

Abstract
Hepatitis B virus (HBV) is one of the major etiological factors responsible for the development of hepatocellular carcinoma (HCC). We used a transgenic mouse, containing HBV sequences, as a model system to unravel the molecular mechanisms of hepatocarcinogenesis induced by HBV. We chose this animal model because it consistently develops liver cancer after intermediate steps that mimic the natural history of HBV infection in humans. In this study, we focus our attention on the early events leading to liver cancer. We compared the gene expression profile of 3-month-old transgenic mice with that of 3-month-old wild-type (wt) animals. In the transgenic mouse, microarray data analysis showed a total of 45 significantly differentially expressed genes, 25 highly expressed (fold change ≥2; P = 0.0025), and 20 downregulated (fold change ≤0.5; P = 0.0025). These genes belong to several different functional categories such as the regulation of immunological response, transcription, intracellular calcium ion mobilization, regulation of cell cycle and proliferation, NF-κb signal transduction cascades, and apoptosis. In particular, the upregulation of the antiapoptotic gene NuprI and the downregulation of the proapoptotic gene Bnip3 were found. This observation was supported by an in vitro apoptosis assay that showed downregulation of apoptosis in hepatocytes of HBV transgenic mouse compared with wt mice treated with staurosporine. In conclusion, our experimental approach allowed identification of new genes modulated by HBV and showed that the apoptotic process was deregulated in transgenic mouse hepatocytes. These data shed light on one possible mechanism by which HBV induces hepatocarcinogenesis.
INTRODUCTION
Hepatocellular carcinoma (HCC) is the most frequent primary solid tumor of the liver. It is a heterogeneous disease in terms of etiology and underlying associations as well as biologic and clinical behavior. HCC develops commonly, but not exclusively, in a setting of liver cell injury, which leads to inflammation, hepatocyte regeneration, liver matrix remodeling, fibrosis, and ultimately, cirrhosis. The vast majority of HCC worldwide (80%) is attributed to hepatitis B virus (HBV) or HCV infection, but other risk factors include dietary exposure to aflatoxin B1, alcohol abuse, hemochromatosis, fatty liver disease, androgenic steroid use, and α-1 antitrypsin deficiency. The mechanisms by which these varied etiologies lead to cirrhosis and HCC are not well understood.
Numerous evidences indicate that HBV is one of the major etiological factors responsible for the development of HCC (1). The HBV genome is a circular, partially double-stranded DNA molecule containing 4 overlapping open-reading frames (ORFs), encoding PreS/S, PreC/C, P, and X proteins (HBx). HBV DNA integrates into the host genome, and this integration is believed, in part, to be carcinogenetic. In fact, the random integration of viral sequences could determine the fortuitous activation of an adjacent cellular oncogene (2). In particular, the integration of HBV genomic DNA encoding HBx is the most frequent viral marker found in HCC (3).
Several evidences indicate that HBx has oncogenic function. This protein is essential for viral replication in vivo (4,5) and can be oncogenic in strains of transgenic mice (2). Furthermore, HBx induces cellular gene expression deregulation through its transactivational activity (6) by interacting with components of the basal transcription machinery and several transcription factors (7–10). HBx also activates several signal transduction pathways including NF-κB, mitogen-activated protein kinase, protein kinase C, and JAK/STAT pathways (11–16), which take part in many aspects of cell regulation. HBx has been shown to promote tumor cell invasion (17), and several studies showed that HBx plays a role in regulating apoptosis (18–22). Collectively, these studies indicate that HBx plays a prominent role in hepatocarcinogenesis induced by HBV infection. It should be remembered, however, that chronic liver injury initiates a cascade of events characterized by elevated cell proliferative activity and compromised cellular detoxification and repair function. These events may lead to accumulation of DNA mutations, probable in replicating cells, which in the long run would favor the selection of cells with a malignant phenotype (23). This cascade of events is consistent with the close correlation between chronic inflammation and carcinogenesis (24) and with the observation of a greater risk of HCC in HBV-infected patients with cirrhosis than in those without cirrhosis.
At present, it still remains uncertain which of the oncogenic functions of HBV are really involved in HCC development and what is the sequence of genomic events responsible for hepatocyte transformation. The HBV transgenic mouse, officially designed as Tg (Alb-1 HBV) Bri44, contains the HBV genomic sequences for pre-S, S, and X proteins. It represents an ideal model for the study of the steps (gene and protein expression alterations) that lead to HCC (25–27). In fact, this transgenic mouse develops progressive hepatocyte damage (25–29) which determines, in the first month of life, degenerative alterations (26), followed by an active inflammatory damage that is characterized by elevated damage-related compensatory proliferative response (27, 29). This is a precancerous condition which is consistently followed, after the eighth to ninth month after birth, by the appearance of dysplastic hepatic lesions (27,28), which become clearly neoplastic after the 12th month (27). Successively, neoplastic lesions progressively grow to macroscopic nodules that can be observed in all animals within the 16th to 18th month of life. Therefore, this transgenic mouse mimics many of the pathological events that occur before the development of HCC in chronic HBV infection in humans, representing a useful model system to study the processes of initiation, development, and progression of HCC upon HBV infection.
In the effort to elucidate the early molecular mechanisms of hepatocarcinogenesis induced by HBV, we investigated the gene expression profiling of HBV transgenic mice compared with that of wt mice. We focused our attention on the gene expression alterations occurring in 3-month-old HBV transgenic mice in the hypothesis that this age represents the start of the multistep process of hepatocarcinogenesis. Our data show the downregulation of apoptotic process in HBV transgenic mouse hepatocytes, indicating deregulation as a possible mechanism by which HBV induces hepatocarcinogenesis.
MATERIALS AND METHODS
Animals
Twelve-week-old male C57BL/6J (wt) mice were purchased from Charles River (Calco, Italy). In addition, 12-week-old male, transgenic mice designed as Tg (Alb-1HBV) Bri 44 were obtained from Jackson Laboratories (Bar Harbor, ME, USA). All mice were kept in temperature-, air-, and light-controlled (light on from 7 a.m. to 7 p.m.) conditions and received food and water ad libitum for at least 1 week before being used for our experiments. All animals received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals.
