

Abstract
Microarray experiments have been used recently in genetical genomics studies, as an additional tool to understand the genetic mechanisms governing variation in complex traits, such as for estimating heritabilities of mRNA transcript abundances, for mapping expression quantitative trait loci, and for inferring regulatory networks controlling gene expression. Several articles on the design of microarray experiments discuss situations in which treatment effects are assumed fixed and without any structure. In the case of two-color microarray platforms, several authors have studied reference and circular designs. Here, we discuss the optimal design of microarray experiments whose goals refer to specific genetic questions. Some examples are used to illustrate the choice of a design for comparing fixed, structured treatments, such as genotypic groups. Experiments targeting single genes or chromosomic regions (such as with transgene research) or multiple epistatic loci (such as within a selective phenotyping context) are discussed. In addition, microarray experiments in which treatments refer to families or to subjects (within family structures or complex pedigrees) are presented. In these cases treatments are more appropriately considered to be random effects, with specific covariance structures, in which the genetic goals relate to the estimation of genetic variances and the heritability of transcriptional abundances.
MICROARRAY technology allows the monitoring of messenger RNA (mRNA) abundance in cells for thousands of genes simultaneously (Schena et al. 1995; Lockhart et al. 1996). Microarray experiments have been used extensively to provide a surrogate measure of gene activity to compare expression levels of treatment groups (within either experimental or observational contexts), such as different tissues, cell types (e.g., disease and normal cell) in a specific tissue, different developmental stages, or experimental conditions (e.g., different drugs or stress conditions).
The application of this technology however, especially for two-color platforms (either cDNA or long oligonucleotide arrays), poses some experimental design challenges. As samples are assayed in a pairwise competitive hybridization fashion on each slide, microarray experiments using two-color systems impose a block structure with blocks of size two. Whenever more than two groups are compared, this defines an incomplete block design, in which estimated treatment effects and differences between slides are partially confounded. In addition, as each sample is labeled with a different dye on each slide, microarray experiments involve an additional blocking factor to be accounted for, which leads to a row–column, or two-way blocking, structure (slide–dye combination).
Many articles on design for two-color microarray experiments make some comparison of efficiency or power (Kerr and Churchill 2001; Yang and Speed 2002; Kerr 2003; Rosa et al. 2005; Steibel and Rosa 2005; Tempelman 2005; Vinciotti et al. 2005) of different designs. Most of them focus on the comparison of a discrete set of designs, such as reference and circular structures. Wit et al. (2005) present a more general approach, searching for optimal designs given specific experimental conditions and optimality criteria. It is shown that the optimal microarray design for a specific optimality criterion may refer to a more complex experimental layout than simple reference or circular structures.
The objective of the experiment guides the choice of the treatment structure, whereas the definition of the experimental units leads to the blocking structure of the experiment. As discussed throughout this article, the treatment choice in microarray experiments is general for any platform (e.g., high-density short oligos, cDNA, or long oligonucleotide technologies), whereas the blocking structure depends on the microarray technology, such as two-color arrays (in which slides refer to a blocking factor) or single-color arrays such as Affymetrix (in which each sample is hybridized to a different slide). The treatment structure (as well as the number of replications) depends also on the practical/logistical constraints of the experiment, such as limited biological samples (limited either by the number of individuals to be assayed or by the amount of mRNA from each individual) or number of slides available. The choice of the experimental unit, which can be an individual (such as a plant or animal, hereinafter denoted as a subject) or a group of subjects (such as a family or mRNA pool), may impose some spatial or genetic structure among units, which should be taken into account when assigning treatments to units.
Microarray experiments have been recently used on genetical genomics studies, as an additional tool to understand the genetic mechanisms governing variation in complex traits, such as disease susceptibility in human medicine or production traits in agriculture. Microarray experiments using related subjects can be utilized, for example, to infer heritabilities of mRNA transcript abundances (Monks et al. 2004). In addition, gene expression profiling can be combined with marker genotype information for mapping expression quantitative trait loci (eQTL; Jansen and Nap 2001; Brem et al. 2002; Darvasi 2003; Schadt et al. 2003) and to infer regulatory networks controlling gene expression (Chesler et al. 2004; Bystrykh et al. 2005).
In this article we discuss the optimal design of microarray experiments whose goals refer to specific genetic questions, such as the comparison of expression levels of different genotypic groups, eQTL studies, or estimation of heritabilities of mRNA transcript abundances. This goes beyond the scope of earlier work on fixed, unstructured treatments.
The experimenters' interest might be in specific treatment comparisons, e.g., referring to additive and dominance effects. Treatments may refer to families or to subjects (within family structures or complex pedigrees) in a given experiment. In these cases treatments are more appropriately considered to be random effects, with specific covariance structures. These covariance structures should then be taken into account when planning microarray experiments, so that the efficiency of broad inferences to the population of interest is maximized.
The high cost of microarrays greatly limits the sample sizes of heritability or gene-mapping studies of mRNA abundance traits (Jin et al. 2004). Therefore, a careful design of such experiments is extremely important to make the most of the limited number of slides that are generally available in a single experiment.
This article is organized as follows. In the next section we review some experimental design concepts, such as treatment choice, treatment-to-unit allocation, design optimality criteria, and some useful methods and results on optimal designs. The third section presents genetic research goals that can motivate microarray studies with two or more treatments, such as genotypic groups. Treatments are considered fixed, but the genetic component that defines the treatments provides a natural structure (which is translated into genetic parameters of interest, such as additive and dominance effects), which should be explored when designing the experiments. In the fourth section, the design of microarray experiments with random treatments is discussed. Treatments in this case may refer to related individuals or families, and the main goal of the experiments relates to inference on genetic variances and heritabilities. Finally, some concluding remarks are presented in the last section.
DESIGN PRINCIPLES AND CRITERIA
The general statistical principles of the design of experiments apply to experiments of any kind, including microarray studies. The general procedure for choosing a good design involves the following steps (see also Bailey 1981, 1997; Mead 1990):
The objectives of the experiment lead to the choice of a set of treatments (groups to compare).
The experimental units (here the combination of slides and dyes) that can be used are identified and any expected patterns of variability among them lead to a choice of blocking structure (here a row–column structure).
Consideration of any restrictions on which treatments can be applied to which units might lead to complex structures such as split-plot designs.
