

Abstract
We examine the interactions between amino acid residues in the context of their secondary structural environments (helix, strand, and coil) in proteins. Effective contact energies for an expanded 60-residue alphabet (20 aa × three secondary structural states) are estimated from the residue–residue contacts observed in known protein structures. Similar to the prototypical contact energies for 20 aa, the newly derived energy parameters reflect mainly the hydrophobic interactions; however, the relative strength of such interactions shows a strong dependence on the secondary structural environment, with nonlocal interactions in β-sheet structures and α-helical structures dominating the energy table. Environment-dependent residue contact energies outperform existing residue pair potentials in both threading and three-dimensional contact prediction tests and should be generally applicable to protein structure prediction.
The main obstacle for protein structure prediction comes from the immense number of possible conformations accessible to a polypeptide chain and the complexity and variety of interactions involved in the folding process. This obstacle makes it a formidable task to carry out protein folding simulations using an all-atom representation of the polypeptide. Coarse-grained protein models, which treat amino acid residues as united interaction sites, offer a more practical approach to tackling the protein folding problem (1). Because such models omit many detailed features of atomic interactions, new energy parameters suitable for representing interactions at low level of resolution are required. These parameters often are derived empirically from the analysis of experimentally observed residue contact preferences in sets of known structures (2). Whether such knowledge-based potentials correctly reflect the actual physical forces stablizing the native structures of proteins remains a subject of debate (3–6). Nevertheless, structure-derived potentials have contributed substantially to the current theoretical studies of protein folding (7).
Over the years, many specifically designed residue pair potentials have been proposed (2, 7, 8), most of which use the 20-aa alphabet and assume a constant energy contribution for a given residue pair regardless of its local structural environment. However, it is known that the folded conformation of a protein is stabilized by a multitude of weak noncovalent interactions and the contribution of each of these interactions depends on its context within the folded structure (9–11). To derive better, more specific potential energy functions, one has to take into account the influence of varied structural circumstances on the specificity of inter-residue interactions. In this study, we examine pairwise amino acid interactions in the context of secondary structural environments (helix, strand, and coil) and report a set of residue contact energies for an expanded 60-residue alphabet (20 aa × three secondary structural states).
The discriminatory capability of the environment-dependent contact energies (ERCE) was tested first in the threading experiments where the secondary structural states of the template protein structures are experimentally determined. Though only the simple nongapped threading protocol was used, the improvement over existing residue pair potentials was already evident as ERCE correctly recognized the native structures for more than 97% of the testing proteins. The applicability of ERCE is greatly extended when combined with secondary structure prediction. The new generation of algorithms for secondary structure prediction benefits from using evolutionary information provided by multiple sequence alignment (12–14). Recently developed structure prediction methods, including both ab initio methods (15–17) and fold recognition-based methods (18–23), all used predicted secondary structures and have achieved an important degree of success. We compared ERCE with several existing residue pair potentials in predicting residue contacts in the native protein structure from the amino acid sequence. A test based on a large number of distinct protein domains showed that the use of ERCE in combination of predicted secondary structures led to a significant improvement in the accuracy of contact prediction. This result has broader implications for protein structure prediction, because an energy function that favors conformations with higher percentage of native contacts has a better chance to guide global optimization toward the native folded state (24–26).
Methods
Protein Structure Set.
A set of representative protein domain structures were extracted from the Protein Date Bank (27) by referring to the scop database (version 1.37) (28). Specifically, we started with the <40% identity set built by the authors of scop and then performed additional sorting. First, we removed composite domains and domains with fewer than 50 residues or more than 300 residues. A single member then was selected from each scop family in the four structural classes, all-α, all-β, α/β, and α+β; wherever applicable, structures that had been determined to the highest crystallographic resolution were chosen. Domains with only Cα-traces or containing large sequence gaps were excluded. The structures thus obtained were inspected at the scop superfamily level. If a superfamily had multiple representatives, those structures with resolution lower than 2.30 Å were deleted. Domains that do not form a compact globular structure (e.g., long α-helical coiled-coils) also were removed. The remaining protein domains were examined by pairwise sequence alignment; when the sequence identity between a pair was higher than 25%, only one structure, typically the one with higher resolution, was kept. The final data set contains 407 protein domains; of those, 80 are all-α, 105 are all-β, 91 are α/β, and 131 are α+β (the complete list is available from the authors on request).
