

Abstract
The cabbage butterflies Pieris rapae and Pieris brassicae have unique enzymes, named pierisin-1 and -2, respectively, that catalyze the ADP-ribosylation of guanine residues of DNA, which has been linked with induction of apoptosis and mutation in mammalian cell lines. In the present study, we identified ADP-ribosylation activity targeting DNA in six kinds of edible clam. Similar to our observations with pierisin-1 and -2, crude extracts from the clams Meretrix lamarckii, Ruditapes philippinarum, and Corbicula japonica incubated with calf thymus DNA and β-NAD resulted in production of N2-(ADP-ribos-1-yl)-2′-deoxyguanosine. The DNA ADP-ribosylating protein in the hard clam M. lamarckii, designated as CARP-1, was purified by column chromatography, and its cDNA was cloned. The cDNA encodes a 182-aa protein with a calculated molecular mass of 20,332. The protein synthesized in vitro from the cDNA in a reticulocyte lysate exhibited the same ADP-ribosylating activity as that of purified CARP-1. Neither the nucleotide nor the deduced amino acid sequence of CARP-1 showed homology with pierisin-1 or -2. However, a glutamic acid residue (E128) at the putative NAD-binding site, conserved in all ADP-ribosyltransferases, was found in CARP-1, and replacement of aspartic acid for this glutamic acid resulted in loss of almost all ADP-ribosylating activity. CARP-1 in the culture medium showed no cytotoxicity against HeLa and TMK-1 cells; however, introduction of this protein by electroporation induced apoptosis in these cells. The finding of clam ADP-ribosylating protein targeting guanine residues in DNA could offer new insights into the biological significance of ADP-ribosylation of DNA.
Keywords: pierisin, N2-(ADP-ribos-1-yl)-2′-deoxyguanosine, NAD-binding site, receptor-binding domain, shellfish
ADP ribosylation is a posttranslational modification in which the ADP-ribose moiety of β-NAD is transferred to acceptor molecules. ADP-ribosyltransferases are classified by their reaction products. Mono-ADP-ribosyltransferases catalyze the transfer of a single ADP-ribose to a target molecule, whereas poly(ADP-ribose) polymerases, termed PARPs, initially transfer ADP-ribose to proteins, then catalyze the polymerization of ADP-ribose residues (1, 2).
Mono-ADP-ribosyltransferases are found in bacteria. Cholera toxin (CT) and pertussis toxin (PT) target the Gs- and Gi-type α-subunits of G proteins, respectively, whereas diphtheria toxin (DT) modifies the diphthamide of elongation factor-2 (3). There are also reports of mono-ADP-ribosyltransferases in mammals and avian species. For example, arginine-specific ADP-ribosyltransferases have been shown to be present as glycosylphosphatidylinositol-anchored or soluble forms; they have been shown to modify target proteins such as integrins and defensins (2, 4–6).
Pierisin-1 was initially identified as a cytotoxic substance from pupae of the cabbage butterfly Pieris rapae (7, 8). Pierisin-1 shares sequence similarities with ADP-ribosyltransferases such as CT and PT in its N-terminal region and with the lectin domain of the ricin superfamily in the C-terminal region (9–11). Unlike other ADP-ribosyltransferases, pierisin-1 targets the N2 amino groups of guanine residues in DNA to yield N2-(ADP-ribos-1-yl)-2′-deoxyguanosine (12). The C-terminal domain binds to glycosphingolipid receptors, such as globotriaosylceramide (Gb3) and globotetraosylceramide (Gb4), and is responsible for internalization of pierisin-1 into cells by its interaction with the lipid at the mammalian cell surface (10, 11). Pierisin-1 is cytotoxic to a variety of cell lines with IC50 values ranging from 0.043 to 270 ng/ml in 13 cell lines (10, 13). The amounts of Gb3 and Gb4 on cells largely determine their sensitivity (11), and apoptosis is induced by pierisin-1 via a mitochondrial pathway involving Bcl-2 and caspases (14). Moreover, mutation of the HPRT gene in Chinese hamster lung cells can be induced with low concentrations of pierisin-1, causing mainly G:C to C:G transversions (15). Interestingly, pierisin-1 mRNA is highly expressed in late-stage larvae, and the protein is accumulated in fat bodies, where it persists during pupation, suggesting that pierisin-1 may function during metamorphosis in P. rapae (16). Pupae of Pieris brassicae and Pieris napi, which belong to the genus Pieris, contain a cytotoxic activity against mammalian cell lines similar to P. rapae (7). Moreover, pierisin-2, with an amino acid sequence 91% identical to that of pierisin-1, has been found in P. brassicae (13). Pierisin-2 also targets DNA, and the structure of the DNA adduct produced by pierisin-2 is the same adduct produced with pierisin-1 (17).
To study the biological importance of DNA ADP-ribosylation, we attempted to identify the distribution of DNA ADP-ribosylating activities in various species, including insects, fish, and mammals. During this screening, we found that six kinds of edible clams have probable DNA ADP-ribosylating activity. In the present study, ADP-ribosylated DNA adducts produced by crude extracts from three kinds of clams were analyzed; the structure of the adduct was determined to be N2-(ADP-ribos-1-yl)-2′-deoxyguanosine, the same as that produced by pierisin-1 and -2. In addition, the DNA ADP-ribosylating protein from the hard clam Meretrix lamarckii was purified, and its cDNA was cloned. The gene encodes a 182-aa protein whose deduced amino acid sequence shares regional sequence similarity with the NAD-binding pocket of other ADP-ribosyltransferases. The possible biological significance of DNA ADP-ribosylation by this clam-derived protein is discussed here.