Given 1–3, a combinatorial, algorithmic or ad hoc method is found to construct a design.
The block structure is already clear and we do not discuss complex structures here, so the two main tasks in developing a design are treatment choice and treatment-to-unit allocation.
Treatment choice:
When the experimental resources are limited, a choice of the treatments that will be represented in the experiment, from a larger set of candidate treatments, might be needed. In some situations particular treatments cannot be produced, or the number of candidate treatments is too big for all of them to be implemented. For example, suppose we want to study isolines for transgenic versions of three genes (A/a, B/b, and C/c) from some parental line. Even restricting each locus to express just the homozygote genotypes, it is unlikely that all seven desired isolines will be obtained by transformation events or any other currently known gene manipulation techniques. Starting from the nontransgenic genotype (AABBCC), for instance, a single mutation may give rise to the genotypes AABBcc, AAbbCC, or aaBBCC; however, two or three mutations are needed to obtain the genotypes AAbbcc, aaBBcc, and aabbCC or aabbcc, respectively. Another and more obvious example is given by partial diallels, in which some crossings cannot be done. We discuss later in this article some multilocus analysis problems. In these cases, we need to define a model for the treatment effects and an optimality criterion related to the variances of the estimated parameters of the model. Then we must find a method of searching for a design that optimizes the criterion, e.g., using the optimal continuous (approximate) design theory developed from Kiefer and Wolfowitz (1960) or a computer algorithm that searches for the optimal exact design, such as those developed from Fedorov (1972). As stated earlier, the treatment choice in microarray experiments is independent of the array platform, so the same procedure can be used for single- or two-color technologies.
Treatment-to-unit allocation:
A given set of treatments (and their respective replication numbers) should be combined in an optimal way within a given experimental setup. In the context of two-color microarray experiments, for example, this setup is given by the number of slides and the two labels (Cy3 and Cy5 dyes). Again, construction of the design might require optimal design theory, this time most likely using a computer search.
The treatment-to-unit allocation applications discussed in this article are specific to two-color array platforms (cDNA or long oligos), in which samples should be allocated in pairs to the slides (blocks). With single-color microarrays (such as Affymetrix), on the other hand, each sample is hybridized to a different slide, so this is not an issue and allocation should be done completely at random.
Linear mixed models for microarray experiments:
The outcomes of a microarray experiment are luminous intensities of thousands of genes, which are proportional to their mRNA transcriptional abundances. After suitable normalization, the response intensities from gene g in a two-color microarray experiment can be represented by the model
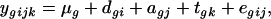 |
(1) |
(Wolfinger et al. 2001), where 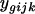 is the normalized intensity of the gene expression measured by the dye (label) i of the array j that received treatment k,
is the normalized intensity of the gene expression measured by the dye (label) i of the array j that received treatment k, 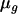 is a common experimental constant,
is a common experimental constant, 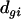 is the effect of dye i (green or red),
is the effect of dye i (green or red), 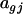 is the effect of array (or slide) j,
is the effect of array (or slide) j, 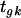 is the effect of the treatment k, and
is the effect of the treatment k, and 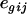 is a residual term. We consider a single gene at a time for design purposes, as the same model is generally used to analyze all genes in a single experiment, and so the subscript g is dropped. Different experimental layouts may need extra terms in the model for the appropriate modeling of the data. With Affymetrix data, for example, there is no dye effect in the model, but a hierarchical structure could be used to model intensities at the probe level, by including the effects of probe sets (clones) and of probe pairs within clones. Model choice should then be closely connected to treatment and block structures of the experiments (Rosa et al. 2005).
is a residual term. We consider a single gene at a time for design purposes, as the same model is generally used to analyze all genes in a single experiment, and so the subscript g is dropped. Different experimental layouts may need extra terms in the model for the appropriate modeling of the data. With Affymetrix data, for example, there is no dye effect in the model, but a hierarchical structure could be used to model intensities at the probe level, by including the effects of probe sets (clones) and of probe pairs within clones. Model choice should then be closely connected to treatment and block structures of the experiments (Rosa et al. 2005).
As additional assumptions of model (1), residual terms can be modeled as being normally distributed with variance 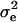 ; likewise, as arrays come from a population of possible assembled arrays, their effects can be modeled as random normal variables centered around zero, with variance
; likewise, as arrays come from a population of possible assembled arrays, their effects can be modeled as random normal variables centered around zero, with variance 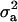 . The term “treatment” here is used in a very broad context, representing either experimental groups (such as genotypic classes) or families (and subjects within families) in quantitative genetics studies to estimate heritabilities. Therefore, treatment effects can be modeled as fixed or random, depending on the objective of the experiment.
. The term “treatment” here is used in a very broad context, representing either experimental groups (such as genotypic classes) or families (and subjects within families) in quantitative genetics studies to estimate heritabilities. Therefore, treatment effects can be modeled as fixed or random, depending on the objective of the experiment.
The usual matrix notation for linear mixed models such as model (1) is
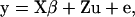 |
(2) |
where y and e are vectors with elements 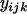 and
and 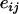 , respectively,
, respectively, 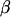 is the vector of fixed effects, X is a known (design) matrix that relates y to
is the vector of fixed effects, X is a known (design) matrix that relates y to 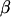 , u is the vector of random effects, and Z is the matrix that relates y to u. An extra assumption is that
, u is the vector of random effects, and Z is the matrix that relates y to u. An extra assumption is that 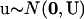 , where U is the covariance matrix of the random effects.
, where U is the covariance matrix of the random effects.
Note that the treatment effects can be taken as being either fixed or random and so they can be contained in either 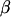 or u, depending on the experimental context. In the examples discussed in this article, whenever treatment effects are considered random, they are assumed normally distributed; i.e.,
or u, depending on the experimental context. In the examples discussed in this article, whenever treatment effects are considered random, they are assumed normally distributed; i.e., 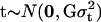 , where G is a matrix of known constants representing, for example, genetic relationships among the elements in t (such as a numerator relationship matrix A; Henderson 1976), and
, where G is a matrix of known constants representing, for example, genetic relationships among the elements in t (such as a numerator relationship matrix A; Henderson 1976), and 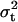 is the variance among treatments, such as an additive genetic variance.
is the variance among treatments, such as an additive genetic variance.
