

Abstract
Cyclic adenosine 3′, 5′ monophosphate (cAMP) is a ubiquitous mediator of intracellular signalling events. It principally acts through stimulation of cAMP-dependent protein kinases (PKA)1, 2 but also activates certain ion-channels and guanine nucleotide exchange factors (Epacs)3. Metabolism of cAMP is catalyzed by phosphodiesterases (PDEs)4, 5. A cAMP-responsive signalling complex maintained by the muscle specific A-kinase anchoring protein (mAKAP) that includes PKA, PDE4D3 and Epac1 was identified. These intermolecular interactions facilitate the dissemination of distinct cAMP signals through each effector protein. Anchored PKA stimulates PDE4D3 to reduce local cAMP concentrations whereas an mAKAP-associated ERK5 module suppresses PDE4D3. PDE4D3 also functions as an adapter protein that recruits Epac1, an exchange factor for the small GTPase Rap-1 to enable cAMP-dependent attenuation of ERK5. Pharmacological and molecular manipulation of the mAKAP complex show that anchored ERK5 can induce cardiomyocyte hypertrophy. Thus, two coupled cAMP-dependent feedback loops are coordinated within the context of the mAKAP complex, suggesting that local control of cAMP signalling by AKAPs is more intricate than previously appreciated.
Keywords: cAMP, Phosphodiesterases (PDEs), Epac1, cAMP-dependent protein kinase (PKA), A-Kinase Anchoring Protein (AKAP), heart
Spatiotemporal control of cAMP flux requires the concerted action of adenylyl cyclases that synthesize cAMP and phosphodiesterases that locally metabolize it into 5′AMP4. PDE binding proteins target distinct isozymes to specific subcellular sites5–7. In cardiomyocytes, mAKAP assembles a negative feedback loop containing PDE4D3 and PKA at the perinuclear membrane8. PKA phosphorylation of serine 13 on PDE4D3 augments mAKAP binding9, whereas phosphorylation of serine 54 enhances catalysis to favour cAMP metabolism10. This configuration may generate local fluctuations in cAMP and pulses of compartmentalized PKA activity11.
Fluorescent reporters of PKA activity were used to test this hypothesis in living cells12. AKAR2 is a chimeric protein consisting of cyan fluorescent protein (CFP), a consensus PKA substrate sequence, a forkhead associated (FHA) domain that binds phosphoamino acids, and the yellow fluorescent protein citrine (Figure 1A). PKA phosphorylation of the consensus site engages the FHA domain, permitting fluorescence resonance energy transfer (FRET) between the fluorescent moieties. Two modified AKAR2 reporters were generated: AKAR-PKA that recruits PKA via a consensus anchoring sequence (Figure 1B) and AKAR-PKA-PDE that recruits PKA and tethers PDE4D3 via a binding site derived from mAKAP (Figure 1C). HeLa cells expressing AKAP-PKA exhibited a rapid and sustained elevation of FRET upon cAMP stimulation (Figure 1B, lower panel &1D, red, n=10, Supplementary Video 1). In contrast, cells expressing AKAR-PKA-PDE exhibited a less robust and transient FRET response (Figure 1C, lower panel & 1D, green, n=13, Supplementary Video 2). The PKA antagonist H89 blocked all cAMP dependent FRET changes (Figure 1D). These dynamic changes in PKA activity were unaffected by the PDE3 inhibitor milrinone (10μM), however, suppression of PKA activity was prevented when the same cells received the PDE4 inhibitor rolipram (1μM) (Figure 1E, n=10, Supplementary Video 3). Thus, recruitment of a PDE4 terminates activation of anchored PKA in the context of the AKAR-PKA-PDE reporter.
Figure 1. Bi-directional control of the mAKAP associated PDE4D3 activity.