Results
Screening for DNA ADP-Ribosylating Activity in Various Shellfish.
Crude extracts from 14 shellfish were incubated with calf thymus DNA and β-[adenylate-32P]NAD (32P-NAD), the DNA was enzymatically digested, and the formation of ADP-ribosylated deoxyribonucleotide adducts was analyzed by TLC. Data for radioactivity of the adduct spots are shown in Table 1, and typical examples of formation of ADP-ribosylated DNA adducts are illustrated in Fig. 1. When crude extracts from M. lamarckii, Ruditapes philippinarum, and Corbicula japonica were reacted with DNA and 32P-NAD, adduct spots were clearly detected on TLC sheets, showing 1,790–6,900 photo-stimulated luminescence (PSL) [the unit of radioactivity for the Bio-Image Analyzer (BAS-2500, Fuji Photo Film)] per microgram of protein; the Rf values of the spots were 0.05, the same as that of the adduct produced by pierisin-1. The Rf values of other deoxyribonucleotides, ADP-ribose, and NAD were from 0.30 to 0.95. When these crude extracts were treated with 50 μg of pronase or 50 μg of proteinase K before the reaction with DNA and 32P-NAD, adduct spots were not detected, suggesting that these adducts were the results of enzymatic reactions. In the case of crude extracts from Mytilus galloprovincialis, Solen strictus, and Spisula sachalinensis, adduct spots were also detected with the same Rf value. These PSL values were 189–450 per microgram of protein. Crude extracts from Buccinum middendorffi, Crassostrea gigas, Atrina pinnata, and Buccinum inclytum gave faint adduct spots with the same Rf value. No detectable signals appeared when Turbo cornutus, Mactra chinensis, Tresus keenae, and Scapharca broughtonii were used as the protein sources.
Table 1.
Incorporation of radioactivity from 32P-NAD into DNA adducts by extracts from shellfish
| Species | Activity |
|---|---|
| M. lamarckii (hard clam) | 6,900 |
| R. philippinarum (littleneck clam) | 2,300 |
| C. japonica (brackishwater clam) | 1,790 |
| My. galloprovincialis (blue mussel) | 450 |
| So. strictus (razor clam) | 190 |
| Sp. sachalinensis (surf clam) | 189 |
| B. middendorffi* (whelk) | 74 |
| Cr. gigas (Pacific oyster) | 25 |
| Atrina pectinata (pen shell) | 18 |
| B. inclytum* (whelk) | 3 |
| T. cornutus* (turban shell) | <1 |
| Ma. chinensis (hen clam) | <1 |
| Tr. keenae (softshell) | <1 |
| Sc. broughtonii (blood clam) | <1 |
Activity was calculated by densitometric analysis of DNA adducts and is expressed as PSL per microgram of protein. Activity of pierisin-1 was ≈10 PSL per picogram of protein.
*Gastropods (all others are bivalves).
Fig. 1.

Detection of ADP-ribosylated DNA adducts formed with crude extracts of clams. Calf thymus DNA was reacted with 32P-NAD in the presence of 1 μg of crude extract protein. The DNA samples were spotted on TLC sheets with and without nuclease digestion, which were developed and subjected to autoradiography. Sample 1, no protein; sample 2, pierisin-1 (10 pg, positive control); sample 3, M. lamarckii; sample 4, R. philippinarum; sample 5, C. japonica; sample 6, My. galloprovincialis. The arrow indicates the Rf value of 0.05. +, with nuclease digestion; −, without nuclease digestion.
Identification of DNA Adducts Formed by Clam Extracts.
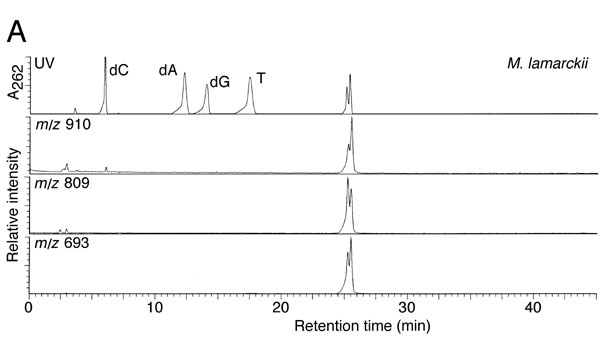
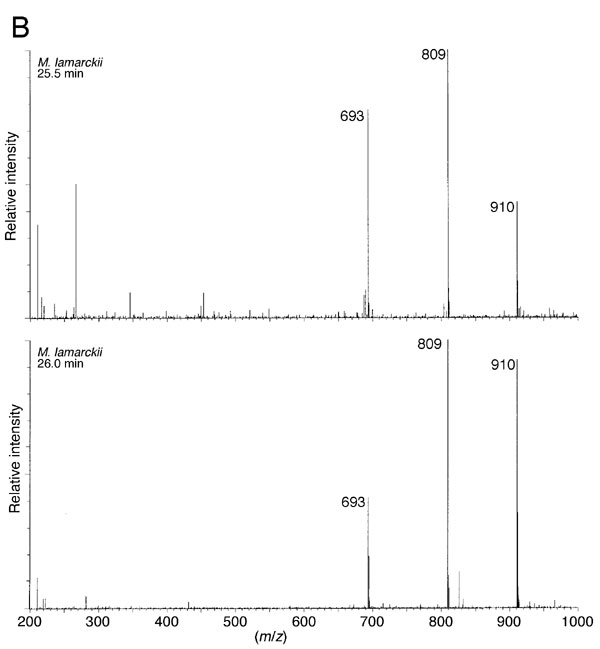
The crude extract from M. lamarckii, which showed the highest activity in the TLC assay, was fractionated by ammonium sulfate precipitation. Then, the adduct generated with the sample was analyzed with HPLC after dephosphorylation by bacterial alkaline phosphatase. As shown in Fig. 2A, the height of the dG peak was decreased; instead, two peaks were detected with retention times of 21.5 and 22.5 min, coincident with those of the α- or β-isoform of N2-(ADP-ribos-1-yl)-2′-deoxyguanosine, formed by pierisin-1. On the other hand, amounts of dC, dA, and T remained unchanged. UV spectra of the peak fractions at retention times of 21.5 and 22.5 min were exactly the same as those for N2-(ADP-ribos-1-yl)-2′-deoxyguanosine (Fig. 2B). Furthermore, liquid chromatography–electrospray ionization–MS analysis showed the compounds in these peak fractions to have a molecular ion peak at m/z 809, an ion peak at m/z 693 arising from the loss of a deoxyribose moiety, and an ion peak at m/z 910 corresponding to a triethylamine addition, derived from the HPLC eluent, to the parent mass at m/z 809 (see Fig. 6, which is published as supporting information on the PNAS web site). Thus, N2-(ADP-ribos-1-yl)-2′-deoxyguanosine was proven to be generated through the reaction of calf thymus DNA and β-NAD in the presence of the M. lamarckii extract. Moreover, N2-(ADP-ribos-1-yl)-2′-deoxyguanosine was shown to be formed by extract samples from R. philippinarum and C. japonica under similar conditions (data not shown).