Design criteria:
The covariance matrix of the parameter estimates for model (2) is the inverse of the information matrix M, where
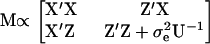 |
Almost any sensible criterion defining design efficiency is a function of the information matrix M.
Costs are important when comparing alternative experimental designs. In this article, however, designs are generally compared for a given number of slides. As pointed out by Yang and Speed (2002), the number of slides is generally the most important limiting factor in microarray experiments. In addition, for two-color microarray platforms, as the variation between slides is generally larger than the variation within slides, blocking by slides is crucial. Since the ratio of between-slide to within-slide variation is unknown, it is usual to design experiments for the worst case, i.e., where this ratio is very large, in which case the design problem becomes equivalent to one with fixed blocks. Note, however, that after the experiment is run, in the mixed-models data analysis, we will estimate the variance components; if their ratio turns out to be large, we will estimate the treatment effects with the precision expected when we chose the design; if their ratio turns out to be small, then the interblock information will give us estimates of treatment effects better than expected when we chose the design.
In this context, Kerr and Churchill (2001) used the so-called A-optimality criterion to compare block designs for microarray studies with simple treatment structures, and Yang and Speed (2002) discussed the efficiency of alternative designs for specific contrasts of interest in factorial and time course experiments. Kerr (2003) later argued in favor of double reference designs as being suboptimal but more robust when compared to loop designs. Glonek and Solomon (2004) studied some alternative designs for factorial treatment structures. More general work on two-level factorial designs in blocks of size two, which has some relevance, has been done by Draper and Guttman (1997), Yang and Draper (2003), Wang (2004), and Kerr (2006).
Similar sorts of experiments are now being used for understanding the genetic basis of mRNA transcript abundances, with a variety of experimental settings. For example, fixed treatment effect models arise in genetic analysis when comparing multilocus genotypic groups, including interaction (epistasis) terms. In addition, random treatment effect models may be used in quantitative genetics to infer genetic variances and heritabilities. No work, however, has been published on optimal design for microarray experiments in situations where the treatments have any of these structures.
In each of the following sections we present different experiments and discuss possible parameter contrasts of interest, as well as the variances of their estimates, sometimes weighted to correct for scale problems. In each situation, alternative optimality criteria are discussed. We assume that all power comparisons have a monotonic (inverse) relation with the variances of the desired comparisons and describe treatment structures in microarray experiments for some common goals in genetic studies. The resulting designs are depicted by a set of arrows representing the slides, with heads and tails denoting alternative dye labeling (Cy3 and Cy5). Arrows (slides) refer always to different biological replications from each experimental group, unless otherwise stated.
GENETIC STUDIES WITH FIXED TREATMENT EFFECTS
Two treatment groups in microarray experiments:
Genetic motivation:
One of the simplest scenarios that results in two-treatment experiments arises with a single candidate gene (with alleles B and b) and a backcross (BC) or double-haploid (DH) experiment. In these circumstances, individuals belong to one of two possible genotypic groups, namely Bb or bb if in a BC experiment and BB or bb if in a DH scenario. In both situations one is interested in the additive effect 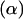 of the candidate gene on the expression of each gene represented in the microarray slide, i.e., how polymorphisms at a specific site of the genome affect transcription activity of other genes in the genome. Such effects are generally referred to as trans-acting factors (Jansen and Nap 2001).
of the candidate gene on the expression of each gene represented in the microarray slide, i.e., how polymorphisms at a specific site of the genome affect transcription activity of other genes in the genome. Such effects are generally referred to as trans-acting factors (Jansen and Nap 2001).
The model (1) for such a simple treatment structure can be written with 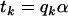 . For the BC situation,
. For the BC situation, 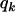 is a design index on genotypes assuming values
is a design index on genotypes assuming values 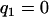 (for genotype bb) and
(for genotype bb) and 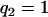 (for genotype Bb). In this situation, one wants to estimate
(for genotype Bb). In this situation, one wants to estimate 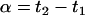 and the variance of this contrast is given by
and the variance of this contrast is given by 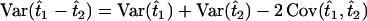 . For the DH situation,
. For the DH situation, 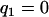 (for genotype bb) and
(for genotype bb) and 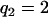 (for genotype BB). In this situation, one wants to estimate
(for genotype BB). In this situation, one wants to estimate 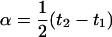 , and the variance of this comparison is given by one-quarter of the right-hand side of the above variance. As these two variances are proportional, the same optimal design minimizes both of them.
, and the variance of this comparison is given by one-quarter of the right-hand side of the above variance. As these two variances are proportional, the same optimal design minimizes both of them.
In a microarray experiment, such a simple two-treatment group situation can arise within a selective phenotyping approach based on a single locus (or small chromosomic region), as proposed by Jin et al. (2004), or within a candidate gene context in which one is interested in the effect of a specific locus (e.g., transgene) on the expression of other genes in the genome (see, for example, Noueiry et al. 2000).
Another example is to define two experimental groups by taking the extremes of the phenotypic distribution of a quantitative trait of interest (Lander and Botstein 1989). By comparing the allelic frequencies of molecular markers between the two groups, candidate regions containing genes contributing to variation of the phenotypic trait can be pinpointed. In the context of microarrays, candidate genes are generated by searching for differential transcript abundances between the two groups. In any situation, the proportions of the selected individuals should be made small enough so that the two phenotypic groups are quite distinct (Darvasi and Soller 1992).
Microarray experiments with two experimental groups define row–column designs obtained from multiple copies of a 2 × 2 Latin square (in the case of two-color systems), for which extensive literature is available.
Three treatment groups in microarray experiments:
Genetic motivation:
In some situations there are three possible genotypes in a segregating population, for example, with F2 populations and codominant markers. The genotypes for each marker can then be described by an additive–dominance model. Here we discuss the case of a single locus (such as a candidate gene situation) and its trans-acting effect on the expression of other genes. The case of multiple loci is discussed later in this article, with factorial and fractional factorial treatment structures.
