

Abstract
We have cloned a human macrophage receptor that binds to apolipoprotein (apo)B48 of dietary triglyceride (TG)-rich lipoproteins. TG-rich lipoprotein uptake by the apoB48R rapidly converts macrophages and apoB48R-transfected Chinese hamster ovary cells in vitro into lipid-filled foam cells, as seen in atherosclerotic lesions. The apoB48R cDNA (3,744 bp) encodes a protein with no known homologs. Its ≈3.8-kb mRNA is expressed primarily by reticuloendothelial cells: monocytes, macrophages, and endothelial cells. Immunohistochemistry shows the apoB48R is in human atherosclerotic lesion foam cells. Normally, the apoB48R may provide essential lipids to reticuloendothelial cells. If overwhelmed, foam cell formation, endothelial dysfunction, and atherothrombogenesis may ensue, a mechanism for cardiovascular disease risk of elevated TG.
Elevated plasma triglyceride (TG) and persistent dietary chylomicrons (CM) and remnants are emergent risk factors for cardiovascular disease (CVD), a major cause of death worldwide (1). CVD involves both endothelial cell (EC) dysfunction and the formation of macrophage-derived lipid-filled foam cells, hallmarks of both early fatty streak and more advanced thrombogenic atherosclerotic lesions. Hypertriglyceridemic very low-density lipoproteins (HTG-VLDL), CM, and their remnants are the only native lipoproteins that cause rapid, receptor (R)-mediated macrophage lipid engorgement in vitro; normal very low-density lipoprotein (VLDL) and LDL do not (2, 3). This also appears to occur in vivo: chylomicronemic humans have foam cells in the bone marrow, spleen, liver, and skin (4). As these triglyceride-rich lipoproteins (TRL) also cause EC cholesterol uptake (5, 6) and fibrinolytic dysfunction in vitro (7), they may be atherothrombogenic in humans with elevated fasting or postprandial plasma TG.
TRL interact with cells by multiple mechanisms. HTG-VLDL and CM remnants bind to the LDLR and related Rs via apolipoprotein (apo)E (8–11). Dietary TRL cannot bind to the LDLR via apoB because apoB48, the apoB species formed in the intestine, lacks the C-terminal half of apoB100 that contains the LDLR-binding domain (12). Although most CM are lipolyzed into remnants that are taken up by hepatocyte LDLRs via apoE (13, 14), animal studies indicate that a significant CM fraction is rapidly taken up by reticuloendothelial cells (15, 16), such as accessible macrophages in bone marrow and spleen, independent of apoE (17).
We identified and characterized an apoE-independent R pathway in murine and human macrophages (18, 19) that binds to apoB48 of dietary TRL or to a like domain of apoB100 in HTG-VLDL (20) that could account, at least in part, for the observed direct reticuloendothelial uptake of CM in vivo, because no other known R binds apoB48. The apoB48R could also be involved in foam cell formation both in humans with elevated TRL (4) and in apoE-null mice, which have elevated apoB48 and spontaneous atherosclerosis (21, 22). The apoB48R mediates the rapid high-affinity uptake of CM, HTG-VLDL, and trypsinized (tryp) VLDL devoid of apoE (a model used to define this pathway), but not normal VLDL or LDL, resulting in lipid engorgement in vitro in both murine and human macrophages (18, 19). We identified two R candidates in human macrophages (19), of apparent Mr of 200 and 235 kDa, that can be reduced into a single active ligand-binding species with intermediate mobility on SDS/PAGE (200R). Each species has the same high-affinity (nanomolar Kd) saturable specific ligand-binding characteristics, restricted cell distribution, and lack of regulation by sterol or state of differentiation that distinguish the apoB48R from the LDLR and the scavenger R families (18, 23).
We purified the apoB48R candidate (200R) from THP-1 monocytes (24) and obtained partial unique amino acid sequence data (25). This allowed us to clone the apoB48R cDNA, characterize its mRNA, express apoB48R activity in Chinese hamster ovary-K1 cells (CHO), and determine that the apoB48R is in human atherosclerotic lesion foam cells, reported here.
Methods
Cloning of the apoB48R.
Standard molecular biology techniques were used (26). Nested degenerate oligonucleotide primers (bp 2,763–2,745, CCNCCNGTNACCATNARNC, 2,048-fold degeneracies and bp 2,754–2,738, ACCATNARNCCYTCNGC, 256-fold degeneracies), based on the unambiguous 10-residue sequence (AEGLMVTGGR) that produced R-specific antibodies (25), and the λgt10 forward primer (AGCAAGTTCAGCCTGGTTAAG) primed nested PCR with a human THP-1 monocyte λgt10 cDNA library (CLONTECH) as template. Based on a 137-bp product, nested PCR with new primers (bp 2,763–2,745 and bp 2,754–2,738) and the λgt10 forward primer gave a 631-bp product (pcr-631). The THP-1 cDNA library was screened with pcr-631 labeled with the Genius digoxigenin (DIG) system (Roche Molecular Biochemicals). Nested PCR by using antisense primers based on the 5′-end sequence of THP-1 clone λ73 (bp 2,203–2,187 and 2,139–2,123) and the λgt10 forward primer and a human placenta λgt10 cDNA library (CLONTECH) gave a 1,465-bp product (clone pcr-4–3), from which the 5′ end was produced as a 722-bp product (pcr-722). Primers based on pcr-722 produced 5′ end cDNA clones from the THP-1 cDNA library and THP-1 genomic DNA by PCR. PCR clones were used to screen both cDNA libraries. Multiple clones from both were sequenced for confirmation.
