

Abstract
Xeroderma pigmentosum (XP) C is involved in the recognition of a variety of bulky DNA-distorting lesions in nucleotide excision repair. Here, we show that XPC plays an unexpected and multifaceted role in cell protection from oxidative DNA damage. XP-C primary keratinocytes and fibroblasts are hypersensitive to the killing effects of DNA-oxidizing agents and this effect is reverted by expression of wild-type XPC. Upon oxidant exposure, XP-C primary keratinocytes and fibroblasts accumulate 8,5′-cyclopurine 2′-deoxynucleosides in their DNA, indicating that XPC is involved in their removal. In the absence of XPC, a decrease in the repair rate of 8-hydroxyguanine (8-OH-Gua) is also observed. We demonstrate that XPC–HR23B complex acts as cofactor in base excision repair of 8-OH-Gua, by stimulating the activity of its specific DNA glycosylase OGG1. In vitro experiments suggest that the mechanism involved is a combination of increased loading and turnover of OGG1 by XPC-HR23B complex. The accumulation of endogenous oxidative DNA damage might contribute to increased skin cancer risk and account for internal cancers reported for XP-C patients.
Keywords: cancer, DNA repair, keratinocytes, oxidative DNA damage, xeroderma pigmentosum
Introduction
Different types of damage require different repair mechanisms but several lines of evidence indicate that functional overlap between pathways occurs frequently to guarantee genomic stability. An example is the repair of oxidative DNA damage, which is a ubiquitous type of damage. Oxidative stress from ionizing radiations (IR) and oxidants, and normal cellular metabolism cause oxidative damage to bases and sugar-phosphates, as well as single- and double-strand breaks in DNA and formation of DNA–protein crosslinks (reviewed in Evans et al, 2004). Base oxidation products are cytotoxic and mutagenic, and several studies suggest that they may contribute to ageing and human pathologies, including neurodegeneration and cancer (reviewed in Evans et al, 2004). Base excision repair (BER) is the major mechanism for repair of DNA base damage by reactive oxygen species (ROS), but other repair pathways like NER, mismatch repair and recombination may be involved too. Specific DNA glycosylases are responsible for modified base removal by BER followed by formation of a single-stranded gap, filling of the gap and ligation by either short- or long-patch BER (reviewed in Fortini et al, 2003). The key role of BER in oxidative DNA damage repair is testified by the high risk of colorectal tumours in individuals carrying inherited mutations of the MYH DNA glycosylase that specifically excises adenine mispaired with 8-OH-Gua (reviewed in Sampson et al, 2005).
The major function of NER is to repair a wide range of lesions, including UV photoproducts, all resulting in large local distortions of the DNA structure. Impaired NER activity is associated with the rare autosomal recessive disorders, like xeroderma pigmentosum (XP), Cockayne syndrome (CS) and tricothiodystrophy (TTD) (reviewed in Lehmann, 2003). XP is a cancer-prone disorder, whereas CS and TTD are multisystemic diseases characterized by developmental and neurological abnormalities and premature ageing. NER operates through two subpathways that differ primarily in the recognition of base damage. In the transcription-coupled repair (TCR), it is the stalling of RNA polymerase II that triggers rapid repair of lesions from the transcribed strand of active genes. In the global genome repair (GGR), specific protein factors bind to damage sites and initiate repair on the genome overall (reviewed in Hoeijmakers, 2001; Hanawalt, 2002). XPC plays a key role as GGR-specific damage recognition protein. The human XPC protein in vivo is a heterotrimeric complex including HR23B and centrin 2 proteins (Araki et al, 2001). This complex binds to various types of NER lesions by recognizing alterations in the DNA structure more than the lesions themselves (Sugasawa et al, 2002). In agreement with its role as structure-specific DNA-binding factor, XPC has been involved in the recognition of structurally unrelated lesions. In vitro experiments have shown that XPC-containing complexes are able to excise oxidative DNA lesions such as free radical-induced 8,5′-cyclopurine 2′-deoxynucleosides (Brooks et al, 2000; Kuraoka et al, 2000) that are then repaired via NER. Recent evidence points to an additional, unexpected role of XPC in BER of endogenous lesions. The XPC–HR23B complex functionally interacts with 3-methyadenine DNA glycosylase (Miao et al, 2000) and thymine DNA glycosylase (Shimizu et al, 2003) that initiate BER of alkylation and deamination products, respectively. It has been suggested that the involvement of XPC in repair of oxidative DNA damage is responsible for the high frequency of spontaneous lung tumours recently described in XPC mutant mice (Hollander et al, 2005). Whether a defect in XPC leads to an altered response to ROS-generating agents in human cells, thus contributing to cancer development, is unknown.
Here, we analysed the role of XPC in the response to oxidizing agents by using human primary keratinocytes and fibroblasts from the same skin biopsy as a model cell system. We show that XPC protects human skin cells from the killing effects of X-rays and potassium bromate (KBrO3). We provide the first in vivo evidence that XPC is involved in the repair of 8,5′-cyclopurine 2′-deoxynucleosides and major oxidized DNA bases, 8-OH-Gua and 8-hydroxyadenine (8-OH-Ade). By in vitro reconstitution experiments, we uncover a new role of XPC as a cofactor for the efficient cleavage of 8-OH-Gua by OGG1. XPC complex might contribute to cancer prevention by participating in BER of 8-OH-Gua and other oxidative DNA lesions.
Results
XP-C keratinocytes and fibroblasts are hypersensitive to the killing effects of oxidizing agents
XP-C primary fibroblasts and keratinocytes were exposed to X-rays and cell sensitivity measured by a clonal assay. As shown in Figure 1A and B, both XP-C cell types were more sensitive to X-rays (approximately 2 fold on the basis of D37) than normal cells. Similarly to what previously reported for UVB (D'Errico et al, 2005), fibroblasts appear to be more sensitive to oxidative damage than keratinocytes. To determine whether X-ray sensitivity originated from oxidatively induced DNA lesions, cells were treated with KBrO3 that is known to produce ROS, lipid peroxidation and oxidative DNA damage (Ballmaier and Epe, 1995). A clear hypersensitivity to the killing effects of KBrO3 was observed in XP-C keratinocytes and fibroblasts when compared with normal cells (Figure 1C and D). This finding suggests that the hypersensitivity to IR of XP-C skin cells involves oxidative DNA damage.
Figure 1.
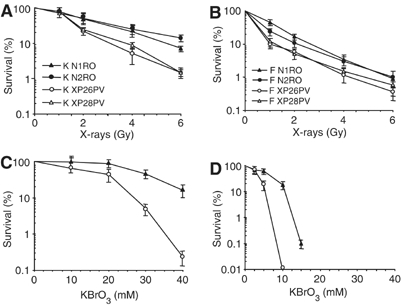
XP-C keratinocytes and fibroblasts are hypersensitive to the killing effects of X-rays and KBrO3. Survival of primary keratinocytes (A, C) and fibroblasts (B, D) from two normal (N1RO and N2RO, closed symbols) and two XP-C (XP26PV and XP28PV, open symbols) donors after X-rays and KBrO3 treatment. (A–B) Survival of keratinocytes and fibroblasts after exposure to X-rays. (C–D) Survival of keratinocytes and fibroblasts after exposure to KBrO3. Survival was determined by colony formation assay. The reported values are the mean of at least two independent experiments, each performed in triplicate with standard errors always <10%.
Lack of XPC is responsible for the hypersensitivity to DNA-oxidizing agents of human fibroblasts
To further investigate the correlation between absence of a functional XPC protein and hypersensitivity to oxidizing agents, normal and XP-C fibroblasts were analysed for their sensitivity to KBrO3 following transfection with an expression vector carrying the human EGFP-XPC fusion cassette (pEGFP-XPC) or an empty vector (pEGFP-C1). The efficiency of transfection was usually lower in XP-C (3–4%) as compared with normal cells (10–12%), independently of the type of vector used. The median EGFP intensity of both normal and XP-C cells expressing the XPC chimera was lower as compared with cells expressing the control EGFP, likely reflecting cytotoxic effects and/or decreased intrinsic fluorescence of the ectopically expressed protein. In each experiment, cell killing by KBrO3 was evaluated as percentage of dead cells 24 h post-treatment by enumerating propidium iodide (PI)-positive cells (i.e. red cells) in at least 100 transfected cells, identified as green cells expressing the EGFP protein (Figure 2A–D). Examples of cells showing nuclear accumulation of the green fluorescent signal of the XPC chimera (Figure 2A and C), and either sensitive (Figure 2B) or resistant (Figure 2D) to KBrO3 are shown. The overexpression of XPC in normal cells was slightly toxic (Figure 2E). At 24 h after treatment with two doses of KBrO3, the XP-C fibroblasts expressing normal XPC showed significant protection from cell killing (approximately 50% recovery; p<0.05) as compared with fibroblasts transfected with the empty vector (Figure 2E).
