol. Biol. 1997;274:353–364. doi: 10.1006/jmbi.1997.1416. [DOI] [PubMed] [Google Scholar]
- Costa M., Christian E.L., Michel F. Differential chemical probing of a group II self-splicing intron identifies bases involved in tertiary interactions and supports an alternative secondary structure model of domain V. RNA. 1998;4:1055–1068. doi: 10.1017/s1355838298980670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa M., Michel F., Westhof E. A three-dimensional perspective on exon binding by a group II self-splicing intron. EMBO J. 2000;19:5007–5018. doi: 10.1093/emboj/19.18.5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels D.L., Michels W.J., Jr., Pyle A.M. Two competing pathways for self-splicing by group II introns: A quantitative analysis of in vitro reaction rates and products. J. Mol. Biol. 1996;256:31–49. doi: 10.1006/jmbi.1996.0066. [DOI] [PubMed] [Google Scholar]
- Davis J.H., Tonelli M., Scott L.G., Jaeger L., Williamson J.R., Butcher S.E. RNA helical packing in solution: NMR structure of a 30 kDa GAAA tetraloop-receptor complex. J. Mol. Biol. 2005;351:371–382. doi: 10.1016/j.jmb.2005.05.069. [DOI] [PubMed] [Google Scholar]
- Dayie K.T. Resolution enhanced homonuclear carbon decoupled triple resonance experiments for unambiguous RNA structural characterization. J. Biomol. NMR. 2005;32:129–139. doi: 10.1007/s10858-005-5093-6. [DOI] [PubMed] [Google Scholar]
- Delaglio F., Grzesiek S., Vuister G., Zhu G., Pfeiffer J., Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- de Lencastre A., Hamil S., Pyle A.M. A single active-site region for a group II intron. Nat. Struct. Mol. Biol. 2005;12:626–627. doi: 10.1038/nsmb957. [DOI] [PubMed] [Google Scholar]
- Dominguez C., Boelens R., Bonvin A.M. HADDOCK: A protein–protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- D'Silva L., Ozdowy P., Krajewski M., Rothweiler U., Singh M., Holak T.A. Monitoring effects of antagonists on protein–protein interactions with NMR spectroscopy. J. Am. Chem. Soc. 2005;127:13220–13226. doi: 10.1021/ja052143x. [DOI] [PubMed] [Google Scholar]
- D'Souza V., Dey A., Habib D., Summers M.F. NMR structure of the 101-nucleotide core encapsidation signal of the Moloney murine leukemia virus. J. Mol. Biol. 2004;337:427–442. doi: 10.1016/j.jmb.2004.01.037. [DOI] [PubMed] [Google Scholar]
- Fersht A. Structure and mechanism in protein science: A guide to enzyme catalysis and protein folding. Freeman; New York: 2000. [Google Scholar]
- Fontaine J.M., Rousvoal S., Leblanc C., Kloareg B., Loiseaux-de Goer S. The mitochondrial LSU rDNA of the brown alga Pylaiella littoralis reveals α-proteobacterial features and is split by four group IIB introns with an atypical phylogeny. J. Mol. Biol. 1995;251:378–389. doi: 10.1006/jmbi.1995.0441. [DOI] [PubMed] [Google Scholar]
- Fontaine J.M., Goux D., Kloareg B., Loiseaux-de Goer S. The reverse-transcriptase-like proteins encoded by group II introns in the mitochondrial genome of the brown alga Pylaiella littoralis belong to two different lineages which apparently coevolved with the group II ribosyme lineages. J. Mol. Evol. 1997;44:33–42. doi: 10.1007/pl00006119. [DOI] [PubMed] [Google Scholar]
- Franzen J.S., Zhang M., Peebles C.L. Kinetic analysis of the 5′ splice junction hydrolysis of a group II intron promoted by domain 5. Nucleic Acids Res. 1993;21:627–634. doi: 10.1093/nar/21.3.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fushman D., Xu R., Cowburn D. Direct determination of changes of interdomain orientation on ligation Use of the orientational dependence of 15N NMR relaxation in Abl SH(32) Biochemistry. 1999;38:10225–10230. doi: 10.1021/bi990897g. [DOI] [PubMed] [Google Scholar]
- Golden B.L., Gooding A.R., Podell E.R., Cech T.R. A preorganized active site in the crystal structure of the Tetrahymena ribozyme. Science. 1998;282:259–264. doi: 10.1126/science.282.5387.259. [DOI] [PubMed] [Google Scholar]
- Golden B.L., Kim H., Chase E. Crystal structure of a phage Twort group I ribozyme-product complex. Nat. Struct. Mol. Biol. 2005;12:82–89. doi: 10.1038/nsmb868. [DOI] [PubMed] [Google Scholar]
