

Abstract
This study explores the ability of regression models, with no knowledge of the underlying physiology, to estimate physiological parameters relevant for metabolism and endocrinology. Four regression models were compared: multiple linear regression (MLR), principal component regression (PCR), partial least-squares regression (PLS) and regression using artificial neural networks (ANN). The pathway of mammalian gluconeogenesis was analyzed using [U−13C]glucose as tracer. A set of data was simulated by randomly selecting physiologically appropriate metabolic fluxes for the 9 steps of this pathway as independent variables. The isotope labeling patterns of key intermediates in the pathway were then calculated for each set of fluxes, yielding 29 dependent variables. Two thousand sets were created, allowing independent training and test data. Regression models were asked to predict the nine fluxes, given only the 29 isotopomers. For large training sets (>50) the artificial neural network model was superior, capturing 95% of the variability in the gluconeogenic flux, whereas the three linear models captured only 75%. This reflects the ability of neural networks to capture the inherent non-linearities of the metabolic system. The effect of error in the variables and the addition of random variables to the data set was considered. Model sensitivities were used to find the isotopomers that most influenced the predicted flux values. These studies provide the first test of multivariate regression models for the analysis of isotopomer flux data. They provide insight for metabolomics and the future of isotopic tracers in metabolic research where the underlying physiology is complex or unknown.
KEYWORDS: systems biology, multivariate regression, stable isotopes, metabolism, gluconeogenesis
Introduction
Complex interactions of genes, proteins and metabolites underlie physiological regulation. A major challenge for physiologists today is to develop practical models reflecting regulatory relationships among system variables. The term systems variables refers to the large sets of data that are now routinely generated in the course of a single experiment. For example, a single microarray chip generates thousands of transcription measurements, while a two dimensional gel produces thousands of bits of proteomic information. With regard to metabolism, the emerging field of metabolomics will generate systems variables in the form of the concentrations of large number of metabolites (Raamsdonk et al., 2001; German et al., 2002). Analyzing these large volumes of data is becoming the main challenge in generating new knowledge from high throughput experiments. There is a clear need for computational methods that can integrate large sets of physiological data into a structured picture. The goal of these models will be to capture the complex relationships that are at the heart of the functioning of living cells and organisms with limited a priori knowledge of the structure of these interactions.
Recently, several data mining algorithms based on projection methods have been successfully applied to the analysis of large amounts of microarray data. Gene clustering, identification of discriminatory genes, and determination of characteristic gene expression patterns are examples of such applications (Misra et al., 2002; Stephanopoulos et al., 2002). The principal component analysis (PCA) projection method is of particular interest as an unsupervised method that can be applied to reveal the true dimensionality of data, identify redundancies and conveniently represent data in a reduced dimensional space. An introduction to PCA for the physiological oriented researcher has been provided by Benigni and Giuliani (Benigni and Giuliani, 1994). On the other hand, regression analysis is the major tool for obtaining models from measured data. Combination of PCA and regression modeling yields predictive models in lower dimensions that capture aspects of the physiology of the system. In this paper we critically evaluate the potential use of three linear regression modeling methods, multiple linear regression (MLR), principal component regression (PCR) and partial least squares regression (PLS), and one non-linear regression model based on artificial neural networks (ANN), for the analysis of data derived from a system with complex underlying structures. Currently PCR and PLS models are increasingly used in medicine and industry for the determination of concentrations of chemical compounds from complex mixtures based on near-infrared spectroscopy data (Irudayaraj and Tewari, 2003). For example, a PLS model has been applied to estimate the concentration of urea in dialysate samples from hemodialysis patients utilizing the near-infrared spectral data of the dialysate (Eddy et al., 2003). Neural network models have been successfully trained to perform complex functions in various fields of application including pattern recognition, speech and image analysis, classification, and control (Bishop, 1996; Haykin, 1998). For example, an ANN was used for prediction of the chemotherapeutic response of human cancer cells from NMR spectroscopy data (El-Deredy et al., 1997). ANN was also recently applied to solve an inverse metabolic problem, that is, to determine kinetic parameters in metabolic models with known structures given steady-state metabolite levels (Mendes and Kell, 1996).
Many uses of multivariate statistical tools have recently appeared, especially for the analysis of microarray data. While these studies are often provocative, they rarely include an objective mechanism to determine how well the model works. Thus, it is difficult to determine if a specific multivariate technique is optimally designed to discover quantitative relationships between gene expression levels and a phenotype such as insulin resistance because we lack detailed knowledge of the quantitative relationship between gene expression and physiological phenotypes. We cannot create a realistic test case for this complex relationship. In contrast, the pathway of mammalian glucose metabolism is much better understood. Metabolic simulations can provide precise data for isotopic labeling of intermediates and for glucose production. This data can serve as a test case. Here, we present the first use of multivariate regression models in mammalian physiology to estimate fluxes from 13C labeling patterns of metabolites and to identify relationships between the labeling patterns of key metabolites and fluxes. We chose a familiar metabolic system, mammalian gluconeogenesis as assessed by constant infusion of [U−13C]glucose. To this end, we first created a metabolic simulation of the gluconeogenic pathway (comprising of key intra-hepatic metabolites and fluxes) to generate data in the form of isotopic metabolite labeling patterns and metabolic fluxes for this system. We then trained various regression models on this data to allow the model to develop quantitative relationships between mass isotopomers and metabolic fluxes. We evaluated the trained regression models for their ability to predict fluxes using new data not part of the training set. The accuracy of predictions was evaluated by comparing the fluxes predicted by the regression model with those from the metabolic simulation. We also evaluated the sensitivity of model predictions to measurement errors and to noise. Finally, we use this example to demonstrate how physiological insight is obtained from the analysis of the relative values of model parameters. The application of multivariate statistics to a metabolic network of isotopic fluxes demonstrated here serves as a model for the broader application of these techniques in the emerging fields of metabolomics and systems biology.
Methods
Notation
We identify mass isotopomers as M0, M1, etc., where the numerical subscript denotes the mass increase over the non-enriched molecule. In keeping with previous conventions, we represent mass isotopomers of glucose as Mi and mass isotopomers of lactate as mi.