Figure 2.
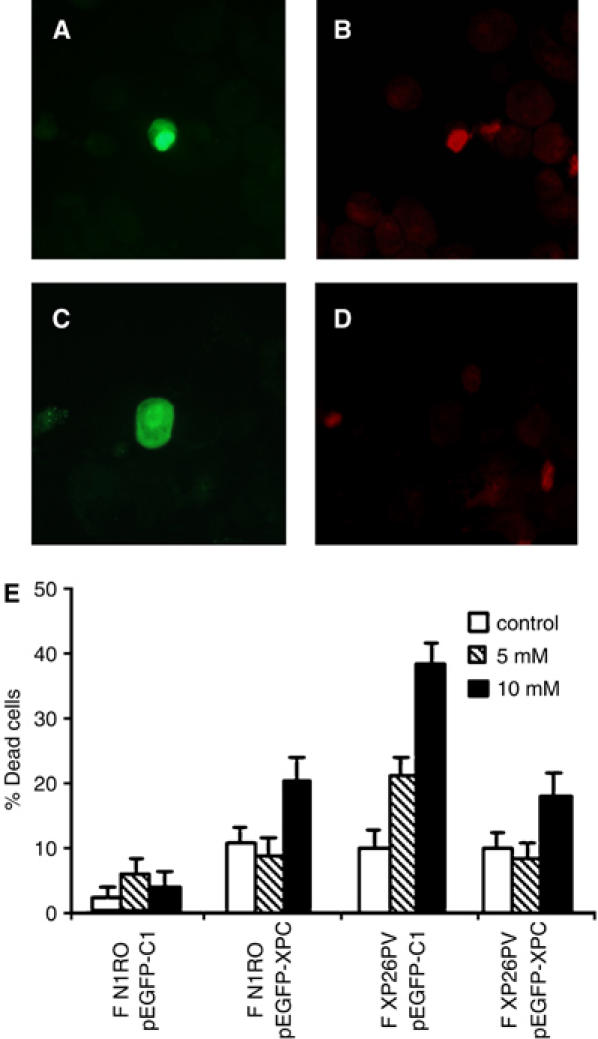
The lack of XPC is responsible for the hypersensitivity to KBrO3 of human fibroblasts. Normal and XP-C fibroblasts were transiently transfected with an expression vector encoding for the EGFP-XPC fusion protein (pEGFP-XPC) or with the empty vector encoding for EGFP only (pEGFP-C1). At 24 h after transfection, cells were either untreated (control) or exposed to different KBrO3 doses (5 and 10 mM). At 48 h after transfection, cells were stained with PI to label in red dead cells, cytocentrifuged on slides and then analysed by fluorescence microscopy. (A–D) The photographs are representative fields of normal fibroblasts expressing the EGFP-XPC chimera (green fluorescence) and stained with PI to evaluate their viability following KBrO3 treatment (red fluorescence). Examples of cells expressing the ectopic protein and either dead (A–B) or alive (C–D). (E) The histograms report the percentages of dead cells following KBrO3 treatment of normal (F N1RO) and XP-C (F XP26PV) cells transfected with either the pEGFP-XPC or the pEGFP-C1 plasmid DNA. The reported values are the mean of at least three independent experiments and standard deviations are indicated.
The restoration of normal sensitivity to KBrO3 in XP-C fibroblasts upon wild-type XPC expression provides evidence that XPC is directly involved in cell protection from the lethal effects of oxidizing agents.
XP-C keratinocytes are defective in the repair of oxidatively induced DNA lesions
The protection by XPC from oxidative stress-induced cell killing strongly suggested an involvement of this protein in the repair of oxidative DNA damage. Oxidative stress generates different kinds of lesions in DNA (reviewed in Evans et al, 2004). A unique reaction of hydroxyl radical-induced sugar radicals leads to the formation of 8,5′-cyclopurine 2′-deoxynucleosides that involves a concomitant damage to the base and sugar moieties of a purine nucleoside in DNA. One of these lesions, that is, (5′S)-8,5′-cyclo-2′-deoxyadenosine [(5′S)-cdA] has been shown to be repaired in vitro by NER (Kuraoka et al, 2000). The induction of (5′S)-cdA, (5′R)-8,5′-cyclo-2′-deoxyguanosine [(5′R)-cdG] and (5′S)-8,5′-cyclo-2′-deoxyguanosine [(5′S)-cdG] by a low dose of X-rays (5 Gy) and their repair was assayed by liquid chromatography/mass spectrometry (LC/MS) or gas chromatography/MS (GC/MS) in primary keratinocytes of two normal subjects (K N1RO, K N2RO) and two XP-C patients (K XP26PV, K XP28PV). Keratinocytes were selected for this analysis because the steady-state level of oxidative DNA base damage in this cell type is significantly lower than in fibroblasts (see below), thus allowing a better estimate of oxidatively induced DNA lesions. As shown in Figure 3A, the steady-state levels of selected 8,5′-cyclopurine 2′-deoxynucleosides were similar in normal and XP-C keratinocytes. Upon irradiation with 5 Gy, (5′S)-cdA, (5′R)-cdG and (5′S)-cdG were induced at statistically significant levels (P<0.05) and then repaired to completion in 2 h in normal keratinocytes, whereas they were not repaired in XP-C cells. This is the first evidence that XPC–NER complexes are required for the repair of 8,5′-cyclopurine 2′-deoxynucleosides in vivo, as previously shown in vitro (Brooks et al, 2000; Kuraoka et al, 2000).
Figure 3.
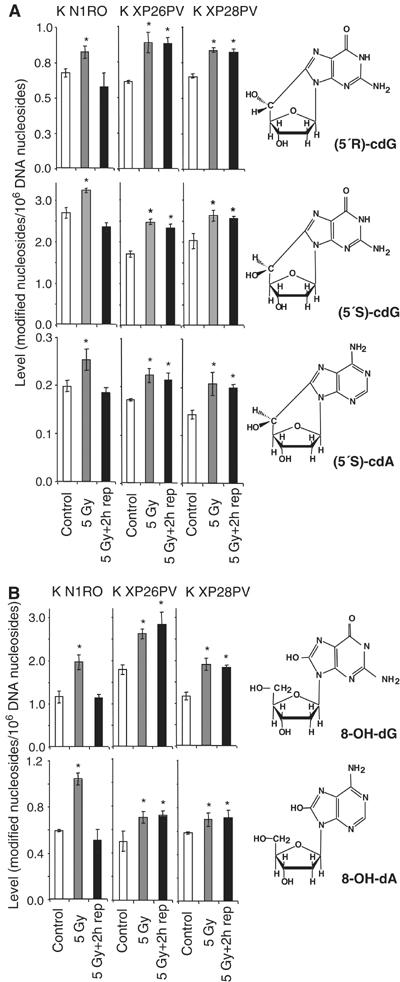
XP-C keratinocytes accumulate cyclopurines and oxidized DNA bases induced by X-rays. Levels of oxidatively modified nucleosides in DNA of keratinocytes from normal (K N1RO) and XP-C (K XP26PV and K XP28PV) donors were measured by LC/MS or GC/MS. For each data point, DNA samples isolated from three independent experiments for cell strain were used. The chemical structures of the modified nucleosides are illustrated. (A) Induction by X-rays and repair of (5′S)-cdA, (5′R)-cdG and (5′S)-cdG. (B) Induction by X-rays and repair of 8-OH-dG and 8-OH-dA. DNA samples were isolated from untreated cells (control), cells exposed to X-rays (5 Gy), and exposed to X-rays and allowed to repair for 2 h (5 Gy+2 h rep). The data represent the mean of three independent experiments and standard deviations are reported. Statistical analysis was performed using one-way analysis of variance. The stars indicate statistically significant differences between control and 5 Gy, or control and 5 Gy+2 h rep with a P<0.05.