RNA Extraction and Quantification
Mice were killed by CO2 asphyxiation. The whole liver was isolated, snap frozen, and stored in liquid nitrogen. Total RNA from liver tissues was prepared using TRIzol Reagent (Invitrogen, Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s protocol. After the ethanol precipitation step in the TRIzol extraction procedure, a cleanup was performed using RNeasy columns (Qiagen, Valencia, CA, USA). Total RNA concentration and purity were assessed by spectrophotometric analysis. The A260/A280 ratio was between 1.9 and 2.1 for each sample. The integrity of total RNA was assessed on standard 1% agarose/formaldehyde gel.
Microarray
The microarray hybridization experiments were performed in triplicate. The cRNA “targets” were generated as described by the Affymetrix Expression Analysis Gene Chip Technical Manual, protocol P/N 900218rev.2, P/N70021rev.3. Briefly, double-stranded cDNA was synthesized using the SuperScript Choice System (Invitrogen) and a primer containing poly(dT) downstream to T7 RNA polymerase promoter sequence (MWG). In vitro transcription using double-stranded cDNA as a template in the presence of biotinylated UTP and CTP was carried out using BioArray High Yield RNA Transcript Labeling Kit (Enzo Diagnostic, Farming-dale, NY, USA). Biotinylated cRNA was purified, fragmented, and hybridized to the Affymetrix MG-U74av2 chips (Affymetrix, Santa Clara, CA, USA), containing 12625 probe sets recognizing murine genes of mostly known function. The cRNA was detected with strepta-vidin-phycoerythrin (Molecular Probes, Eugene, OR, USA), and analysis was completed by using a Hewlett-Packard Gene Array Scanner (Affymetrix). The 20x eukaryotic hybridization control kit was included, as recommended by the manufacturer (Affymetrix Data Analysis Fundamentals manual). The signal for each probe set was quantified using Micro Array Suite 5.0 software (Affymetrix). The recommended internal controls and quality checks were performed: briefly, average background and noise were verified to be comparable between different experiments; the percentages of probe sets scored as present were similar in the replicates; the ratios of 3′ and 5′ probe sets for GAPDH and actin were less than 3; finally the scaling/normalization factors were comparable between different arrays. Default parameters were used for the statistical algorithm and for probe set scaling using Microarray Suite 5.0 (with a target intensity of 100).
Analysis of Data
Potential clusters corresponding to subgroups were visualized with a 2-step approach. In the first step, the data were scaled from each array to a target intensity value of 100 (Microarray Suite 5.0) to enable interarray comparisons. Data were filtered so that the absolute value of the fold change was 2 or more. Additionally, genes that were scored as absent in experimental and baseline files were removed. The statistical significance in the comparison analyses was calculated using Wilcoxon signed rank test as described in the Affymetrix Data Analysis Fundamentals manual. The default cut off P value of 0.0025 was used. The sorting for change (2 fold) was chosen to remove false positives and to detect changes that were biologically more relevant. In the second step, all data were permutated 100 cycles using the multiclass response parameter of the Significance Analysis of microarrays algorithm (SAM) (30), a statistical method for identifying significant changes in gene expression that takes into account multiple testing. For a user-defined threshold, based on the number of expected false-positive regulated genes, a list with the significantly regulated genes is generated.
The total set of 12,600 genes was reduced to the significant differentially expressed genes. The reduced set was prepared for biological interpretation.
Annotations about individual genes were obtained in the Net Affx Analysis Center (www.affymetrix.com) and the NCBI site (ncbi.nlm.nih.gov: OMIM and PubMed).
RT-PCR
The results obtained by microarray analysis were validated by semiquantitative reverse transcription (RT) followed by polymerase chain reaction (PCR). The RNAs used for these analyses were not the same as those used for gene chip analysis, but derived from independent extraction. Total RNA (1 μg), previously treated with RNase-free DNase I (Roche Diagnostics S.p.a., Milano, Italy), was used for semiquantitative RT-PCR. Reverse transcription of mRNAs was carried out with the Superscript III reverse transcriptase kit (Invitrogen) using oligo(dT) as primer. Single-stranded cDNAs in 1 μL of a 20-μL reaction mixture were amplified by PCR using the following conditions: 25 pmol of each primer, 25 nmol of each deoxyribonucleoside triphosphate, 3 U Taq Gold DNA polymerase (Applied Biosystems, Branchberg, NJ, USA), 5 μL 10× PCR buffer in a final volume of 50 μL. Oligonucleotides were designed using the PRIMER3 program (http://www.genome.wi.mit.edu). The nucleotide sequences of primers and corresponding annealing temperatures used are available on request. β-Actin mRNA was amplified as an internal control for the reverse transcription reaction. Further controls were performed with RNA preparations amplified in absence of reverse transcriptase to verify the absence of DNA contamination.
Western Blot
Liver tissues were resuspended in the lysis buffer containing 10 mM Tris-HCl buffer (pH 7.5) and protease inhibitors (PIs) (2 μg/mL leupeptin, 2 μg/mL apro-tinin, 1 μg/mL pepstatin, 100 μg/mL phenylmethylsulfonyl fluoride [Roche Diagnostics S.p.a., Milano, Italy], and 2 mM EDTA]. Liver tissues were homogenized using the Dunce homogenizer, and homogenates were centrifuged at 14000g for 10 min. The supernatants were collected and stored frozen at −80 °C. All procedures were carried out at 4 °C. Protein concentration was determined by the Bradford assay (Bio-Rad). Total proteins (50 μg) were diluted in Laemmli sample buffer, resolved by 12% SDS-PAGE, and transferred onto nitrocellulose membranes (Schleicher and Schuell, Dassel, Germany). The membranes were incubated with polyclonal rabbit antibody against HMGB2 (PharMingen, Becton Dickinson Biosciences, San Diego, CA, USA) at 1:750 dilution and with antitubulin antibody (Chemicon International, Temecula, CA, USA) at 1:1000 dilution. Immunodetection was realized using enhanced chemiluminescence reagents (Cell Signaling Technology, New England Biolabs, Ipswich, MA, USA).