Fig. 2.

Analysis of reaction products formed by DNA, clam protein, and β-NAD. (A) HPLC elution patterns of hydrolysate of DNA incubated with pierisin-1 (Top), with partially purified protein from M. lamarckii (Middle), or without protein (Bottom). These DNA samples were enzymatically hydrolyzed to deoxyribonucleosides and injected into a Develosil RPAQUEOUS column, and the eluate was monitored by measuring its UV absorbance at 262 nm. Arrows indicate the peaks coincident with N2-(ADP-ribos-1-yl)-2′-deoxyguanosine. (B) UV absorption spectra with a photodiode array detector of the compounds (I–IV) in the peak fractions at retention times of 21.5 and 22.5 min presented in A.
Purification of DNA ADP-Ribosylating Protein from the Hard Clam M. lamarckii.
Table 2 summarizes purification steps for DNA ADP-ribosylating protein from the hard clam M. lamarckii. The extract was prepared from the soft tissue of five clams (150 g), excluding gills, mantles, and digestive glands, which have much less DNA adduct formation activity. Most of the activity was detected in the precipitate obtained at 60–90% saturation of ammonium sulfate. Adduct formation activity of this fraction was observed at almost the same level at pH from 7.5 to 8.5 at 37°C. The activity increased in a time-dependent manner up to 4 h. The active fraction was then purified on a CM52 column. Fifteen percent of the activity of the starting material was eluted with 20 mM sodium phosphate buffer (pH 6.0), and the remaining 43% of the activity was subsequently recovered with a linear gradient of 0–400 mM NaCl in 20 mM sodium phosphate buffer. The active fractions, eluted with ≈80 mM NaCl in 20 mM sodium phosphate buffer, were further purified on Sephacryl and Mono-S columns. SDS/PAGE and an activity gel assay of the purified protein revealed a single predominant band, corresponding to a molecular mass of 20 kDa (Fig. 3). At the steps of Sephacryl and Mono-S column chromatography, adduct formation activity was observed only in fractions containing the M. lamarckii 20-kDa protein, but not in other fractions. From five clams, 0.48 mg of purified protein were obtained, and the adduct formation activity was increased 870-fold as compared with the extract. Moreover, the adduct formed by the purified protein was identical to N2-(ADP-ribos-1-yl)-2′-deoxyguanosine by HPLC, UV spectrum, and liquid chromatography–MS analyses (data not shown). These data indicate that the purified protein was the major active component present in the clam extract.
Table 2.
Purification of ADP-ribosylating protein from the hard clam M. lamarckii
| Step | Total protein, mg | Recovery of activity, % | Purification, fold |
|---|---|---|---|
| Extract | 3,175 | 100 | 1 |
| Ammonium sulfate fractionation | 439 | 80 | 6 |
| Carboxymethyl-cellulose chromatography | 14.5 | 43 | 96 |
| Sephacryl chromatography | 1.82 | 34 | 592 |
| Mono-S chromatography | 0.48 | 11 | 870 |
Recovery and purification values were calculated from the results of an assay measuring incorporation of radioactivity from 32P-NAD into DNA; all assays were performed in duplicate. The purification was repeated six times, and these experiments gave almost the same results. A typical example is shown.
Fig. 3.

SDS/PAGE of purified CARP-1. One microgram of purified protein was separated by SDS/PAGE by using 15% gels, which were stained with Coomassie brilliant blue R-250 (A) or assessed for activity by using the activity gel assay (B).
Because it is likely that clams commonly contain DNA ADP-ribosylating activity, we propose here that the protein purified from M. lamarckii be named CARP (clam ADP-ribosylating protein)-1.
Nucleotide Sequence, Cloning, and Expression of the CARP-1 Gene.