The levels of other major oxidatively induced DNA bases induced by X-rays, 8-OH-Gua and 8-OH-Ade, were also measured by LC/MS as their nucleosides 8-hydroxy-2′-deoxyguanosine (8-OH-dG) and 8-hydroxy-2′-deoxyadenosine (8-OH-dA), respectively. As shown in Figure 3B, the steady-state levels were similar in normal and XP-C keratinocytes. A significant increase in the level of these products was detected immediately after X-ray exposure in both normal and XP-C keratinocytes. Normal keratinocytes repaired to completion both modified DNA bases within 2 h after treatment, as expected on the basis of their repair rate in vivo (Tuo et al, 2003). In contrast, the repair of both lesions was defective in XP-C keratinocytes.
The repair kinetics of 8-OH-dG was investigated in keratinocytes and fibroblasts following treatment with KBrO3 by using high-performance liquid chromatography/electrochemical detection (HPLC-ED) methodology. This technique was suited to estimate oxidatively induced 8-OH-dG in both cell types. As shown in Figure 4A and B, the steady-state level of 8-OH-dG was significantly higher (P<0.05) in fibroblasts than in keratinocytes. This effect is associated with a higher antioxidant capacity of keratinocytes as compared with fibroblasts (D'Errico et al, 2006). It should be noted that the background levels of 8-OH-dG measured in keratinocytes by HPLC-ED differ from those measured by LC/MS. This is not unexpected as two different methods have been used; however, the observed values are both within the range of the internationally accepted background levels of 8-OH-dG in cells (ESCODD, 2003; Collins et al, 2004). Therefore, the comparison of the results obtained by these two techniques can only be qualitative but not quantitative. Upon treatment with KBrO3 (40 mM), a statistically significant increase in DNA levels of 8-OH-dG was induced in both cell types (P<0.001) (Figure 4A and B), in agreement with a significant production of this damaged base by this agent (Ballmaier and Epe, 1995). At this high oxidant dose, the levels of induced 8-OH-dG in the two cell types were similar although the steady-state levels were significantly different. At lower KBrO3 doses (up to 20 mM), higher induced levels of 8-OH-dG were detected in fibroblasts than in keratinocytes (data not shown) in agreement with an additive effect of the background and induced levels. It is likely that under massive ROS production (high oxidant doses), the cellular antioxidant defence mechanisms are saturated, and the genetic background of the cell system does not influence the DNA oxidation levels anymore. When cells were allowed to repair after DNA damage, 8-OH-dG levels decreased significantly during the time course both in normal and XP-C keratinocytes and fibroblasts (P<0.001) (Figure 4A and B). However, in the absence of XPC, the levels of 8-OH-dG at each repair time were significantly higher than in normal cells in both cell types (P<0.001). At 2 h post-treatment, when most lesions were repaired in normal cells, residual damage was still present in XP-C cells. When the levels of 8-OH-dG at different repair times were expressed as percentage of the levels immediately after treatment (Figure 4C and D), the repair rate of XP-C keratinocytes and fibroblasts was slower than that of their normal counterparts at early repair times. This is remarkable in the case of keratinocytes that repaired 30% of 8-OH-dG lesions within the first 10 min after treatment but, when XPC is defective, they did not show any repair of this oxidized base within the same time frame (Figure 4C).
Figure 4.
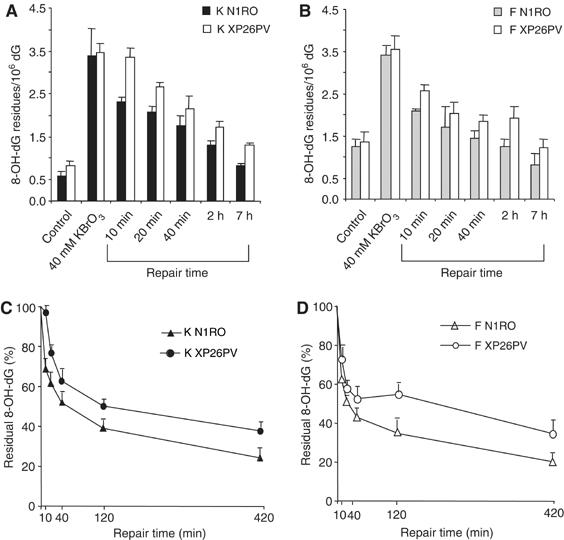
Repair of 8-OH-Gua is slower in XP-C keratinocytes and fibroblasts as compared with normal cells. 8-OH-dG levels were analysed in normal (N1RO) and XP-C (XP26PV) human fibroblasts and keratinocytes after exposure to 40 mM KBrO3 (1 h). Aliquots of cells were taken at the indicated times and the levels of 8-OH-Gua were measured by HPLC-ED. (A–B) 8-OH-dG levels in keratinocytes (A) and fibroblasts (B). (C–D) 8-OH-dG levels present at different repair times in keratinocytes (C) and fibroblasts (D) expressed as percentage of amounts at time 0.The data represent the mean of three independent experiments, and standard deviations (A–B) or standard errors (C–D) of the means are reported. Statistical analysis was performed using one- and two-way analyses of variance.
The contribution of XPC to 8-OH-Gua repair was, therefore, confirmed in both cell types. In addition, the analysis of the repair kinetics allowed estimating the relative contribution of the XPC-dependent mechanism to the overall repair of 8-OH-Gua. This discloses a role for XPC as cofactor in the repair of this oxidized base.
A deficiency in XPC leads to a reduction in cleavage of 8-OH-Gua-containing oligonucleotides by keratinocyte extracts
It is well known that 8-OH-Gua is repaired via BER pathway initiated by a specific DNA glycosylase, OGG1 (reviewed in Fortini et al, 2003; Evans et al, 2004). To gain insights into the mechanism of oxidative DNA base damage accumulation in XP-C keratinocytes, we examined the incision of 8-OH-Gua-containing duplex oligonucleotides by nuclear extracts of normal and XP-C keratinocytes. The incision activity was estimated by measuring the relative yield of uncleaved and cleaved products by PAGE analysis. A significant reduction (approximately 50%, P<0.05) in 8-OH-Gua cleavage was recorded when XP-C extracts were compared with normal extracts (Figure 5A). To test whether the XP-C defect was indeed responsible for the reduced cleavage efficiency of XP-C extracts, the assay was repeated in the presence of increasing concentrations of purified recombinant XPC–HR23B complex. As shown in Figure 5B, the correction of the defect was achieved in both cell extracts by adding 80 fmol of XPC–HR23B. The addition of the same amount of XPC–HR23B to wild-type extracts did not change the cleavage efficiency (data not shown).
Figure 5.
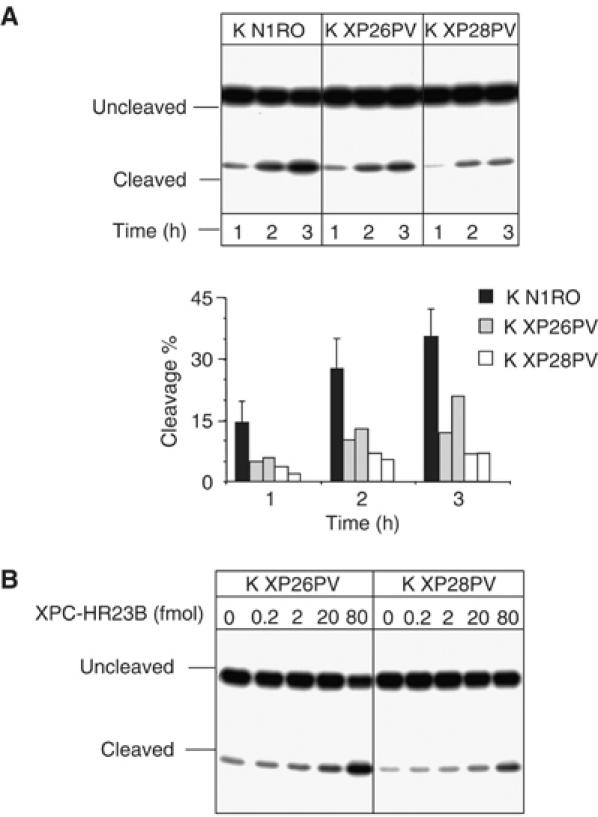
Cell extracts from XP-C keratinocytes are defective in 8-OH-Gua cleavage, but addition of purified XPC–HR23B restores normal cleavage activity. 30-mer duplex oligonucleotides (50 fmol) containing 8-OH-Gua were incubated with nuclear extracts (5 μg) of XP-C keratinocytes at 37°C for different periods of time as indicated. The 5′ end-labelled oligonucleotide was the 8-OH-Gua-containing strand. The products were separated by denaturing 20% PAGE. (A) Keratinocyte extracts from normal (K N1RO) and XP-C (K XP26PV and K XP28PV) donors. The relative percentages of 8-OH-Gua cleavage were obtained by electronic autoradiography of the gel (Instant Imager, Packard). Two or three independent extracts per cell strain were tested. (B) The 8-OH-Gua cleavage reaction by XP-C keratinocyte extracts was performed in the presence of varying concentrations of XPC–HR23B as indicated.