PCR used the GeneAmp kit with AmpliTaq polymerase (Perkin–Elmer) or the Advantage-GC kit with KlenTaq polymerase (CLONTECH). The cDNA clones were sequenced directly or first subcloned into EcoRI-digested pBluescript II SK (Stratagene). PCR-generated clones were ligated into the TA cloning vector (PCR 2.1, Invitrogen). Both strands of all clones were sequenced by using the Sequenase 2.0 kit (United States Biochemicals) or the Dye Termination Cycle Sequencing kit (Perkin–Elmer) in the automated sequencing cores at the University of Alabama at Birmingham (UAB).
ApoB48R mRNA Characterization.
Northern analysis: pcr-631 was labeled by random hexamer priming or by T7 polymerase transcription (riboprobe) by using Genius DIG (Roche Molecular Biochemicals) with hybridization, wash, and detection per Genius kit instructions. Chemiluminescence was recorded on Xmat film and digitized by a Hewlett–Packard optical scanner. Probes for glyceraldehyde-3-phosphate dehydrogenase (GPDH) mRNA were controls.
Reverse Transcription–PCR.
Total RNA was isolated by a single-step protocol (26). First-strand cDNA synthesis used 5 μg total RNA, the antisense primer (bp 2,756–2,736) and Superscript II reverse transcriptase (Life Technologies, Grand Island, NY). PCR used 10 pmol antisense primer, 10 pmol sense primer (bp 2,185–2,204), and 20 μl of the first-strand cDNA; products were electrophoresed on 1% agarose gel.
Chromosomal Localization.
Normal human metaphase and interphase nuclei were prepared from phytohaemagglutinin-stimulated cultured blood lymphocytes by standard procedures. ApoB48R cDNA (bp 1 to 2,614) was labeled with Cy3-dUTP (Amersham) by nick translation for localization by fluorescence in situ hybridization, as described with slight modification (27). Cy3-labeled cDNA (200 ng) coprecipitated with human Cot-1 DNA (1 μg; Life Technologies) was hybridized to nuclei and counterstained with 4′,6-diamidine-2′-phenylindole dihydrochloride for chromosome identification. Fluorescent images were captured by digitized image microscopy (28).
Transfection Studies.
These studies used a THP-1 monocyte apoB48R cDNA or a minigene consisting of the cDNA with the first intron, made by ligation of a 2,454-bp PCR-generated genomic clone (bp 2–2,095 of cDNA) that includes the first intron (see supplemental data on the PNAS web site, www.pnas.org) with the λ73 clone after digestion with EagI. The minigene was removed from pBluescriptII KS (−) by EcoRI/NotI digestion and ligated into pcDNA 3.1 (−) with Ready-to-go T4 ligase (Amersham Pharmacia) per the manufacturer's protocol. Clones were isolated by standard procedures and verified by partial DNA sequencing.
CHO (American Type Culture Collection) were transfected by using lipofectamine (Life Technologies) per the manufacturer's protocol and selected in medium containing G418 (0.5 mg/ml; Fisher Scientific) for 2–4 weeks. Cells were trypsinized and subcultured onto coverslips for visualization or into 6-well plates for quantification of cellular TG after incubation with lipoproteins as specified. Lipoproteins were isolated and VLDL trypsinized as described (20, 29).
Antibodies to the apoB48R.
Glutathione S-transferase (GST) fusion proteins of three domains encoded by clones pcr-4–3, λ73–3, and pcr-631 were ligated into the EcoRI site of the pGEX-5X-3 vector (Amersham Pharmacia) per the manufacturer's instruction. Transformants were screened for production of the appropriate fusion product and sequenced to verify correct orientation and reading frame. Fusion protein was produced by batch culture, purified per the manufacturer's protocol on glutathione-sepharose, and used as immunogens to generate polyclonal antibodies in rabbits by standard techniques. IgG were precipitated from antisera with (NH4)2 SO4, reconstituted in PBS, and were specific for all forms of the apoB48R (200 kDa, 235 kDa, and 200R) by immunoblotting.
Immunohistochemical Localization of the apoB48R in Atherosclerotic Lesions.