Treatment choice:
An additive–dominance treatment effect of a candidate gene in an F2 population can be expressed as 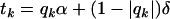 , where
, where 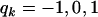 , for genotypes bb, Bb, and BB, respectively. Denote by rk the proportion of the n observations that represent each genotypic group (k = 1, 2, 3). The model can be rewritten as
, for genotypes bb, Bb, and BB, respectively. Denote by rk the proportion of the n observations that represent each genotypic group (k = 1, 2, 3). The model can be rewritten as 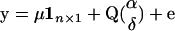 , where 1n×1 is a column vector of ones and Q gives the incidences of genotypes bb, Bb, and BB as
, where 1n×1 is a column vector of ones and Q gives the incidences of genotypes bb, Bb, and BB as
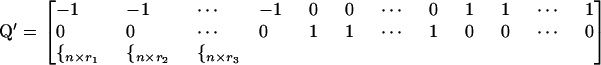 |
Both parameters can be optimally estimated using the same proportion for each homozygous genotype (bb and BB), in which case r1 = r3 = r and r2 = 1 − 2r. The information matrix is then given by
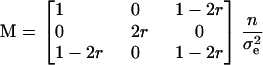 |
and the covariance matrix is
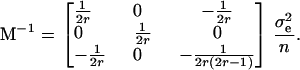 |
Figure 1 plots changes in the variances of the parameter estimates with the proportion (r) of each homozygous group. If one is interested solely in the (trans-acting) additive effects, just homozygotes should be considered in the design. To estimate (trans-acting) dominance only, half of the observations should be taken from the heterozygotes and one-quarter from each of the homozygotes. To estimate additive and dominance effects simultaneously, it is necessary to define an optimality criterion. Throughout this article, we use the A-efficiency on this scale, i.e., the mean of the variances of the estimates, but in any real experiment careful thought should be given to whether a weighted mean of the variances would be a more appropriate criterion. The A-optimal proportion of each homozygote is the irrational number 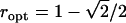 . As no possible number of arrays satisfies this optimum exactly, an approximation is needed. It is reasonable to use ∼0.293 of the observations for each homozygote and the remaining 0.414 of the observations for the heterozygote group.
. As no possible number of arrays satisfies this optimum exactly, an approximation is needed. It is reasonable to use ∼0.293 of the observations for each homozygote and the remaining 0.414 of the observations for the heterozygote group.
Figure 1.—

Average variances of the estimates of genetic parameters for a single-locus experiment as a function of the proportion of each homozygous group (r), when the experiment goal is to estimate the additive effect (A), the dominance effect (B), or both additive and dominance effects simultaneously (C).
Suppose, for example, that we are interested in the best design that can be made with only 10 slides using a two-color microarray platform. We proceed as follows. To estimate just additive effects, each homozygous genotype (BB or bb) will have 10 replications. The heterozygote group, in this case, is not included in the experiment. To estimate only dominance effects there will be 5 replications of each homozygous genotype and 10 replications of the heterozygote group. Finally, for estimation of both additive and dominance effects, the A-optimal design has 6 replications for each homozygote and the remaining 8 replications for the heterozygote.
Treatment-to-unit allocation:
After the treatments have been chosen (i.e., the number of replications for each genotypic group), the next step in planning the microarray experiment is the treatment-to-unit allocation, i.e., describing which treatment should be applied to each unit, when array and color effects are in the model. The optimal design for estimating just additive effects with 10 slides is shown in Figure 2A, in which all slides have the two homozygous genotypes only, with alternating directions of the Cy3 and Cy5 labeling. If one is concerned with estimating dominant effects, all of the slides should have one of the homozygous genotypes cohybridized with the heterozygous genotype, as shown in Figure 2B. Finally, if both additive and dominance effects are to be estimated, combinations of the two types of slide should be used (Figure 2C). It is worth mentioning that the designs of Figure 2, A–C, are, respectively, ∼100, 50, and 9% more efficient than the classical loop structure. When compared to the reference design, their relative efficiencies are 6.6, 4.7, and 3.4, respectively.
Figure 2.—



Optimal designs for estimating trans-acting additive effects (A), dominance effects (B), and both additive and dominance effects (C) from one gene, with 10 slides in a two-color microarray experiment.
Generalization of these results for multiple loci:
The effect of multiple candidate genes on the overall genome transcriptional activity can be investigated at once in a single microarray study. With simple line-crossing experiments, each gene (locus) may have either two or three levels (possible genotypes), depending on the mating system used (BC or DH and F2, respectively). Therefore, the treatment structure for L loci is a factorial of the series 2L or 3L for two or three possible genotypes in each locus, respectively. A suitable model for the expression of the treatments in such a system, including epistasis, is given by
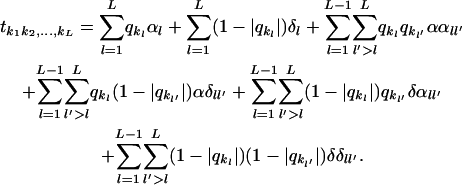 |
If necessary, higher-order interloci interactions (additional epistatic effects) can also be included in the model. A parsimonious version of this model can be obtained by reducing the order of the estimable epistatic effects. For example, in a situation with two loci with three possible genotypes each, only five parameters would be needed to model phenotypes (i.e., transcriptional profiling) from the nine possible genotypes if no epistasis is to be estimated.
A matrix representation of a two-locus version (alleles A/a and B/b, respectively) without epistasis is given by
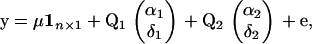 |
where 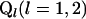 is a representation of the design matrix for each locus. If total sampling control of the population of genotypes is possible, equal representation of each homozygote genotype for each locus could be considered. Let rjl represent the proportion of genotype j for locus l. In this case, assuming equal proportions of homozygotes within each locus, we have
is a representation of the design matrix for each locus. If total sampling control of the population of genotypes is possible, equal representation of each homozygote genotype for each locus could be considered. Let rjl represent the proportion of genotype j for locus l. In this case, assuming equal proportions of homozygotes within each locus, we have  and
and 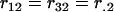 . Table 1 presents the number of observations for each genotype and the expected phenotypic values.
. Table 1 presents the number of observations for each genotype and the expected phenotypic values.
TABLE 1.