- Gordon P.M., Piccirilli J.A. Metal ion coordination by the AGC triad in domain 5 contributes to group II intron catalysis. Nat. Struct. Biol. 2001;8:893–898. doi: 10.1038/nsb1001-893. [DOI] [PubMed] [Google Scholar]
- Gordon P.M., Sontheimer E.J., Piccirilli J.A. Metal ion catalysis during the exon-ligation step of nuclear pre-mRNA splicing: Extending the parallels between the spliceosome and group II introns. RNA. 2000;6:199–205. doi: 10.1017/s1355838200992069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon P.M., Fong R., Deb S.K., Li N.S., Schwans J.P., Ye J.D., Piccirilli J.A. New strategies for exploring RNA's 2′-OH expose the importance of solvent during group II intron catalysis. Chem. Biol. 2004;11:237–246. doi: 10.1016/j.chembiol.2004.02.011. [DOI] [PubMed] [Google Scholar]
- Griffin E.A., Jr., Qin Z., Michels W.J., Jr., Pyle A.M. Group II intron ribozymes that cleave DNA and RNA linkages with similar efficiency, and lack contacts with substrate 2′-hydroxyl groups. Chem. Biol. 1995;11:761–770. doi: 10.1016/1074-5521(95)90104-3. [DOI] [PubMed] [Google Scholar]
- Gumbs O.H., Campbell A.M., Weil P.A. High-affinity DNA binding by a Mot1p–TBP complex: Implications for TAF-independent transcription. EMBO J. 2003;22:3131–3141. doi: 10.1093/emboj/cdg304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschlag D., Cech T.R. Catalysis of RNA cleavage by the Tetrahymena thermophila ribozyme. I. Kinetic description of the reaction of an active RNA substrate complementary to the active site. Biochemistry. 1990;29:10159–10171. doi: 10.1021/bi00496a003. [DOI] [PubMed] [Google Scholar]
- Hoopes B.C., LeBlanc J.F., Hawley D.K. Kinetic analysis of yeast TFIID-TATA box complex formation suggests a multi-step pathway. J. Biol. Chem. 1992;67:11539–11547. [PubMed] [Google Scholar]
- Jarrell K.A., Peebles C.L., Dietrich R.C., Romiti S.L., Perlman P.S. Group II intron self-splicing: Alternative reaction conditions yield novel products. J. Biol. Chem. 1988;263:3432–3439. [PubMed] [Google Scholar]
- Jones J.P., III, Kierlin M.N., Coon R.G., Perutka J., Lambowitz A.M., Sullenger B.A. Retargeting mobile group II introns to repair mutant genes. Mol. Ther. 2005;11:687–694. doi: 10.1016/j.ymthe.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Juneau K., Podell E., Harrington D.J., Cech T.R. Structural basis of the enhanced stability of a mutant ribozyme domain and a detailed view of RNA–solvent interactions. Structure. 2001;9:221–231. doi: 10.1016/s0969-2126(01)00579-2. [DOI] [PubMed] [Google Scholar]
- Kao C., Zheng M., Rudisser S. A simple and efficient method to reduce nontemplated nucleotide addition at the 3′ terminus of RNAs transcribed by T7 RNA polymerase. RNA. 1999;5:1268–1272. doi: 10.1017/s1355838299991033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly A.E., Ou H.D., Withers R., Dotsch V. Low-conductivity buffers for high-sensitivity NMR measurements. J. Am. Chem. Soc. 2002;124:12013–12019. doi: 10.1021/ja026121b. [DOI] [PubMed] [Google Scholar]
- Konarska M.M., Grabowski P.J., Padgett R.A., Sharp P.A. Characterization of the branch site in lariat RNAs produced by splicing of mRNA precursors. Nature. 1985;313:552–557. doi: 10.1038/313552a0. [DOI] [PubMed] [Google Scholar]
- Konforti B.B., Liu Q., Pyle A.M. A map of the binding site for catalytic domain 5 in the core of a group II intron ribozyme. EMBO J. 1998;17:7105–7117. doi: 10.1093/emboj/17.23.7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz J.R. Principles of fluorescence spectroscopy. 2d ed. Kluwer Academic/Plenum Publishers; New York: 1999. pp. 291–366. [Google Scholar]
- Li-Pook-Than J., Bonen L. Multiple physical forms of excised group II intron RNAs in wheat mitochondria. Nucleic Acids Res. 2006;34:2782–2790. doi: 10.1093/nar/gkl328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukavsky P.J., Kim I., Otto G.A., Puglisi J.D. Structure of HCV IRES domain II determined by NMR. Nat. Struct. Biol. 2003;10:1033–1038. doi: 10.1038/nsb1004. [DOI] [PubMed] [Google Scholar]