Immunohistochemical staining used a labeled streptavidin-biotin kit (Nichirei, Tokyo) per manufacturer's protocol. Paraffin sections were treated with xylene, then ethanol, and then immersed in 0.3% H2O2 in methanol for 45 min to block endogenous peroxidase activity. Specimens were rinsed with PBS (pH 7.4) and incubated with 10% normal goat serum for 12 h at 4°C. The anti-apoB48R polyclonal rabbit IgG (1:1,000) used is directed against the GST fusion protein linked to amino acids 223 to 710 of the apoB48R. Preimmune rabbit IgG was used as a negative control. A monoclonal antibody against human macrophages (HAM56; Dako) identified lesion macrophages. The specimens were incubated overnight with each primary antibody, rinsed three times with PBS, and incubated with biotinylated horse anti-mouse or anti-rabbit IgG for 30 min at room temperature. The specimens were rinsed three times with PBS, incubated with peroxidase-conjugated streptavidin for 30 min, visualized by using a 3-amino-9-ethylcarbazole substrate (Nichirei), and stained with hematoxylin.
Results and Discussion
ApoB48R cDNA Sequences.
The full-length THP-1 monocyte cDNA (GenBank accession no. AF141332) is 3,744 bp with a Kozak start site (bp 5–14), a 3,264-bp ORF beginning at bp 11, and a stop codon (TGA) at bp 3,275 (see Fig. 8, which is published as supplemental material). The 3′ untranslated region contains an Alu-like sequence, two polyadenylation signals (AATAAA), and a poly(A) tail. Sequencing multiple clones from both sources spanning the full-length sequence indicated the human THP-1 monocyte and placenta cDNA (GenBank accession no. AF141334) sequences differ at six bases (mutations or gene polymorphisms); three of these predict identical amino acids (b 520, A to G; b 574, A to G; b 1084, A to T), one is in the 3′ untranslated region (b 3446, T to G), one causes a conservative amino acid change (b 1434, G to A; R→K, amino acid residue 475), and one introduces a premature stop codon at amino acid 1,077 (b 3239, G to T) that would cause a 12-residue truncation at the C terminus in the placental R.
Chromosome Location.
The apoB48R gene (GenBank accession no. AF141333) is located on chromosome 16p11, as determined by fluorescence in situ hybridization by using a cDNA probe lacking the 3′ untranslated region to eliminate the Alu-like sequence (see Fig. 9, which is published as supplemental material). As only one chromosomal site was identified, there appear to be no closely related or duplicated apoB48R genes elsewhere in the human genome. PCR cloning and sequencing of human THP-1 genomic DNA identified three small introns within the coding sequence (Table 1; see supplemental data).‡‡
Characteristics of the Predicted apoB48R Protein.
The deduced monocyte apoB48R sequence of 1,088-aa residues contains three internal sequences previously obtained by microsequence analysis of tryptic digests of the purified R (Fig. 1). Immunoblotting data and transfection studies presented below indicate this cDNA is sufficient to encode a functional apoB48R. The sequence is unique; moreover, it is not closely related to any known protein. Unlike other lipoprotein Rs, which have functionally important cysteine-rich domains, there are only eight cysteines distributed throughout the apoB48R sequence. These can form internal disulfides, giving rise to the observed microheterogeneity of unreduced apoB48R extracts observed on SDS/PAGE that disappears on reduction (19, 23). The reduced and nonreduced forms of the apoB48R have full ligand-binding activity (23), indicating that the cysteines are not directly involved in ligand binding. The apoB48R protein is highly polar, with 242 negative and 122 positive amino acids. How the protein is anchored to the plasma membrane is unclear. There are two hydrophobic regions that are potential membrane-spanning or lipid-interacting domains. The first, amino acids 1 to 30, is predicted to be helical by some but not all analyses, contains a putative leader sequence, a leucine zipper motif (amino acids 8–29), and a hydrophobic domain encompassing three-fourths of the helical face. The other hydrophobic domain (amino acids 751–773) is 23 residues long with a high helical potential, consistent with known transmembrane domains, but with an atypically lower hydrophobicity. Or, the R may be anchored via interaction with an integral membrane protein, consistent with the observed 235-kDa receptor species, which is a ≈35 kDa protein(s) that, with the ≈200 kDa apoB48R, constitutes the observed 235 kDa ligand-binding species. The apoB48R does not contain a tyrosine-based internalization signal like that in the LDLR family. Rather, it contains tyrosine-independent di-leucine motifs (ExxxLL), which signal coated pit localization and internalization in immune cells (30).
Figure 1.

The deduced protein sequence of the THP-1 monocyte apoB48R is 1,088 aa. The 10-residue tryptic peptide (amino acids 910–919) determined by microsequence analysis (25) and used to design the oligonucleotide primers is double underlined, as are two other sequenced tryptic peptides. The putative 23-aa transmembrane domain (amino acid 751–773) is single underlined. Cysteines are bold and underlined (C); potential coiled-coil interactive domains are bold and italicized (amino acids 155–189, 473–486, and 520–533).