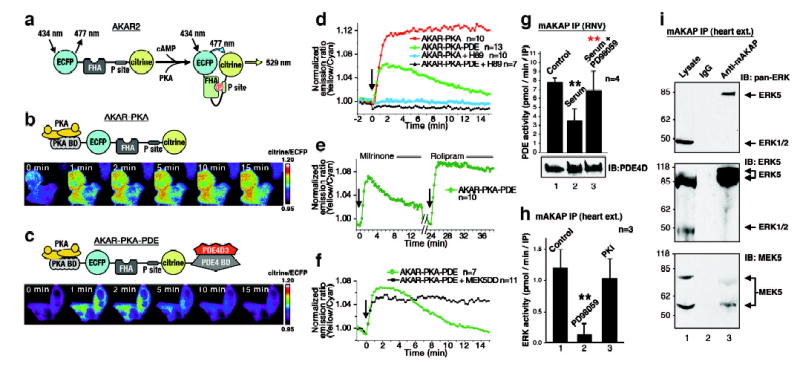
A) Diagram depicting the modular composition and action of AKAR2. B) Diagram of AKAR2-PKA. Bottom) Pseudo-coloured images of FRET changes in HeLa cells stimulated with cAMP over a 15 min time course. C) Diagram of AKAR2-PKA-PDE. Bottom) Pseudo-coloured images of FRET changes in HeLa cells stimulated with cAMP over a 15 min time course. D) Amalgamated FRET measurements for AKAR-PKA (Red, n=10), AKAR-PKA-PDE (Green, n=13), AKAR-PKA + H89 (Blue, n=10) and AKAR-PKA-PDE + H89 (Black, n=7) for 15 minutes after cAMP stimulation with forskolin (arrow). E) Amalgamated FRET traces (n=10) using the AKAR-PKA-PDE reporter after application of the PDE3 inhibitor milrinone (0–12 min) and the PDE4 inhibitor rolipram (24–36 min). Stimulation with cAMP was at times 0 and 24 min (arrows). F) FRET measurements from HeLa cells expressing the AKAR-PKA-PDE reporter in the presence (black, n=11) or absence (green, n=7) of a dominant active MEK5 for 15 minutes after cAMP stimulation with forskolin (arrow). G) Phosphodiesterase activity (n=4, error bars show S.E.M.) in mAKAP immune complexes isolated from RNVs. Treatment conditions are indicated above each column. H) ERK activity in the mAKAP complex from heart extracts (n=3, error bars show S.E.M.). Treatments with kinase inhibitors are indicated. I) Co-precipitated ERK was detected by immunoblot using (top) pan-ERK, (middle) ERK5 specific and (bottom) MEK5 specific antibodies. P values <0.01 (**) are indicated relative to control (black) and sample (red).
ERK kinases phosphorylate PDE4D3 on serine 579 to suppress phosphodiesterase activity13. Therefore, mAKAP might incorporate an ERK kinase module to counterbalance the anchored PDE4D3. Accordingly, sustained elevation of FRET upon cAMP stimulation was observed in HeLa cells expressing AKAR-PKA-PDE plus constitutively active MEK, an upstream kinase for ERKs (Figure 1F, black, n=11). Thus ERK activity suppresses anchored phosphodiesterase activity. Complementary studies were performed in cultured rat neonatal ventriculocytes (RNVs). Treatment with 10% serum activated ERK prior to immunoprecipitation of the mAKAP complex. The associated PDE activity was attenuated 52 ± 9 % (n = 4, p<0.0002) compared to unstimulated controls (Figure 1G, top, column 2). The MEK inhibitor PD98059 (20μM) blocked this effect (Figure 1G, top, column 3, p<0.002). Equivalent amounts of PDE4D3 co-purified with the anchoring protein under each experimental condition suggesting that the reduction in phosphodiesterase activity is not a consequence of displacing the enzyme from the mAKAP complex (Figure 1G, bottom). ERK activity was also measured in mAKAP immune complexes (Figure 1H, column 1), and an increase of 3.9 ± 0.7 fold (n=3, p<0.0007) over IgG control was observed (data not shown). PD98059 (2μM) blocked the mAKAP-associated ERK activity (Figure 1H, column 2, p<0.0001). The PKA inhibitor PKI 5–24 peptide (50nM) had no significant effect on ERK activity (Figure 1H, column 3). Thus, mAKAP-associated ERK activity suppresses anchored PDE4D3 activity.
To determine which ERK family members may regulate mAKAP associated PDE4D3, we performed Western analysis on mAKAP immunoprecipitates from heart extract. An 80 kDa protein that co-purified with mAKAP was detected with an antibody that recognizes all ERK family members and with an ERK5 specific antibody (Figure 1I, top & middle, lane 3). ERK5 was enriched in mAKAP immune complexes rather than the 45 kDa ERK 1/2 (Figure 1I, top, lane 1). The upstream kinase MEK514, 15 also co-precipitated with mAKAP (Figure 1I, bottom). Immunofluorescence detection of mAKAP and ERK5 at the perinuclear membranes of hypertrophic RNVs confirmed the in situ assembly of this signaling complex (Supplemental Figure S1)16.