Using RNA from the M. lamarckii foot muscle and DNA probes corresponding to internal amino acid sequences of digested peptides, we obtained a cDNA encoding the entire CARP-1. The nucleotide sequence of the isolated cDNA is shown in Fig. 4. The full-length cDNA consisted of 853 bp accompanied by a 5′ anchor sequence and a polyadenylate stretch. The first ATG began at nucleotide position 65 and ended with a TAG termination codon at nucleotide position 611. A possible polyadenylation signal (AATAAA) was found at nucleotides 834–839, close to the poly(A) sequence, suggestive of a eukaryotic sequence. The putative protein encoded by the ORF comprised 182 aa with a calculated molecular weight of 20,332. Moreover, the nine peptide sequences of digests of purified clam ADP-ribosylating protein were found to be identical to parts of the deduced amino acid sequence from the ORF (Fig. 4). From these results, the ORF in the isolated cDNA was concluded to be the coding region of the CARP-1 gene.
Fig. 4.

Nucleotide and deduced amino acid sequences of the CARP-1 cDNA determined by PCR direct sequencing. The ORF is located between nucleotides 65 and 613. All determined internal amino acid sequences of the purified CARP-1 protein are indicated by underlining. A possible polyadenylation signal is indicated by #. Possible conserved amino acids are shown in bold type.
To obtain a clone, a PCR-amplified CARP-1 coding sequence was inserted into the pMAL-p2x plasmid vector. The product of the CARP-1 gene would be highly toxic to Escherichia coli, as expected from our studies of pierisin-1 and -2, and we inserted the PCR-amplified sequence in the vector in an opposite direction to the tac promoter. Ten clones were sequenced, and no clone had any nucleotide substitution, so one was used for CARP-1 expression in vitro. The molecular mass of the in vitro expressed protein was ≈20 kDa on SDS/PAGE, the same as that of purified native CARP-1. When the expressed protein was reacted with DNA and 32P-NAD, an adduct spot was clearly detected on the TLC sheet with an Rf value of 0.05, the same as that of the adduct produced by purified native protein. Moreover, when the DNA adduct was further analyzed by HPLC after dephosphorylation by bacterial alkaline phosphatase, most radioactivity was recovered at retention times of 21.0–23.0 min, coincident with those of N2-(ADP-ribos-1-yl)-2′-deoxyguanosine (see Fig. 7, which is published as supporting information on the PNAS web site). Thus, the expressed protein apparently produced N2-(ADP-ribos-1-yl)-2′-deoxyguanosine residues. The results indicate that functionally active protein was expressed in vitro from the isolated cDNA clone.
Although the purified native protein did not show any cytotoxic effects to HeLa cells at 20 μg/ml in the culture medium, the cell proliferation rate was clearly decreased by 77%, 58%, and 26% when 3, 5, and 12.5 μg/ml protein was introduced into cells by electroporation, respectively. Similarly, in vitro expressed CARP-1 protein exhibited cytotoxicity against HeLa cells with, but not without, electroporation. These electroporated cells exhibited nuclear condensation and DNA fragmentation at 18 h after electroporation, suggesting that cell death was by apoptosis, as shown for the N-terminal domain of pierisin-1 (10). Similar results were obtained with TMK-1 cells.
Although overall homology of the CARP-1 protein was not detectable with other proteins in the DNA Data Bank of Japan by BLAST and FASTA search, Arg-12, Ser–Thr–Thr at a position of 46–48, and Glu-128 were detected in the CARP-1 sequence; these amino acids comprise a motif for ADP-ribosyltransferase activity (Fig. 5).
Fig. 5.

Alignment of the deduced amino acid sequence of the homologous region of CARP-1 with pierisin-1 and ADP-ribosylating toxins. PT-S1, the S1 subunit of PT (GenBank accession no. P04977); pierisin-1 (9); CT-A, the A subunit of CT (GenBank accession no. 1001196A). The conserved arginine, Ser–Thr–Ser/Thr motif, and glutamic acid residues are boxed. Completely conserved amino acids are marked by asterisks.
To examine whether ADP-ribosylation is involved in the formation of the DNA adducts, the glutamic acid residues at position 127 or 128 were replaced with aspartic acid (E127D or E128D, respectively) by site-directed mutagenesis, and the mutated or nonmutated clones were transcribed and translated in vitro. The efficiencies of protein expression from the nonmutated control clone and mutated clone were almost the same, as judged by SDS/PAGE after translation with [35S]methionine. While the E127D mutant had 94% of the activity of the wild-type protein, the E128D mutant had only 1% of original activity, suggesting that Glu-128 is the essential glutamic acid for the DNA ADP-ribosylating activity.
Discussion
In the present study, we investigated the presence of DNA ADP-ribosylating activity in 14 shellfish that are commonly consumed in many countries, and we found that six clams contain probable DNA ADP-ribosylating activity. Among these, M. lamarckii, R. philippinarum, and C. japonica were demonstrated to possess activity to yield N2-(ADP-ribos-1-yl)-2′-deoxyguanosine residues from β-NAD and DNA, as in the case of pierisin-1 and -2 from the cabbage butterfly. A 20-kDa protein, named CARP-1, purified from M. lamarckii, and in vitro translated CARP-1 obtained from cloned cDNA both exhibited DNA ADP-ribosylating activity. The DNA ADP-ribosylating activity of crude extract from M. lamarckii is 6,900 PSL per microgram, and this value is ≈1/2 and ≈1/20th of those from larva and pupa of P. rapae, respectively. The DNA ADP-ribosylating activity of CARP-1 is ≈1/20th of pierisin-1. The deduced amino acid sequence of CARP-1 shows little homology with pierisin-1 and -2. Site-directed mutagenesis at a putative NAD-binding site, Glu-128, further demonstrated that CARP-1 is a DNA ADP-ribosylating protein but not a homologue of pierisins, although their targets and the resultant adduct structure are the same. The DNA ADP-ribosylating activity of CARP-1 was not observed with ADP-ribose, and NADase activity was not detected with CARP-1 (data not shown).