Expected phenotypic value and number of observations for each genotype in a two-locus experiment with an F2 population
| Genotype | Expected phenotypic value | No. of observations |
|---|---|---|
| AABB | 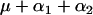 |
r.1r.2n |
| AABb | 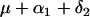 |
r.1(1 − 2r.2)n |
| AAbb | 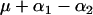 |
r.1r.2n |
| AaBB | 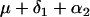 |
(1 − 2r.1)r.2n |
| AaBb | 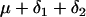 |
(1 − 2r.1)(1 − 2r.2)n |
| Aabb | 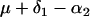 |
(1 − 2r.1)r.2n |
| aaBB | 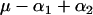 |
r.1r.2n |
| aaBb | 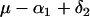 |
r.1(1 − 2r.2)n |
| aabb | 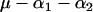 |
r.1r.2n |
The information matrix for this model is given by
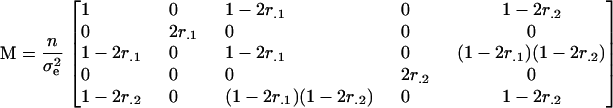 |
and so the covariance matrix of the estimated model parameters is
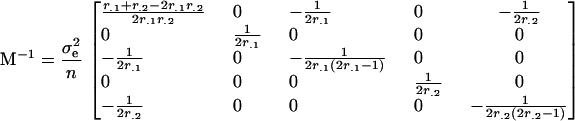 |
The optimal designs for this situation are a straightforward extension of the optimal proportions for each genotype: 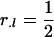 ,
, 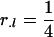 , and
, and 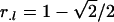 , if one is interested in estimating additive, dominance, or both effects for each locus l, respectively.
, if one is interested in estimating additive, dominance, or both effects for each locus l, respectively.
As an example, Table 2 gives the optimal number of replications of each genotype in a two-locus experiment with 10 slides. For estimating additive effects only, the treatments compose a 22 factorial set, equally replicated. For estimating dominance or both additive and dominance effects, the treatments correspond to 32 factorial designs with different replications in the two cases. It can be seen that the design obtained by continuous techniques can be directly implemented only for additive effects; in the other two situations an approximation is needed. For dominant effects, the continuous optimal design gives 0.0625 × 20 = 1.25 and 0.0125 × 20 = 2.5, which were approximated by 1 and 3, respectively. This reflects a decrease in the relative importance of homozygote combinations as compared to values for the optimal large sample design. On the other hand, the genotype AaBb has only four replicates (instead of 0.25 × 20 = 5). This specific treatment choice does not allow a straightforward application of the continuous optimal design theory and the design chosen might not actually be optimal. The same problem happens when estimating both additive and dominant effects: either the approximation of 0.0858 × 20 = 1.716 ≈ 2 or the truncation of 0.1213 × 20 = 2.426 ≈ 2 forces the final treatment choice away from the large-sample optimal genotype proportions.
TABLE 2.
Optimal number of replications of each genotype in a two-locus experiment with no epistasis, using 20 experimental units
| Parameters of interest
|
||||
|---|---|---|---|---|
| Genotype | Optimal proportions | α's | δ's | (α and δ)'s |
| AABB | r.1r.2 | 0.25n = 5 | 0.0625n ≈ 1 | ≈0.0858n ≈ 2 |
| AABb | r.1(1 − 2r.2) | 0n = 0 | 0.125n ≈ 3 | ≈0.1213n ≈ 2 |
| AAbb | r.1r.2 | 0.25n = 5 | 0.0625n ≈ 1 | ≈0.0858n ≈ 2 |
| AaBB | (1 − 2r.1)r.2 | 0n = 0 | 0.125n ≈ 3 | ≈0.1213n ≈ 2 |
| AaBb | (1 − 2r.1)(1 − 2r.2) | 0n = 0 | 0.25n ≈ 4 | ≈0.1716n ≈ 4 |
| Aabb | (1 − 2r.1)r.2 | 0n = 0 | 0.125n ≈ 3 | ≈0.1213n ≈ 2 |
| aaBB | r.1r.2 | 0.25n = 5 | 0.0625n ≈ 1 | ≈0.0858n ≈ 2 |
| aaBb | r.1(1 − 2r.2) | 0n = 0 | 0.125n ≈ 3 | ≈0.1213n ≈ 2 |
| aabb | r.1r.2 | 0.25n = 5 | 0.0625n ≈ 1 | ≈0.0858n ≈ 2 |
After the treatment choice is performed, the design of the experiment is completed with the treatment-to-unit allocation step. Optimal two-color microarray designs (with 10 slides) for inferring additive, dominance, or both effects are shown in Figure 3. Similarly to the situation with a single locus, a clear pattern of allowing only the important contrasts within each slide arises from these pictures as well. It can be seen that all the comparisons for estimating additive effects are done among homozygotes for each locus. For estimating dominance effects, all slides bring a comparison of a homozygous with the heterozygous genotype, for both loci. Moreover, for the simultaneous estimation of additive and dominance effects, the two types of comparison are needed. The relative efficiencies of the designs of Figure 3, A–C, are, respectively, 1.5, 2.9, and 2.1 when compared with the loop design and ∼5.1 compared with the reference design.
Figure 3.—



Optimal designs for estimating trans-acting additive effects (A), dominance effects (B), and both additive and dominant effects (C) of two nonepistatic genes, with 10 slides in a two-color microarray experiment. Dashed arrows connect the homozygous genotypes in the corners.
Note that these designs are still optimal for estimating the 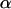 - and
- and 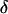 -effects for one locus (say for gene A/a) even if the other locus considered (say B/b) has no effect at all! Similar conclusions can be generalized to situations with more than two loci and larger numbers of slides.
-effects for one locus (say for gene A/a) even if the other locus considered (say B/b) has no effect at all! Similar conclusions can be generalized to situations with more than two loci and larger numbers of slides.
It is interesting to note how different the designs of Figure 3, which explore a natural structure on treatment groups, are from the optimal designs for two-color microarray experiments involving unstructured treatments, which tend toward symmetry whenever possible. For example, Figure 4 depicts two experiments, one involving four treatments and 10 slides (Figure 4A) and the other with nine treatments and 9 slides (Figure 4B). The experiment with nine treatments is an example of the so-called loop (or circular) design commonly found in the literature, whereas the experiment with four treatments consists of two loops with reverse-dye labeling plus two additional interwoven connections.
Figure 4.—


Optimal designs for two-color microarray experiments with four (A) and nine (B) unstructured treatments of fixed effects and 10 and 9 slides, respectively.