Metabolic system
As a familiar metabolic system for this analysis we chose mammalian gluconeogenesis at metabolic and isotopic steady state evaluated by constant infusion of [U−13C]glucose (figure 1). The infusion of [U−13C]glucose under gluconeogenic conditions leads to recycling of the tracer to plasma glucose that generates a distinct metabolite labeling pattern that can be detected by GC/MS. While the infused glucose is comprised of the fully labeled, M6 isotopomer, the isotope is diluted in the pathway, and the process of gluconeogenesis produces newly synthesized glucose that is labeled in one of the two triose moieties. Thus, newly synthesized glucose is comprised of glucose isotopomers containing zero to three enriched atoms, M0 through M3. The glucose to glucose pathway diagramed in figure 1 represents an idealized case that does not include all relevant fluxes in vivo. Among the missing fluxes are the contributions to gluconeogenesis of glycerol and of amino acids not equilibrated with plasma lactate. Indeed, the failure to consider such fluxes has led Landau and colleagues to conclude that the [U−13C]glucose method leads to underestimates of gluconeogenesis in vivo (Landau et al., 1998). Despite these limitations we utilized the [U−13C]glucose protocol to explore the use of multivariate regression models in the analysis of isotopic flux data using pathways familiar to metabolic researchers. In this idealized model, the label distribution of all metabolites in the system is strongly dependent on the specific values of fluxes in the metabolic system. Different flux distributions result in significant tracer redistribution and yield different metabolite labeling patterns. We have constructed a mathematical model that describes the relationship between metabolite labeling patterns and fluxes (see Appendix A). This model allows us to simulate the labeling patterns of all metabolites in the network for any set of steady state fluxes. We quantify the labeling of metabolites in the pathway in terms of fractional abundances of isotopomers, where the sum of all isotopomers of a specific metabolite is 1. Under isotopic steady state condition, isotopomer balances describe labeling distribution in metabolites as a function of fluxes. Equation (1) illustrates the type of relationship that can be written for a particular isotopomer of plasma glucose:
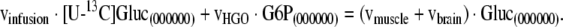 |
1 |
Figure 1.
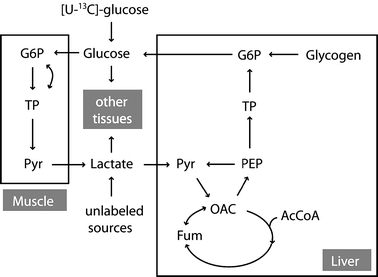
Schematic representation of mammalian glucose metabolism evaluated by constant [U−13C]glucose infusion. Abbreviations of metabolites: G6P, glucose-6-phosphate; Pyr, pyruvate; OAC, oxaloacetate; Fum, fumarate; AcCoA, acetyl coenzyme A; PEP, phosphoenolpyruvate; TP, triose phosphates.
In equation (1), [U−13C]Gluc(000000) refers to the fractional abundance of one particular positional isotopomer of infused glucose, in this case the isotopomer with no enriched carbon atoms. In general, 2N of such equations are constructed for an N-carbon atom metabolite, one equation for each positional isotopomer (i.e. each possible labeling pattern of the carbon atoms of the metabolite). For example, there are 64 (=26) model equations for plasma glucose isotopomers. Our metabolic simulation consists of the complete set of isotopomer balance equations for all metabolites in the system. Isotopomer mapping matrices were used to create a model that properly considers all isotopomer conversions in the system (Zupke and Stephanopoulos, 1994; Schmidt et al., 1997). These models are non-linear because the full set of equations contains product terms of fluxes with isotopomers and product terms of isotopomers with isotopomers due to linear and condensation reactions in the system. Recently, an elegant solution algorithm was introduced by Wiechert et al. (Wiechert et al., 1999) that greatly facilitates the derivation of the unique solution for this non-linear problem. For a given set of fluxes, the non-linear model was solved using Wiechert’s approach to yield the positional isotopomer fractions for all compounds. Mass isotopomer fractional abundances were then obtained by a linear transformation from the positional isotopomer fractions. To simplify calculations, the model assumes that all data have been corrected for natural isotope abundances.
Data generation
The metabolic system in figure 1 contains 9 independent fluxes and a total of 29 independent mass isotopomer fractional abundances (tables 1, 2). We simulated 2500 random sets of fluxes that satisfy the steady state condition. For each set of fluxes the isotopomer balances were then solved to yield the corresponding metabolite labeling patterns from which we generated the corresponding 2500 sets of GC/MS data of mass isotopomer abundances. The simulated data was divided into a training set for the calibration of regression models (1000 simulations), a validation set to check the calibration (500 simulations), and a test set to determine the prediction accuracy of the models (1000 simulations). Table 1 summarizes the ranges of flux values that were used to generate the random fluxes. The TCA flux was arbitrarily set to 1. The other 8 fluxes are expressed as fluxes relative to the TCA flux. The mass isotopomer data was corrupted with random noise of a standard deviation of 0.05 mol% enrichment, reflecting the detection limit of GC/MS measurements. The simulated data was collected into matrices X (with isotopic data in columns) and Y (with fluxes in columns). Each row in X and Y contains data collected from one simulation.
Table 1.
Range of flux values used for the generation of random fluxes
| Flux | Range |
|---|---|
| TCA Cycle | 1 |
| Gluconeogenesis | 0.4–0.7 |
| Glycogenolysis | 0.1–1.7 |
| Pyruvate carboxylase (y) | 0.5–2.5 |
| Cori cyclea | 0.3–2.0 |
| Label scrambling in muscle due to pentose pathway | 0.1–1.0 |
| Label scrambling in liver due to fumarase | 1.0–4.0 |
| Tracer infusion rate | 0.05–0.3 |
| Plasma lactate dilution | 0.7–1.5 |
aCori cycle refers to flux from plasma glucose to plasma lactate.
Table 2.