Posttranslational glycosylation or other modification may account for the disparity between the deduced (≈114.8 kDa) and the apparent Mr determined by SDS/PAGE (≈200 kDa). A potential glycosaminoglycan attachment site is at amino acid 118, a potential N-glycosylation site is at amino acid 617, and 4 possible O-glycosylation sites are at amino acids 265, 514, 565, and 942. However, treatment of partially purified R preparations with various glycosidases failed to affect significantly the mobility of the apoB48R on SDS/PAGE or its ligand-binding properties (data not shown), suggesting that it contains little, if any, functionally significant carbohydrate. Repeated attempts to dissociate polymeric species failed to shift the mobility of the apoB48R from ≈200 kDa, including boiling and reducing (23). There are, however, three potential coiled-coil domains in the predicted protein sequence that could promote homodimerization of the apoB48R and account for the size discrepancy (Fig. 1). Homodimerization is supported by results with a GST-fusion protein containing amino acids 223–710 that migrates as a dimer when analyzed on SDS/PAGE, suggesting spontaneous dimerization of this domain. In contrast, GST fusion proteins from more C-terminal domains of the apoB48R that lack potential coiled-coil domains (amino acids 629–1,088 and 714–917) do not dimerize and migrate as expected by their predicted Mrs (data not shown).
Other than the motifs noted above, the apoB48R protein has no common homologies to known proteins represented in GenBank. The significance of the internal unique repeating element (three repeats of the nine-residue sequence GGEEAETAS and eight nonexact repeats) is not known.
ApoB48R mRNA.
Northern analysis of mRNA isolated from THP-1 monocytes and from human placenta revealed an apoB48R mRNA of ≈3.8 kb, in close agreement with the cDNA size, indicating we had sequenced the full-length cDNA and that alternatively spliced mRNAs were not detected (Fig. 2A). Probing of a multiple immune tissue Northern blot also revealed apoB48R mRNA of the same size in peripheral blood leukocytes > bone marrow = spleen > lymph node, and only faintly visible in appendix and thymus (Fig. 2B). PCR screening of a human tissue cDNA panel with gene-specific primers identified the R cDNA in the brain, heart, kidney, liver, lung, pancreas, and placenta but not in skeletal muscle (Fig. 3). Expression appeared to be greatest in lung and placenta (the only positive tissues after 25 PCR cycles). Control reactions with GPDH probes or primers verified approximately equal representation of transcripts in the mRNA and cDNA panels. Not shown are reverse transcription–PCR data indicating the expression of the apoB48R mRNA in human bloodborne, THP-1, and U-937 monocyte–macrophages; in murine P388D1 macrophages and NIH 3T3 cells (thought to be of EC origin); and in human umbilical vein EC. Consistent with previous studies (18, 19, 25) CHO, human fibroblasts, and HepG2s were negative, indicating the apoB48R is expressed primarily by cells of RE origin.
Figure 2.

(A) Human THP-1 monocytes and placenta mRNA (2 μg/lane) were prepared from total RNA by using oligo-dT cellulose chromatography. ApoB48R mRNA was detected with DIG-pcr-631-riboprobe. (B) The DIG-pcr-631 cDNA probe was hybridized to a multiple tissue Northern blot (MTN, CLONTECH) containing 2 μg/lane mRNA (Left to Right): spleen, lymph node, thymus, appendix, peripheral blood leukocytes, and bone marrow. The filters were then probed with a human DIG-GPDH cDNA (see Methods).
Figure 3.

A multiple-tissue cDNA panel (MTC, CLONTECH), containing normalized first-strand cDNA (Left to Right) brain, heart, kidney, liver, lung, pancreas, placenta, skeletal muscle, positive control (combined tissues), no DNA control, and GPDH control, was PCR amplified with apoB48R sequence-specific primers overlapping introns 2 and 3 (see supplemental Table 1) to detect only mRNA. The expected 455-bp product was found in all but skeletal muscle, with most in lung and placenta (seen after 25 cycles) and least in brain and heart (seen after 35 cycles). Control PCR analyses for the GPDH cDNA verified normalization of mRNA levels.
Transfection Studies.
Transfection of apoB48R cDNA constructs (with and without the first intron) into CHO conferred apoB48R expression and activity, whereas transfection with vector alone or inverted cDNA insert did not. Ligand and Western blotting experiments performed as previously described (19, 20, 25) document expression of a single apoB48R species in the R-transfected cells, but not in vector-transfected cells, that exhibits coincident ligand-binding activity and apoB48R immunoreactivity (Fig. 4). The apparent Mr of ≈190 kDa in apoB48R-transfected CHO is slightly lower than the active forms in THP-1s (200 and 235 kDa). The Mr differences and the presence of a single rather than two ligand-binding species may be because of processing differences in CHO cells vs. macrophages. Of note, however, the apparent Mr of the R in the apoB48R-transfected CHO approaches twice that predicted by the cDNA, consistent with homodimerization of the R via protein interacting motifs previously noted.