It was important to know if anchored PKA and ERK5 facilitate the bi-directional regulation of the mAKAP-associated phosphodiesterase and, hence, control localized cAMP metabolism. GST pulldowns from heart extracts established the formation of a mAKAP-PDE4D3-ERK5 ternary complex (Supplemental Figure S2). Cellular binding studies demonstrated that PDE4D3 acts as an adapter protein linking ERK5 to the mAKAP complex. This was confirmed when a mutant PDE4D3form unable to bind ERKs (KIM/FQF double mutant 17) could not recruit ERK5 to mAKAP (Supplemental Figures S3–S5). Thus, ERK5 is well positioned to suppress PDE4D3 activity and influence localized accumulation of cAMP.
cAMP modulates ERK signaling in a context specific manner18. In neuronally derived cells lines cAMP activates ERK, yet it inhibits this kinase in certain fibroblasts and kidney cells19, 20. Cross-talk between mAKAP anchored PKA and ERK5 was evaluated in RNVs. ERK was activated with 10% serum prior to immunoprecipitation of the mAKAP complex. The mAKAP-associated ERK activity was increased 2.9 ± 0.6 fold (n=3, p<0.0003) over controls (Figure 2A, column 2). However, pretreatment with forskolin (20μM) to elevate intracellular cAMP prevented the serum-dependent activation of the mAKAP-associated ERK5 pool (89 ± 8%, n=3, p<0.0004; Figure 2A, column 3). Surprisingly, the PKA antagonist H89 (10μM) did not overcome the cAMP dependent suppression of ERK5 activity (Figure 2A, column 4). Similar results were obtained using two other well-characterized PKA inhibitors, KT5720 (1μM) and Rp-cAMPs (1mM; Figure 2A, columns 5 & 6). Thus, the cAMP-dependent block of ERK5 activity was independent of PKA. Controls demonstrate that this effect was not a consequence of displacing the ERK5 from the mAKAP complex (Figure 2A, bottom). Therefore, another cAMP effector within the mAKAP complex may function to suppress ERK5 activation.
Figure 2. Epac1 suppresses mAKAP associated ERK5 activity.
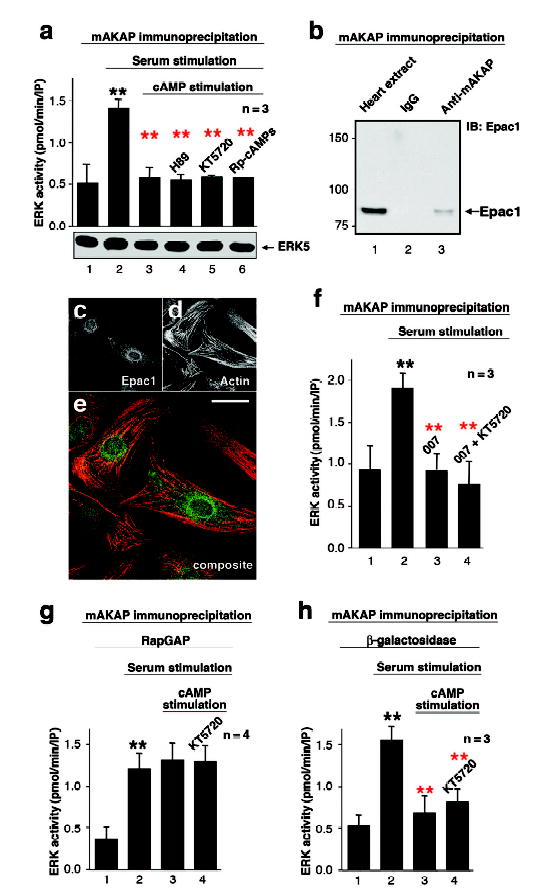
A) Serum stimulated mAKAP associated ERK5 activity (n=3, error bars show S.E.M.) in parallel cultures pretreated with forskolin to elevate cAMP (columns 2&3) or in the presence of the PKA inhibitors H89, KT5720, or Rp-cAMPs (columns 4–6). The amount of ERK5 was detected in each sample. B) Immunoblot detection of Epac1 in mAKAP immune complexes from rat heart extracts. C–E) Fluorescent staining of hypertrophic RNV with antibody for Epac1 (C) and Alexa 568 phalliodin for the actin cytoskeleton (D). Composite image (E) shows the distribution of Epac1 (green) and actin (red, scale bar =20μm). F) The mAKAP complex was immunoprecipitated from cultured RNV following serum stimulation to activate ERK. Parallel cultures were pretreated with either the Epac-selective activator 007 or KT5720 prior to serum stimulation (n=3, error bars show S.E.M.). G & H) Serum stimulated mAKAP associated ERK activity in cells expressing of constitutively active RapGAP (J, n=4, error bars show S.E.M.) or control β-galactosidase (K, n=3, error bars show S.E.M.). Stimulation of intracellular cAMP or treatment with the kinase inhibitor KT5720 is indicated. P values <0.01 (**) are indicated relative to control (black) and sample (red).