Independent mass isotopomers in the metabolic system.
| Metabolite | Number of independent mass isotopomersa |
|---|---|
| Plasma glucose | 6 |
| Plasma lactate | 3 |
| Hepatic pyruvate | 3 |
| Hepatic oxaloacetate | 4 |
| Hepatic fumarate | 4 |
| Hepatic phosphoenolpyruvate | 3 |
| Hepatic glucose-6-phosphate | 6 |
| Total | 29 |
aMass isotopomers values are modeled as fractional abundances. For each metabolite one isotopomer is not independent but known as 1 – sum of all other isotopomers.
Model training can be made more efficient if certain preprocessing steps are performed on the raw data. In regression analysis it is customary to normalize the mean and standard deviation of the training set, especially if the variables have different (or arbitrary) units and scales. By transforming variables in this way all variables are treated equally, thus preventing any bias towards variables with large numerical values and large variances. For our analysis all variables were mean-centered and variance scaled, also known as autoscaling: the average value for each variable was calculated and then subtracted from each corresponding variable; scaling was accomplished by dividing all values for a particular variable by the standard deviation for that variable, so that the variance for each variable is one.
Regression modeling
The main goal of any regression model is to predict the dependent (response) variables y from independent (predictor) variables x. Typically, independent variables are routine measurements that are easily available and provide a low resolution description of the state of the system. Dependent variables, on the other hand, are usually harder to obtain and have higher information content. In the example used here, mass isotopomer fractional abundances are the independent variables that we might obtain experimentally and fluxes are the dependent variables with a higher information content that we want to predict from isotopic data. Regression analysis consists of two steps. First, a mathematical model for the behavior of the system is proposed. Next, optimal values for model parameters are determined based on training samples. This is the training or calibration step. Note that for the training step both the independent and dependent variables are required. In the second step the trained model is used to predict values of dependent variables, given the values of independent variables for one or more new samples. This is the prediction step. For the prediction step only independent variables are required as input.
Multiple linear regression (MLR)
Suppose we can measure values for m predictor variables xi (i=1...m) and one response variable y1, then the simplest model we can propose assuming no prior knowledge of the structure of the system is a linear (or first-order) relationship:
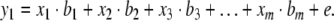 |
2a |
In terms of the model used here an example might be:
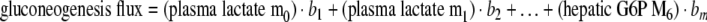 |
2b |
In equation (2) bi (i=1...m) are the sensitivities or model parameters, and e is the modeling error or residual. This equation describes the multilinear dependencies for one sample with one response variable. For k response variables and n number of samples equation (2) may be written in the following matrix form:
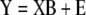 |
3 |
here, Y is an n × k matrix, X is an n × m matrix, B is an m × k matrix and E is an n × k matrix. Each row in matrices X and Y contains data from one particular sample. In our model system each row of the Y matrix contains all 8 independent fluxes and each row of the matrix X contains all 29 isotopomer abundances. The best regression model is the one that minimizes the modeling errors in matrix E. We find the best model by choosing appropriate values for the model variables in matrix B based on the training data. Note that there are a total of m·k model variables that need to be determined. In our example this is 29·8=232. Each sample provides k relations of the form of equation (2), one such relation for each response variable yj (j=1...k). Therefore, in order to determine all model parameters we require at least m number of samples for the training step. If n<m then equation (3) is underdetermined and infinite number of solutions minimize the residuals in matrix E. Thus, MLR cannot work when the number of variables exceeds the number of samples. For n ≥ m we obtain the following familiar least-squares estimate for model parameters:
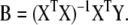 |
4 |
Once the optimal values for the model parameters have been determined we can apply equation (2) to predict the values of response variables given values for predictor variables from a new sample. In our system we would estimate fluxes from isotopomer abundances using equation (2b). A major concern with the application of MLR is the large number of samples required for the training step. In many cases the number of independent variables is much greater than the number of samples. For example, consider the measurement of a few thousand transcription levels as predictors. In order to train the MLR model we would require at least as many calibration samples which may not be feasible. Another frequent problem with MLR is that the inverse of XTX in equation (4) may not exist. This occurs when two or more variables behave in very similar fashion, a problem known as collinearity. Reduced space regression models provide a practical solution to the above problems and this leads us to principal component analysis.
Principal component regression (PCR)
When measuring m independent variables, we obtain an m-dimensional description of the state of the system. However, some variables may be interrelated or in fact contain exactly the same information. The amount of redundancy is likely to be large in a sizable data set. Therefore, an equally satisfactory description of the data may be possible with fewer dimensions. One particular data reduction technique called principal component analysis (PCA) is used to reveal the true dimensionality of a data set. PCA defines a new lower-dimensional space spanned by variables that are linear combinations of the original variables and account for as much of the original total variation as possible. The new variables are called latent variables or principal components. The PCA projection of matrix X is represented as follows:
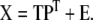 |
5 |
Here, matrix T (size n × d) is called the scores matrix and matrix P (size m × d) is called the loadings matrix, where d is the number of principal components. Matrix E is the residuals matrix. PCA is a stepwise optimization procedure where the successive principal components are extracted in such a way that they are uncorrelated with each other and account for successively smaller amounts of the total variation. It is possible to extract as many principal components as there are original variables, however, in most PCA applications the goal is to account for most of the total variation with as few principal components as possible.
The main goal of PCA with regard to regression analysis is to reduce the dimensionality of matrix X from m initial variables to a (significantly) smaller number d. The principal component regression (PCR) model then considers the linear (or first-order) relationship between the response variables summarized in matrix Y and the scores matrix T:
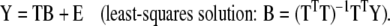 |
6 |
Note the similarity between the MLR model (equation (3)) and the PCR model (equation (6)). The significant difference is the reduced number of model parameters, which allows reduction of the number of experiments required for model training. PCR also solves the collinearity problem by guaranteeing an invertible (TTT) in equation (6). For new unknown samples the value for any response variable is predicted with:
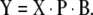 |
7 |
Partial least-squares regression (PLS)
PLS is closely related to PCR, with the addition that now both the independent matrix X and dependent matrix Y are decomposed into lower dimensional space:
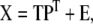 |
8 |
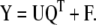 |
9 |
Equations (8) and (9) are called the outer relations. There is also a linear inner relationship constructed between the scores matrices U and T. The PLS model is established as the combined or mixed relation given by:
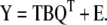 |
10 |
Thus, equation (10) captures the relationship between the response and independent variables in the lower dimensional spaces defined by equations (8) and (9). It has been suggested that PLS is a good alternative to PCR that yields more robust model parameters, i.e. model parameters that do not change very much when new calibration samples are included in the training set (Geladi and Kowalski, 1986).