- Maschhoff K.L., Padgett R.A. The stereochemical course of the first step of pre-mRNA splicing. Nucleic Acids Res. 1993;21:5456–5462. doi: 10.1093/nar/21.23.5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel F., Umesono K., Ozeki H. Comparative and functional anatomy of group II catalytic introns—A review. Gene. 1989;82:5–30. doi: 10.1016/0378-1119(89)90026-7. [DOI] [PubMed] [Google Scholar]
- Michels W.J., Jr., Pyle A.M. Conversion of a group II intron into a new multiple-turnover ribozyme that selectively cleaves oligonucleotides: Elucidation of reaction mechanism and structure/function relationships. Biochemistry. 1995;34:2965–2977. doi: 10.1021/bi00009a028. [DOI] [PubMed] [Google Scholar]
- Milligan J.F., Uhlenbeck O.C. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol. 1989;180:51–62. doi: 10.1016/0076-6879(89)80091-6. [DOI] [PubMed] [Google Scholar]
- Moore M.J., Sharp P.A. Evidence for two active sites in the spliceosome provided by stereochemistry of pre-mRNA splicing. Nature. 1993;365:364–368. doi: 10.1038/365364a0. [DOI] [PubMed] [Google Scholar]
- Nilsen T.W. The spliceosome: The most complex macromolecular machine in the cell? Bioessays. 2003;25:1147–1149. doi: 10.1002/bies.10394. [DOI] [PubMed] [Google Scholar]
- Padgett R.A., Podar M., Boulanger S.C., Perlman P.S. The stereochemical course of group II intron self-splicing. Science. 1994;266:1685–1688. doi: 10.1126/science.7527587. [DOI] [PubMed] [Google Scholar]
- Panchuk-Voloshina N., Haugland R.P., Bishop-Stewart J., Bhalgat M.K., Millard P.J., Mao F., Leung W.Y., Haugland R.P. Alexa dyes, a series of new fluorescent dyes that yield exceptionally bright, photostable conjugates. J. Histochem. Cytochem. 1999;47:1179–1188. doi: 10.1177/002215549904700910. [DOI] [PubMed] [Google Scholar]
- Peebles C.L., Perlman P.S., Mecklenburg K.L., Petrillo M.L., Tabor J.H., Jarell K.A., Cheng H.L. A self-splicing RNA excises an intron lariat. Cell. 1986;44:213–223. doi: 10.1016/0092-8674(86)90755-5. [DOI] [PubMed] [Google Scholar]
- Peebles C.L., Zhang M., Perlman P.S., Franzen J.S. Catalytically critical nucleotide in domain 5 of a group II intron. Proc. Natl. Acad. Sci. 1995;92:4422–4426. doi: 10.1073/pnas.92.10.4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pley H.W., Flaherty K.M., McKay D.B. Model for an RNA tertiary interaction from the structure of an intermolecular complex between a GAAA tetraloop and an RNA helix. Nature. 1994;372:68–74. doi: 10.1038/372111a0. [DOI] [PubMed] [Google Scholar]
- Podar M., Perlman P.S., Padgett R.A. Stereochemical selectivity of group II intron splicing, reverse splicing, and hydrolysis reactions. Mol. Cell. Biol. 1995;15:4466–4478. doi: 10.1128/mcb.15.8.4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podar M., Perlman P.S., Padgett R.A. The two steps of group II intron self-splicing are mechanistically distinguishable. RNA. 1998;4:890–900. doi: 10.1017/s1355838298971643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyle A.M., Green J.B. Building a kinetic framework for group II intron ribozyme activity: Quantitation of interdomain binding and reaction rate. Biochemistry. 1994;33:2716–2725. doi: 10.1021/bi00175a047. [DOI] [PubMed] [Google Scholar]
- Qin P.Z., Pyle A.M. The architectural organization and mechanistic function of group II intron structural elements. Curr. Opin. Struct. Biol. 1998;8:301–308. doi: 10.1016/s0959-440x(98)80062-6. [DOI] [PubMed] [Google Scholar]
- Qin P.Z., Butcher S.E., Feigon J., Hubbell W.L. Quantitative analysis of the isolated GAAA tetraloop/receptor interaction in solution: A site-directed spin labeling study. Biochemistry. 2001;40:6929–6936. doi: 10.1021/bi010294g. [DOI] [PubMed] [Google Scholar]
- Riek R., Pervushin K., Wuthrich K. TROSY and CRINEPT: NMR with large molecular and supramolecular structures in solution. Trends Biochem. Sci. 2000;25:462–468. doi: 10.1016/s0968-0004(00)01665-0. [DOI] [PubMed] [Google Scholar]