Hepatocyte Isolation
All liver perfusions were carried out at noon. Hepatocytes were isolated as previously described (31). Viability was determined by trypan blue exclusion assay, and only preparations having a viability higher than 80% were used. The purity of the preparations was more than 98% as demonstrated by the staining for glucose-6-phosphatase performed as previously described (31). Hepatocytes, resuspended in minimum essential medium (Eagle) with Earle’s salts and supplemented with glucose (1.8 g/L), pyruvate (0.11 g/L), aspartate (0.022 g/L), serine (0.021 g/L), proline (0.030 g/L), glutamine (0.584 g/L), HEPES (4.75 g/L), hydrocortisone (50 nM), insulin (10–7 M) and 10% FCS, were plated at a density of 1.2 × 106 cells/4.5 mL medium per 60-mm Falcon dish, and maintained at 37 °C and 5% humidified atmosphere. After 4 h (attachment period), medium was replaced with FCS-free medium (incubation medium), and hepatocytes were maintained for additional 12 or 16 h in the presence or in absence of 10 μM staurosporine, an inducer of apoptosis.
Thiazolyl Blue (MTT) Assay
After the exposure to the proapoptotic agent staurosporine, the number of untreated and treated cells was determined by MTT assay. The MTT assay was performed in 3 dishes for each experimental time point (12 and 16 h, in the presence or absence of staurosporine) as described by the manufacturer (Sigma, Milano, Italy; product no. M 5655).
DNA Ladder Assay
For the evaluation of DNA fragmentation, a modification of the method described by Cifone et al. was used (32). At the end of the incubation period, the medium was removed (dead cells in suspension were not collected because their number was very low) and adherent hepatocytes were collected by scraping and resuspended in 1× PBS. After centrifugation (5 min at 450g), the cells were resuspended in 300 μL lysis buffer (10 mM Tris-HCl pH 8.0, 25 mM EDTA, 100 mM NaCl, 0.5% SDS, 0.1 mg/mL proteinase K) and incubated overnight at 37 °C. Genomic DNA was extracted by phenol/chloroform (1:1, vol/vol), precipitated with ethanol, resuspended in TE 1× (Tris-HCl 10 mM, pH 8.0, 1 mM EDTA) and treated with RNase (final concentration 1 μg/mL) at 37 °C for 1 h. Finally, genomic DNA was analyzed on a 1.8% agarose gel using ethidium bromide staining for the detection.
RESULTS
Gene Expression Profile Analysis
To unravel the early molecular events of hepatocarcinogenesis induced by HBV, we performed a gene-profiling approach on the liver total RNAs of 3-month-old wt and HBV transgenic mice. Genes that were statistically differently expressed were grouped in categories according to subcellular localization, biological process, or molecular function as revealed by the gene ontology (GO) biological process description. The genes either increased or decreased in transgenic mouse are classified in Tables 1 and 2 based on the GO biological term.
Table 1.
Upregulated genes in 3-month-old HBV transgenic mice.
| Gene Function and GenBank Acc. No. | Gene Description | Gene Symbol | Fold Change |
|---|---|---|---|
| Immunological function | |||
| M13018 | Cysteine-rich protein 1 (intestinal) | Crip1 | 33.58 |
| Z80112 | Chemokine orphan receptor 1 | Cmkor1 | 4.91 |
| X52643 | Histocompatibility 2, class II antigen A, alpha | H2-Aa | 17.80 |
| M21932 | Histocompatibility 2, class II antigen A, beta 1 | H2-Ab1 | 8.01 |
| U16985 | Lymphotoxin B | Ltb | 8.76 |
| X00496 | Ia-associated invariant chain | Ii | 7.38 |
| J04170 | CD72 antigen | Cd72 | 4.98 |
| Ubiquitination | |||
| AI788534 | Ring finger protein 8 | Rnf8 | 11.65 |
| NF-κB signal transduction cascades | |||
| M84487 | Vascular cell adhesion molecule 1 | Vcam1 | 5.06 |
| AF000236 | Chemokine (C-X-C motif) receptor 4 | Cxcr4 | 5.04 |
| One-carbon compound metabolism | |||
| M25944 | Carbonic anhydrase 2 | Car2 | 6.51 |
| Regulation of transcription | |||
| X67668 | High mobility group box 2 | Hmgb2 | 5.44 |
| AF087434 | Nuclear factor of activated T-cells, cytoplasmic 1 | Nfatc1 | 18.56 |
| AI852641 | Nuclear protein 1 | Nupr1 | 72.44 |
| AK217100 | Activating transcription factor 3 | Atf3 | 3.8 |
| Intracellular calcium ion mobilization | |||
| L36860 | Guanylate cyclase activator 1a (retina) | Guca1 | 15.14 |
| AJ222586 | Nucleobindin 2 | Nucb2 | 9.69 |
| AF035645 | Protein tyrosine phosphatase 4a3 | Ptp4a3 | 4.99 |
| U20159 | Lymphocyte cytosolic protein 2 | Lcp2 | 4.43 |
| Regulation of cell cycle and proliferation | |||
| U19597 | Cyclin-dependent kinase inhibitor 2D (p19, inhibits CDK4) | Cdkn2d | 3.71 |
| AF099973 | Schlafen 2 | Slfn2 | 9.76 |
| Actin cytoskeleton organization and biogenesis | |||
| AI852553 | Thymosin, beta 10 | Tmsb10 | 4.27 |
| M60474 | Myristoylated alanine rich protein kinase C substrate | Marcks | 3.66 |
| Protein modification | |||
| X61232 | Carboxypeptidase E | Cpe | 4.63 |
| Intracellular signalling cascade | |||
| AA189555 | RIKEN cDNA F630107H02 gene | F630107H02Rik | 8.13 |
Table 2.