Optimal number of components
For the construction of reduced space models the optimal number of principal components (or the optimal dimensionality of the new space) needs to be determined from available calibration data. Using too few components results in significant information loss. On the other hand, since measured data is never noise free, some components will only describe noise. Therefore, using too many dimensions will cause overfitting of data and yield inaccurate predictions as well. A number of criteria have been proposed for the rational selection of the optimal number of principal components; a cross-validation method is the preferred choice for the construction of predictive models. In this approach, each sample is in turn omitted from the training set and the model is trained using the remaining n−1 samples. The trained model is then used to predict the values of the response variables in the sample that was left out, and residuals are calculated as the difference between the actual observed values and the predicted values. The prediction residual sum of squares (PRESS) is then calculated as the sum of all squared residuals. This PRESS value is determined for varying number of components (i.e. dimensions), as one searches for the number of components that gives the minimum PRESS value. However, the location of the minimum is not always well defined and models with varying number of components may yield similar magnitude PRESS values. In this study, the optimal number of principal components was defined as the fewest number of components yielding a PRESS value within 5% of the minimal observed PRESS value. Figure 2 gives an example plot of the PRESS value against the number of components for the training of a PLS model used in this study. The optimal number of dimensions in this case was 10.
Figure 2.
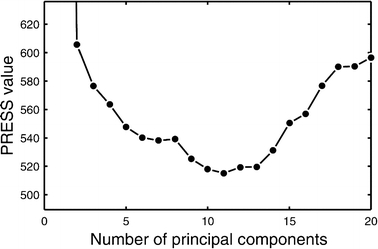
Determination of the optimal number of principal components by the leave-one-out cross-validation method. The optimal number of principal components is defined as the fewest number of components yielding a PRESS value within 5% of the minimal observed PRESS value; in this case 10 principal components.
An alternative method for optimizing the number of components is to use a separate validation set to check the calibration. Note that this validation set has to be independent of the training set; as such, this method may be less practical when the number of samples is limited. Since we can generate enough samples in this study, we applied both methods for optimizing the number of components and compared the results. Here, we used 500 independent simulations as the validation set.
Artificial neural networks (ANN)
Neural networks are a general method of modeling non-linear systems. They are composed of simple computational elements (i.e. neurons) operating in parallel. In short, each neuron produces one output through a transfer function, typically a sigmoid function, which takes the weighted sum of the input arguments and a constant term called the bias (Haykin, 1998). The inputs and outputs may be to and from external variables, or other neurons. Multiple neurons are combined into a layer, and a particular network can contain one or more (hidden) layers. ANN have been demonstrated to fit any arbitrary function given enough neurons in the hidden layers (White, 1992). In this study, we have applied fully connected feedforward networks with one or more hidden layers, updated via the backpropagation algorithm. The inputs to the neural network were component scores from PCA analysis of matrix X, and the outputs were fluxes. The optimal number principal components, i.e. the number of inputs to the neural network, was determined as described above. In the base case we considered two network architectures: ANN with one hidden layer with 20 neurons, and ANN with two hidden layers with 10 neurons in each layer. For both topologies the following sigmoidal transfer function was used in the hidden nodes:
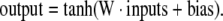 |
11 |
Alternatively, we used radial basis functions in the hidden layer, as indicated in the text. For the output nodes we used a linear transfer function, which is recommended to prevent artifacts introduced by sigmoidal transfer functions in the output layer (Mendes and Kell, 1996). Thus for example, when the number of principle components was 9, we considered the following two neural network architectures: ANN with a 9-20-8 topology, i.e. with 9 input nodes, 20 hidden neurons, and 8 output neurons; and ANN with 9-10-10-8 topology (9 inputs, 10 nodes on the first hidden layer, 10 nodes on the second hidden layer, and 8 outputs). Finally, we also considered ANNs with a single output node (one ANN for each flux), i.e. 8×ANNs with 9-20-1, or 9-10-10-1 topology. We used Matlab 6.5 and Matlab Neural Network Toolbox to train the neural networks. For the training we used backpropagation training (Matlab’s trainlm function), and applied an early stopping technique to prevent overfitting of the neural network model, which is the default setting in the Matlab Neural Network Toolbox.
Calculation methods
The commercially available PLS Toolbox 2.1 (Eigenvector Research Inc.) and Matlab Neural Network Toolbox (Mathworks Inc.) were used for all calculations. The above discussion on linear regression and reduced space regression modeling was not meant to be comprehensive. See Dillon and Goldstein (Dillon and Goldstein, 1984), Geladi and Kowalski (Geladi and Kowalski, 1986) and several good books (Chatfield and Collins, 1981; Causton, 1987; Martens and Naes, 1989; Manly, 1994; Tabachnick and Fidell, 2001) for a more complete treatment of these subjects.
Results and discussion
Model training
The first step in our analysis is the training of the regression models to correlate fluxes summarized in matrix Y with isotopic labeling data similarly summarized in matrix X assuming no prior knowledge of the structural connection between data in the X block and Y block. We are particularly interested in the influence of the size of the training set on the accuracy of model predictions, and the impact of measurement errors on precision. It is naturally expected that larger training sets will produce better models. To determine the minimal number of samples required to obtain acceptable predictions from the models we varied the size of the training data set between 10 and 1000 samples when performing the training step. Note that training of the MLR regression model required a minimum of 29 samples since there are 29 mass isotopomers variables in our model. The PCR, PLS and ANN regression models did not have this limitation due to the reduced number of dimensions.