Thus, the defect in 8-OH-Gua cleavage is likely to account for its accumulation in the genome of XP-C keratinocytes.
XPC–HR23B stimulates the activity of OGG1
To determine the effect of XPC–HR23B on OGG1-catalysed 8-OH-Gua excision, a limiting amount of OGG1 (60 fmol) was incubated with an 8-OH-Gua-containing duplex oligonucleotide in the presence of varying amounts of XPC–HR23B. In this assay, the OGG1-processed DNA was treated with alkali to convert AP sites to nicks before denaturing PAGE. As shown in Figure 6A, the activity of OGG1 increased as a function of the concentration of XPC–HR23B (lanes 2 and 3). A three-fold stimulation of OGG1 activity was observed after 30 min incubation in the presence of 200 fmol of XPC–HR23B. Purified XPC–HR23B by itself showed no detectable DNA glycosylase activity on the 8-OH-Gua-containing oligomer (lane 4). No stimulation of OGG1 activity was detected upon addition to the cleavage reaction of another NER protein, XPA (lanes 8 and 9), or following addition of bovine serum albumin (BSA) (data not shown). To examine which subunit of the complex was responsible for the stimulation, HR23B was added alone. This subunit had a minor effect on OGG1 activity (lanes 5 and 6) and no relation with the dose was detected. The involvement of XPC in OGG1 stimulation was further demonstrated by the similar level of stimulation by XPC alone compared to that induced by the XPC–HR23B complex (Figure 6C, lanes 2 and 3). These findings clearly indicate that XPC is the subunit of the complex responsible for the stimulation of the OGG1 activity. The use of the alkali treatment does not allow to test which is the activity of OGG1 that is stimulated by XPC–HR23B. However, as the AP lyase activity of OGG1 is very weak, it is likely that the stimulation involves the DNA glycosylase activity of OGG1.
Figure 6.
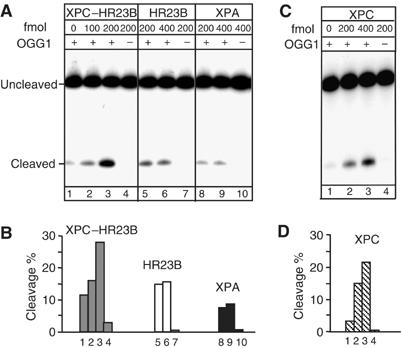
XPC–HR23B stimulates the activity of OGG1. DNA fragments (210 bp) containing a single 8-OH-Gua lesion were incubated with purified proteins, as indicated, at 37°C for 30 min. The 5′ end-labelled strand was the 8-OH-Gua-containing strand. The products were subjected to alkali treatment and separated by denaturing 8% PAGE. (A) The nicking assay was conducted in 10 μl (final volume) in the presence of 60 fmol of OGG1 and various concentrations of XPC–HR23B (lanes 2 and 3) or XPC-HR23B alone (lane 4); various concentrations of HR23B (lanes 5 and 6) or HR23B alone (lane 7); various concentrations of XPA (lanes 8 and 9) or XPA alone (lane 10), as indicated. (B) The amount of cleavage in each lane of (A) was evaluated by densitometric scanning of the autoradiography and expressed in arbitrary units following subtraction of the background. One representative experiment is shown. (C) The nicking assay was conducted in 10 μl (final volume) in the presence of 60 fmol of OGG1 and various concentrations of XPC (lanes 2 and 3) or XPC alone (lane 4). (D) The amount of cleavage in each lane of (C) was evaluated as in (B).
To investigate the mechanism by which the XPC complex stimulates OGG1 activity, the following experiment was performed. OGG1 was allowed to react with the 8-OH-Gua-containing oligonucleotides for 120 min, which is the time required to achieve a plateau in the cleavage reaction (Figure 7A, lanes 1–4). After this time frame, increasing concentrations of XPC–HR23B complex were added and incubated further for the indicated periods of time (lanes 5–16). As shown in Figure 7B, addition of XPC–HR23B drastically enhanced OGG1 activity and the stimulation was dependent on the concentration of XPC–HR23B complex added. It is well known that OGG1 remains sequestered by the resulting AP site following excision of 8-OH-Gua. These findings suggest that XPC–HR23B stimulates OGG1 activity by promoting its dissociation from the DNA substrate (likely the abasic site), thus allowing OGG1 to react with other 8-OH-Gua lesions.
Figure 7.
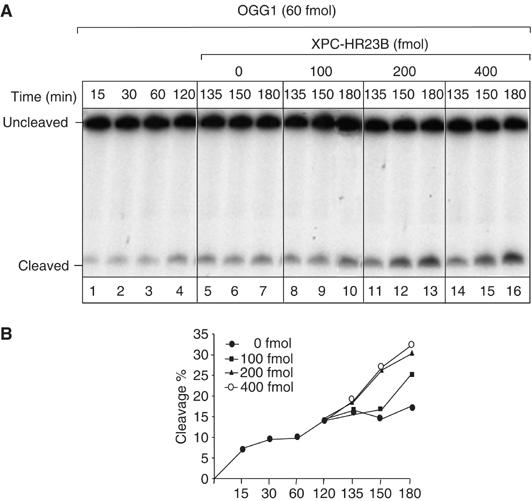
XPC–HR23B stimulates the turnover of OGG1 from its DNA substrate. 5′ end-labelled 210 bp fragments containing 8-OH-Gua were incubated with OGG1 (60 fmol) at 37°C for different periods of time, as indicated (lanes 1–4). After 120 min, different amounts of XPC–HR23B were added at the indicated concentrations and incubated further for 15, 30 or 60 min (lanes 5–16). The products were subjected to alkali treatment and separated by denaturing 8% PAGE. (A) Autoradiography of the gel. The incubation times indicated include the 120 min with OGG1 alone and the following periods in the presence of XPC–HR23B. (B) The amount of cleavage in each lane of (A) was evaluated by densitometric scanning of the autoradiography and expressed in arbitrary units following subtraction of the background. One representative experiment is shown.
XPC–HR23B stimulates binding of OGG1 to damaged DNA
The stimulatory effect of XPC might rely on an interaction between XPC and OGG1 and/or between XPC and the DNA base lesion. A series of experiments was performed to attempt to demonstrate a direct interaction between the two proteins. No indication for such a protein–protein interaction was obtained either by coimmunoprecipitation of OGG1 with XPC–HR23B using antibodies against OGG1 or by pull-down of OGG1 with histidine-tagged XPC–HR23B on nickel agarose beads (data not shown). As a last attempt to verify the interaction between XPC and OGG1, Far Western analysis was performed with purified proteins applied directly to nitrocellulose using a dot-blot apparatus. In this experiment, XPC was bound to the membrane and probed with OGG1. Interaction between OGG1 and XPC was monitored using a polyclonal antibody against OGG1 or XPC. The results shown in Figure 8 indicate that OGG1 binds directly to XPC–HR23B. Negative binding controls were BSA and buffer alone. OGG1 was applied as positive control for antibody specificity. It should be noted that the XPC–HR23B–OGG1 interaction was rather weak (80 mM KCl wash) and this might explain why it was not revealed by the pull-down assay where the antibody and the tag of the protein may further impede the interaction.
Figure 8.
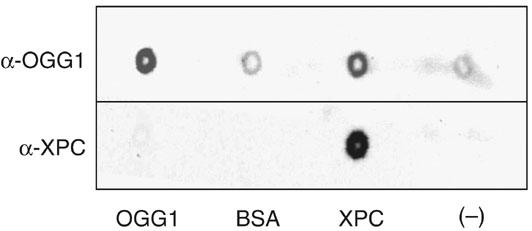
Far Western analysis of OGG1 binding to XPC–HR23B. Purified proteins were spotted onto nitrocellulose and incubated with OGG1. OGG1 binding to proteins was tested by probing either with anti-XPC or anti-OGG1 polyclonal antibodies and visualization was performed by chemiluminescence. BSA and buffer alone were used as negative controls. Purified OGG1 was applied as positive control for antibody specificity.