The Definition of Secondary Structural States.
The experimentally determined (real) secondary structural states (helix, strand, and coil) of residues in these structures were extracted from the dssp database (29). A simple mapping scheme was used where only H states in dssp were mapped to helix, E states mapped to strand, and all other states mapped to coil. The predicted secondary structural states were obtained by running the psi-pred program (14). This program uses the position-specific scoring matrices generated by psi-blast (30) as input. To prepare for these matrices, we performed a psi-blast search for each of the 407 protein domains against a nonredundant sequence database NRDB90 (31). The default E-value cutoff (0.0001) was used, and the maximum number of iterations was set to seven. The overall accuracy of secondary structure prediction for 407 protein domains is 79.5%.
Extracting Contact Energies from Known Protein Structures.
The combination of 20 aa and three secondary structural states gives rise to 60 residue types. We applied the Miyazawa–Jernigan procedure (MJ) (32) with a different definition of the reference state (33) to derive contact energies for this expanded residue alphabet from the 407 protein domain structures. Specifically, amino acid residues were represented by the centroids of their side chains, and two residues were considered to be in contact if the distance between their centroids fell within RC = 6.5 Å. The numbers of contacts formed between two residues, i and j, and between them and the solvent molecules (represented by 0) were related to the contact energy by a hypothetical chemical reaction
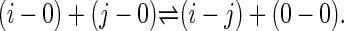 |
1 |
The effective contact energy (eij) is defined as the negative logarithm of the equilibrium constant of the reaction (32)
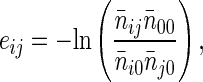 |
2 |
where n̄ij, 2n̄i0, 2n̄j0, and n̄00 are the number of residue i-residue j contacts, the number of residue i-solvent contacts, the number of residue j-solvent contacts, and the number of solvent-solvent contacts, respectively. In practice, eij was estimated by
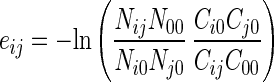 |
3 |
to minimize the bias from amino acid compositional heterogeneity and polypeptide chain connectivity, where Nij, Ni0, Nj0, and N00 are the contact numbers observed in known structures, and Cij, Ci0, Cj0, and C00 are the corresponding quantities expected in a reference state.
The calculations of Nij = Σpnij;p and Ni0 = Σpni0;p require the knowledge of the contact numbers nij;p and ni0;p in individual proteins (represented by p). Whereas nij;p was counted directly from the structure, ni0;p was derived from
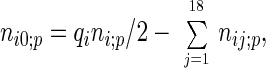 |
4 |
where qi is the precalculated coordination number of residue i (Table 1) and ni;p is the number of residue i in protein p.
Table 1.
Coordination numbers of amino acids in different secondary structural environments for RC = 6.5 Å
| Amino acid | α-Helix (α) | β-Strand (β) | Coil (C) |
|---|---|---|---|
| ALA | 6.59 (1.38) | 6.16 (1.26) | 6.18 (1.31) |
| ARG | 6.07 (1.34) | 6.04 (1.33) | 6.13 (1.25) |
| ASN | 6.35 (1.40) | 6.26 (1.10) | 6.13 (1.34) |
| ASP | 6.33 (1.22) | 6.16 (1.28) | 6.12 (1.28) |
| CYS | 6.51 (1.22) | 6.26 (1.24) | 6.26 (1.35) |
| GLN | 6.17 (1.26) | 6.30 (1.28) | 6.19 (1.23) |
| GLU | 6.22 (1.17) | 6.24 (1.22) | 6.18 (1.26) |
| GLY | 6.41 (1.46) | 5.90 (1.34) | 5.80 (1.30) |
| HIS | 6.15 (1.24) | 6.33 (1.26) | 6.01 (1.26) |
| ILE | 6.38 (1.35) | 5.99 (1.31) | 5.97 (1.26) |
| LEU | 6.48 (1.37) | 6.27 (1.24) | 6.27 (1.28) |
| LYS | 6.07 (1.17) | 6.07 (1.27) | 6.02 (1.50) |
| MET | 6.31 (1.28) | 6.12 (1.19) | 6.21 (1.25) |
| PHE | 6.13 (1.32) | 6.15 (1.27) | 6.07 (1.37) |
| PRO | 5.95 (1.15) | 5.93 (1.27) | 5.84 (1.32) |
| SER | 6.39 (1.33) | 6.05 (1.39) | 5.98 (1.24) |
| THR | 6.58 (1.36) | 6.05 (1.20) | 6.17 (1.27) |
| TRP | 5.72 (1.26) | 5.76 (1.38) | 5.61 (1.28) |
| TYR | 6.17 (1.29) | 6.15 (1.24) | 6.05 (1.28) |
| VAL | 6.59 (1.38) | 6.15 (1.28) | 6.19 (1.30) |
The coordinate number qi (i = 1, 60) is calculated as the number of non-nearest neighbor contacts formed by a buried residue of type i averaged over 407 protein domain structures. A residue is considered buried if more than 95% of its surface area becomes inaccessible to solvent when the protein is folded. Values in parentheses are the SDs.