One candidate is Epac1 (exchange protein directly activated by cAMP), a cAMP dependent guanine nucleotide exchange factor for the small G-protein Rap1 21, 22. Epac1 co-purified with mAKAP from heart extracts, but was not detected in the IgG control (Figures 2B). Immunofluorescence techniques detected Epac1 at the perinuclear membranes of hypertrophic RNVs where mAKAP is also found (Figure 2C–E).
The analog 8-CPT-2′-O-Me-cAMP was used to assess if Epac1 is the cAMP effector that inhibits mAKAP-associated ERK5. This compound, also called 007, is a potent and selective activator for Epacs with up to 300-fold selectivity over the R subunits of PKA23. Treatment of RNVs with 007 alone resulted in the inhibition of ERK5 activity in mAKAP immune complexes (Figure 2F, column 3, n=3, p<0.0004). Importantly, KT5720 did not augment 007’s effect (Figure 2F, column 4), emphasizing that PKA does not have an additive effect on cAMP-dependent suppression of ERK5. Assorted biochemical experiments demonstrated that PDE4D3 alone recruits Epac1 to the mAKAP complex (Supplemental data S6–S8). This permits firm control of cAMP concentrations surrounding Epac1, thereby regulating its guanine nucleotide exchange activity.
Epac1 is an effector protein for Rap1, a member of the Ras family of small G-proteins21, 22. Rap1 signaling is attenuated by the GTPase activating protein, RapGAP24. Adenoviral expression of the constitutively active RapGAP in heart cells blocked Rap1 and, consequently, cAMP-mediated suppression of ERK5 activity (Figure 2G, columns 1–3, n=4, p<0.007). Treatment with the PKA inhibitor KT5720 did not reverse this effect (Figure 2G, column 4). Other controls demonstrated that adenoviral expression of β-galactosidase did not affect cAMP dependent suppression of the anchored ERK5 activity (Figure 2H, n=3, p<0.009). Collectively, data in figure 2 suggest that cAMP-dependent activation of Epac1 mobilizes a Rap1 signaling pathway, leading to inhibition of ERK5 in the mAKAP complex.
Cytokines, including leukemia inhibitory factor (LIF) activate ERK5 in RNVs to induce eccentric cardiac hypertrophy, an increase in cell size and length25. LIF treatment for 24 h enhanced RNV cell size by 1.6 fold over controls (Figure 3A–C, n=11 independent experiments). This effect was blocked when ERKs were inhibited by PD98059 or if Epac1 was activated by 007, and it was partially blocked upon PDE4 inhibition with rolipram (Figure 3C, columns 3–5). Plasmid-based RNA interference of mAKAP also suppressed LIF mediated changes in RNV size compared to controls (Figure 3D, & Supplementary Figure S9). Importantly, the LIF response was rescued in RNVs expressing an mAKAP form refractory to the shRNA (Figure 3D, column 3). Finally, displacement of mAKAP from the perinuclear membrane with an inducible, competing fragment (residues 585–1286) blocked LIF-induced RNV growth (Figure 3E & Supplementary Figure S10). Collectively, these data imply that mAKAP organizes a cAMP responsive network of PDE4D3, Epac1 and ERK5 that modulate cardiomyocyte size. Similar results were observed when ANF expression was monitored as an independent index for hypertrophy (Supplemental Figures S11 & S12).
Figure 3. The mAKAP complex facilitates cytokine induced cardiac hypertrophy.