We then employed band-shift assays (Figure 9) to evaluate whether we could detect a ternary complex between XPC–HR23B and OGG1-bound DNA. To increase the stability of OGG1 on the substrate during electrophoresis, a mutant OGG1 protein was used that retains similar binding affinity for 8-OH-Gua as the wild-type protein, but is deprived of any enzymatic activity (M Bjoras, unpublished data). Under these conditions, a shifted OGG1–DNA complex was detected (lane 2). The addition of increasing amounts of XPC–HR23B produced substantially increased amounts of shifted OGG1–DNA complex (lanes 3 and 4), but OGG1–DNA complex was not supershifted in the presence of XPC–HR23B. At 160 fmol of XPC (lane 3), an approximately two-fold stimulation of OGG1 binding was detected. XPC–HR23B alone did not induce a detectable shift on this DNA substrate (lane 6) as previously reported (Kusumoto et al, 2001). Overall, these results indicate that XPC–HR23B promotes binding of OGG1 to 8-OH-Gua containing DNA without the formation of a stable ternary complex with OGG1-DNA.
Figure 9.
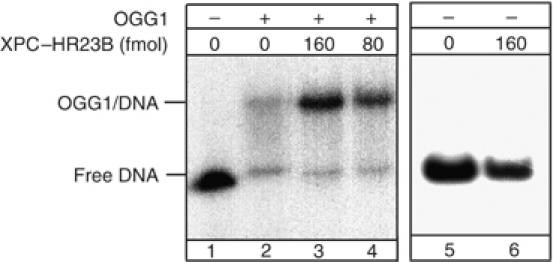
The binding of OGG1 to 8-OH-Gua lesions is improved in the presence of XPC–HR23B. Electrophoretic mobility shift assay: mutant OGG1 (13 pmol), catalytically inactive, was incubated with a 250 bp fragment containing a single 8-OH-Gua residue. All the reactions mixtures were supplemented with BSA and undamaged plasmid DNA as described in Materials and methods. XPC–HR23B was added as indicated.
All together, these results show a novel physical and functional interaction between XPC–HR23B and OGG1.
Discussion
A role for XPC in the repair of oxidative DNA damage
In this study, we provide the first evidence that XPC is involved in the protection of human cells from lethality by oxidizing agents. Oxidants exert their cytotoxic effects via ROS reaction with several targets, including lipids, proteins, and nuclear and mitochondrial DNA. The steady-state and X-ray-induced levels of DNA products are not affected by a defect in XPC. However, upon oxidant exposure, XP-C cells fail to remove the lesions normally, so the levels of 8,5′-cyclopurine 2′-deoxynucleosides and modified DNA bases, namely 8-OH-Gua and 8-OH-Ade, are higher than in wild-type cells. The persistence of these distinct types of oxidative DNA damage in XP-C cells relies upon different mechanisms. Whereas XPC is involved in recognition/excision of 8,5′-cyclopurine 2′-deoxynucleosides likely as a NER component, its participation in 8-OH-Gua removal is as cofactor in OGG1-initiated BER. Accordingly, the kinetics of 8-OH-Gua removal indicates that the contribution of XPC to the overall repair of 8-OH-Gua is relatively small.
The dual role of XPC in the removal of oxidative DNA damage disclosed by our study might lead to different consequences at the cellular level. The persistence of DNA lesions can result in cell death by impeding progression of replication forks. One of the 8,5′-cyclopurine 2′-deoxynucleosides, (5′S)-cdA has been shown to block DNA and RNA polymerases (Brooks et al, 2000; Kuraoka et al, 2000, 2001) in vitro and to prevent transcription factor binding to cognate recognition sequences (Marietta et al, 2002). In contrast, modified DNA bases, such as 8-OH-Gua, are unlikely to be involved in the stalling of replication forks, suggesting that their impact on cell survival is likely to be limited.
The most biologically relevant property of 8-OH-Gua is its miscoding ability. This lesion is preferentially repaired by BER, but several proteins belonging to other repair pathways have been shown to operate either in back-up repair systems or as cofactors to improve the efficiency of the BER reaction. The repair of 8-OH-Gua in human cells has recently been shown to require a functional CSB product (Tuo et al, 2003). Interestingly, the lack of CSB affects the cleavage activity of 8-OH-Gua (Dianov et al, 1999; Tuo et al, 2003) via a mechanism that waits to be clarified. Here, we show that XPC–HR23B complex significantly stimulates the activity of OGG1 in human cell extracts as well as in a reconstituted repair reaction with purified proteins. Interaction between XPC–HR23B and OGG1 was detected (by Far Western analysis only). We were unable to see the formation of a stable ternary complex between XPC–HR23B and OGG1-bound DNA, but enhanced binding of mutant OGG1 to damaged DNA was observed in the presence of XPC complex. To explain DNA lesion recognition by many repair proteins, a model has been proposed where sampling of flexible hinges (owing to poor base stacking at DNA lesions) by a DNA repair protein could facilitate loading of another by direct protein–protein interaction or DNA-mediated interactions (Yang, 2006). Accordingly to this model, XPC–HR23B might bend DNA at sites of damage and thus facilitate loading of OGG1 by direct protein–protein interaction or by competition with the DNA substrate. OGG1 is known to bind tightly the AP site generated by its glycosylase activity (Hill et al, 2001; Vidal et al, 2001), and XPC-HR23B might facilitate its release by competing for binding at AP sites (Shimizu et al, 2003). Evidence for stimulation of BER protein activity by NER proteins has been previously reported. XPG, another NER protein, greatly improves the DNA-binding and catalytic activity of the DNA glycosylase hNTH1 that removes oxidized pyrimidines (Klungland et al, 1999). XPC has been shown to directly interact with thymine DNA glycosylase (TDG), which initiates BER of spontaneous deamination products (Shimizu et al, 2003), and facilitates product (AP site) release from TDG. Interestingly, Lan et al (2004) have recently described an early accumulation of XPC, but not of other NER components, at oxidative damage generated at restricted nuclear regions in mammalian cells. This accumulation might reflect the involvement of XPC–HR23B in recognition/cleavage of oxidized bases revealed by this study.
A link between repair of oxidative DNA damage and clinical features
The main feature of XP-C patients is the high incidence of skin cancer. Keratinocytes are the target cells for sunlight-induced skin cancer. Mutational spectra of p53 (Giglia et al, 1998) in skin tumours from XP-C patients showed the UV signature. However, mutations that are compatible with 8-OH-Gua mutagenesis (i.e. G → T transversions) have recently been reported in the basal layer of human squamous tumours from repair proficient donors (Agar et al, 2004), indicating that oxidative DNA damage may also contribute to skin cancer development in the general population. Interestingly, a novel XPC-dependent DNA repair mechanism involved in removal of nonpyrimidine dimer base damage has been hypothesized on the basis of an XPC-specific p53 mutational hot spot detected in UV-irradiated XPC mutant mice (Nahari et al, 2004).We are tempted to speculate that the increased predisposition to UVB-induced skin cancer in XPC+/− and XPC−/− model mice could be owing not only to reduced NER but also to impaired BER.
Because of XPC involvement in oxidative/endogenous damage, we can expect that XPC deficiency may lead to an increase in mutations also without UV exposure. A significantly increased level of hprt spontaneous mutations, mainly G → T transversions, have been reported in lymphocytes from 12 month-old XPC mutant mice as compared with wild-type mice (Wijnhoven et al, 2000). More recently, a high frequency of spontaneous lung tumours has been reported in XPC−/− mice, suggesting an involvement of XPC in repair of endogenous oxidatively induced lesions in lungs (Hollander et al, 2005). Despite the limited number of reports of tumours at sites other than skin in XP patients, p53 mutations found in primary internal cancers from three XP patients (two belonging to XP-C) were different from those found in XP skin tumours and were compatible with oxidative damage mutagenesis (Giglia et al, 1998).