We assume that the reference state of a protein exhibits the same compactness as the native folded state but with randomly arranged residue-residue and residue-solvent contacts (for details see ref. 33). Under this assumption, we have
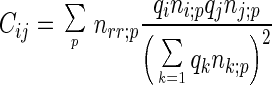 |
5 |
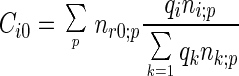 |
6 |
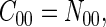 |
7 |
where nrr;p = ΣiΣjnij;p and nr0;p = Σini0;p.
Testing the Derived Energies.
To assess whether ERCE offer better discriminatory power than the existing residue pair potentials, we first tested these potentials by using a nongapped threading protocol (34–36). The sequences of proteins with 200 or fewer residues in the data set were threaded through the structures of all proteins of the same or larger size at all possible positions. When the energy of the native structure of a protein is lower than that of any threaded model, we consider that the sequence of the protein successfully recognizes its structure.
To assess the potential in a more realistic setting where only predicted secondary structure information can be obtained, we applied ERCE to the prediction of the three-dimensional contacts in protein structures. In particular, we concentrated on nonlocal contacts, i.e., contacts formed by residues not closely associated by the polypeptide chain. For contacts between on-chain neighbors, short-range interactions are probably more important. A minimum of four residue separations were required for a contact to be considered. For the purpose of comparing energy functions, we used a simple method for contact prediction where any residue pair that had energy lower than a specified cutoff was predicted to be in contact. By varying the cutoff value, the tradeoff between the completeness (or coverage) of the prediction (how many native contacts are predicted) and the accuracy of the prediction (how many predicted contacts correspond to actually observed contacts) can be examined. The actual three-dimensional contacts were identified from protein structures by using the criterion that two residues in contact must form at least four atom–atom contacts (two atoms are considered in contact if the distance between them is less than 6.0 Å). The results were essentially the same when a more (or less) stringent definition of contact was used.
Results
The Influence of Secondary Structural Environments on Inter-Residue Interactions.
The calculated contact energies are given in Fig. 1. The 60-by-60 ERCE parameters are separated into six groups with respect to the secondary structural environments of the interacting pair: α-α, β-β, α-β, β-C, β-C, and C-C (where α, β, and C represent helix, strand, and coil, respectively). Similar to the contact energies determined by MJ (32), the energies derived here are dominated by hydrophobic interactions. The correlation coefficients of the contact energies from different groups with each other and with those determined by MJ (32) are summarized in Table 2.
Figure 1.

Secondary structural ERCE (in RT units). The energy parameters are divided into six groups, α-α, β-β, α-β, α-C, β-C, and C-C, where α, β, and C represent helix, strand, and coil states, respectively.
Table 2.