The reaction mechanisms of ADP-ribosyltransferases have been studied mostly for CT, PT, diphtheria toxin, Pseudomonas aeruginosa exotoxin A, and E. coli heat-labile enterotoxin (3, 18). X-ray crystallography of these toxins has revealed that they share similar structures in their reaction centers. The glutamic acid residue, which is conserved and necessary in all known ADP-ribosyltransferases, serves as an NAD-binding site. A highly conserved arginine residue and a Ser–Thr–Ser/Thr motif also exist among CT, PT, and heat-labile enterotoxin. The arginine residue is thought to maintain the structure of the reaction pocket, and the Ser–Thr–Ser/Thr motif is considered to construct a β-strand–α-helix structure and maintain the reaction cavity (19, 20). The presence in CARP-1 of Arg-12, Ser–Thr–Thr at positions 46–48, and Glu-128 supports conservation of the catalytic site motif of ADP-ribosyltransferases like CT, PT, and heat-labile enterotoxin. Moreover, the fact that CARP-1 and pierisin-1 or -2 share little distinct amino acid homology suggests that CARP-1 and pierisins are not derived from any common ancestor gene. The existence of evolutionarily different DNA ADP-ribosylating proteins suggests that other kinds of DNA ADP-ribosylating proteins might also be present in various organisms.
Another characteristic of CARP-1 is the probable lack of receptor-binding domains. Bacterial ADP-ribosylating toxins are classified into three groups depending on the existence of receptor-binding domains. A/B-toxins consist of catalytically active portions (A) and receptor-binding portions (B) in the same molecules, whereas binary toxins have catalytically active portions and receptor-binding portions in separate molecules. A-only toxins have no receptor-binding domains and need an incorporation mechanism for target cells. Pierisin-1 has a typical A/B-toxin structure. The C-terminal 71-kDa domain of this protein shares homology with the lectin domain of the ricin superfamily. Although the in vitro synthesized N-terminal 27-kDa domain itself shows no cytotoxicity against HeLa cells, it is toxic when the N-terminal portion is transferred into HeLa cells by electroporation. In the case of CARP-1, the cDNA sequence suggested that the protein is an ADP-ribosyltransferase with no receptor-binding domain. The estimated molecular mass by Sephacryl S-200 gel filtration was ≈20 kDa, suggesting the absence of a receptor-binding subunit in the native form of this protein. In addition, neither the crude extract from M. lamarckii nor purified CARP-1 showed cytotoxicity against various mammalian cell lines. However, purified and in vitro translated CARP-1 induced apoptosis when they were incorporated into HeLa and TMK-1 cells by electroporation, under the same conditions, as shown for the N-terminal domain of pierisin-1. We also found DNA ADP-ribosylating activity in a flow-through fraction on CM52 column chromatography, but gel filtration experiments showed that the molecular mass of the active protein in this flow-through fraction was almost the same as that of purified CARP-1. Our data thus indicate that M. lamarckii has NAD+:DNA (guanine-N2-)-ADP-ribosyltransferase, which lacks a receptor-binding domain needed to enter into mammalian cells.
Because the cDNA isolated in this study showed a typical eukaryotic structure with a polyadenylation signal and a poly(A) stretch, it is unlikely that the isolated CARP-1 gene was harbored in the genome of marine prokaryotic microorganisms. Moreover, DNA adduct formation activity of crude extracts prepared from gills, mantles, and digestive gland, which are expected to contain marine microorganisms, was much less than other soft tissues, suggesting that CARP-1 is produced by the clam itself when necessary. The probable lack of a receptor-binding domain in the protein suggests that CARP-1 might act on intracellular DNA to induce cell death of the clam itself. There is also a possibility that CARP-1 may enter into other cells with an associating protein(s) and thereby exert DNA-damaging activity. It is important to study the presence of CARP-1 in various stages of the clam, including oocytes, planktonic larva, and juvenile shellfish. It is also crucial to detect ADP-ribosylated deoxyguanosine residue of DNA in the clam itself. The biological significance of DNA ADP-ribosylating proteins in clams remains to be elucidated.
The presence of CARP-1-like activity in a variety of clams also offers clues to understanding the nature of DNA ADP-ribosylating activity in the clam. At the same time, we should also point out the potential health concerns with eating these clams. Some species, including mussels and oysters, are eaten without being cooked in many countries. It is essential to study the in vivo toxicity of CARP-1 in animals as is the case of pierisin-1.
Materials and Methods
Materials.
All of the shellfish, collected on the Pacific coast of Japan, were obtained from commercial markets in Tokyo and transported alive to the laboratory. For mRNA extraction, transported M. lamarckii were kept alive in sterile artificial sea water until use. Phosphodiesterase II and micrococcal nuclease were purchased from Worthington (Lakewood, NJ); calf thymus DNA, bacterial alkaline phosphatase, and β-NAD were from Sigma (St. Louis, MO); and 32P-NAD was from PerkinElmer (Boston, MA). ExTaq polymerase was from Takara Bio (Ohtsu, Japan), and KOD-plus polymerase was from Toyobo (Osaka, Japan). Trypsin and oligonucleotides were purchased from Invitrogen (Carlsbad, CA), and CEL 300 PEI polyethyleneimine-cellulose TLC sheet was from Macherey-Nagel (Düren, Germany). Trypsin-treated pierisin-1 was prepared as previously described (21). All other materials were from Wako Pure Chemical (Osaka, Japan) unless otherwise noted.