Multiple loci with epistatic effects:
Suppose that in this situation (two loci with three genotypes each and 10 slides of a two-color microarray system), we would like also to estimate trans-acting epistasis (i.e., how the joint effect of two loci changes the expression of other genes in the genome) with the following model:
 |
In selecting an optimal design, careful thought must be given to which classes of designs are considered as candidates. In this situation, it is reasonable to insist on choosing a treatment set that is symmetrical with respect to the two gene loci, i.e., that is genewise balanced. The best genewise balanced design is given in Figure 5. Note that in all slides one locus has its genotype repeated while the other varies. This allows us to infer each genomic expression and interaction independently but at the expense of decreasing precision for the main effects parameters previously of interest (additive and dominance effects).
Figure 5.—

Optimal design for estimating trans-acting epistatic effects between two genes, with 10 slides in a two-color microarray experiment.
GENETIC STUDIES WITH RANDOM TREATMENT EFFECTS
Multiple-treatment microarray experiments:
Apart from those situations in which 2L or 3L factorials arise from targeting L genes, there are other genetic objectives that need many treatments. One common goal in genetic studies is to obtain good estimates of genetic variances and heritabilities. Within a genetical genomics context this means studying the heritability of the mRNA abundance of thousands of genes, having only a sample of n subjects (with a specific relationship structure among them) to look at.
In genetic models with random treatments, the treatment effects are often the subject effects. This is the case, for example, with the so-called animal models (Lynch and Walsh 1998), which have been extensively used in animal breeding and more recently in plant breeding (mainly in forestry). For a comprehensive presentation of modern analytical techniques for such models refer to Sorensen and Gianola (2002).
For these situations we have the problem of finding good designs to obtain estimates using the best linear unbiased predictors of breeding values, given a known pedigree or family structure. The best designs are very specific for each pedigree and estimation objective, as discussed below.
Design criterion:
Bueno Filho and Gilmour (2003) argued that a suitable criterion for selecting among related treatments can be obtained as a weighted average of the variances of all pairwise contrasts. This criterion has a broad relevance and estimating genetic variance components and heritabilities is one of the possible goals. In the following examples we apply this criterion to two small experimental situations to illustrate some features of optimal designs. In Table 3, two situations are presented, involving nine animals coming from either a hierarchical or a factorial mating system.
TABLE 3.
Examples of hierarchical and factorial mating systems with nine progeny
| Hierarchical
|
Factorial
|
|||
|---|---|---|---|---|
| Progeny | Dam | Sire | Dam | Sire |
| 1 | 1 | 1 | 1 | 1 |
| 2 | 2 | 1 | 2 | 1 |
| 3 | 3 | 1 | 3 | 1 |
| 4 | 4 | 2 | 1 | 2 |
| 5 | 5 | 2 | 2 | 2 |
| 6 | 6 | 2 | 3 | 2 |
| 7 | 7 | 3 | 1 | 3 |
| 8 | 8 | 3 | 2 | 3 |
| 9 | 9 | 3 | 3 | 3 |
These two different pedigrees result in the additive (numerator) relationship matrices
 |
and
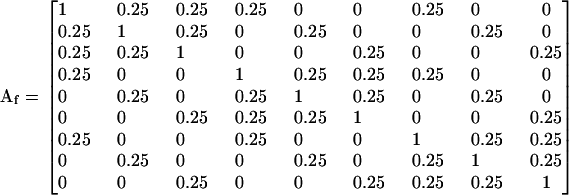 |
(Lynch and Walsh 1998), where the indices h and f refer to the hierarchical and factorial mating systems, respectively.
Restricting our discussion to the estimation of additive variance components, it can be seen that in such cases we need to find both the best replication for each treatment and the best treatment-to-unit allocation. It is worth noting that in this case “treatments and replications” can refer either to families and sibs within families or to biological individuals and technical samples from the same individual.
As an illustration, consider two replications of each treatment (individual). For this kind of group-divisible treatment set, it is suggested (Bueno Filho and Gilmour 2003) that the best designs are in the class of regular graph designs (e.g., loop designs), but the allocation of treatments to treatment labels is important. The best design is one that avoids relatives (in this case, paternal or maternal half-sibs) in the same array, as much as possible. Therefore, the resulting optimal designs are more restrictive for the factorial than for the hierarchical setup. In Figure 6 optimal designs for these two situations are depicted.
Figure 6.—


Optimal design for a microarray experiment with nine treatments, having random effects, representing nine progenies from hierarchical (A) and factorial (B) mating systems (Table 3). (A) Different circle shadings refer to different paternal half-sibs. (B) Each shape and shading denote a different dam and sire, respectively. In both cases, each geometric figure represents a subject (biological replication), which is assayed in two slides each (technical replication).
More complex situations can be found in practice, in which larger numbers of individuals can be considered and some other important measures can be taken. Lynch and Walsh (1998) discuss simple pedigrees in which analytical results are possible and suggest the best number of families and individuals within families to use in the experiment. For more complex pedigrees, however, a general search algorithm should be utilized, as sometimes it is the only way to get a useful design. The algorithm we have used is a simple modification of the exchange algorithm of Fedorov (1972). This has the following general form:
List the possible candidate treatments.
Choose a random set of n treatments, which becomes the current design.
If the current design does not allow the model to be fitted, return to step 2.
Systematically exchange each treatment in the current design with each (different) treatment in the candidate set. Accept any exchanges that improve the criterion to get a new current design, continuing until no exchange improves the design.
When no improvement is possible, the current design becomes the final design returned by the algorithm.
It is common practice to restart the algorithm with several random starting designs and to choose the best design found overall.
As an illustration, consider a treatment choice problem with a microarray experiment with a limited number of slides, for which a subset of 40 subjects needs to be chosen among a population of 80 animals with the pedigree given in Figure 7. An R function was developed to search for optimal designs when treatment effects are random, with any covariance structure among them (i.e., any pedigree setup), such as in this situation. The treatments (animals) selected in the resulting design are indicated by solid circles in Figure 7. Such a design is good only for this very specific situation and it is not possible to produce deep generalizations. The only clear message with messy pedigrees is that a search algorithm may be the only way to reach a good (if not optimal) design for these experiments.
Figure 7.—

Three-generation pedigree with 80 individuals in two genetically disconnected groups, from which 40 individuals (solid circles) were selected for a microarray experiment to infer mRNA abundance heritabilities.