Comparisons between energy parameters
| Average energy* | Pairwise correlation
coefficients
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MJ | α-α | β-β | α-β† | α-β‡ | α-C† | α-C‡ | β-C† | β-C‡ | C-C | ||
| MJ | −2.93 (1.45) | — | |||||||||
| α-α | −0.74 (0.99) | 0.86 | — | ||||||||
| β-β | −1.66 (0.93) | 0.80 | 0.94 | — | |||||||
| α-β† | −0.10 (1.33) | 0.83 | 0.92 | 0.89 | — | ||||||
| α-β‡ | −0.04 (1.33) | 0.84 | 0.92 | 0.89 | 0.88 | — | |||||
| α-C† | 0.23 (0.93) | 0.89 | 0.92 | 0.87 | 0.89 | 0.90 | — | ||||
| α-C‡ | 0.18 (0.94) | 0.91 | 0.92 | 0.89 | 0.90 | 0.92 | 0.90 | — | |||
| β-C† | 0.01 (0.87) | 0.80 | 0.89 | 0.87 | 0.86 | 0.90 | 0.91 | 0.90 | — | ||
| β-C‡ | −0.14 (0.88) | 0.81 | 0.91 | 0.90 | 0.89 | 0.89 | 0.89 | 0.91 | 0.83 | — | |
| C-C | 0.25 (0.70) | 0.86 | 0.91 | 0.88 | 0.87 | 0.89 | 0.95 | 0.93 | 0.92 | 0.91 | — |
The three asymmetric energy groups, α-β, β-C, and β-C, are subdivided into lower and upper off-diagonal triangular components and represented by † and ‡, respectively. MJ, ref. 32.
*In RT units. Values in parentheses are the SDs.
Despite the high correlations, the average values of contact energies from different groups differ significantly (Table 2). The α-α and β-β contacts are generally more favorable than other types of interactions. This result is understandable considering that the cores of most proteins consist of closely packed regular secondary structural elements where helices tend to gather into bundles and β-strands often appear in assembled β-sheets. The β-β contact energies are on average nearly one RT unit lower than the α-α contact energies. Because most of the tertiary contacts formed by β residues are cross-β-bridge contacts, this result suggests that there may be a fundamental difference between the structural and energetic principles governing β-strand register and α-helix packing. Among the energy groups, the α-β contact energies show the largest variations, and therefore, highest specificity, consistent with the unique geometric feature of α-β packing observed in protein structures (37, 38).
Threading Experiments.
There are 316 protein domains in our data set that contain 200 or fewer residues; the number of conformations generated by threading ranges from 4,000 (for a 200-residue protein) to 40,000 (for a 50-residue protein). As shown in Table 3, ERCE consistently outperformed MJ in the treading tests in all four structural categories, with an overall 6% improvement in success rate. For 88 all-β proteins and 49 α/β proteins, perfect recognition was achieved. In seven of the nine cases where ERCE failed to identify the native structures, the native structures were among the five most favorable conformations. MJ failed three times more; in half of those cases, the native structures were ranked below five.
Table 3.
Results of the threading experiments
| all-α (71) | all-β (88) | α/β (49) | α+β (108) | Overall (316) | |
|---|---|---|---|---|---|
| MJ | 87.3% | 94.3% | 93.9% | 90.7% | 91.4% |
| ERCE | 90.1% | 100% | 100% | 98.1% | 97.2% |
The numbers in parentheses are the number of proteins in each category. MJ, ref. 32.
Contact Prediction.
Fig. 2 shows the performance of ERCE in contact prediction for 407 proteins. Both predicted and real secondary structures were used, and the prediction based on real secondary structures in conjunction with ERCE (ERCE-Real) is noticeably better. However, a significant improvement over MJ in terms of prediction accuracy was obtained by both methods. The encouraging result achieved by ERCE in combination with predicted secondary structures (ERCE-Pred) adds a practical value to ERCE. We also have applied other published residue pair potentials for the same task; of those, the only potential that outperformed MJ was one recently reported by Skolnick and coworkers (JS) (5). Interestingly, a recent update to MJ performed slightly worse than the original energy table (data not shown), although the updated energy table was derived from a much larger set of protein structures (36).
Figure 2.