Downregulated genes in 3-month-old HBV transgenic mice.
| Gene Function and GenBank Acc. No. | Gene Description | Gene Symbol | Fold Change |
|---|---|---|---|
| Metabolism of cholesteryl | |||
| AW260404 | PDZ domain containing 1 | Pdzk1 | −1.92 |
| Extra matrix structure component | |||
| U24703 | Reelin | Reln | −2.26 |
| D50586 | Tissue factor pathway inhibitor 2 | Tfpi2 | −1.80 |
| Enzymes | |||
| AF026073 | N-sulfotransferase | Sultn | −1.72 |
| X17069 | FK506 binding protein 4 | Fkbp4 | −2.15 |
| Blood coagulation | |||
| M23109 | Coagulation factor IX | F9 | −2.01 |
| U60473 | CD59a antigen | Cd59a | −169 |
| Cell adhesion | |||
| M77196 | CEA-related cell adhesion molecule 1 | Ceacam1 | −2.03 |
| Receptor activity | |||
| U15012 | Growth hormone receptor | Ghr | −2.25 |
| X99347 | Lipopolysaccharide binding protein | Lbp | −2.13 |
| AF05175 | v-erb-b2 erythroblastic leukaemia viral oncogene homolog 3 (avian) | Erbb3 | −2.1 |
| Electron transport | |||
| U90535 | Flavin containing monooxygenase 5 | Fmo5 | −2.12 |
| AB016248 | Sterol-C5-desaturase (fungal ERG3, delta-5-desaturase) | Sc5d | −2.23 |
| Homolog (S. cerevisiae) | |||
| AF031170 | Hydroxysteroid dehydrogenase-6, delta<5>3-beta | Hsd3b6 | −2.15 |
| Water transport | |||
| AA967194 | Aquaporin 9 | Aqp9 | −2.22 |
| Oxidoreduttase activity | |||
| A1787269 | RIKEN cDNA 1200014D15 gene | 1200014D15Rik | −2.17 |
| Apoptosis | |||
| AF041054 | BCL2/adenovirus E1B 19kDa-interacting protein 1 | Bnip3 (NIP3) | −1.69 |
| Amino acid metabolism | |||
| U28016 | Phosphotriesterase related protein | Pter | −3.46 |
| Unknown | |||
| AA183875 | ESTs | −1.80 | |
| AW125273 | Plastin 3 (T-isoform) | Pls3 | −2.00 |
Upregulated Genes
Table 1 shows the list of 25 liver cell transcripts that were upregulated (fold change ≥ 2, P = 0.0025) in transgenic mice. The most common GO functional category was immunological function. We also observed an overexpression of genes involved in NF-κb signal transduction (Vcam1 and Cxcr4), regulation of transcription (Hmgb2, Nfatc1, Nupr1, and Atf3), and regulation of cell cycle and proliferation (Cdkn2d and Slfn2). In addition, we observed upregulation of some genes involved in intracellular calcium ion mobilization (Guca1, Nucb2, Ptp4a3, Lcp2) and actin cytoskeleton organization and biogenesis (Tmsb10, Marcks).
Downregulated Genes
A total of 20 genes were found to be downregulated (fold change ≤ 0.5, P = 0.0025) in transgenic mice (Table 2). The GO functional categories represented were 1) cholesterol metabolism, 2) amino acid metabolism, 3) blood coagulation, 4) extra matrix structure component, 5) cell adhesion, and 6) water transport. We also found downregulated genes encoding for the receptors involved in the control of proliferation (Ghr, Erbb3) and for the Bnip3 proapoptotic gene.
Validation of Microarray Data by Semiquantitative RT-PCR
We checked the microarray data, analyzing the expression level of the statistically different genes in 3-month-old HBV transgenic and wt mouse livers. Semiquantitative RT-PCR experiments were carried out on several randomly selected differentially expressed genes (data not shown), which were shown to be modulated in HBV transgenic mouse liver as demonstrated in microarray analysis. In particular, Figure 1 shows the semiquantitative RT-PCR analysis performed on the Nupr1, Cdkn2d, and Car2 genes. All 3 genes were clearly overexpressed in the transgenic versus the wt mice.
Figure 1.

Semiquantitative RT-PCR analysis. Semiquantitative RT-PCR analysis was performed on total RNAs prepared from livers of 3-month-old wt (control) and HBV transgenic mice. Nupr1 (nuclear protein 1), Cdkn2d (p19), and Car2 (carbonic anhydrase) expression levels were analyzed. β-Actin mRNA amplification was performed as an internal control. The reaction without cDNAs was the negative control. The results were concordant to microarray data.
Western Blot Analysis
We performed a Western blot analysis to correlate the increase in Hmgb2 gene expression to its protein expression. As shown in Figure 2, the analysis of liver total extracts, obtained from transgenic and wt mice, demonstrated that Hmgb2 protein was expressed at very low level in controls, whereas its expression was significantly increased in the transgenic mice (Figure 2).
Figure 2.
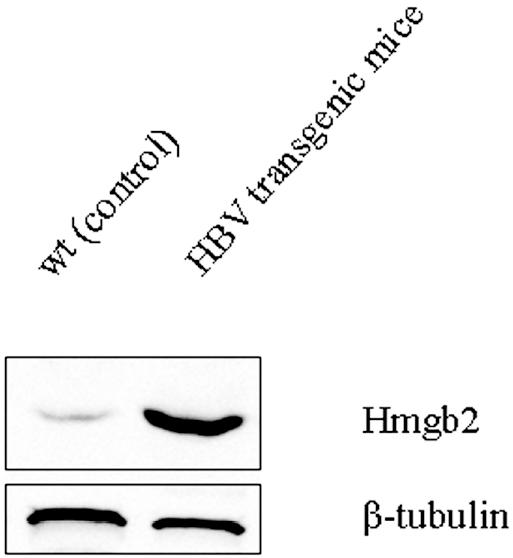
Hmgb2 protein expression in wt and HBV transgenic mice livers. Total protein extracts (50 μg of total protein/lane) were prepared from livers isolated from wt (control) and HBV transgenic mice at 3 months of age. All samples were analyzed by SDS-PAGE and immunoblotting with anti-Hmgb2 and anti–β-tubulin antibodies.