If the underlying model for the relationship between X and Y is a linear model, then the number of principal components needed to describe the system equals the number of degrees of freedom for that system. However, non-linear models are expected to require extra dimensions to describe non-linearities. In figure 3, the optimal number of principal components for the constructed PCR, PLS and ANN models is plotted against the size of the training set. Here, the number of components was determined using the leave-one-out method. We also optimized the number of components using an independent validation set. Virtually the same number of components were predicted by both methods (results not shown); we used the leave-one-out method for all subsequent examples in this study. The optimal number of principal components increased with the size of the training set, but eventually reached a maximum of 17, 16, and 9 principal components for the PCR, PLS and ANN models, respectively. The ANN model typically required fewer components to capture the same amount of information as the PCR and PLS models. When comparing different ANN topologies we did not find significant differences between ANNs with one or two hidden layers, and ANNs with radial basis functions. The same number of input components were predicted. Note that the number of principal components constituted a significant reduction from the original 29 mass isotopomer variables. This result indicates that redundant information is present in mass isotopomer data.
Figure 3.
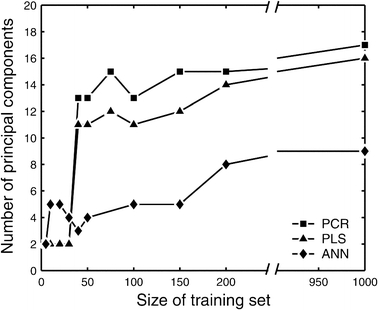
Optimal number of principal components in the PCR and PLS models for varying sizes of training data set. Larger training sets allow more principal components to be included in the model to capture the finer details of the system. ANN models typically required fewer principal components than PCR and PLS models.
Model testing
In the testing phase of our analysis, the trained MLR, PCR, PLS and ANN models were used to predict specific fluxes given a new set of simulated mass isotopomers as described in the Methods section. Note that this data was not included in the training stage, but used only to test the predictive power of models. We calculated the correlation coefficient R2 as an indicator of the accuracy of predictions. The R2 value indicates the fraction of variation that is accounted for by regression. Figure 4 summarizes the calculated correlation coefficients as a function of the size of training set for all regression models. This plot shows a strong influence of the number of training samples on the prediction accuracy. For smaller training sizes the PCR and PLS models perform better than the ANN (with one hidden layer) model, and significantly better than the MLR model. The former two regression models have a R2 of 0.58 for training data of 10 samples, while the ANN and MLR models required at least 35 and 75 training samples, respectively, to allow predictions of similar quality. However, for training data consisting of 50 or more samples it was the ANN model that produced the best predictions. For training sizes larger than 200 samples we found no significant difference in prediction accuracy between the three linear regression models (MLR, PCR and PLS). As expected, larger training sets yielded better predictions, but after a certain point the correlation coefficient reached a maximum value of 0.95 for the ANN model, and 0.75 for the linear MLR, PCR and PLS models, indicating that 95% and 75% of the variability in the gluconeogenic flux is captured by the models. The 20% difference is caused by the inherent limitation of linear models to capture non-linearities. The maximum correlation coefficients for the other fluxes in the system varied between 0.59 for the pyruvate carboxylase flux, and 0.99 for the tracer infusion flux (Table 3). Thus, the regression models clearly performed well for some fluxes, but not for all of them. The accuracy of prediction was similar for ANNs with one and two hidden layers. i.e. the additional hidden layer did not improve predictions. The number of neurons in the hidden layer could be reduced to 5 without affecting the quality of predictions, i.e. a 9-5-8 ANN yielded similar results as 9-20-8 ANN. For the two layer topology the number of neurons could be reduced to 3 in each hidden layer, i.e. a 9-3-3-8 ANN yielded similar results as 9-10-10-8 ANN. ANNs with a single output node, i.e. one ANN for each flux, performed only slightly better than one neural network with 8 output nodes (less than 3% improvement). Furthermore, ANNs with radial basis functions performed slightly worse than ANNs with sigmoidal basis functions, for example, the maximum correlation coefficient for gluconeogenesis flux was 0.93 when radial basis functions were used. In this study, autoscaling did not improve the results, i.e. the accuracy of predictions was the same with and without autoscaling of the data. This was true for MLR, PCR, PLS and ANN models.
Figure 4.
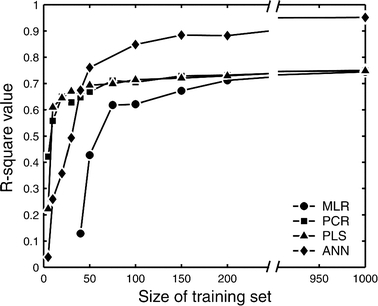
Observed prediction accuracy of the gluconeogenesis flux as a function of the number of samples used in training. For small number of training samples the PCR and PLS models produce the best predictions; for larger number of training samples ANN yields the best predictions.
Table 3.
Correlation coefficients and most sensitive flux predictors based on the PLS model trained with 1000 samples
| Flux | R2 | Most sensitive mass isotopomer predictorsa |
|---|---|---|
| Gluconeogenesis | 0.75 | PEP M3 (0.772), M1 (−0.368); Gluc M3 (−0.721), M1 (0.462), M2 (0.439); OAC M3 (0.454) |
| Glycogenolysis | 0.76 | PEP M3 (−0.755); Gluc M3 (0.724), M0 (0.507), M6 (−0.488), M1 (−0.439), M2 (−0.407); OAC M3 (−0.444) |
| Pyruvate carboxylase (y) | 0.59 | Pyr M3 (−1.781), M3 (1.032); PEP M3 (0.955); Gluc M1 (0.858), M3 (−0.652) |
| Cori cycle | 0.69 | Gluc M6 (−0.822), M3 (0.771), M0 (0.685); PEP M3 (−0.517) |
| Labeling scrambling in muscle | 0.68 | Gluc M3 (−1.500); PEP M3 (−1.279); Lact M1 (0.848) |
| Labeling scrambling in TCA | 0.66 | Gluc M3 (−1.135), M2 (0.936); G6P M3 (−0.787), M2 (0.761) |
| Tracer infusion rate | 0.99 | Gluc M6 (0.665), M0 (−0.461) |
| Plasma lactate dilution | 0.73 | Gluc M6 (1.338), M0 (−0.968); Fum M1 (−0.532); PEP M1 (−0.443); OAC M1 (−0.443) |
Values between parentheses are the model sensitivities obtained from the matrix multiplication P×B (see equation (7)).
aMetabolite abbreviations as in figure 1.