The new function of XPC in BER that we demonstrate in this study strongly suggests that a defect in the repair of 8-OH-Gua and other oxidatively induced DNA lesions might play a major role in internal cancer development in XP-C patients. In these patients, whose median age of onset for first skin neoplasm is 8 years, the role of XPC in tumorigenesis at sites other than skin might be obscured by the multistep nature and long latency of cancer. This is indeed what occurs in the mutant XPC mice that develop multiple spontaneous lung tumours, of which only a few progress to malignant cancers, indicating that additional events are required for cancer progression (Hollander et al, 2005).
Following the discovery that NER removes in vitro 8,5′-cyclopurine 2′-deoxynucleosides (Kuraoka et al, 2000), it has been hypothesized that the persistence of these lesions might be responsible for neuronal death in XP by blocking neuronal gene expression (reviewed in Brooks, 2002). We demonstrate that XP-C cells are defective in the removal of 8,5′-cyclopurine 2′-deoxynucleosides. However, XP-C patients commonly do not show neurological alterations. Our findings indicate that the impairment of GGR of oxidative DNA damage per se is not sufficient to lead to as dramatic effects, in terms of neurodegeneration, as those observed in XP-A patients (defective in both GGR and TCR). However, the drastic enhancement of the neurological phenotype of CSB and CSA knockout mice, when the XPA gene or the XPC gene are additionally inactivated (reviewed in Friedberg and Meira, 2004), suggests that these genes have at least in part an additive role in neuronal development and brain function. The common function may be the prevention of accumulation of endogenous oxidative DNA damage, which is produced at high rate in the brain.
An important implication of the newly identified function of XPC in the repair of oxidatively induced DNA lesions is that alterations in the XPC function in the general population (e.g. haploinsufficiency, polymorphisms) might be involved as predisposing factors in cancer development. Functional polymorphisms of the XPC gene and reduced levels of XPC mRNA have been associated with increased cancer risk (Shen et al, 2001; Marin et al, 2004; Khan et al, 2006). Future research should thoughtfully address this important issue.
Materials and methods
Case reports, cells culture and treatment conditions
The study was performed on two XP patients belonging to the XP-C group (XP26PV and XP28PV). Clinical, cellular and molecular data on XP26PV are reported by Chavanne et al (2000). XP28PV was referred to us at the age of 28 years when she had already developed several basal and squamous cell carcinoma in the photoexposed areas of the face. In both XP-C patients, nonsense mutations resulting in severely truncated proteins and splicing abnormalities leading to a null product were observed (Chavanne et al, 2000; Nardo, Botta and Stefanini, unpublished results).
Experiments were performed on primary fibroblast and keratinocyte cultures established according to previously described procedures (D'Errico et al, 2005) from biopsies from clinically unaffected skin areas of the two patients and two age-matched healthy donors (N1RO and N2RO). Cells were treated with X-rays (Radgil, Gilardoni, Milan, Italy) at a dose rate of 90 cGy/min. The treatment with KBrO3 (Sigma-Aldrich S.r.l. Milan, Italy) was performed in PBS 1 × -Hepes, 20 mM for 1 h at 37°C.
Plasmids and transfections
The pEGFP-C1 plasmid expressing the EGFP protein, and its derivative pEGFP-XPC expressing the XPC-EGFP fusion protein have been previously described (Santagati et al, 2001).
Cells (2 × 105) were plated onto 30 mm Petri dishes and transfected by lipofectamine 2000 method (Invitrogen Life Technologies S.r.l., Milan, Italy) with 4 μg of pEGFP-C1 or pEGFP-XPC DNA. At 24 h after transfection, cells were treated with different doses of KBrO3 (5 and 10 mM) and 24 h post-treatment, cells were trypsinized and washed with PBS. Cell pellets were resuspended in PBS containing 50 mg/ml PI and incubated for 40 min to label dead cells. Cells (1 × 104) were cytocentrifuged on slides and analysed by fluorescence microscopy. At least 100 cells expressing the ectopic protein (green fluorescence) from two independent experiments were evaluated for PI staining (red fluorescence) to calculate the percentage of dead cells.
Cell survival assay
Cell survival was determined by analysing the colony-forming ability, as described previously (Santagati et al, 2001). Briefly, cells were exposed to X-rays (1–6 Gy) or KBrO3 for 1 h (2.5–40 mM) and then plated at increasing density as a function of doses. Colonies were fixed 14 days later. The number of colonies in treated cells was expressed as a percentage of that in untreated cells.
Measurement of modified nucleosides by LC/MS and GC/MS
DNA was isolated as previously described (Tuo et al, 2003). The internal standards 8-hydroxy-2′-deoxyadenosine-15N5, (5′S)-8,5′-cyclo-2′-deoxyadenosine-15N5, (5′R)-8,5′-cyclo-2′-deoxyguanosine-15N5 and (5′S)-8,5′-cyclo-2′-deoxyguanosine-15N5 were prepared as described (Birincioglu et al, 2003; Jaruga et al, 2004). Custom-synthesized 8-hydroxy-2′-deoxyguanosine-15N5 was purchased from Cambridge Isotope Laboratories (Cambridge, MA). 8-OH-dG, 8-OH-dA and (5′S)-cdA were measured by LC/MS with isotope dilution as described (Jaruga et al, 2002). GC/MS with isotope dilution was used to measure (5′R)-cdG and (5′S)-cdG as described (Jaruga et al, 2002).
Measurement of 8-OH-dG by HPLC-ED
8-OH-dG repair was determined by HPLC-ED according to established procedures (Cappelli et al, 2000). Briefly, enzymatic digestion of DNA was accomplished at 37°C with nuclease P1 (Boehringer Mannheim) and alkaline phosphatase (Boehringer Mannheim) for 2 h. Aliquots of the DNA hydrolysate were analysed in an LC18-DB column (250 mm by 46 mm, 5-μm; Supelco, Sigma) equipped with a C18 μ guard column. The separation was isocratic with 50 mM ammonium acetate, pH 5.5, with 10% methanol, at a flow rate of 1.0 ml/min. HPLC (Beckman, System Gold) was equipped with a UV detector (256 nm) and an Electrochemical detector (Coulochem II; ESA Inc.) with applied potentials at 150 and 300 mV for E1 and E2, respectively. 2′-Deoxyguanosine was measured in the same run of corresponding 8-OH-dG and the results are expressed as the number of 8-OH-dG residues/106 Gua residues.
In vitro incision of 8-OH-Gua-containing oligonucleotides
Nuclear extracts from HeLa cells were prepared as described in (McGoldrick et al, 1995). The protocol was adapted to a pellet of 6 × 107 cells. The recombinant (His)6–XPC–HR23B complex was affinity purified from insect cells (Sugasawa et al, 1996), using Ni2+-NTA chelate agarose beads (Qiagen, Valencia, CA, USA). Recombinant human HR23B was purified from insect cells as described before (Sugasawa et al, 1996). Purification of recombinant human XPA was carried out as described by Tapias et al (2004). Purified human OGG1 was purchased from Trevigen (Tema Ricerca S.r.l., Bologna, Italy). An oligodeoxyribonucleotide containing a single 8-OH-Gua residue, 5′-GATCCTCTAGAG(8-OH-Gua)CGACCTGCAG GCATGCA-3' (Eurogentec, Angers, France), was 5′ end-labeled with 32P and then annealed with the complementary oligonucleotide. The incision reaction (final volume 50 μl) contained 50 fmol of duplex oligonucleotides, 25 mM Tris–HCl pH 7.6, 1 mM EDTA, 50 mM NaCl and 5 μg of nuclear extracts. After incubation at 37°C for increasing times, samples were electrophoresed in 20% denaturing polyacrylamide gels. The incision products were visualized by autoradiography and quantified by electronic autoradiography (Instant Imager, Packard).
To test the stimulatory effect of XPC, we used a gel purified 210 bp DNA fragment (36 pmol) containing a single 8-OH-Gua lesion, dephosphorylated and 5′ end-labeled with 32P. This DNA fragment was generated by enzymatic restriction (Bsu36I/ApaI) of a single 8-OH-Gua-containing plasmid (Frit et al, 2002).The incision reaction was performed as described above, except that the nuclear extracts were replaced by 60 fmol of recombinant OGG1 and amount of recombinant XPC–HR23B complex as indicated, in a final volume of 10 μl. The incision products were subjected to alkali treatment and then electrophoresed in 8% denaturing PAGE. The quantification of the autoradiograms was done using GENEQUANT software (Molecular Dynamics)/or Instant Imager.