Evaluation of Apoptosis in Cultured Hepatocytes
The DNA-laddering assay was performed on hepatocytes isolated from 3-month-old transgenic and wt mice and incubated for 12 or 16 h in presence of staurosporine (Figure 3). DNA fragmentation was observable in hepatocytes isolated from wt mice 12 h after incubation with staurosporine; this phenomenon was even more evident after 16 h of incubation. On the other hand, in hepatocytes isolated from transgenic mice, the phenomenon of DNA-laddering was less evident both 12 and 16 h after incubation with staurosporine (Figure 3). To exclude that this difference between transgenic and wt hepatocyte DNA-laddering could be related to a different cellular mass (number of functional active hepato-cytes), this parameter was evaluated by MTT assay. Similar results were obtained in hepatocytes isolated from transgenic and wt mice (data not shown).
Figure 3.

Apoptotic assay. The DNA-laddering assay was performed on primary cultured hepatocytes isolated from 3-month-old wt (lanes 2–5) and HBV transgenic (lanes 6–9) mice in the presence or absence of 10 μM staurosporine.
DISCUSSION
HCC is a heterogeneous disease in terms of etiology and biologic and clinical behavior. Very little is known about how many genes concur at the molecular level for tumor development, progression, and aggressiveness.
As in other solid tumors, development, progression, and metastasis of HCC are multistep processes and are believed to be caused by the accumulation of genetic alterations, including chromosomal aberrations, oncogene activation, and inactivation of tumor suppressor genes (33–37). Recent studies have identified numerous epigenetic changes, such as promoter CpG island methylation, that are responsible for inactivation of tumor suppressor genes (38–41). Gene expression profiles of HCCs with different etiology show different molecular signatures (42,43), suggesting that heterogeneous hepatocarcinogenetic pathways, involved in cell proliferation, cell cycle, apoptosis, and angiogenesis, exist and are deregulated in hepatocarcinogenesis.
Despite efforts to unravel the molecular mechanisms of hepatocarcinogenesis, the biologic heterogeneity and multiple etiologies of HCC result in an incomplete understanding of the key molecular changes leading to HCC development. In particular, gene expression profile analysis, using DNA microarray technique, has paved a way to better understand molecular mechanisms in the development of HCC (42,43). Among the enormous number of deregulated genes, however, we still need to identify the “critical” genes that play a pivotal role in initiation and/or promotion of the development of HCC and distinguish them from “bystander” genes as a result of epigenetic interaction.
Current knowledge about the molecular pathogenesis of HCC is a result of investigations of fully developed HCC, and much less is known about the genetic basis of preneoplastic lesions, namely macroregenerative nodules and/or dysplastic nodules. This is the most critical issue to be addressed to get a clearer picture of the early molecular events for the HCC progression and to identify gene markers for developing effective therapeutic options. To address this issue, we used the HBV transgenic mouse as a model to study the processes of initiation and progression of HCC related to the HBV infection. Although laboratory mice have represented a powerful experimental system for understanding the intricacies of human cancer pathogenesis, there are important differences between mice and humans that influence the way in which cancer develops (44–46). Among the main differences is the number of genetic events involved in cancer development. Fewer genetic, epigenetic, or gene-expression altering events are required to induce a malignant transformation in murine cells compared with human cells (44,45), and studies have shown that several important signaling pathways seem to function differently in human and rodent models of transformation. In spite of these differences, our animal model appears to be a useful tool to study human HCC development because it mimics many of the pathological events that occur before the development of HCC in chronic HBV infection in humans.
Using cDNA microarray technology, we have identified a number of genes differently expressed in normal and pathological specimens during the murine hepatocarcinogenesis process. This allowed us to obtain, for the first time, an almost complete picture of the early genetic alterations that are directly or indirectly involved in the development of HCC. The data obtained from gene expression profiling will allow us to acquire insights on molecular mechanisms of hepatocarcinogenesis and to identify specific genes (or gene products) that can be used for early molecular diagnosis, risk analysis, prognosis prediction, and development of new therapies. Therefore, the transgenic mouse represents a useful model system not only for basic research, but also for clinical research applied to therapeutic drug discovery.
In this article, we focused our attention on the early molecular events that occurred in HBV transgenic mice and probably represent the earliest ones that start the multistep process of hepatocarcinogenesis. By analyzing gene expression profile, we identified 25 upregulated and 20 downregulated genes in the HBV transgenic mouse, suggesting a direct effect of HBV in the modulation of these genes.
We confirmed previously reported data on Atf3 overexpression due to HBV (47). GO analysis showed that several differently expressed genes are involved in several cellular functions. In particular, a large number of upregulated genes were involved in the modulation of immunological functions, suggesting that the accumulation in the hepatocytes of viral proteins yielded cellular damage followed by an immune response (23,27,29). In addition, several upregulated transcripts have already been correlated to cellular dysplasia and tumor malignancy in different types of cancer (48–59). The microarray data showed the upregulation of NuprI, a transcription factor whose overexpression is inversely correlated with apoptosis in pancreatic cancer (60), and the downregulation of the proapoptotic gene Bnip3 (61,62). This finding supports the conclusion that the deregulation of apoptosis, facilitating the escape of “abnormal” cells from death, could be a mechanism by which HBV promotes HCC development. To confirm the influence of HBV on apoptosis, we analyzed DNA fragmentation, a hallmark of apoptosis, upon staurosporine treatment in hepatocytes isolated from 3-month-old transgenic and wt mice using an in vitro apoptotic assay. Our results showed a significant downregulation of apoptosis in hepatocytes isolated from HBV transgenic mice compared with controls, in spite of comparable cell viability. Therefore, the downregulation of the apoptotic process may represent a possible mechanism by which HBV induces malignant transformation. The role of HBx in regulating apoptosis is controversial because previous reports, indicating an abrogation of p53-induced apoptosis (18), were probably refuted by later data indicating a positive regulation of the apoptotic process (19–22).