To analyze the effect of measurement errors on prediction accuracy, the above analysis was repeated with data corrupted with random errors with a 10% coefficient of variation. The prediction accuracy of the MLR, PCR and PLS models was only slightly reduced compared to noise-free data. The ANN on the other hand showed a larger reduction in prediction accuracy. The correlation coefficient for the predicted gluconeogenesis flux reached a maximum value of 0.80 for the ANN model and 0.70 for the MLR, PCR and PLS models, compared to 0.95 and 0.75, respectively, for noise-free data. Similar reduction in R2 values were observed for the other fluxes (results not shown). The relatively larger reduction in prediction accuracy of ANNs is due to inherent limitations of estimating fluxes with noisy data. Even a fully deterministic flux model, i.e. the model that was used to generate our data, estimated gluconeogenesis flux with a correlation coefficient of 0.83 for noisy data, and 1.00 for noise-free data, i.e. perfect predictions (results not shown). Thus, the R2 value of 0.80 indicates that the ANN model performed well with noisy data.
In most studies a number of the measured variables will only describe noise and may not necessarily be relevant as predictor variables. Therefore, we tested the effect of having noisy irrelevant variables as part of the training set. To this end, we added 29 randomly generated variables to the 29 mass isotopomers as predictor variables in each sample, and repeated the analysis. The correlation coefficient for the predicted gluconeogenesis flux was reduced to 0.86 for the ANN model and 0.74 for the MLR, PCR and PLS models. Thus, all models were robust with respect to noisy data. Finally, we tested the robustness of results with respect to incomplete data by randomly leaving out half of the predictor variables from the data set. No significant differences were found, which indicates that significant redundancy is present in the isotopomer data.
Model interpretation
To obtain physiological insight from these models we evaluated the relative values of model parameters in the PLS model. In this study, model parameters are sensitivities of fluxes with respect to isotopomer data (see equation (2a)). Sensitivity values for PCR and PLS models were obtained from the matrix multiplication P×B (see equation (7)). Here, we compared for each flux the relative values of these sensitivities. High values indicate significant correlation between a flux and a particular mass isotopomers suggesting a structural connection. Table 3 lists for each flux the mass isotopomers with the highest sensitivities obtained from the PLS model trained on 1000 samples. When this analysis was conducted including the 29 random variables in addition to the model-generated variables, the random variables were consistently lowest in this ranking. This clearly indicates that the PLS model was able to differentiate between informative variables and irrelevant data. The rankings in Table 3 generally reflect the expected importance of various isotopomers in determining flux. For example, the tracer infusion flux correlates with the M0 and M6 isotopomers of plasma glucose. It is clear that these isotopomers are directly affected by infusion of [U−13C]glucose tracer. However, an interesting point is that the gluconeogenic flux is mainly determined by hepatic phosphoenolpyruvate mass isotopomers M1, M3 and plasma glucose mass isotopomers M1, M2 and M3. A number of algebraic expressions have been proposed for the estimation of gluconeogenesis upon constant infusion of [U−13C]glucose (Tayek and Katz, 1997; Landau et al., 1998; Kelleher, 1999; Haymond and Sunehag, 2000). In these studies the gluconeogenic flux is calculated based on measurements of the accessible isotopomers, plasma glucose mass isotopomers M1, M2, M3, and M6, and plasma lactate mass isotopomers m0, m1, m2 and m3. Our results, indicating that hepatic phosphoenolpyruvate isotopomers are more sensitive predictors of gluconeogenesis than plasma lactate isotopomers may reflect the inherent limitations of gluconeogenic predictions models based on plasma lactate rather than a more direct intrahepatic precursor, phosphoenolpyruvate.
PLS and other linear models are widely used today for the analysis of gene expression and other “omics” data. Part of the attraction of these models is the ability to quantify the relationship between the independent and dependent variables. In contrast with linear models, the internal workings of ANN are typically hard to decipher. One cannot easily ascertain how they produce their results. Rule extraction in neural networks is a growing scientific field that deals with the opacity problem of neural networks by casting network weights in symbolic terms, and several types of methods have been proposed to achieve this goal (Ishikawa, 2000; Saito and Nakano, 2002). In this study, we did not attempt to extract physiological meaning from the trained neural networks. The relationship between isotopomers and fluxes, like many relationships in regulatory biology, is highly non-linear. The superior performance of ANN (figure 4) suggests that non-linear modeling approaches merit more attention as multivariate modeling efforts are developed for physiological processes.
The use of regression models described here represents a novel application of stable isotope labeling data. Our analysis with simulated data indicates that stable isotope tracer data may provide a rich resource for the estimation of physiologically relevant metabolic dependent variables. However, a regression model is not required to estimate a parameter such as gluconeogenesis, which can be defined by a mathematical relationship among the variables. Looking to the future we envision the application isotopic flux data regression models in situations where no known algebraic relationship exists between isotopic labeling data (X variables) and physiologically relevant Y variables. Consider Y variables such as insulin resistance, ketosis or hyperlipidemia. These variables are clearly dependent on metabolic fluxes but they are currently estimated by techniques not involving isotopes, for example, the glucose clamp for insulin resistance. If isotopic data is collected simultaneously with the standard measurement of these physiologically important dependent variables, a regression models could be constructed as described here. This regression model could be used to enhance our understanding of metabolic physiology. For example one could evaluate the true dimensionality of the relationship between isotopic labeling and the Y variables by extracting principal components as shown in figure 3. Additionally, by determining the sensitivities of the isotopomers to the Y variables one could find those isotopomers that are highly correlated to the physiological parameter as demonstrated in Table 3. These analyses may lead to new insights about the fluxes underlying the physiology. To move in this direction will require a sizable amount of data (100 data sets or more) as shown in figure 4. Just as databases of gene expression profiles are valued today because they may be mined with multivariate techniques, perhaps we are fast approaching a time when investigators will value databases of metabolomics (Jenkins et al., 2004) and metabolic isotopic labeling. Probing these databases with multivariate models may provide a new opportunity to supplement our understanding of the complexities regulating carbon fluxes in metabolic pathways. Our study using simulated data and a well-defined pathway serves as an example to show the potential of this approach.