Screening for DNA ADP-Ribosylating Activity in Extracts from 14 Shellfish.
The whole soft tissue of each specimen was homogenized with a mortar and pestle on ice in three volumes of 50 mM Tris·HCl (pH 7.5) supplemented with 1 mM DTT and then sonicated for 1 min. This homogenate was centrifuged at 14,000 × g for 5 min, and the supernatant, termed the crude extract, was stored at −80°C. Five micrograms of calf thymus DNA, 18.5 kBq of 32P-NAD, and 10 μM β-NAD were incubated with 1–50 μg of protein of crude extract from shellfish, equivalent to 0.03–1.5 mg of wet weight of the original soft tissue, in 50 μl of reaction buffer (50 mM Tris·HCl, pH 7.5/1 mM EDTA/50 mM NaCl/1 mM DTT) for 30 min at 37°C. Then 1 μg of proteinase K (Wako Pure Chemical) was added to the reaction, which was incubated for additional 30 min at 37°C. DNA was recovered by using phenol extraction, chloroform extraction, and ethanol precipitation with ammonium acetate salt. Recovered DNA was dissolved in 20 mM Bis-Tris·HCl (pH 6.5) containing 5 mM CaCl2, 2 units of micrococcal nuclease, and 0.02 units of phosphodiesterase II in a volume of 10 μl. Three microliters of the mixture were spotted immediately onto a TLC sheet to detect the background noise. The remainder of the DNA mixture was digested to deoxyribonucleoside 3′-phosphate by incubation at 37°C for 12 h, and 3-μl aliquots of the mixture were spotted onto the TLC sheet. After development using 6 M acetic acid, 0.1 M LiCl, and 3 M urea, the TLC sheet was exposed to a Fuji Imaging Plate (Fuji Photo Film, Tokyo, Japan), and the DNA adduct was detected with a Bio-Image Analyzer.
Analysis of Reaction Products Formed by Incubation of DNA and β-NAD in the Presence of Clam Extract.
Ammonium sulfate was added to crude extracts from M. lamarckii, R. philippinarum, and C. japonica to give 60–90% saturation; the precipitate was recovered by centrifugation. Ten milligrams of the ammonium sulfate-precipitated samples after dialysis against 50 mM Tris·HCl (pH 7.5) was incubated with 1 mg of calf thymus DNA and 20 μmol of β-NAD in 50 mM Tris·HCl (pH 7.5) (pH 8.5 for R. philippinarum), 1 mM EDTA, and 50 mM NaCl in a volume of 10 ml for 4 h at 37°C. DNA was recovered and digested to deoxyribonucleoside 3′-phosphate by incubation with 200 units of micrococcal nuclease, 2 units of phosphodiesterase II, and 5 mM CaCl2 in 2 ml of 20 mM Bis-Tris·HCl (pH 6.5) for 4 h at 37°C. The sample was further digested to deoxyribonucleosides by additional bacterial alkaline phosphatase (final concentration 1.5 units/ml), Tris base (40 mM), and ZnCl2 (5 mM) and incubation for 4 h at 37°C.
After centrifugation, aliquots of the reaction mixture were subjected to HPLC with a LC-10A system (Shimadzu, Kyoto, Japan) equipped with a SPD 10Avp photodiode array detector (Shimadzu) and a Develosil RPAQUEOUS column (4.6 × 250 mm; Nomura Chemical, Seto, Japan) at a flow rate of 1 ml/min at 40°C. The solvent system was as follows: 3.5% acetonitrile in 0.25% triethylamine-acetic acid (pH 7.0) from 0 to 15 min, and then a linear gradient to 16.8% acetonitrile in 0.25% triethylamine-acetic acid (pH 7.0) to 45 min.
Aliquots of the reaction mixture were also analyzed by electrospray ionization–MS by using a ZQ 2000 instrument (Micromass, Manchester, U.K.) equipped with an HP 1000 HPLC system (Hewlett–Packard, Palo Alto, CA).
Purification of DNA ADP-Ribosylating Protein from the Hard Clam M. lamarckii.
Crude extract, prepared from the soft tissue (150 g) excluding the gills, mantles, and digestive gland of five individuals of M. lamarckii, obtained from Kashima Shore, Japan, was fractionated with ammonium sulfate precipitation as described above. The precipitate was then dialyzed against 20 mM sodium phosphate buffer (pH 6.0) and applied to a CM52 carboxymethyl-cellulose column (25 × 150 mm; Whatman, Kent, U.K.). The column was eluted with 20 mM sodium phosphate buffer, and retained proteins were then eluted with 1 liter of linear gradient of 0–400 mM NaCl in 20 mM sodium phosphate buffer at a flow rate of 80 ml/h. Fractions containing ADP-ribosylating activity (286–319 ml) were combined, concentrated with Amicon Ultra-10k (Millipore, Bedford, MA), and applied to a Sephacryl S-200 gel filtration column (16 × 600 mm; Amersham Pharmacia, Uppsala, Sweden). Proteins were separated with 20 mM sodium phosphate buffer containing 150 mM NaCl at a flow rate of 30 ml/h, and fractions containing ADP-ribosylating activity (64–74 ml) were combined and concentrated with Amicon Ultra-10k, then subjected to FPLC equipped with a Mono-S HR 5/5 (Amersham Pharmacia) at a flow rate of 1 ml/min. The solvent system was as follows: linear gradient from 0 to 1 M NaCl over 50 min in 20 mM sodium phosphate buffer (pH 6.0). Active fractions (20–21 ml) were pooled and concentrated to 0.2 ml with Amicon Ultra-10k. All of the above procedures were carried out below 4°C, and purified protein was stored at −80°C.