In conclusion, gene expression profile is a useful tool to acquire new clues and generate new fields of research. Our experimental setup allowed us to acquire new insights on early molecular events of hepatocarcinogenesis induced by HBV, and in particular on possible mechanisms by which HBV induces HCC development. Further investigations are in progress in transgenic mice studied at different ages to confirm and expand these findings and clarify their role in more advanced stages of the disease.
ACKNOWLEDGMENTS
We thank Francioso Doriana for technical assistance. This work was supported in part by MIUR funds (grant 2003069954), FIST project (C.T.), and AIRC regional funds 2005.
Footnotes
Online address: http://www.molmed.org
REFERENCES
- 1.Beasley RP. Hepatitis B virus—the major etiology of hepatocellular carcinoma. Cancer. 1998;61:1942–56. doi: 10.1002/1097-0142(19880515)61:10<1942::aid-cncr2820611003>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 2.Singh M, Kumar V. Transgenic mouse models of hepatitis B virus-associated hepatocellular carcinoma. Rev Med Virol. 2003;13:243–53. doi: 10.1002/rmv.392. [DOI] [PubMed] [Google Scholar]
- 3.Paterlini P, Poussin K, Kew M, Franco D, Brechot C. Selective accumulation of the X transcript of hepatitis B virus in patients negative for hepatitis B surface antigen with hepatocellular carcinoma. Hepatology. 1995;21:313–21. [PubMed] [Google Scholar]
- 4.Zoulim F, Saputelli J, Seeger C. Woodchuck hepatitis virus X protein is required for viral infection in vivo. J Virol. 1994;68:2026–30. doi: 10.1128/jvi.68.3.2026-2030.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bouchard MJ, Wang LH, Schneider RJ. Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science. 2001;294:2376–8. doi: 10.1126/science.294.5550.2376. [DOI] [PubMed] [Google Scholar]
- 6.Andrisani OM, Barnabas S. The transcriptional function of the hepatitis B virus X protein and its role in hepatocarcinogenesis (Review) Int J Oncol. 1999;15:373–9. doi: 10.3892/ijo.15.2.373. [DOI] [PubMed] [Google Scholar]
- 7.Qadri I, Maguire HF, Siddiqui A. Hepatitis B virus transactivator protein X interacts with the TATA-binding protein. Proc Natl Acad Sci U S A. 1995;92:1003–7. doi: 10.1073/pnas.92.4.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maguire HF, Hoeffler JP, Siddiqui A. HBV X protein alters the DNA binding specificity of CREB and ATF-2 by protein-protein interactions. Science. 1991;252:842–4. doi: 10.1126/science.1827531. [DOI] [PubMed] [Google Scholar]
- 9.Lin Y, Nomura T, Cheong J, Dorjsuren D, Iida K, Murakami S. Hepatitis B virus X protein is a transcriptional modulator that communicates with transcription factor IIB and the RNA polymerase II subunit 5. J Biol Chem. 1997;272:7132–9. doi: 10.1074/jbc.272.11.7132. [DOI] [PubMed] [Google Scholar]
- 10.Shamay M, Barak O, Doitsh G, Ben-Dor I, Shaul Y. Hepatitis B virus pX interacts with HBXAP, a PHD finger protein to coactivate transcription. J Biol Chem. 2002;277:9982–8. doi: 10.1074/jbc.M111354200. [DOI] [PubMed] [Google Scholar]
- 11.Pan J, Duan LX, Sun BS, Feitelson MAJ. Hepatitis B virus X protein protects against anti-Fas-mediated apoptosis in human liver cells by inducing NF-kappa B. Gen Virol. 2001;82:171–82. doi: 10.1099/0022-1317-82-1-171. [DOI] [PubMed] [Google Scholar]
- 12.Kekule AS, Lauer U, Weiss L, Luber B, Hofschneider PH. Hepatitis B virus transactivator HBx uses a tumor promoter signaling pathway. Nature. 1993;361:742–5. doi: 10.1038/361742a0. [DOI] [PubMed] [Google Scholar]
- 13.Luber B, Lauer U, Weiss L, Hohne M, Hofschneider PH, Kekule AS. The hepatitis B virus transactivator HBx causes elevation of diacylglycerol and activation of protein kinase C. Res Virol. 1993;144:311–21. doi: 10.1016/s0923-2516(06)80047-6. [DOI] [PubMed] [Google Scholar]
- 14.Benn J, Schneider RJ. Hepatitis B virus HBx protein activates Ras-GTP complex formation and establishes a Ras, Raf, MAP kinase signaling cascade. Proc Natl Acad Sci U S A. 1994;91:10350–4. doi: 10.1073/pnas.91.22.10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee YH, Yun Y. HBx protein of hepatitis B virus activates Jak1-STAT signaling. J Biol Chem. 1998;273:25510–5. doi: 10.1074/jbc.273.39.25510. [DOI] [PubMed] [Google Scholar]
- 16.Waris G, Huh KW, Siddiqui A. Mitochondrially associated hepatitis B virus X protein constitutively activates transcription factors STAT-3 and NF-kappa B via oxidative stress. Mol Cell Biol. 2001;21:7721–30. doi: 10.1128/MCB.21.22.7721-7730.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lara-Pezzi E, et al. The hepatitis B virus X protein promotes tumor cell invasion by inducing membrane-type matrix metalloproteinase-1 and cyclooxygenase-2 expression. J Clin Invest. 2002;110:1831–8. doi: 10.1172/JCI200215887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huo TI, et al. Hepatitis B virus X mutants derived from human hepatocellular carcinoma retain the ability to abrogate p53-induced apoptosis. Oncogene. 2001;20:3620–8. doi: 10.1038/sj.onc.1204495. [DOI] [PubMed] [Google Scholar]