Figure 4.

ApoB48R Western and ligand-blotting activity are detected in apoB48R-transfected CHOs, but not in vector-transfected control CHOs. The blots have electrophoresed extracts of, from outer to inner lanes, THP-1 monocytes (positive control), vector-transfected CHO, and apoB48R minigene-transfected CHO. (Left) The ligand was tryp-VLDL detected with an anti-apoB antibody (20). (Right) The apoB48R protein was detected with rabbit IgGs against apoB48R (amino acids 223–710) GST fusion protein (25).
ApoB48R-mediated TRL uptake by transfected CHO was determined by three alternative methods: first, by TRL-induced increases in cellular TG mass (Fig. 5), a sensitive, quantitative endpoint because TG is the predominant lipid in the lipoprotein and thus the predominant lipid accumulated by cells (2, 3, 18); second, by rapid and massive TRL-induced accumulation of large Oil red O-positive cytoplasmic lipid droplets that form after lysosomal hydrolysis and reesterification (2, 18) (Fig. 6); and third (not shown), by uptake of TRL labeled with a lipid-soluble fluorescent dye. CHO were transfected with vector containing an apoB48R cDNA or minigene or, as controls, vector alone or vector containing inverted apoB48R cDNA, and stable populations obtained. When transfected cells are incubated with increasing concentrations of tryp-VLDL (29) for 4 h at 37°C (Fig. 5A), apoB48R-transfected cells show a rapid and curvilinear TG accumulation, indicating saturable apoB48R-mediated uptake like the apoB48R-mediated uptake in macrophages (19). In contrast, vector-transfected cells show only linear, nonspecific uptake, as seen in cells where no apoB48R activity is present. Similar results were obtained in multiple experiments with tryp-VLDL devoid of apoE, CM containing apoB48 as the only apoB species (20), and HTG-VLDL containing apoB48 and apoB100, but not with normal VLDL containing apoB100 as the only detectable apoB species (Fig. 5B). Studies with CHOs transfected with an apoB48R minigene containing the first intron, but not CHOs containing vector alone, show massive, rapid accumulation of large Oil red O-positive lipid droplets when incubated for ≤3 h with tryp-VLDL devoid of apoE or CM that contain apoB48 but not apoB100, consistent with the ligand specificity of the apoB48R (Fig. 6). These experiments (each replicated three to ten times) indicate by three alternative endpoints that the protein encoded by this cDNA is sufficient to confer apoB48R activity on the transfected cells like that observed in human macrophages in terms of both ligand specificity and kinetics of uptake (19, 20). These transfection studies demonstrate unequivocally that the apoB48R binds and internalizes TRL when out of the context of the macrophage, i.e., in the absence of other macrophage-specific proteins such as apoE or lipoprotein lipase that can enhance macrophage uptake of lipoproteins. Thus, the apoB48-specific R activity, previously documented by both cell and ligand blotting studies in human bloodborne and THP-1 monocytes and macrophages, can indeed be attributed to this apoB48R protein encoded by this cDNA and not to a combination of other macrophage processes.
Figure 5.

(A) G418-selected, stable apoB48R-transfected, and vector-transfected control CHO were incubated with tryp-VLDL 100–400 Sf at the concentrations indicated for 4 h at 37°C and the cells processed to measure TG mass as previously reported (18). The upper curve (closed squares; apoB48R transfected) reflects a rapid, curvilinear accumulation of TG with increasing levels of tryp-VLDL. The lower curve (open squares; cells transfected with pcDNA 3.1 vector plus inverted insert) shows linear accumulation, representing low-affinity nonspecific uptake. Values are averages from duplicate dishes that differ by <10%; This represents one of four experiments with the apoB48R cDNA and four with the apoB48R minigene containing the first intron. (B) Transfections were as described in Fig. 5A. After a 2-h incubation with the indicated lipoprotein at 37°C, the cells were processed as above. The bar graph shows the amount of TG that accumulated relative to a buffer control, which was subtracted from the level for each lipoprotein. Like human THP-1 macrophages, the apoB48R-transfected cells accumulate significant TG when exposed to apoB48 only containing CM Sf 1,100–3,200 S (CMII) and to the model ligand, tryp-VLDL Sf 100–400 (t-V1), but not to VLDL Sf 100–400 from a donor with normal plasma TG levels (N-V1). The experiment was repeated twice with identical results. Concurrent experiments with control vector-transfected CHO showed no significant TG accumulation under the same experimental conditions (data not shown).
Figure 6.