In another very common situation in animal experimentation, related individuals are subjected to different experimental procedures (such as different diets, drugs, etc.). In these circumstances both random and fixed treatment effects are present in the experiment. Such experiments may have multiple objectives and variance component (or heritability) estimation can be one of them. One should be aware, however, that designs that are optimal for some of these objectives might not be so for others. An example of such a situation is given in Table 4. The experiment consists of 20 pigs from five full-sib families of size four, to be arrayed on a limited set of 10 two-color system slides. The four individuals in each family (litter) refer to two male and two female sibs that were selected at random. One animal from each sex was submitted to one of two experimental treatments (placebo and drug). Two different objectives of the study were considered: (a) estimation of heritability and (b) estimation of the fixed effects of drug and sex, as well as the interaction between them. Optimal designs for each of these situations for a specific configuration of variance components are presented in Figure 8.
TABLE 4.
Litter, treatment (drug), and sex of 20 piglets available for microarray experiments for inferring the effects of sex, drug, and the interaction between sex and drug, as well as the heritability
| Progeny | Litter | Drug | Sex |
|---|---|---|---|
| 1 | 1 | Yes | F |
| 2 | 1 | Yes | M |
| 3 | 1 | No | F |
| 4 | 1 | No | M |
| 5 | 2 | Yes | F |
| 6 | 2 | Yes | M |
| 7 | 2 | No | F |
| 8 | 2 | No | M |
| 9 | 3 | Yes | F |
| 10 | 3 | Yes | M |
| 11 | 3 | No | F |
| 12 | 3 | No | M |
| 13 | 4 | Yes | F |
| 14 | 4 | Yes | M |
| 15 | 4 | No | F |
| 16 | 4 | No | M |
| 17 | 5 | Yes | F |
| 18 | 5 | Yes | M |
| 19 | 5 | No | F |
| 20 | 5 | No | M |
Figure 8.—


Optimal designs for the estimation of heritability (A) and fixed effects of drug and sex and their interaction (B), in a two-color microarray experiment with five full-sib swine families of two males (squares) and two females (circles), submitted either to a placebo (open squares and circles) or to a drug treatment (shaded squares and circles).
It can be seen that in the first situation (estimation of heritability) the search algorithm converged to a design in which all slides connected individuals from different sibs. Also, all slides have individuals of the same sex and/or same experimental group (placebo or drug). Conversely, if the experiment goal is to infer the effects of sex, drug, and their interaction, the genetic covariance among sibs becomes another blocking factor, which the search algorithm tended to confound or combine with the blocking effect of arrays. In this case, the slides now tend to assay genetically related individuals. As expected, to infer the main effects of drug and sex, some of the slides compare individuals of the same sex, but different experimental groups (such as those of family 5), while others compare individuals of the same group, but different sex (such as those of family 3). In addition, a third set of slides compares individuals of different sex and experimental groups (e.g., family 1) to infer the interaction between sex and drug effects.
CONCLUDING REMARKS
This article discusses the design of microarray experiments whose goals refer to a variety of genetic questions, such as the comparison of expression levels of different genotypic groups, eQTL studies, or estimation of heritabilities of mRNA transcript abundances.
The general approach adopted for choosing a good design consists of a first step of treatment choice, in which subjects (as well as their experimental group labels) are selected to be included in the experiment. This first step is general and common to any microarray experiment, regardless of platform. In the case of two-color microarray systems, however, the design choice is followed by treatment-to-unit allocation, in which mRNA samples are assigned in an optimal way to slides and dye labeling.
It has been shown how the design choice should be intimately related to the goal of the experiment, such as the contrast between fixed effects (e.g., the comparison of alternative drugs or the expected phenotypic value of different genotypes), or inference regarding variance components (such as genetic variances), or heritability of transcriptional levels.
Sometimes it is possible to have an analytical solution for the optimality problem, but in many situations a numerical solution is needed. Whenever a numerical solution is implemented, a complete search (comparing all possible designs) guarantees the optimal finding. A complete search, however, is often unattainable due to the size of the search problem involved. In these situations algorithms are needed that can find optimal or near-optimal designs. R functions were developed for searching and comparing the different designs discussed in this article; they can be obtained directly from the authors.
As a final remark, it is important to note that this article considers a single gene at a time for design purposes, as the same model is generally used to analyze all genes in a single experiment. This is not an issue for fixed-effects models (such as for the examples comparing genotypic groups) as the optimal design criteria for fixed-effects models depend only on the design matrix. This is not true in mixed- or random-effects models, as in these cases optimal designs depend on both the design matrix and the variance components parameters. Since the variance parameters vary among genes, the optimal design for one gene may be different from that for another gene. As a consequence, caution is needed before we simplify the optimal design for the microarray to an optimal design for a single gene if random- or mixed-effects models are considered.
This is a very interesting topic, which deserves further research. A possible approach to this issue would be to use an empirical distribution for the genetic variances or heritabilities to weight the decision function that yields the design criterion. Alternatively, “robust” designs could be sought by focusing on the high heritability genes. This could be achieved by putting more weight on the higher heritabilities than suggested by an empirical distribution or by even ignoring the region of low heritability when searching for good designs. An extreme version of this approach would be by taking genetic effects as fixed, but in general this would be an inefficient strategy. Moreover, while modeling genetic effects as fixed may be simple with experiments involving family structures such as half- or full-sibs, it is not clear how this approach could be easily extended for complex pedigrees.
Acknowledgments
This work was supported by the Michigan State University Intramural Research Grant Program, by grant 2004-33120-15204 from the United States Department of Agriculture to G.J.M.R., and by grant 474787/2004-4 from the Conselho Nacional de Desenvolvimento Científico e Tecnológico-Brazil to J.S.S.B.F.