Acknowledgments
We acknowledge the support of NIH Grant DK58533 and the DuPont-MIT Alliance.
Appendix A
Metabolic system and model equations
The first step in developing the simulation model is to write the list of reactions and corresponding carbon transformations in the metabolic system. Table A1 lists all reactions in figure 1, including corresponding carbon transformations represented using a letter code. For example, in reaction 3 glucose-6-phosphate (G6P) is split into two triose phosphate (TP) moieties. The first three carbon atoms of G6P (atoms abc) become the first TP moiety (atoms cba), i.e. the first C-atom of G6P becomes the third C-atom of TP, the second C-atom of G6P becomes the second C-atom of TP, and the third C-atom of G6P becomes the first C-atom of TP; the last three carbon atoms of G6P (atoms def) become the second TP moiety. Note that in our model we do not require the labeling of muscle pyruvate to depend only on the labeling of plasma glucose. Reaction 4 has been included to describe exchange of 13C with 12C in muscle metabolism. Multiple pathways may be responsible for exchange of isotopes, for example the pentose phosphate pathway and TCA cycle. The transketolase reaction (reaction 4) was used here to model the combined effect of all these pathways.
The metabolic system has several sources and sinks for labeled and unlabeled mass. The infusion of [U−13C]glucose is the input of tracer into the system (reaction 1), while naturally labeled mass enters the system from unlabeled sources of lactate (reaction 7), through the breakdown of glycogen (reaction 16), and through carboxylation in reaction 10. The exit points of mass are reactions 8, 18 and the TCA cycle reaction 11. Reversible reactions 4 and 12 are modeled as separate forward and backward fluxes. There are a total of 20 reactions in the metabolic model. Under steady state condition fluxes around 11 intermediate metabolites are balanced, which leads to 9 (=20−11) independent fluxes in the system (Table 1).
Table A1.
Carbon transformations for reactions in the metabolic system
| Reaction number | Reaction | Carbon transformationsa |
|---|---|---|
| 1 | [U−13C]Gluc > Gluc | abcdef > abcdef |
| 2 | Gluc > G6P[M] | abcdef > abcdef |
| 3 | G6P[M] > TP[M] + TP[M] | abcdef > cba + def |
| 4 | G6P[M] + TP[M] <> E4P[M] + R5P[M] | abcdef + ABC <> cdef + abABC |
| 5 | TP[M] > Pyr[M] | acb > abc |
| 6 | Pyr[M] > Lact | abc > abc |
| 7 | Lact[O] > Lact | abc > abc |
| 8 | Lact > other | abc > abc |
| 9 | Lact > Pyr[L] | abc > abc |
| 10 | Pyr[L] + CO2[L] > OAC[L] | abc + A > abcA |
| 11 | OAC[L] + AcCoA[L] > Fum[L] + 2 CO2 | abcd + AB > ABbc + a + d |
| 12 | Fum[L] <> OAC[L] | (1/2 abcd + 1/2 dcba) <> abcd |
| 13 | OAC[L] > PEP[L] + CO2 | abcd > abc + d |
| 14 | PEP[L] > Pyr[L] | abc > abc |
| 15 | PEP[L] + PEP[L] > G6P[L] | abc + ABC > cbaABC |
| 16 | Glycogen[L] > G6P[L] | abcdef > abcdef |
| 17 | G6P[L] > Gluc | abcdef > abcdef |
| 18 | Gluc > other | abcdef > abcdef |
aFor each compound carbon atoms are identified using lower case letters to represent successive carbon atoms of each compound. Uppercase letters represent a second compound in the reaction. Because fumarate is a rotationally symmetric molecule no distinction can be made between carbon atoms 1 and 4, and carbon atoms 2 and 3.
The mathematical model used for isotopic simulations consists of the complete set of isotopomer balances, which were derived using a matrix based method. First, atom mapping matrices (AMMs) were constructed for each reaction as described by Zupke and Stephanopoulos (Zupke and Stephanopoulos, 1994), followed by the construction of corresponding isotopomer mapping matrices (IMMs) using the algorithm by Schmidt et al. (Schmidt et al., 1997). IMMs describe the transformation of isotopomers of one molecule into another. For example, IMMLact>Pyr is the transformation matrix that describes how the isotopomers of lactate are transformed into isotopomers of pyruvate. Isotopomer distribution vectors (IDV) collect fractional abundances of all isotopomers for the metabolites in the system. The order of isotopomers in IDVs matches the order of isotopomers in IMMs. Conventionally, ordering based on the binomial description of labeling patterns of isotopomers is applied. It has been previously described how IMMs and IDVs can be applied to derive all isotopomer balances for a given metabolic system (Schmidt et al., 1997). The complete set of isotopomer balances for our network is given below. Each expression represents the set of isotopomer balances for a particular metabolite in the system. For example, the first expression represents 64 isotopomer balance equations for plasma glucose. If written out in full, the first of these 64 expressions is the balance equation for the unlabeled plasma glucose isotopomer, shown above in equation (1).
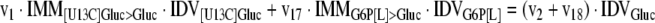 |
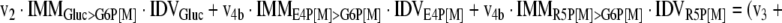 |
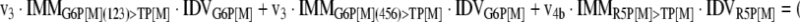 |
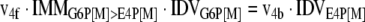 |
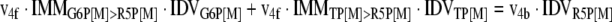 |
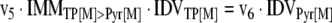 |
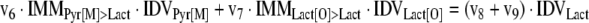 |
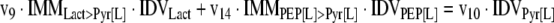 |
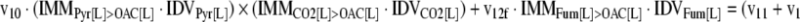 |
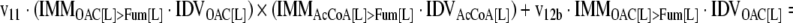 |
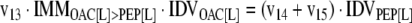 |
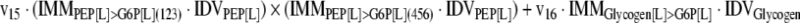 |
A consequence of the balances and the matrix representation is that the entire model describing all isotopomers and fluxes is represented simply as the above set of 12 relationships.
Footnotes
We acknowledge the support of NIH Grant DK58533 and the DuPont-MIT Alliance.