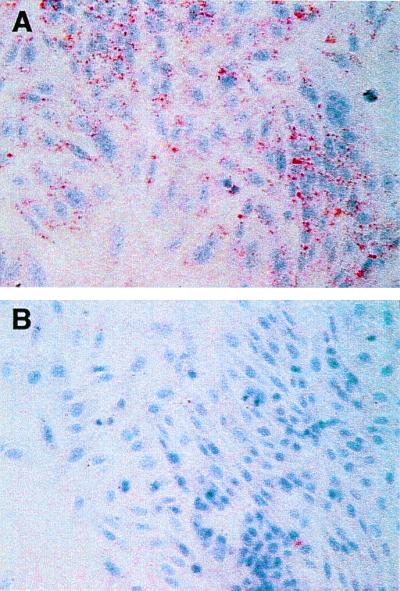
(A) ApoB48R-transfected CHO were incubated at 37° for 3 h with CM Sf >400, with apoB48 as the only apoB species, at 100 μg TG/ml RPMI and stained with Oil red O to detect cytoplasmic neutral lipid droplets (2). (B) Vector only-transfected CHOs were incubated with CM under identical conditions and stained with Oil red O. R- and vector-transfected cells treated with buffer had no lipid staining inclusions (not shown).
Identification of the apoB48R in Human Atherosclerotic Lesions.
The transfection studies coupled with our previously published studies in human macrophages (19, 20, 23, 25) suggest that excessive uptake of TRL by the apoB48R may promote atherosclerosis in vivo. Indeed, immunohistochemical data support this possibility. Polyclonal antibodies produced against a domain of the apoB48R (amino acids 223–710) that specifically identify the apoB48R by immunoblotting (Fig. 4) demonstrate immunohistochemically the in vivo expression of the apoB48R by macrophages and foam cells within human atherosclerotic lesions. Fig. 7 shows representative serial sections of a carotid artery atheroma. The macrophage-specific monoclonal antibody HAM56 identifies macrophages and macrophage foam cells around the lipid core. Anti-apoB48R IgGs also bind to these macrophages and foam cells. In contrast, preimmune IgGs do not bind to any cells in the lesion. Foam cells in early aortic fatty streak lesions as well as in more advanced coronary lesions were also apoB48R-positive (data not shown), supporting evidence that the apoB48R may contribute to foam cell formation and atherogenesis in vivo.
Figure 7.

Serial sections of carotid arteries obtained by endarterectomy were processed as described in Methods. Rabbit polyclonal antibodies produced against an apoB48R-specific domain (amino acids 223–710) GST-fusion protein were used to determine apoB48R expression and location. (A) Hematoxylin-stained section of an advanced lesion; a lipid core surrounded by foam cells is seen to the right and center (×100). (B) A macrophage-specific monoclonal antibody (HAM56) identifies the macrophage-derived foam cells around the lipid core (×100). (C) Preimmune IgG from the rabbit used to generate the anti-apoB48R IgGs did not bind to lesion cells (negative control) (×80). (D) Anti-apoB48R IgGs specifically bind to lesion macrophages and foam cells (×100), indicating these cells express the apoB48R protein.
The restricted cell distribution and unique ligand specificity of the apoB48R suggest that its normal role may be to ensure efficient delivery of essential dietary lipids and lipid-soluble vitamins and other nutrients to RE cells, particularly monocytes and accessible macrophages of the immune system, and to the placenta. The apoB48R's ligand specificity would ensure cellular uptake while CM are lipid and nutrient rich, before extensive lipolysis and the transfer of apoE into CM remnants that diverts remnants to hepatocytes for uptake by apoE-dependent LDLRs and related Rs (13, 14).
In addition, studies in apoE-deficient mice have demonstrated unequivocally that apoE, which is necessary for the binding of TRL to LDLR family members (8–11), is not required either for normal embryologic development or for foam cell formation and atherosclerosis (21, 22). ApoE-deficient mice initially develop normally but later acquire extensive atherosclerosis even in the absence of dietary fat excess, presumably partly because of the excessive levels of circulating atherogenic lipoproteins containing apoB48 (21, 22), observations that strongly implicate the apoB48R in atherogenesis in apoE-deficient mice. In contrast, apoB is essential for embryologic development, as homozygous deficiency results in early fetal death, presumably because of its necessity in the transport of essential lipid components from the yolk sac visceral endodermal cells to the developing mouse embryo (31, 32). ApoB48 expression is sufficient to rescue normal development of mice even in the absence of apoE (33). This rescue may involve the apoB48R reported here, because it is the only known R for apoB48, it is apoE-independent, and it is expressed in murine reticuloendothelial cells (2, 18, 25). Here we document the expression of the apoB48R in the human placenta as well as in reticuloendothelial cells and atheroma, suggesting it, like its ligand, may play a role in early development. When overwhelmed, as in disease states with elevated plasma TG and/or persistent dietary TRL, the apoB48R may contribute to foam cell formation, atherogenesis, and EC dysfunction, even when apoE is absent or defective. The apoB48R pathway thus provides a unique cellular mechanism to explain, at least in part, the cardiovascular disease risk of elevated plasma TG and dietary TRL.