Contact prediction using three different potentials: MJ, JS, and ERCE. ERCE predictions based on predicted (ERCE-Pred) and real secondary structures (ERCE-Real) both are shown. The x axis indicates the fraction of experimentally determined contacts that are correctly predicted (defined here as the coverage). Each coverage value x corresponds to an energy cutoff such that the percentage of native contacts whose energies are lower than the cutoff happens to be x. There are other residue pairs that do not form contacts in the native protein structure but also have energies lower than the cutoff. The fraction of predicted contacts that correspond to actually observed contacts define the accuracy. Here we compare different prediction methods with a common standard: the random prediction. In theory, the accuracy of a random prediction averaged over many trials equals the ratio of native contacts to the number of all possible residue pairs in a protein. This value varies with protein size and contact density. For each method, the y axis indicates how much more accurate the prediction is relative to that from a random method.
More detailed comparisons of different prediction methods are shown in Table 4. The 407 testing proteins were divided into three size categories and four structural classes. Because the fraction of residue pairs that form tertiary contacts in a larger protein is less than that in a smaller protein, the level of difficulty in predicting contact rises as the size of the protein increases. This trend is clearly seen in the case of a random prediction (Table 4). Although the use of potential energy function improves the chance of detecting native contacts, the basic trend of prediction accuracy versus protein size persists. ERCE-based predictions are consistently better than other residue pair potentials for all three size categories.
Table 4.
Comparisons of the accuracy of contact prediction by different methods
| Coverage* | Method/potential† | Size
categories
|
Structural classes
|
Overall | |||||
|---|---|---|---|---|---|---|---|---|---|
| 50–114 | 115–179 | 180–299 | All-α | All-β | α/β | α+β | |||
| Random prediction | 3.72 | 2.97 | 2.36 | 2.49 | 4.00 | 2.21 | 3.30 | 3.08 | |
| 20% | MJ | 15.03 | 8.87 | 5.41 | 10.05 | 12.28 | 7.00 | 10.78 | 10.18 |
| JS | 15.43 | 9.11 | 5.55 | 10.31 | 12.73 | 7.15 | 11.01 | 10.45 | |
| ERCE-Pred | 18.60 | 11.32 | 6.58 | 9.91 | 15.42 | 9.83 | 14.19 | 12.69 | |
| ERCE-Real | 19.61 | 12.07 | 7.20 | 10.17 | 16.87 | 10.10 | 15.20 | 13.50 | |
| 50% | MJ | 9.17 | 5.47 | 3.39 | 6.00 | 7.50 | 4.45 | 6.68 | 6.26 |
| JS | 9.66 | 5.85 | 3.66 | 6.31 | 8.15 | 4.65 | 7.04 | 6.65 | |
| ERCE-Pred | 11.56 | 6.82 | 4.01 | 6.13 | 9.81 | 5.36 | 8.86 | 7.79 | |
| ERCE-Real | 12.75 | 7.49 | 4.32 | 6.06 | 11.16 | 5.70 | 9.95 | 8.55 | |
The analysis of prediction performance with respect to structural classes is more revealing. Compared with the random prediction, the use of residue pair potentials such as MJ and JS yields the maximal gain in contact prediction for all-α proteins. The added value of using ERCE is that it also improves predictions on other three structural classes (all-β, α/β, and α+β) such that the overall improvement ratios relative to a random prediction are comparable for all four structural classes.
Discussion
Long-range interactions play an important role in determining the tertiary structures of proteins. However, computational simulations of such interactions have encountered great difficulties in the past. Coarse-grained reside pair potentials attempt to strike a balance between the accuracy of energy representation and the computational expediency by harnessing the rich amount of information about intramolecular interactions provided by experimentally solved structures. In this study, we extended the contact energy formulation (32) to include the influence of secondary structural environments on the specificity of residue interactions, taking advantage of the amount and quality of structural data currently available. Other ways to divide up the data to extract features usable in various categories of protein simulations also have been reported (10, 39–41).
The added value of ERCE over existing residue pair potentials was explored in both threading and three-dimensional contact prediction experiments. The merit of using contact prediction to test energy functions is that it is independent of the conformational search algorithms and can be readily applied to a large set of protein structures. Because the objectives of two-dimensional contact map prediction and three-dimensional structure prediction are essentially the same (16, 42–44), the combined power of ERCE and secondary structure prediction illustrated here should be of general interest to protein structure prediction.