For identification of DNA ADP-ribosylating activity and estimation of the molecular mass of the purified protein, an activity gel assay was carried out as previously described (21). Electroporation of the purified protein into human cervical carcinoma HeLa cells and human gastric carcinoma TMK-1 cells was performed as previously described (10). No cell damage was detected without the purified protein under these conditions. Cell damage was examined with WST-1 assay (10).
PCR Amplification, Sequencing, and Cloning of cDNA Encoding the Clam Protein.
Twenty micrograms of purified clam protein was digested with trypsin, and the digested peptides were separated by reverse-phase HPLC with a TSKgel ODS-80Ts column (2.0 × 250 mm; Tosoh, Shunan, Japan). Amino acid sequences of the isolated peptides were determined by a Procise 494 HT protein sequencer (PerkinElmer). Total RNA was extracted from the foot muscle of M. lamarckii using Isogen (Nippon Gene, Toyama, Japan). Full-length cDNA was synthesized from the total RNA preparation (2 μg) using a GeneRacer Kit (Invitrogen). The synthesized cDNA was then subjected to PCR amplification of clam peptide-specific sequences by ExTaq DNA polymerase using 1 μM sense and antisense degenerate primers corresponding to the clam peptides HAAQAFYWLSVK and EIHLAALTDTESSSEGYKENDYDVDT, or sense degenerate primers and 1 μM 3′ anchor primer (5′-CGCTACGTAACGGCATGACAGTG). To obtain a 5′-terminal cDNA fragment, PCR was performed with 1 μM each of the 5′ anchor primer (5′-GGACACTGACATGGACTGAAGGAGTA) and an antisense primer (5′-GCTTTTGTTCTCTTTTATCAGTGTATCT); the sequence was chosen from the internal cDNA fragment. The resulting 5′ cDNA fragment was subjected to PCR direct sequencing. Full-length clam protein cDNA was obtained by PCR amplification using a 5′ primer (5′-AGCGTTTACTTCCTCTTTCTCTTT), which is the sequence adjacent to the 5′ anchor primer, and a 3′ primer (5′-GACTATCGTCGTTGTTTATTTTGA), the sequence adjacent to the poly(A) stretch, with KOD-plus DNA polymerase. The amplified 0.9-kb fragment was subjected to PCR direct sequencing to determine the full-length cDNA sequence.
To construct a clone, this 0.9-kb PCR fragment was used as a template. The coding region of the clam protein gene, with 217 bp of 3′-flanking region, was amplified, and HindIII and EcoRI restriction sites (underlined) were introduced into the 5′ and 3′ ends, respectively, by using the primer pair 5′-CCTAAGCTTATGCCTGCTGGAAAATACCTATAC and 5′-GGAGAATTCGACTATCGTCGTTGTTTATTTTGA. The amplified fragment was inserted into the HindIII–EcoRI site of pMAL-p2x (New England Biolabs, Beverly, MA) in the opposite direction to the tac promoter.
In Vitro Transcription and Translation of Cloned cDNA.
In vitro transcription and translation of cDNA for the ADP-ribosylating protein was performed as previously described (13). Briefly, a 5′ primer containing a T7 promoter sequence at the 5′ end of the coding region (5′-TAATACGACTCACTATAGGGAGACCACCATGCCTGCTGGAAAATACCTATACCG) and the 3′ primer (5′-GACTATCGTCGTTGTTTATTTTGA) were used for PCR to introduce a T7 promoter sequence upstream of the coding sequence. The amplified fragment of the cDNA with the T7 promoter sequence was transcribed by MEGAscript (Ambion, Austin, TX) and then translated with a rabbit reticulocyte lysate (Ambion).
Site-Directed Mutagenesis.
A DNA fragment containing the altered sequence at the desired position was amplified from an intact cDNA subclone of the clam protein with the overlap extension PCR technique. To obtain overlapped 5′ and 3′ fragments, two separate PCRs were carried out using a 5′ primer containing a T7 promoter sequence at the 5′ end of the coding region (5′-TAATACGACTCACTATAGGGAGACCACCATGCCTGCTGGAAAATACCTATACCG) and a 3′ primer (primer A, 5′-CCTTAACCAAAACGTCTTCATATT) for the 5′ fragment, and a 5′ primer (primer B, 5′-AATATGAAGACGTTTTGGTTAAGG) and a 3′ primer (5′-GACTATCGTCGTTGTTTATTTTGA) for the 3′ fragment. Primers A and B are complementary to each other and contain mutations at the nucleotide corresponding to Glu128Asp. After purification using Recochip (Takara Bio), the purified 5′ and 3′ fragments were mixed together and used as the template for a second round of PCR to obtain a full-length mutated DNA fragment. This PCR product was sequenced to confirm the appropriate mutation. To obtain a DNA fragment containing a mutation at the nucleotide corresponding to Glu127Asp, the same protocol described above was used except the sequences of primers A and B were 5′-AACCAAAACTTCGTCATATTTTGC and 5′-GCAAAATATGACGAAGTTTTGGTT, respectively. The resultant DNA fragments were used as the template for the in vitro expression system described here.
Supplementary Material
Acknowledgments
We thank Dr. Kazuto Nishio (National Cancer Center Research Institute) for kindly performing cytotoxicity analysis of shellfish extract against various cell lines. T.N., M.Y., S.E., and Y.M. were the recipients of a Research Resident Fellowships from the Foundation for Promotion of Cancer Research in Japan during the performance of this work. This study was supported by a Grant-in-Aid for Cancer Research from the Ministry of Health, Labour, and Welfare, Japan.