Supplementary Material
Acknowledgments
Microsequence analysis and mass spectrometry were done by Dr. Kristine M. Swiderek, Director, City of Hope Core Facility, Beckman Research Institute, Duarte, CA. Blood for lipoprotein isolation was obtained from volunteers after informed consent under guidelines of the UAB Institutional Review Board, in the General Clinical Research Center (National Institutes of Health, Division of Research Resources Grant RR00032). Rabbits were managed by the professional staff of the UAB Animal Resources Program according to institutional guidelines. We gratefully acknowledge advice from Drs. Jeff Kudlow, Andrew Patterson, and Jeff Engler of UAB, Dr. Daniel Pinkel of the University of California at San Francisco for help with fluorescence in situ hybridization, Dr. Elliot Lefkowitz at UAB for help with sequence analyses, Caryl Reese and Dana Stinson for technical assistance, and Marilyn Robinson for editorial assistance and preparation of the manuscript. We thank Drs. Jan L. Breslow, Suzanne G. Eskin, Richard J. Havel, Ronald M. Krauss, and Jonathan D. Smith for reading and providing helpful comments on the manuscript. This research was supported by National Institutes of Health Grant HL44480 and by National Institutes Research Service Awards T32 HL-07703, HL-07631–06A1, and a fellowship from the Juvenile Diabetes Foundation (M.L.B.).
Abbreviations
- apo
apolipoprotein
- CHO
Chinese hamster ovary-K1 cells
- CM
chylomicrons
- DAPI
4′,6, diamidine-2′-phenylindole dihydrochloride
- DIG
digoxigenin
- EC
endothelial cells
- GST
glutathione-S-transferase
- HTG-VLDL
hypertriglyceridemic very low density lipoprotein
- R
receptor
- REC
reticuloendothelial cells
- TG
triglycerides
- TRL
triglyceride-rich lipoproteins
- tryp
trypsinized
- UAB
University of Alabama at Birhimgham
- GPDH
glyceraldehyde-3-phosphate dehydrogenase
Footnotes
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. AF141332, AF141333, AF141334, and AC002544).
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.120184097.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.120184097
After completion of all sequencing and chromosomal location studies, a corresponding gene sequence was reported as a “sequence in progress” in the High Throughput Genomic Sequencing [htgs] data bank from a human BAC clone at chromosome 16p11.2 (accession no. AC002544). A recently revised sequence of the BAC clone differs from the THP-1 cDNA and genomic sequences we determined at 4 b [T to G b 1028 of cDNA, changing amino acid 340 from S to A; T to C at b 1,165 (silent); A to G, b 2,068 (silent); C to G, b 3,495 in the 3′ untranslated region]. These differences could be mutants, gene polymorphisms, or sequencing errors, but the revised htgs sequence is more like the THP-1 genomic sequence we determined than the placental cDNA sequence and confirms its chromosomal location.
References
- 1.Hokanson J E, Austin M A. J Cardiovasc Risk. 1996;3:213–219. [PubMed] [Google Scholar]
- 2.Gianturco S H, Bradley W A, Gotto A M, Jr, Morrisett J D, Peavy D L. J Clin Invest. 1982;70:168–178. doi: 10.1172/JCI110590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown M S, Goldstein J L. Annu Rev Biochem. 1983;52:223–261. doi: 10.1146/annurev.bi.52.070183.001255. [DOI] [PubMed] [Google Scholar]
- 4.Fredrickson D S, Goldstein J L, Brown M S. In: The Metabolic Basis of Inherited Diseases. Stanbury J G, Wyngaarden M F, Fredrickson D S, editors. New York: McGraw–Hill; 1978. pp. 604–655. [Google Scholar]
- 5.Fielding C J, Vlodavsky I, Fielding P E, Gospodarowicz D. J Biol Chem. 1979;254:8861–8868. [PubMed] [Google Scholar]
- 6.Gianturco S H, Eskin S H, Navarro L T, Lahart C J, Smith L C, Gotto A M., Jr Biochim Biophys Acta. 1980;618:143–152. doi: 10.1016/0005-2760(80)90061-2. [DOI] [PubMed] [Google Scholar]