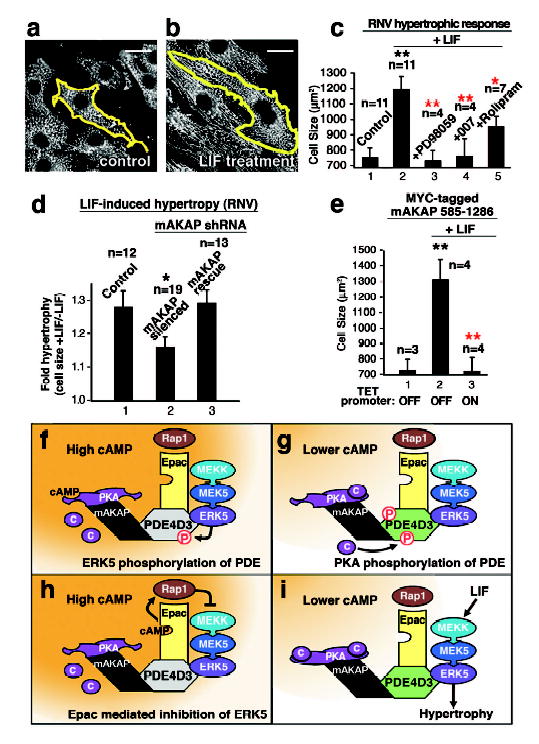
A–C) Leukemia inhibitory factor (LIF) induced changes in RNV size. The outline of RNV from control (A) and LIF treated (B) samples are presented, scale bar =20μm. C) Quantitation of cell size (μm2) in control (1) and LIF treated RNV (2–5). Pharmacological manipulation of ERK5 (PD98095), Epac1 (007) or PDE4 (rolipram) activity is indicated (error bars show S.E.M.). The number of experiments is indicated above each column. D) Expression of mAKAP was suppressed by RNA interference. Quantitation of LIF induced hypertrophy (cell size +LIF/−LIF) in control (1), mAKAP silenced (2) and RNV rescued with an mAKAP form resistant to the shRNA (3, error bars show S.E.M.). E) Displacement of mAKAP from the nuclear membrane was achieved by overexpression of the mAKAP targeting domain fragment using the TET OFF inducible promoter. Quantitation of cell size (μm2) from control (1), from LIF-stimulated RNV controls (2, TET promoter OFF), and from LIF-stimulated cells expressing the mAKAP 585–1286 fragment (3, TET promoter ON, error bars show S.E.M.). F–I) Schematics depicting the major findings of this study. F) ERK5 phosphorylation of PDE4D3 shuts down cAMP metabolism. G) PKA phosphorylation of PDE4D3 enhances cAMP metabolism. H) Activation of Epac mobilizes Rap1 to suppress ERK5 activation, and I) low cAMP represses the Epac mediated block of ERK5 allowing cardiac hypertrophy. P values <0.01 (**) and p values <0.05 (*) are indicated relative to control (black) and sample (red).
Beta-adrenergic agonists that elevate intracellular cAMP control the strength, duration, and frequency of heart contraction. Elegant cell imaging experiments in cardiomyocytes revealed transient microdomains of cAMP where PKA becomes active26, 27. Our FRET data show that co-localization of PKA and a phosphodiesterase generate localized pulses of cAMP. Thus, a variety of cAMP responsive events of differing durations, and which are responsive to different thresholds of cAMP, can emanate from the same microdomain. This is particularly relevant in mAKAP signaling complexes where three functionally distinct, cAMP-dependent enzymes (PKA, PDE4D3 and Epac1) reside. PKA responds to nanomolar cAMP and would become active early in a second messenger response. However, PDE4D3 (Km 1–4μM) and Epac1 (Kd 4μM) activities would only commence when cAMP accumulated to micromolar levels4. Conversely, inactivation of PDE4D3 and Epac1 would precede PKA holoenzyme reformation as cAMP levels decline. Furthermore, Epac1 suppresses mAKAP-associated ERK5 activity in a PKA-independent manner. This is consistent with the notion that Epacs provide the cAMP-dependent link to ERK signalling events through Rap122, 23, 28.
In conclusion, mAKAP and PDE4D3 create an integrated and internally regulated signaling network. mAKAP anchors PKA and directs the cellular localization of the complex while PDE4D3 serves as the adapter protein for Epac1 and ERK5. The key regulatory enzyme is PDE4D3. ERK phosphorylation of PDE4D3 decreases the phosphodiesterase activity, thereby favoring local accumulation of cAMP and subsequent PKA activation (Figure 3F) and Epac1 activation (Figure 3H). Conversely, PKA phosphorylation of PDE4D3 increases its affinity for mAKAP9 and its Vmax for cAMP10 to decrease localized cAMP levels (Figure 3G). This ultimately sustains ERK5 activity by repressing Epac1 mediated inhibition of the kinase (Figure 3I). This novel configuration creates a cohesive unit where transduction enzymes from distinct pathways are spatially organized to exploit the temporal constraints of cAMP action.