References
- Benigni R., Giuliani A. (1994) Quantitative modeling and biology: the multivariate approach. Am. J. Physiol 266:R1697–R1704 [DOI] [PubMed]
- Bishop, C.M. (1996). Neural Networks for Pattern Recognition. Oxford University Press.
- Causton D.R. (1987). A Biologist’s Advanced Mathematics. London, Allen & Unwin
- Chatfield C., Collins A.J. (1981). Introduction to Multivariate Analysis. London, Chapman & Hall
- Dillon W.R., Goldstein M. (1984) Multivariate Analysis Methods and Applications. Wiley, New York
- Eddy C.V., Flanigan M., Arnold M.A. (2003) Near-infrared spectroscopic measurement of urea in dialysate samples collected during hemodialysis treatments. Appl. Spectrosc. 57:1230–1235 [DOI] [PubMed]
- El-Deredy W., Ashmore S.M., Branston N.M., Darling J.L., Williams S.R., Thomas D.G. (1997). Pretreatment prediction of the chemotherapeutic response of human glioma cell cultures using nuclear magnetic resonance spectroscopy and artificial neural networks. Cancer Res. 57:4196–4199 [PubMed]
- Geladi P., Kowalski B.R. (1986). Partial least-squares regression: a tutorial. Anal. Chim. Acta 185:1–17 [DOI]
- German J.B., Roberts M.A., Fay L., Watkins S.M. (2002). Metabolomics and individual metabolic assessment: the next great challenge for nutrition. J. Nutr. 132:2486–2487 [DOI] [PubMed]
- Haykin, S.S. (1998) Neural Networks: A Comprehensive Foundation. Prentice Hall.
- Haymond M.W., Sunehag A.L. (2000). The reciprocal pool model for the measurement of gluconeogenesis by use of [U-(13)C]glucose. Am. J. Physiol. Endocrinol. Metab. 278:E140–E145 [DOI] [PubMed]
- Irudayaraj J., Tewari J. (2003) Simultaneous monitoring of organic acids and sugars in fresh and processed apple juice by Fourier transform infrared-attenuated total reflection spectroscopy. Appl. Spectrosc. 57:1599–1604 [DOI] [PubMed]
- Ishikawa M. (2000). Rule extraction by successive regularization. Neural Netw. 13:1171–1183 [DOI] [PubMed]
- Jenkins H., Hardy N., Beckmann M., Draper J., Smith A.R., Taylor J., Fiehn O., Goodacre R., Bino R.J., Hall R., Kopka J., Lane G.A., Lange B.M., Liu J.R., Mendes P., Nikolau B.J., Oliver S.G., Paton N.W., Rhee S., Roessner-Tunali U., Saito K., Smedsgaard J., Sumner L.W., Wang T., Walsh S., Wurtele E.S., Kell D.B. (2004) A proposed framework for the description of plant metabolomics experiments and their results. Nat. Biotechnol. 22:1601–1606 [DOI] [PubMed]
- Kelleher J.K. (1999) Estimating gluconeogenesis with [U-13C]glucose: molecular condensation requires a molecular approach. Am. J. Physiol. 277:E395–E400 [DOI] [PubMed]
- Landau B.R., Wahren J., Ekberg K., Previs S.F., Yang D., Brunengraber H. (1998) Limitations in estimating gluconeogenesis and Cori cycling from mass isotopomer distributions using [U-13C6]glucose. Am. J. Physiol. 274:t-61 [DOI] [PubMed]
- Manly B.F.J. (1994) Multivariate Statistical Methods: A Primer. London, Chapman & Hall
- Martens H., Naes T. (1989). Multivariate Calibration. Chichester, John Wiley
- Mendes P., Kell D.B. (1996). On the analysis of the inverse problem of metabolic pathways using artificial neural networks. Biosystems 38:15–28 [DOI] [PubMed]
- Misra J., Schmitt W., Hwang D., Hsiao L.L., Gullans S., Stephanopoulos G., Stephanopoulos G. (2002) Interactive exploration of microarray gene expression patterns in a reduced dimensional space. Genome Res. 12:1112–1120 [DOI] [PMC free article] [PubMed]
- Raamsdonk L.M., Teusink B., Broadhurst D., Zhang N., Hayes A., Walsh M.C., Berden J.A., Brindle K.M., Kell D.B., Rowland J.J., Westerhoff H.V., van Dam K., Oliver S.G. (2001). A functional genomics strategy that uses metabolome data to reveal the phenotype of silent mutations. Nat. Biotechnol. 19:45–50 [DOI] [PubMed]
- Saito K., Nakano R. (2002) Extracting regression rules from neural networks. Neural Netw. 15:1279–1288 [DOI] [PubMed]
- Schmidt K., Carlsen M., Nielsen J., Villadsen J. (1997) Modeling isotopomer distributions in biochemical networks using isotopomer mapping matrices. Biotechnol. Bioeng. 55:831–840 [DOI] [PubMed]
- Stephanopoulos G., Hwang D., Schmitt W.A., Misra J., Stephanopoulos G. (2002) Mapping physiological states from microarray expression measurements. Bioinformatics 18:1054–1063 [DOI] [PubMed]
- Tabachnick B.G., Fidell L.S. (2001) Using Multivariate Statistics. Boston, Allyn and Bacon
- Tayek J.A., Katz J. (1997) Glucose production, recycling, Cori cycle, and gluconeogenesis in humans: relationship to serum cortisol. Am. J. Physiol. 272:E476–E484 [DOI] [PubMed]
- White H. (1992) Artificial Neural Networks: Approximation and Learning Theory. Oxford, Blackwell
- Wiechert W., Mollney M., Isermann N., Wurzel M., De Graaf A.A. (1999) Bidirectional reaction steps in metabolic networks: III. Explicit solution and analysis of isotopomer labeling systems. Biotechnol. Bioeng. 66:69–85 [DOI] [PubMed]
- Zupke C., Stephanopoulos G. (1994) Modeling of isotope distributions and intracellular fluxes in metabolic networks using atom mapping matrices. Biotechnol. Prog. 10:489–498 [DOI]