It should be noted that we used the same set of 407 structures to derive and test ERCE. It has been noticed before that contact energies derived by the MJ formulation is relatively insensitive to the changes in the database size and content (36, 40). Because our data set contains a large number of nonredundant structures, a Jackknife test produces essentially the same results (data not shown). In fact, we also used an independent set of 91 protein chains (33) to derive an ERCE table, and the parameters thereof were highly correlated with those given in Fig. 1 and achieved comparable accuracy in threading and contact prediction experiments.
Acknowledgments
We thank Inna Dubchak and Bob Jernigan for critical reading of the paper. We also thank David Jones for communicating the psi-pred program. This work was supported by grants from the Department of Energy (DE-AC03-76SF00098), the National Science Foundation (97–23352), and the National Institutes of Health (CA78406).
Abbreviations
- ERCE
environment-dependent residue contact energies
- MJ
Miyazawa–Jernigan procedure
- JS
J. Skolnick et al. potential
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.040573597.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.040573597
References
- 1.Levitt M. J Mol Biol. 1976;104:59–107. doi: 10.1016/0022-2836(76)90004-8. [DOI] [PubMed] [Google Scholar]
- 2.Jernigan R L, Bahar I. Curr Opin Struct Biol. 1996;6:195–209. doi: 10.1016/s0959-440x(96)80075-3. [DOI] [PubMed] [Google Scholar]
- 3.Godzik A, Kolinski A, Skolnick J. Protein Sci. 1995;4:2107–2117. doi: 10.1002/pro.5560041016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thomas P D, Dill K A. J Mol Biol. 1996;257:457–469. doi: 10.1006/jmbi.1996.0175. [DOI] [PubMed] [Google Scholar]
- 5.Skolnick J, Jaroszewski L, Kolinski A, Godzik A. Protein Sci. 1997;6:676–688. doi: 10.1002/pro.5560060317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang C. Proteins Struct Funct Genet. 1998;31:299–308. [Google Scholar]
- 7.Hao M-H, Scheraga H A. Curr Opin Struct Biol. 1999;9:184–188. doi: 10.1016/s0959-440x(99)80026-8. [DOI] [PubMed] [Google Scholar]
- 8.Rooman M, Gilis D. Eur J Biochem. 1998;254:135–143. doi: 10.1046/j.1432-1327.1998.2540135.x. [DOI] [PubMed] [Google Scholar]
- 9.Minor D L J, Kim P S. Nature (London) 1994;371:264–267. doi: 10.1038/371264a0. [DOI] [PubMed] [Google Scholar]
- 10.Cootes A P, Curmi P M G, Cunningham R, Donnelly C, Torda A E. Proteins Struct Funct Genet. 1998;32:175–189. [PubMed] [Google Scholar]
- 11.Hutchinson E G, Sessions R B, Thornton J M, Woolfson D N. Protein Sci. 1998;7:2287–2300. doi: 10.1002/pro.5560071106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rost B, Sander C. J Mol Biol. 1993;232:584–599. doi: 10.1006/jmbi.1993.1413. [DOI] [PubMed] [Google Scholar]
- 13.Cuff J A, Barton G J. Proteins Struct Funct Genet. 1999;34:508–519. doi: 10.1002/(sici)1097-0134(19990301)34:4<508::aid-prot10>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 14.Jones D T. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 15.Skolnick J, Kolinski A, Ortiz A R. J Mol Biol. 1997;265:217–241. doi: 10.1006/jmbi.1996.0720. [DOI] [PubMed] [Google Scholar]
- 16.Ortiz A R, Kolinski A, Skolnick J. Proc Natl Acad Sci USA. 1998;95:1020–1025. doi: 10.1073/pnas.95.3.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang E S, Samudrala R, Ponder J W. J Mol Biol. 1999;290:267–281. doi: 10.1006/jmbi.1999.2861. [DOI] [PubMed] [Google Scholar]