Methods
Antibodies
Primary antibodies were: monoclonal pan-ERK (Transduction Labs), monoclonal MEK5 (Transduction Labs); monoclonal pan-PDE4D (ICOS); rabbit ERK5 (StressGen); monoclonal 9E10-Myc (Upstate); rabbit mAKAP (VO54); rabbit Epac1 (Santa Cruz). Mouse α-actinin (Sigma), rabbit anti-rat ANF (USB). Secondary antibodies were: donkey HRP-conjugated (Jackson ImmunoResearch) and Alexa conjugated (Molecular Probes).
AKAR-PKA & AKAR-PKA-PDE
Plasmids (pcDNA4) encoding AKAR-PKA or AKAR-PKA-PDE were derived from the original AKAR2 construct provided by Roger Tsien. A DNA fragment encoding the consensus RII binding peptide from AKAPis following a Kozak sequence was generated by PCR. This fragment was ligated at the 5′ end of AKAR2 to generate AKAR-PKA. The AKAR-PKA-PDE reporter was constructed by ligating a fragment encoding residues 1286–1831 of mAKAP to the 3′ end of AKAR-PKA coding region.
Cell Culture
Preparation of primary RNVs and treatment to induce hypertrophy was as previously described16.
Live Cell Imaging
HeLa cells grown on glass coverslips were transfected with vectors encoding AKAR-PKA or AKAR-PKA-PDE using Lipofectamine 2000. After incubation at 37°C for 18–48 h, cells were washed twice with Hank’s Balanced Salt Solution, mounted in a Ludin chamber (Life Imaging Services) and imaged using the Leica AS MDW workstation. Images were acquired on a Leica DM IRE2 microscope equipped with a CoolSNAP-HQ charge-coupled device camera (Roper Photometrics). This device was equipped with a Dual-View module (Optical Insights), D465/30 and HQ535/30 emission filters (Chroma) and a 505dcxr dichroic mirror (Chroma). CFP and YFP images were acquired simultaneously, aligned, background-corrected, and analyzed using Leica’s FRET Applicator software. Exposure time was from 250–1000 ms, and images were taken every 15 s. Treatment with PKA inhibitor H89 or isoform-specific PDE inhibitors preceded cAMP stimulation with forskolin by 10 min.
Expression constructs
Plamids encoding wildtype PDE4D3 and mutant PDE4D3 (K455A/K456A/F597A/Q598A/F599A) forms were from Miles Houslay (University of Glasgow). pcDNA3 expression vectors encoding Myc-tagged ERK5 and Epac1 and a RAP-GAP adenovirus were provided by Philip Stork (Vollum Institute). For GST -PDE4D3 and GST-ERK5, cDNAs were subcloned into PGEX-4T2. shRNA vectors incorporated double-stranded hairpin oligonucleotides based upon rat mAKAP mRNA sequence (NCBI GI:5070430, base pairs 7210–7228) under the direction of the U6 promoter: mAKAP shRNA (sense strand), 5′-GACGAACCTTCCTTCCGAATTCAAGAGATTCGGAAGGAAGGTTCGTCTTTTT-3′; control shRNA (sense strand) – 5′ GACGAACCCCTGTTCCGAATTCAAGAGATTCGGAACAGGGGTTCGTCTTTTT-3′. Underlined base pairs in the control shRNA are different from the wildtype (WT) mAKAP shRNA. Rescue experiments were performed with a full-length MYC-His rat mAKAP, lacking the shRNA recognition sequence that resides in the 3′ untranslated region of the mAKAP message.
Enzyme Assays
PDE activity was measured by the method of Beavo29, and PKA activity was measured as previously described8. ERK assays were performed on immunoprecipitates from heart extract and RNVs. Immunoprecipitates were washed 3X in HSE buffer before the addition of inhibitors, 25 μl H2O, ERK assay buffer 25 μl [80 mM HEPES pH 7.4, 80 mM MgCl2, 0.2 μM ATP, 2 mM sodium orthovanadate, 20 mM NaF, 1 μg tyrosine hydroxylase peptide (BioSource), and 10 μCi γ-32-ATP]. After 30 min incubation at 30°C, the reaction mixture was spotted onto phosphocellulose strips and extensively washed in 75mM phosphoric acid. Filters were washed in 95% ethanol, air-dried and counted by liquid scintillation.
RNV were serum starved for 16 h before the addition of either 20 μM PD98059 for 1 hour, the PKA inhibitors H-89 (10 μM), KT5720 (1 μM), Rp-cAMPs (1 mM), followed by the addition of 20 μM forskolin and 75 μM IBMX for 20 min. In the case of 8-CPT-2′-O-Me-cAMP “007” (50 μM, the Epac-selective cAMP analog) cells were not exposed to forskolin. Cells were stimulated with 10% FCS for 10 min, lysed in 1 ml HSE buffer containing 50 mM NaF, 1 mM sodium orthovanadate, 0.1 μM okadaic acid, 0.1 μM cyclosporine, and immunoprecipitations were performed as described above. H9C2 heart cells were incubated with adenoviral constructs of Rap1GAP1 or β-galactosidase at a concentration of 50 MOI for 48 hours. The cells were treated as described above before the addition of forskolin/IBMX and prior to serum-stimulation.