Gel retardation assay
Recombinant human OGG1 was purified as previously described (Bjørås et al, 1997). This protein presents two mutations, K249A and D268A, that abolish completely the cleavage activity.
For the preparation of the radioactive probe, covalently closed circular DNA containing a single 8-OH-Gua was synthesized in vitro as previously described (Fortini et al, 1999) using as a primer the same oligonucleotide used in the in vitro incision assay. The modified plasmid was than digested with BstNI and PvuII to yield a 250 bp fragment containing the lesion. This fragment was purified with the QIAEX II Gel extraction kit and then 32P-labeled as described by Sugasawa et al (2001). Binding reactions were carried out as described by Sugasawa et al (2001). Briefly, 13 pmol of OGG1 and the indicated amount of recombinant XPC–HR23B complex were incubated at 30°C for 30 min in mixtures (10 μl) containing 20 mM sodium phosphate (pH 7.4), 1 mM EDTA, 70 mM NaCl, 1 mM DTT, 5% glycerol, *BSA (1 μg), 32P-labeled probe DNA (7.5 fmol) and unmodified covalently closed circular plasmid DNA (0.5 ng). The reactions were then chilled on ice and the resulting DNA–protein complexes were fixed by addition of 1 μl of 2.5% gluteraldehyde. The mixtures were directly loaded on 4% nondenaturing polyacrylamide gels (acrylamide: bis-acrylamide, 37.5:1) containing 2.5% glycerol and TGE buffer (25 mM Tris, 192 mM glycine and 1 mM EDTA) and electrophoresed at 11 V/cm in a sequence gel electrophoresis apparatus (Biorad). After electrophoresis, the gel was visualized by autoradiography and quantified by electronic autoradiography (Instant Imager, Packard).
Far Western
Purified XPC–HR23B (10 pmol) in blotting buffer (0.5 × PBS) was applied to nitrocellulose using a dot-blot apparatus (Bio-Rad). The membrane was blocked with 5% nonfat milk in blotting buffer for 2 h and then incubated with OGG1 (4 μg/ml) in the same buffer for an additional 2 h. The membrane was probed either with anti-XPC or anti-OGG1 polyclonal antibody (Santa-Cruz OGG1/2 (L-19) sc-12075). Signals were visualized using a chemiluminescence detection kit (NEN Life Science Products). BSA (18 pmol), OGG1 (17 pmol) and blotting buffer alone were used as controls.
Statistical analysis
A nonparametric test (Kruskal–Wallis rank test) was used to compare the cell killing data by KBrO3 in XPC cells expressing the XPC chimera with those obtained in cells transfected with the empty vector. Mean values of modified nucleosides were compared by one-way analysis of variance. Nonparametric tests for trend across repair times were performed in order to analyse the repair kinetics of 8-OH-dG in different cell types. Two-way analysis of variance was performed to compare the repair kinetics in normal and XP-C cells. All analyses were carried out with the STATA statistical package release 8.1 (Stata, College Station, TX).
Acknowledgments
We are grateful to Nicolas Charlet-Berguerand (IGBMC, Illkirch, France) for sharing experimental data and useful suggestions. We thank Ivano Iavarone (Istituto Superiore di Sanità, Rome) for statistical analysis of the data. BMBJ is supported by a Marie Curie Research Training Network Contract (number MRTN-CT-2003-503618). Grant support: Associazione Italiana per la Ricerca sul Cancro (AIRC), MIUR/FIRB (RBNE01RNN7), Fondazione Cariplo, Italian Ministry of Health (Ricerca Finalizzata e Corrente). Certain commercial equipment or materials are identified in this paper in order to specify adequately the experimental procedure. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the materials or equipment identified are necessarily the best available for the purpose.
References
- Agar NS, Halliday GM, Barnetson RS, Ananthaswamy HN, Wheeler M, Jones AM (2004) The basal layer in human squamous tumors harbors more UVA than UVB fingerprint mutations: a role for UVA in human skin carcinogenesis. Proc Natl Acad Sci USA 101: 4954–4959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki M, Masutani C, Takemura M, Uchida A, Sugasawa K, Kondoh J, Ohkuma Y, Hanaoka F (2001) Centrosome protein centrin 2/caltractin 1 is part of the xeroderma pigmentosum group C complex that initiates global genome nucleotide excision repair. J Biol Chem 276: 18665–18672 [DOI] [PubMed] [Google Scholar]
- Ballmaier D, Epe B (1995) Oxidative DNA damage induced by potassium bromate under cell-free conditions and in mammalian cells. Carcinogenesis 16: 335–342 [DOI] [PubMed] [Google Scholar]
- Birincioglu M, Jaruga P, Chowdhury G, Rodriguez H, Dizdaroglu M, Gates KS (2003) DNA base damage by the antitumor agent 3-amino-1,2,4-benzotriazine 1,4-dioxide (tirapazamine). J Am Chem Soc 125: 11607–11615 [DOI] [PubMed] [Google Scholar]
- Bjørås M, Luna L, Johnsen B, Hoff E, Haug T, Rognes T, Seeberg E (1997) Opposite base-dependent reactions of a human base excision repair enzyme on DNA containing 7,8-dihydro-8-oxoguanine and abasic sites. EMBO J 16: 6314–6322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks PJ (2002) DNA repair in neural cells: basic science and clinical implications. Mutat Res 509: 93–108 [DOI] [PubMed] [Google Scholar]
- Brooks PJ, Wise DS, Berry DA, Kosmoski JV, Smerdon MJ, Somers RL, Mackie H, Spoonde AY, Ackerman EJ, Coleman K, Tarone RE, Robbins JH (2000) The oxidative DNA lesion 8,5′-(S)-cyclo-2′-deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. J Biol Chem 275: 22355–22362 [DOI] [PubMed] [Google Scholar]
- Cappelli E, Degan P, Thompson LH, Frosina G (2000) Efficient repair of 8-oxo-7,8-dihydrodeoxyguanosine in human and hamster xeroderma pigmentosum D cells. Biochemistry 39: 10408–10412 [DOI] [PubMed] [Google Scholar]
- Chavanne F, Broughton BC, Pietra D, Nardo T, Browitt A, Lehmann AR, Stefanini M (2000) Mutations in the XPC gene in families with xeroderma pigmentosum and consequences at the cell, protein, and transcript levels. Cancer Res 60: 1974–1982 [PubMed] [Google Scholar]
- Collins AR, Cadet J, Moller L, Poulsen HE, Vina J (2004) Are we sure we know how to measure 8-oxo-7,8-dihydroguanine in DNA from human cells? Arch Biochem Biophys 423: 57–65 [DOI] [PubMed] [Google Scholar]
- D'Errico M, Lemma T, Calcagnile A, Proietti De Santis L, Dogliotti E (2006) Cell type and DNA damage specific response of human skin cells to environmental agents. Mutat Res, (in press) [DOI] [PubMed] [Google Scholar]
- D'Errico M, Teson M, Calcagnile A, Nardo T, De Luca N, Lazzari C, Soddu S, Zambruno G, Stefanini M, Dogliotti E (2005) Differential role of transcription-coupled repair in UVB-induced response of human fibroblasts and keratinocytes. Cancer Res 65: 432–438 [PubMed] [Google Scholar]
- Dianov G, Bischoff C, Sunesen M, Bohr VA (1999) Repair of 8-oxoguanine in DNA is deficient in Cockayne syndrome group B cells. Nucleic Acids Res 27: 1365–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Standards Committee on Oxidative DNA Damage (ESCODD) (2003) Measurement of DNA oxidation in human cells by chromatographic and enzymic methods. Free Radic Biol Med 34: 1089–1099 [DOI] [PubMed] [Google Scholar]
- Evans MD, Dizdaroglu M, Cooke MS (2004) Oxidative DNA damage and disease: induction, repair and significance. Mutat Res 567: 1–61 [DOI] [PubMed] [Google Scholar]
- Fortini P, Parlanti E, Sidorkina OM, Laval J, Dogliotti E (1999) The type of DNA glycosylase determines the base excision repair pathway in mammalian cells. J Biol Chem 274: 15230–15236 [DOI] [PubMed] [Google Scholar]