References
- Bailey, R. A., 1981. A unified approach to design of experiments. J. R. Stat. Soc. A 144: 214–223. [Google Scholar]
- Bailey, R. A., 1997. Statistics and mathematics: the appropriate use of mathematics within statistics (with discussion). Statistician 47: 261–271, 273–290. [Google Scholar]
- Brem, R. B., G. Yvert, R. Clinton and L. Kruglyak, 2002. Genetic dissection of transcriptional regulation in budding yeast. Science 296: 752–755. [DOI] [PubMed] [Google Scholar]
- Bueno Filho, J. S. S., and S. G. Gilmour, 2003. Planning incomplete block experiments when treatments are genetically related. Biometrics 59: 375–381. [DOI] [PubMed] [Google Scholar]
- Bystrykh, L., E. Weersing, B. Dontje, S. Sutton, M. T. Pletcher et al., 2005. Uncovering regulatory pathways affecting hematopoietic stem cell function using “genetical genomics.” Nat. Genet. 37: 225–232. [DOI] [PubMed] [Google Scholar]
- Chesler, E. J., L. Lu, J. Wang, R. W. Williams and K. F. Manly, 2004. WebQTL: rapid exploratory analysis of gene expression and genetic networks for brain and behavior. Nat. Neurosci. 7: 485–486. [DOI] [PubMed] [Google Scholar]
- Darvasi, A., 2003. Gene expression meets genetics. Nature 422: 269–271. [DOI] [PubMed] [Google Scholar]
- Darvasi, A., and M. Soller, 1992. Selective genotyping for determination of linkage between a marker locus and a quantitative trait locus. Theor. Appl. Genet. 85: 353–359. [DOI] [PubMed] [Google Scholar]
- Draper, N. R., and I. Guttman, 1997. Two-level factorial and fractional factorial designs in blocks of size two. J. Qual. Technol. 29: 71–75. [Google Scholar]
- Fedorov, V. V., 1972. Theory of Optimal Experiments. Academic Press, New York.
- Glonek, G. F. V., and P. J. Solomon, 2004. Factorial and time course designs for cDNA microarray experiments. Biostatistics 5: 89–111. [DOI] [PubMed] [Google Scholar]
- Henderson, C. R., 1976. Simple method for computing inverse of a numerator relationship matrix used in prediction of breeding values. Biometrics 32: 69–83. [Google Scholar]
- Jansen, R. C., and J. Nap, 2001. Genetical genomics: the added value from segregation. Trends Genet. 17: 388–391. [DOI] [PubMed] [Google Scholar]
- Jin, C., H. Lan, A. D. Attie, G. A. Churchill, D. Bulutoglu et al., 2004. Selective phenotyping for increased efficiency in genetic mapping studies. Genetics 168: 2285–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr, M. K., 2003. Design considerations for efficient and effective microarray studies. Biometrics 59: 822–828. [DOI] [PubMed] [Google Scholar]
- Kerr, M. K., 2006. 2^k factorials in blocks of size 2, with application to two-color microarray experiments. UW Biostatistics Working Paper Series No. 227 (http://www.bepress.com/uwbiostat/paper227).
- Kerr, M. K., and G. A. Churchill, 2001. Statistical design and the analysis of gene expression microarray data. Genet. Res. 77: 123–128. [DOI] [PubMed] [Google Scholar]
- Kiefer, J. C., and J. Wolfowitz, 1960. The equivalence of two extremum problems. Can. J. Math. 12: 363–366. [Google Scholar]
- Lander, E. S, and D. Botstein, 1989. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121: 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhart, D. J., H. L. Dong, M. C. Byrne, M. T. Follettie, M. V. Gallo et al., 1996. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat. Biotech. 14: 1675–1680. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998. Genetics and Analysis of Quantitative Traits. Sinauer, Sunderland, MA.
- Mead, R., 1990. The nonorthogonal design of experiments (with discussion). J. R. Stat. Soc. A 153: 151–201. [Google Scholar]
- Monks, S. A., A. Leonardson, H. Zhu, P. Cundiff, P. Pietrusiak et al., 2004. Genetic inheritance of gene expression in human cell lines. Am. J. Hum. Genet. 75: 1094–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noueiry, A. O., J. Chen and P. Ahlquist, 2000. A mutant allele of essential, general translation initiation factor DED1 selectively inhibits translation of a viral mRNA. Proc. Natl. Acad. Sci. USA 97: 12985–12990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa, G. J. M., J. P. Steibel and R. J. Tempelman, 2005. Reassessing design and analysis of two-color microarray experiments using mixed effects models. Comp. Funct. Genomics 6: 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt, E. E., S. A. Monks, T. A. Drake, A. J. Lusis, N. Che et al., 2003. Genetics of gene expression surveyed in maize, mouse and man. Nature 422: 297–302. [DOI] [PubMed] [Google Scholar]
- Schena, M., D. Shalon, R. W. Davis and P. O. Brown, 1995. Quantitative monitoring of gene-expression patterns with a complementary-DNA microarray. Science 270: 467–470. [DOI] [PubMed] [Google Scholar]
- Sorensen, D., and D. Gianola, 2002. Likelihood, Bayesian, and MCMC Methods in Quantitative Genetics. Springer, New York.
- Steibel, J. P., and G. J. M. Rosa, 2005. On reference designs for microarray experiments. Stat. Appl. Gen. Mol. Biol. 4(1): Article 36 (http://www.bepress.com/sagmb/vol4/issl/art36/). [DOI] [PubMed] [Google Scholar]
- Tempelman, R. J., 2005. Assessing statistical precision, power, and robustness of alternative experimental designs for two color microarray platforms based on mixed effects models. Vet. Immunol. Immunopathol. 105: 175–186. [DOI] [PubMed] [Google Scholar]
- Vinciotti, V., R. Khanin, D. D'Alimonte, X. Liu, N. Cattini et al., 2005. An experimental evaluation of a loop versus a reference design for two-channel microarrays. Bioinformatics 21: 492–501. [DOI] [PubMed] [Google Scholar]
- Wang, P. C., 2004. Designing two-level fractional factorial experiments in blocks of size two. Sankhya 66: 327–342. [Google Scholar]
- Wit, E., A. Nobile and R. Khanin, 2005. Near-optimal designs for dual channel microarray studies. Appl. Stat. 54: 817–830. [Google Scholar]
- Wolfinger, R. D., G. Gibson, E. D. Wolfinger, L. Bennett, H. Hamadeh et al., 2001. Assessing gene significance from cDNA microarray expression data via mixed models. J. Comp. Biol. 8: 625–637. [DOI] [PubMed] [Google Scholar]
- Yang, Y. H., and T. Speed, 2002. Design issues for cDNA microarray experiments. Nat. Rev. Genet. 3: 570–588. [DOI] [PubMed] [Google Scholar]
- Yang, Y. J., and N. R. Draper, 2003. Two-level factorial and fractional factorial designs in blocks of size two. J. Qual. Technol. 35: 294–305. [Google Scholar]