Immunocytochemistry and Microscopy
Fixation and staining of RNVs was as previously described8. To determine the effect of shRNA expression on cellular hypertrophy, myocytes were co-transfected with pTRE-U6 mAKAP shRNA or control shRNA plasmid, pEGFPN3 green fluorescent protein (GFP)-expression plasmid (Clontech) and either pBluescript carrier DNA or pcDNA3.1 MYC-His mAKAP expression vector for rescue experiments. Cells were stained with primary and Alexa conjugated specific-secondary antibodies. Anti-α-actinin stained sarcomeric Z-disks to distinguish myocytes from contaminating fibroblasts. Images were acquired using a Leica DMRA Fluorescent Microscope. Cell size was measured using the method of Kodama30. Data are presented as the average mean cell area +/− S.E.M. P-values were determined using a two-tailed Student’s t-Test.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH (DK54441, J.D.S.; HL075398, M.S.K.) and AHA (0435452N K.L.D-K.). The authors wish to thank Nicki Mayer, Daniel Bleckinger, and Robert Mouton for excellent technical assistance, and Roger Tsien, Miles Houslay Jack E. Dixon and Philip Stork for expression constructs.
Footnotes
Supplementary Information accompanies the paper on www.nature.com/nature.
Competing Interests statement The authors declare that there are no competing financial interests.
References
- 1.Walsh DA, Perkins JP, Krebs EG. An adenosine 3′,5′-monophosphate-dependent protein kinase from rabbit skeletal muscle. J Biol Chem. 1968;243:3763–3765. [PubMed] [Google Scholar]
- 2.Su Y, et al. Regulatory subunit of protein kinase A: structure of deletion mutant with cAMP binding proteins. Science. 1995;269:807–813. doi: 10.1126/science.7638597. [DOI] [PubMed] [Google Scholar]
- 3.Bos JL. Epac: a new cAMP target and new avenues in cAMP research. Nat Rev Mol Cell Biol. 2003;4:733–8. doi: 10.1038/nrm1197. [DOI] [PubMed] [Google Scholar]
- 4.Beavo JA, Brunton LL. Cyclic nucleotide research -- still expanding after half a century. Nat Rev Mol Cell Biol. 2002;3:710–8. doi: 10.1038/nrm911. [DOI] [PubMed] [Google Scholar]
- 5.Houslay MD, Adams DR. PDE4 cAMP phosphodiesterases: modular enzymes that orchestrate signalling cross-talk, desensitization and compartmentalization. Biochem J. 2003;370:1–18. doi: 10.1042/BJ20021698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perry SJ, et al. Targeting of cyclic AMP degradation to β2-adrenergic receptors by β-arrestins. Science. 2002;298:834–836. doi: 10.1126/science.1074683. [DOI] [PubMed] [Google Scholar]
- 7.Verde I, et al. Myomegalin is a novel protein of the golgi/centrosome that interacts with a cyclic nucleotide phosphodiesterase. J Biol Chem. 2001;276:11189–98. doi: 10.1074/jbc.M006546200. [DOI] [PubMed] [Google Scholar]
- 8.Dodge KL, et al. mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. EMBO J. 2001;20:1921–1930. doi: 10.1093/emboj/20.8.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carlisle Michel JJ, et al. PKA phosphorylation of PDE4D3 facilitates recruitment of the mAKAP signaling complex. Biochem J. 2004;381:587–92. doi: 10.1042/BJ20040846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sette C, Conti M. Phosphorylation and activation of a cAMP-specific phosphodiesterase by the cAMP-dependent protein kinase. J Biol Chem. 1996;271:16526–16534. doi: 10.1074/jbc.271.28.16526. [DOI] [PubMed] [Google Scholar]
- 11.Wong W, Scott JD. AKAP Signalling complexes: Focal points in space and time. Nature Reviews Molecular Cell biology. 2004;5:959–71. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- 12.Zhang J, Ma Y, Taylor SS, Tsien RY. Genetically encoded reporters of protein kinase A activity reveal impact of substrate tethering. Proc Natl Acad Sci U S A. 2001;98:14997–5002. doi: 10.1073/pnas.211566798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffmann R, Baillie GS, MacKenzie SJ, Yarwood SJ, Houslay MD. The MAP kinase ERK2 inhibits the cyclic AMP-specific phosphodiesterase HSPDE4D3 by phosphorylating it at Ser579. Embo J. 1999;18:893–903. doi: 10.1093/emboj/18.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou G, Bao ZQ, Dixon JE. Components of a new human protein kinase signal transduction pathway. J Biol Chem. 1995;270:12665–9. doi: 10.1074/jbc.270.21.12665. [DOI] [PubMed] [Google Scholar]