- Fortini P, Pascucci B, Parlanti E, D′Errico M, Simonelli V, Dogliotti E (2003) The base excision repair: mechanisms and its relevance for cancer susceptibility. Mutat Res 531: 127–139 [DOI] [PubMed] [Google Scholar]
- Friedberg EC, Meira LB (2004) Database of mouse strains carrying targeted mutations in genes affecting biological responses to DNA damage (Version 6). DNA Repair (Amst) 3: 1617–1638 [DOI] [PubMed] [Google Scholar]
- Frit P, Kwon K, Coin F, Auriol J, Dubaele S, Salles B, Egly JM (2002) Transcriptional activators stimulate DNA repair. Mol Cell 10: 1391–1401 [DOI] [PubMed] [Google Scholar]
- Giglia G, Dumaz N, Drougard C, Avril MF, Daya-Grosjean L, Sarasin A (1998) p53 mutations in skin and internal tumors of xeroderma pigmentosum patients belonging to the complementation group C. Cancer Res 58: 4402–4409 [PubMed] [Google Scholar]
- Hanawalt PC (2002) Subpathways of nucleotide excision repair and their regulation. Oncogene 21: 8949–8956 [DOI] [PubMed] [Google Scholar]
- Hill JW, Hazra TK, Izumi T, Mitra S (2001) Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Res 29: 430–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers JH (2001) Genome maintenance mechanisms for preventing cancer. Nature 411: 366–374 [DOI] [PubMed] [Google Scholar]
- Hollander MC, Philburn RT, Patterson AD, Velasco-Miguel S, Friedberg EC, Linnoila RI, Fornace AJ (2005) Deletion of XPC leads to lung tumors in mice and is associated with early events in human lung. Proc Natl Acad Sci USA 102: 13200–13205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaruga P, Birincioglu M, Rodriguez H, Dizdaroglu M (2002) Mass spectrometric assays for the tandem lesion 8,5′-cyclo-2′-deoxyguanosine in mammalian DNA. Biochemistry 41: 3703–3711 [DOI] [PubMed] [Google Scholar]
- Jaruga P, Theruvathu J, Dizdaroglu M, Brooks PJ (2004) Complete release of (5′S)-8,5′-cyclo-2′-deoxyadenosine from dinucleotides, oligodeoxynucleotides and DNA, and direct comparison of its levels in cellular DNA with other oxidatively induced DNA lesions. Nucleic Acids Res 32: e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SG, Oh KS, Shahlavi T, Ueda T, Busch DB, Inui H, Emmert S, Imoto K, Muniz-Medina V, Baker CC et al. (2006) Reduced XPC DNA repair gene mRNA levels in clinically normal parents of xeroderma pigmentosum patients. Carcinogenesis 27: 84–94 [DOI] [PubMed] [Google Scholar]
- Kuraoka I, Bender C, Romieu A, Cadet J, Wood RD, Lindahl T (2000) Removal of oxygen free-radical-induced 5′,8-purine cyclodeoxynucleosides from DNA by the nucleotide excision-repair pathway in human cells. Proc Natl Acad Sci USA 97: 3832–3837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuraoka I, Robins P, Masutani C, Hanaoka F, Gasparutto D, Cadet J, Wood RD, Lindahl T (2001) Oxygen free radical damage to DNA. Translesion synthesis by human DNA polymerase eta and resistance to exonuclease action at cyclopurine deoxynucleoside residues. J Biol Chem 276: 49283–49288 [DOI] [PubMed] [Google Scholar]
- Kusumoto R, Masutani C, Sugasawa K, Iwai S, Araki M, Uchida A, Mizukoshi T, Hanaoka F (2001) Diversity of the damage recognition step in the global genomic nucleotide excision repair in vitro. Mutat Res 485: 219–227 [DOI] [PubMed] [Google Scholar]
- Lan L, Nakajima S, Oohata Y, Takao M, Okano S, Masutani M, Wilson SH, Yasui A (2004) In situ analysis of repair processes for oxidative DNA damage in mammalian cells. Proc Natl Acad Sci USA 101: 13738–13743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann AR (2003) DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie 85: 1101–1111 [DOI] [PubMed] [Google Scholar]
- Marietta C, Gulam H, Brooks PJ (2002) A single 8,5′-cyclo-2′-deoxyadenosine lesion in a TATA box prevents binding of the TATA binding protein and strongly reduces transcription in vivo. DNA Repair (Amst) 1: 967–975 [DOI] [PubMed] [Google Scholar]
- Marin MS, Lopez-Cima MF, Garcia-Castro L, Pascual T, Marron MG, Tardon A (2004) Poly (AT) polymorphism in intron 11 of the XPC DNA repair gene enhances the risk of lung cancer. Cancer Epidemiol Biomarkers Prev 13: 1788–1793 [PubMed] [Google Scholar]
- McGoldrick JP, Yeh YC, Solomon M, Essigmann JM, Lu AL (1995) Characterization of a mammalian homolog of the Escherichia coli MutY mismatch repair protein. Mol Cell Biol 15: 989–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao F, Bouziane M, Dammann R, Masutani C, Hanaoka F, Pfeifer G, O′Connor TR (2000) 3-Methyladenine-DNA glycosylase (MPG protein) interacts with human RAD23 proteins. J Biol Chem 275: 28433–28438 [DOI] [PubMed] [Google Scholar]
- Nahari D, McDaniel LD, Task LB, Daniel RL, Velasco-Miguel S, Friedberg EC (2004) Mutations in the Trp53 gene of UV-irradiated Xpc mutant mice suggest a novel Xpc-dependent DNA repair process. DNA Repair (Amst) 1: 379–386 [DOI] [PubMed] [Google Scholar]
- Sampson JR, Jones S, Dolwani S, Cheadle JP (2005) MutYH (MYH) and colorectal cancer. Biochem Soc Trans 33: 679–683 [DOI] [PubMed] [Google Scholar]
- Santagati F, Botta E, Stefanini M, Pedrini AM (2001) Different dynamics in nuclear entry of subunits of the repair/transcription factor TFIIH. Nucleic Acids Res 29: 1574–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Sturgis EM, Khan SG, Qiao Y, Shahlavi T, Eicher SA, Xu Y, Wang X, Strom SS, Spitz MR, Kraemer KH, Wei Q (2001) An intronic poly (AT) polymorphism of the DNA repair gene XPC and risk of squamous cell carcinoma of the head and neck: a case–control study. Cancer Res 61: 3321–3325 [PubMed] [Google Scholar]
- Shimizu Y, Iwai S, Hanaoka F, Sugasawa K (2003) Xeroderma pigmentosum group C protein interacts physically and functionally with thymine DNA glycosylase. EMBO J 22: 164–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugasawa K, Masutani C, Uchida A, Maekawa T, van der Speck PJ, Bootsma D, Hoeijmakers JHJ, Hanaoka F (1996) HHR23B, a human Rad23 homolog, stimulates XPC protein in nucleotide excision repair in vitro. Mol Cell Biol 16: 4852–4861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugasawa K, Okamoto T, Shimizu Y, Masutani C, Iwai S, Hanaoka F (2001) A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev 15: 507–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugasawa K, Shimizu Y, Iwai S, Hanaoka F (2002) A molecular mechanism for DNA damage recognition by the xeroderma pigmentosum group C protein complex. DNA Repair (Amst) 1: 95–107 [DOI] [PubMed] [Google Scholar]
- Tapias A, Auriol J, Forget D, Enzlin JH, Scharer OD, Coin F, Coulombe B, Egly JM (2004) Phosphorylation of XPB helicase regulates TFIIH nucleotide excision repair activity. J Biol Chem 279: 19074–19083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuo J, Jaruga P, Rodriguez H, Bohr VA, Dizdaroglu M (2003) Primary fibroblasts of Cockayne syndrome patients are defective in cellular repair of 8-hydroxyguanine and 8-hydroxyadenine resulting from oxidative stress. FASEB J 17: 668–674 [DOI] [PubMed] [Google Scholar]
- Vidal AE, Hickson ID, Boiteux S, Radicella JP (2001) Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Res 29: 1285–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijnhoven SW, Kool HJ, Mullenders LH, van Zeeland AA, Friedberg EC, van der Horst GT, van Steeg H, Vrieling H (2000) Age-dependent spontaneous mutagenesis in Xpc mice defective in nucleotide excision repair. Oncogene 19: 5034–5037 [DOI] [PubMed] [Google Scholar]
- Yang W (2006) Poor base stacking at DNA lesions may initiate recognition by many repair proteins. DNA Repair (Amst) 5: 654–666, E-pub: 29 March 2006 [DOI] [PubMed] [Google Scholar]