- Sashital D.G., Cornilescu G., McManus C.J., Brow D., Butcher S. U2-U6 RNA folding reveals a group II intron-like domain and a four-helix junction. Nat. Struct. Mol. Biol. 2004;11:1237–1242. doi: 10.1038/nsmb863. [DOI] [PubMed] [Google Scholar]
- Schmidt U., Riederer B., Morl M., Schmelzer C., Stahl U. Self-splicing of the mobile group II intron of the filamentous fungus Podospora anserina (COI I1) in vitro. EMBO J. 1990;9:2289–2298. doi: 10.1002/j.1460-2075.1990.tb07400.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt U., Podar M., Stahl U., Perlman P.S. Mutations of the two-nucleotide bulge of D5 of a group II intron block splicing in vitro and in vivo: Phenotypes and suppressor mutations. RNA. 1996;11:1161–1172. [PMC free article] [PubMed] [Google Scholar]
- Seetharaman M., Eldho N.V., Padgett R.A., Dayie K.T. Structure of a self-splicing group II intron catalytic effector domain 5: Parallels with spliceosomal U6 RNA. RNA. 2006;12:235–247. doi: 10.1261/rna.2237806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla G.C., Padgett R.A. A catalytically active group II intron domain 5 can function in the U12-dependent spliceosome. Mol. Cell. 2002;9:1145–1150. doi: 10.1016/s1097-2765(02)00505-1. [DOI] [PubMed] [Google Scholar]
- Sigel R.K., Sashital D.G., Abramovitz D.L., Palmer A.G., Butcher S.E., Pyle A.M. Solution structure of domain 5 of a group II intron ribozyme reveals a new RNA motif. Nat. Struct. Mol. Biol. 2004;11:187–192. doi: 10.1038/nsmb717. [DOI] [PubMed] [Google Scholar]
- Smith D., Pace N.R. Multiple magnesium ions in the ribonuclease P reaction mechanism. Biochemistry. 1993;32:5273–5281. doi: 10.1021/bi00071a001. [DOI] [PubMed] [Google Scholar]
- Stringer J.A., Bronnimann C.E., Mullen C.G., Zhou D.H., Stellfox S.A., Li Y., Williams E.H., Rienstra C.M. Reduction of RF-induced sample heating with a scroll coil resonator structure for solid-state NMR probes. J. Magn. Reson. 2005;173:40–48. doi: 10.1016/j.jmr.2004.11.015. [DOI] [PubMed] [Google Scholar]
- Su L.J., Brenowitz M., Pyle A.M. An alternative route for the folding of large RNAs: Apparent two-state folding by a group II intron ribozyme. J. Mol. Biol. 2003;334:639–652. doi: 10.1016/j.jmb.2003.09.071. [DOI] [PubMed] [Google Scholar]
- Su L.J., Waldsich C., Pyle A.M. An obligate intermediate along the slow folding pathway of a group II intron ribozyme. Nucleic Acids Res. 2005;33:6674–6687. doi: 10.1093/nar/gki973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullenger B.A., Gilboa E. Emerging clinical applications of RNA. Nature. 2002;418:252–258. doi: 10.1038/418252a. [DOI] [PubMed] [Google Scholar]
- Swisher J., Duarte C.M., Su L.J., Pyle A.M. Visualizing the solvent-inaccessible core of a group II intron robozyme. EMBO J. 2001;20:2051–2061. doi: 10.1093/emboj/20.8.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Veen R., Arnberg A.C., van der Horst G., Bonen L., Tabak H.F., Grivell L.A. Excised group II introns in yeast mitochondria are lariats and can be formed by self-splicing in vitro. Cell. 1986;44:225–234. doi: 10.1016/0092-8674(86)90756-7. [DOI] [PubMed] [Google Scholar]
- van der Veen R., Kwakman J.H., Grivell L.A. Mutations at the lariat acceptor site allow self-splicing of a group II intron without lariat formation. EMBO J. 1987;6:3827–3831. doi: 10.1002/j.1460-2075.1987.tb02719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss N.R., Gerstein M. Calculation of standard atomic volumes for RNA and comparison with proteins: RNA is packed more tightly. J. Mol. Biol. 2005;346:477–492. doi: 10.1016/j.jmb.2004.11.072. [DOI] [PubMed] [Google Scholar]
- Wuthrich K. NMR studies of structure and function of biological macromolecules (Nobel lecture) Angew. Chem. Int. Ed. Engl. 2003;42:3340–3363. doi: 10.1002/anie.200300595. [DOI] [PubMed] [Google Scholar]
- Zhang L., Doudna J.A. Structural insights into group II intron catalysis and branch-site selection. Science. 2002;295:2084–2088. doi: 10.1126/science.1069268. [DOI] [PubMed] [Google Scholar]