- 15.English JM, Vanderbilt CA, Xu S, Marcus S, Cobb MH. Isolation of MEK5 and differential expression of alternatively spliced forms. J Biol Chem. 1995;270:28897–902. doi: 10.1074/jbc.270.48.28897. [DOI] [PubMed] [Google Scholar]
- 16.Kapiloff MS, Schillace RV, Westphal AM, Scott J. D mAKAP: an A-kinase anchoring protein targeted to the nuclear membrane of differentiated myocytes. J Cell Sci. 1999;112:2725–36. doi: 10.1242/jcs.112.16.2725. [DOI] [PubMed] [Google Scholar]
- 17.MacKenzie SJ, Baillie GS, McPhee I, Bolger GB, Houslay MD. ERK2 mitogen-activated protein kinase binding, phosphorylation, and regulation of the PDE4D cAMP-specific phosphodiesterases. The involvement of COOH-terminal docking sites and NH2-terminal UCR regions. J Biol Chem. 2000;275:16609–17. doi: 10.1074/jbc.275.22.16609. [DOI] [PubMed] [Google Scholar]
- 18.Pearson GW, Cobb MH. Cell Condition-dependent Regulation of ERK5 by cAMP. J Biol Chem. 2002;277:48094–8. doi: 10.1074/jbc.M208535200. [DOI] [PubMed] [Google Scholar]
- 19.Cook SJ, McCormick F. Inhibition by cAMP of ras-dependent activation of raf. Science. 1993;262:1069–1072. doi: 10.1126/science.7694367. [DOI] [PubMed] [Google Scholar]
- 20.Vaillancourt RR, Gardner AM, Johnson GL. B-Raf-dependent regulation of the MEK-1/mitogen-activated protein kinase pathway in PC12 cells and regulation by cyclic AMP. Mol Cell Biol. 1994;14:6522–6530. doi: 10.1128/mcb.14.10.6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Rooij J, et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–7. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- 22.Kawasaki H, et al. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998;282:2275–9. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- 23.Enserink JM, et al. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol. 2002;4:901–6. doi: 10.1038/ncb874. [DOI] [PubMed] [Google Scholar]
- 24.Rubinfeld B, et al. Molecular cloning of a GTPase activating protein specific for the Krev-1 protein p21rap1. Cell. 1991;65:1033–42. doi: 10.1016/0092-8674(91)90555-d. [DOI] [PubMed] [Google Scholar]
- 25.Nicol RL, et al. Activated MEK5 induces serial assembly of sarcomeres and eccentric cardiac hypertrophy. Embo J. 2001;20:2757–67. doi: 10.1093/emboj/20.11.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zaccolo M, Pozzan T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science. 2002;295:1711–5. doi: 10.1126/science.1069982. [DOI] [PubMed] [Google Scholar]
- 27.Mongillo M, et al. Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ Res. 2004;95:67–75. doi: 10.1161/01.RES.0000134629.84732.11. [DOI] [PubMed] [Google Scholar]
- 28.Laroche-Joubert N, Marsy S, Michelet S, Imbert-Teboul M, Doucet A. Protein kinase A-independent activation of ERK and H,K-ATPase by cAMP in native kidney cells: role of Epac I. J Biol Chem. 2002;277:18598–604. doi: 10.1074/jbc.M201868200. [DOI] [PubMed] [Google Scholar]
- 29.Beavo JA, Bechtel PJ, EG K. Preparation of homogenous cyclic AMP-dependent protein kinase(s) and its subunits from rabbit skeletal muscle. Methods of Enzymology. 1974;38:299–308. doi: 10.1016/0076-6879(74)38046-9. [DOI] [PubMed] [Google Scholar]
- 30.Kodama H, et al. Significance of ERK cascade compared with JAK/STAT and PI3-K pathway in gp130-mediated cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2000;279:H1635–44. doi: 10.1152/ajpheart.2000.279.4.H1635. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.