- 19.Takada S, Shirakata Y, Kaneniwa N, Koike K. Association of hepatitis B virus X protein with mitochondria causes mitochondrial aggregation at the nuclear periphery, leading to cell death. Oncogene. 1999;18:6965–73. doi: 10.1038/sj.onc.1203188. [DOI] [PubMed] [Google Scholar]
- 20.Su F, Schneider RJ. Hepatitis B virus HBx protein sensitizes cells to apoptotic killing by tumor necrosis factor alpha. Proc Natl Acad Sci U S A. 1997;94:8744–9. doi: 10.1073/pnas.94.16.8744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bergametti F, Prigent S, Luber B, Benoit A, Tiollais P, Sarasin A, Transy C. The proapoptotic effect of hepatitis B virus HBx protein correlates with its transactivation activity in stably transfected cell lines. Oncogene. 1999;18:2860–71. doi: 10.1038/sj.onc.1202643. [DOI] [PubMed] [Google Scholar]
- 22.Shirakata Y, Koike K. Hepatitis B virus X protein induces cell death by causing loss of mitochondrial membrane potential. J Biol Chem. 2003;278:22071–8. doi: 10.1074/jbc.M301606200. [DOI] [PubMed] [Google Scholar]
- 23.Nakamoto Y, Guidotti LG, Kuhlen V, Fowler P, Chisari FV. Immune pathogenesis of hepatocellular carcinoma. J Exp Med. 1998;188:341–50. doi: 10.1084/jem.188.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dolberg DS, Hollingsworth R, Hertle M, Bissell MJ. Wounding and its role in RSV-mediated tumor formation. Science. 1985;230:676–8. doi: 10.1126/science.2996144. [DOI] [PubMed] [Google Scholar]
- 25.Chisari FV, Pinkert CA, Milich DR, Filippi P, McLachlan A, Palmiter RD, Brinster RL. A transgenic mouse model of the chronic hepatitis B surface antigen carrier state. Science. 1985;230:1157–60. doi: 10.1126/science.3865369. [DOI] [PubMed] [Google Scholar]
- 26.Chisari FV, et al. Structural and pathological effects of synthesis of hepatitis B virus large envelope polypeptide in transgenic mice. Proc Natl Acad Sci U S A. 1987;84:6909–13. doi: 10.1073/pnas.84.19.6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dunsford HA, Sell S, Chisari FV. Hepatocarcinogenesis due to chronic liver cell injury in hepatitis B virus transgenic mice. Cancer Res. 1990;50:3400–7. [PubMed] [Google Scholar]
- 28.Toshkov I, Chisari FV, Bannasch P. Hepatic preneoplasia in hepatitis virus transgenic mice. Hepatology. 1994;20:1162–72. [PubMed] [Google Scholar]
- 29.Huang SN, Chiari FV. Strong, sustained hepatocellular proliferation precedes hepatocarcinogenesis in hepatitis B surface antigen transgenic mice. Hepatology. 1995;21:620–6. [PubMed] [Google Scholar]
- 30.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barone M, et al. Modulation of rat hepatocyte proliferation by bile salts: in vitro and in vivo studies. Hepatology. 1996;23:1159–66. doi: 10.1053/jhep.1996.v23.pm0008621149. [DOI] [PubMed] [Google Scholar]
- 32.Cifone MG, D’Alo S, Parroni R, Millimaggi D, Biordi L, Martinotti S, Santoni A. Inter-leukin-2-activated rat natural killer cells express inducible nitric oxide synthase that contributes to cytotoxic function and interferon-gamma production. Blood. 1999;93:3876–84. [PubMed] [Google Scholar]
- 33.Wang G, et al. Allelic loss and gain, but not genomic instability, as the major somatic mutation in primary hepatocellular carcinoma. Genes Chromosomes Cancer. 2001;31:221–7. doi: 10.1002/gcc.1138. [DOI] [PubMed] [Google Scholar]
- 34.Zhu GN, Zuo L, Zhou Q, Zhang SM, Zhu HQ, Gui SY, Wang Y. Loss of heterozygosity on chromosome 10q22-10q23 and 22q11.2-22q12.1 and p53 gene in primary hepatocellular carcinoma. World J Gastroenterol. 2004;10:1975–8. doi: 10.3748/wjg.v10.i13.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jou YS, et al. Clustering of minimal deleted regions reveals distinct genetic pathways of human hepatocellular carcinoma. Cancer Res. 2004;64:3030–6. doi: 10.1158/0008-5472.can-03-2320. [DOI] [PubMed] [Google Scholar]
- 36.Suriawinata A, Xu R. An update on the molecular genetics of hepatocellular carcinoma. Semin Liver Dis. 2004;24:77–88. doi: 10.1055/s-2004-823102. [DOI] [PubMed] [Google Scholar]
- 37.Zhang SH, Cong WM, Xian ZH, Wu MC. Clinicopathological significance of loss of heterozygosity and microsatellite instability in hepatocellular carcinoma in China. World J Gastroenterol. 2005;11:3034–9. doi: 10.3748/wjg.v11.i20.3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park WS, et al. Hypermethylation of the RUNX3 gene in hepatocellular carcinoma. Exp Mol Med. 2005;37:276–81. doi: 10.1038/emm.2005.37. [DOI] [PubMed] [Google Scholar]