Abbreviations
- CT
cholera toxin
- PT
pertussis toxin
- PSL
photo-stimulated luminescence.
Footnotes
Conflict of interest statement: No conflicts declared.
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AB266110).
References
- 1.Sugimura T. Prog Nucleic Acid Res Mol Biol. 1973;13:127–151. doi: 10.1016/s0079-6603(08)60102-6. [DOI] [PubMed] [Google Scholar]
- 2.Seman M, Adriouch S, Haag F, Koch-Nolte F. Curr Med Chem. 2004;11:857–872. doi: 10.2174/0929867043455611. [DOI] [PubMed] [Google Scholar]
- 3.Krueger KM, Barbieri JT. Clin Microbiol Rev. 1995;8:34–47. doi: 10.1128/cmr.8.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okazaki IJ, Moss J. Annu Rev Nutr. 1999;19:485–509. doi: 10.1146/annurev.nutr.19.1.485. [DOI] [PubMed] [Google Scholar]
- 5.Zhao Z, Gruszczynska-Biegala J, Zolkiewska A. Biochem J. 2005;385:309–317. doi: 10.1042/BJ20040590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paone G, Stevens LA, Levine RL, Bourgeois C, Steagall WK, Gochuico BR, Moss J. J Biol Chem. 2006;281:17054–17060. doi: 10.1074/jbc.M603042200. [DOI] [PubMed] [Google Scholar]
- 7.Koyama K, Wakabayashi K, Masutani M, Koiwai K, Watanabe M, Yamazaki S, Kono T, Miki K, Sugimura T. Jpn J Cancer Res. 1996;87:1259–1262. doi: 10.1111/j.1349-7006.1996.tb03141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sugimura T. Biochem Biophys Res Commun. 2002;296:1037–1038. doi: 10.1016/s0006-291x(02)02011-9. [DOI] [PubMed] [Google Scholar]
- 9.Watanabe M, Kono T, Matsushima-Hibiya Y, Kanazawa T, Nishisaka N, Kishimoto T, Koyama K, Sugimura T, Wakabayashi K. Proc Natl Acad Sci USA. 1999;96:10608–10613. doi: 10.1073/pnas.96.19.10608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kanazawa T, Watanabe M, Matsushima-Hibiya Y, Kono T, Tanaka N, Koyama K, Sugimura T, Wakabayashi K. Proc Natl Acad Sci USA. 2001;98:2226–2231. doi: 10.1073/pnas.051628898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsushima-Hibiya Y, Watanabe M, Hidari KI, Miyamoto D, Suzuki Y, Kasama T, Koyama K, Sugimura T, Wakabayashi K. J Biol Chem. 2003;278:9972–9978. doi: 10.1074/jbc.m212114200. [DOI] [PubMed] [Google Scholar]
- 12.Takamura-Enya T, Watanabe M, Totsuka Y, Kanazawa T, Matsushima-Hibiya Y, Koyama K, Sugimura T, Wakabayashi K. Proc Natl Acad Sci USA. 2001;98:12414–12419. doi: 10.1073/pnas.221444598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsushima-Hibiya Y, Watanabe M, Kono T, Kanazawa T, Koyama K, Sugimura T, Wakabayashi K. Eur J Biochem. 2000;267:5742–5750. doi: 10.1046/j.1432-1327.2000.01640.x. [DOI] [PubMed] [Google Scholar]
- 14.Kanazawa T, Kono T, Watanabe M, Matsushima-Hibiya Y, Nakano T, Koyama K, Tanaka N, Sugimura T, Wakabayashi K. Biochem Biophys Res Commun. 2002;296:20–25. doi: 10.1016/s0006-291x(02)00815-x. [DOI] [PubMed] [Google Scholar]
- 15.Totsuka Y, Kawanishi M, Nishigaki R, Matsukawa K, Yagi T, Takamura-Enya T, Watanabe M, Sugimura T, Wakabayashi K. Chem Res Toxicol. 2003;16:945–952. doi: 10.1021/tx034052o. [DOI] [PubMed] [Google Scholar]
- 16.Watanabe M, Nakano T, Shiotani B, Matsushima-Hibiya Y, Kiuchi M, Yukuhiro F, Kanazawa T, Koyama K, Sugimura T, Wakabayashi K. Comp Biochem Physiol. 2004;139A:125–131. doi: 10.1016/j.cbpb.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 17.Takamura-Enya T, Watanabe M, Koyama K, Sugimura T, Wakabayashi K. Biochem Biophys Res Commun. 2004;323:579–582. doi: 10.1016/j.bbrc.2004.08.132. [DOI] [PubMed] [Google Scholar]
- 18.Aktories K, Just I. Bacterial Protein Toxins. Berlin: Springer; 2000. [Google Scholar]
- 19.Domenighini M, Rappuoli R. Mol Microbiol. 1996;21:667–674. doi: 10.1046/j.1365-2958.1996.321396.x. [DOI] [PubMed] [Google Scholar]
- 20.Pallen MJ, Lam AC, Loman NJ, McBride A. Trends Microbiol. 2001;9:302–307. doi: 10.1016/s0966-842x(01)02074-1. discussion 308. [DOI] [PubMed]
- 21.Watanabe M, Enomoto S, Takamura-Enya T, Nakano T, Koyama K, Sugimura T, Wakabayashi K. J Biochem. 2004;135:471–477. doi: 10.1093/jb/mvh062. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}