- 18.Russel R B, Copley R R, Barton G J. J Mol Biol. 1996;259:349–365. doi: 10.1006/jmbi.1996.0325. [DOI] [PubMed] [Google Scholar]
- 19.Di Francesco V, Garnier J, Munson P J. J Mol Biol. 1997;267:446–463. doi: 10.1006/jmbi.1996.0874. [DOI] [PubMed] [Google Scholar]
- 20.Rice D W, Eisenberg D. J Mol Biol. 1997;267:1026–1038. doi: 10.1006/jmbi.1997.0924. [DOI] [PubMed] [Google Scholar]
- 21.Rost B, Schneider R, Sander C. J Mol Biol. 1997;270:471–480. doi: 10.1006/jmbi.1997.1101. [DOI] [PubMed] [Google Scholar]
- 22.Aurora R, Rose G D. Proc Natl Acad Sci USA. 1998;95:2818–2823. doi: 10.1073/pnas.95.6.2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grigoriev I V, Kim S-H. Proc Natl Acad Sci USA. 1999;96:14318–14323. doi: 10.1073/pnas.96.25.14318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Onuchic J N, Luthey-Schulten Z, Wolynes P G. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 25.Dill K A, Chan H S. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 26.Liwo A, Lee J, Ripoll D R, Pillardy J, Scheraga H A. Proc Natl Acad Sci USA. 1999;96:5482–5485. doi: 10.1073/pnas.96.10.5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berstein F C, Koetzle T F, Williams G J B, Meyer E F, Jr, Brice M D, Rodgers J R, Kennard O, Shimanouchi T, Tasumi M. J Mol Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 28.Murzin A, Brenner S E, Hubband T, Chothia C. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 29.Kabsch W, Sander C. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 30.Altschul S F, Madden T L, Schaffer A A, Zhang J, Zhang Z, Miller W, Lipman D J. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holm L, Sander C. Bioinformatics. 1998;14:423–429. doi: 10.1093/bioinformatics/14.5.423. [DOI] [PubMed] [Google Scholar]
- 32.Miyazawa S, Jernigan R L. Macromolecules. 1985;18:534–552. [Google Scholar]
- 33.Zhang C, Vasmatzis G, Cornette J L, DeLisi C. J Mol Biol. 1997;267:707–726. doi: 10.1006/jmbi.1996.0859. [DOI] [PubMed] [Google Scholar]
- 34.Hendlich M, Lackner P, Weitckus S, Floeckner H, Froschauer R, Gottsbacher K, Casari G, Sippl M J. J Mol Biol. 1990;216:167–180. doi: 10.1016/S0022-2836(05)80068-3. [DOI] [PubMed] [Google Scholar]
- 35.Kocher J P, Rooman M J, Wodak S J. J Mol Biol. 1994;235:1598–1613. doi: 10.1006/jmbi.1994.1109. [DOI] [PubMed] [Google Scholar]
- 36.Miyazawa S, Jernigan R L. J Mol Biol. 1996;256:623–644. doi: 10.1006/jmbi.1996.0114. [DOI] [PubMed] [Google Scholar]
- 37.Cohen F E, Sternberg M J E, Taylor W R. J Mol Biol. 1982;156:821–862. doi: 10.1016/0022-2836(82)90144-9. [DOI] [PubMed] [Google Scholar]
- 38.Reddy B V B, Nagarajaram H A, Blundell T L. Protein Sci. 1999;8:573–586. doi: 10.1110/ps.8.3.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bahar I, Jernigan R L. Folding Des. 1996;1:357–370. doi: 10.1016/S1359-0278(96)00051-X. [DOI] [PubMed] [Google Scholar]
- 40.Bahar I, Jernigan R L. J Mol Biol. 1997;266:195–214. doi: 10.1006/jmbi.1996.0758. [DOI] [PubMed] [Google Scholar]
- 41.Miyazawa S, Jernigan R L. Proteins Struct Funct Genet. 1999;15:347–356. [PubMed] [Google Scholar]
- 42.Gobel U, Sander C, Schneider R, Valencia A. Proteins Struct Funct Genet. 1994;18:309–317. doi: 10.1002/prot.340180402. [DOI] [PubMed] [Google Scholar]
- 43.Shindyalov I N, Kolchanov N A, Sander C. Protein Eng. 1994;7:349–358. doi: 10.1093/protein/7.3.349. [DOI] [PubMed] [Google Scholar]
- 44.Olmea O, Rost B, Valencia A. J Mol Biol. 1999;293:1221–1239. doi: 10.1006/jmbi.1999.3208. [DOI] [PubMed] [Google Scholar]