- 39.Chu HJ, et al. Detection of aberrant p16INK4A methylation in sera of patients with liver cirrhosis and hepatocellular carcinoma. J Korean Med Sci. 2004;19:83–6. doi: 10.3346/jkms.2004.19.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qiu W, et al. Hypermethylation of growth arrest DNA damage-inducible gene 45 beta promoter in human hepatocellular carcinoma. Am J Pathol. 2004;165:1689–99. doi: 10.1016/s0002-9440(10)63425-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lei PP, Zhang ZJ, Shen LJ, Li JY, Zou Q, Zhang HX. Expression and hypermethylation of p27 kip1 in hepatocarcinogenesis. World J Gas-troenterol. 2005;11:4587–91. doi: 10.3748/wjg.v11.i29.4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iizuka N, et al. Molecular signature in three types of hepatocellular carcinoma with different viral origin by oligonucleotide microarray. Int J Oncol. 2004;24:565–74. [PubMed] [Google Scholar]
- 43.Zhang LH, Ji JF. Molecular profiling of hepatocellular carcinomas by cDNA microarray. World J Gastroenterol. 2005;11:463–8. doi: 10.3748/wjg.v11.i4.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hahn WC, Weinberg RA. Modelling the molecular circuitry of cancer. Nat Rev Cancer. 2002;2:331–41. doi: 10.1038/nrc795. [DOI] [PubMed] [Google Scholar]
- 45.Rangarajan A, Weinberg RA. Opinion: Comparative biology of mouse versus human cells: modeling human cancer in mice. Nat Rev Cancer. 2003;3:952–9. doi: 10.1038/nrc1235. [DOI] [PubMed] [Google Scholar]
- 46.Anisimov VN, Ukraintseva SV, Yashin AI. Cancer in rodents: does it tell us about cancer in humans? Nat Rev Cancer. 2005;5:807–19. doi: 10.1038/nrc1715. [DOI] [PubMed] [Google Scholar]
- 47.Tarn C, Bilodeau ML, Hullinger RL, Andrisani OM. Differential immediate early gene expression in conditional hepatitis B virus pX-transforming versus nontransforming hepatocyte cell lines. J Biol Chem. 1999;274:2327–36. doi: 10.1074/jbc.274.4.2327. [DOI] [PubMed] [Google Scholar]
- 48.Line A, Stengrevics A, Slucka Z, Li G, Jankevics E, Rees RC. Serological identification and expression analysis of gastric cancer-associated genes. Br J Cancer. 2002;86:1824–30. doi: 10.1038/sj.bjc.6600321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Iovanna JL. Expression of the stress-associated protein p8 is a requisite for tumor development. Int J Gastrointest Cancer. 2002;31:89–98. doi: 10.1385/IJGC:31:1-3:89. [DOI] [PubMed] [Google Scholar]
- 50.Ito Y, et al. Expression and cellular localization of p8 protein in thyroid neoplasms. Cancer Lett. 2003;201:237–44. doi: 10.1016/j.canlet.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 51.Mohammad HP, Seachrist DD, Quirk CC, Nilson JH. Reexpression of p8 contributes to tumorigenic properties of pituitary cells and appears in a subset of prolactinomas in transgenic mice that hypersecrete luteinizing hormone. Mol Endocrinol. 2004;18:2583–93. doi: 10.1210/me.2004-0163. [DOI] [PubMed] [Google Scholar]
- 52.Jiang WG, Watkins G, Douglas-Jones A, Mokbel K, Mansel RE, Fodstad O. Expression of Com-1/P8 in human breast cancer and its relevance to clinical outcome and ER status. Int J Cancer. 2005;117:730–7. doi: 10.1002/ijc.21221. [DOI] [PubMed] [Google Scholar]
- 53.Takano T, Hasegawa Y, Miyauchi A, Matsuzuka F, Yoshida H, Kuma K, Amino N. Quantitative analysis of thymosin beta-10 messenger RNA in thyroid carcinomas. Jpn J Clin Oncol. 2002;32:229–32. doi: 10.1093/jjco/hyf054. [DOI] [PubMed] [Google Scholar]
- 54.Alldinger I, et al. Gene expression analysis of pancreatic cell lines reveals genes over-expressed in pancreatic cancer. Pancreatology. 2005;5:370–9. doi: 10.1159/000086537. [DOI] [PubMed] [Google Scholar]
- 55.Sardi I, et al. Molecular profiling of high-risk neuroblastoma by cDNA array. Int J Mol Med. 2002;9:541–5. [PubMed] [Google Scholar]
- 56.Saha S, et al. A phosphatase associated with metastasis of colorectal cancer. Science. 2001;294:1343–6. doi: 10.1126/science.1065817. [DOI] [PubMed] [Google Scholar]
- 57.Mackay A, et al. cDNA microarray analysis of genes associated with ERBB2 (HER2/neu) overexpression in human mammary luminal epithelial cells. Oncogene. 2003;22:2680–8. doi: 10.1038/sj.onc.1206349. [DOI] [PubMed] [Google Scholar]
- 58.Ruiz-Ballesteros E, et al. Splenic marginal zone lymphoma: proposal of new diagnostic and prognostic markers identified after tissue and cDNA microarray analysis. Blood. 2005;106:1831–8. doi: 10.1182/blood-2004-10-3898. [DOI] [PubMed] [Google Scholar]
- 59.Boon K, Edwards JB, Eberhart CG, Riggins GJ. Identification of astrocytoma associated genes including cell surface markers. BMC Cancer. 2004;4:39. doi: 10.1186/1471-2407-4-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Su SB, et al. Overexpression of p8 is inversely correlated with apoptosis in pancreatic cancer. Clin Cancer Res. 2001;7:1320–4. [PubMed] [Google Scholar]
- 61.Ray R, et al. BNIP3 heterodimerizes with Bcl-2/Bcl-X(L) and induces cell death independent of a Bcl-2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. J Biol Chem. 2000;275:1439–48. doi: 10.1074/jbc.275.2.1439. [DOI] [PubMed] [Google Scholar]
- 62.Bruick RK. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci U S A. 2000;97:9082–7. doi: 10.1073/pnas.97.16.9082. [DOI] [PMC free article] [PubMed] [Google Scholar]