- 7.Li X, Koons J C, Benza R L, Parks M, Varma V K, Bradley W A, Gianturco S H, Taylor K B, Grammer J R, Tabengwa E M, et al. Biochemistry. 1996;35:6080–6088. doi: 10.1021/bi952032i. [DOI] [PubMed] [Google Scholar]
- 8.Gianturco S H, Gotto A M, Jr, Hwang S C, Karlin J B, Lin A H, Prasad S C, Bradley W A. J Biol Chem. 1983;258:4526–4533. [PubMed] [Google Scholar]
- 9.Beisiegel U, Weber W, Ihrke G, Herz J, Stanley K. Nature (London) 1989;341:162–164. doi: 10.1038/341162a0. [DOI] [PubMed] [Google Scholar]
- 10.Kowal R C, Herz J L, Goldstein J L, Esser V, Brown M S. Proc Natl Acad Sci USA. 1989;86:5810–5814. doi: 10.1073/pnas.86.15.5810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bradley W A, Hwang S C, Karlin J B, Lin A H, Prasad S C, Gotto A M, Jr, Gianturco S H. J Biol Chem. 1984;259:14728–14735. [PubMed] [Google Scholar]
- 12.Yang C, Chen S, Gianturco S H, Bradley W A, Sparrow J T, Tanimura M, Li W, Sparrow D A, DeLoof H, Rosseneu M, et al. Nature (London) 1986;323:738–742. doi: 10.1038/323738a0. [DOI] [PubMed] [Google Scholar]
- 13.Havel R J. Arteriosclerosis. 1985;5:569–580. doi: 10.1161/01.atv.5.6.569. [DOI] [PubMed] [Google Scholar]
- 14.Brown M S, Goldstein J L. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 15.Ross C A, Zilversmit D B. J Lipid Res. 1977;18:169–181. [PubMed] [Google Scholar]
- 16.Nagata Y, Zilversmit D B. J Lipid Res. 1987;28:684–692. [PubMed] [Google Scholar]
- 17.Hussain M M, Mahley R W, Boyles J K, Fainaru M, Brecht W J, Lindquist P A. J Biol Chem. 1989;264:9571–9582. [PubMed] [Google Scholar]
- 18.Gianturco S H, Lin A H, Hwang S C, Young J, Brown S A, Via D P, Bradley W A. J Clin Invest. 1988;82:1633–1643. doi: 10.1172/JCI113775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gianturco S H, Ramprasad M P, Lin A, Song R, Bradley W A. J Lipid Res. 1994;35:1674–1687. [PubMed] [Google Scholar]
- 20.Gianturco S H, Ramprasad M P, Song R, Li R, Brown M L, Bradley W A. Arterioscler Thromb Vasc Biol. 1998;18:968–976. doi: 10.1161/01.atv.18.6.968. [DOI] [PubMed] [Google Scholar]
- 21.Plump A S, Smith J D, Hayek T, Aalto-Setala K, Walsh A, Verstuyft J G, Rubin E M, Breslow J L. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- 22.Zhang S H, Reddick R L, Piedrahita J A, Maeda N. Science. 1992;258:468–471. doi: 10.1126/science.1411543. [DOI] [PubMed] [Google Scholar]
- 23.Ramprasad M P, Li R, Bradley W A, Gianturco S H. Biochemistry. 1995;34:9126–9135. doi: 10.1021/bi00028a023. [DOI] [PubMed] [Google Scholar]
- 24.Ramprasad M P, Li R, Gianturco S H, Bradley W A. Biochem Biophys Res Comm. 1995;210:491–497. doi: 10.1006/bbrc.1995.1687. [DOI] [PubMed] [Google Scholar]
- 25.Bradley W A, Brown M L, Ramprasad M P, Li R, Song R, Gianturco S H. J Lipid Res. 1999;40:744–752. [PubMed] [Google Scholar]
- 26.Ausubel M, Brent R, Kingston R E, Moore D D, Smith J A, Seidman J G, Struhl K, editors. Current Protocols in Molecular Biology. New York: Wiley; 1999. [Google Scholar]
- 27.Stokke T, Collins C, Kuo W -L, Kowbel D, Shadravan F, Tanner M, Kalliioniemi A, Kallioniemi O -P, Pinkel D, Deaven L, et al. Genomics. 1995;26:134–137. doi: 10.1016/0888-7543(95)80092-z. [DOI] [PubMed] [Google Scholar]
- 28.Sakamoto M, Pinkel D, Mascio L, Sudar D, Peters D, Kuo W -L, Yamakawa K, Nakamura Y, Drabkin H, Jericevic Z, et al. Cytometry. 1995;19:60–69. doi: 10.1002/cyto.990190108. [DOI] [PubMed] [Google Scholar]
- 29.Gianturco S H, Bradley W A. In: Methods in Enzymology. Segrest J P, Albers J J, editors. New York: Academic; 1986. pp. 319–344. [DOI] [PubMed] [Google Scholar]
- 30.Mellman I. Annu Rev Cell Dev Biol. 1996;12:575–625. doi: 10.1146/annurev.cellbio.12.1.575. [DOI] [PubMed] [Google Scholar]
- 31.Huang L S, Voyiaziakis E, Markenson D P, Sokol K A, Hayek T, Breslow J L. J Clin Invest. 1995;96:2152–2161. doi: 10.1172/JCI118269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Farese R V, Ruland S L, Flynn L M, Stokowski R P, Young S G. Proc Natl Acad Sci USA. 1995;92:1774–1778. doi: 10.1073/pnas.92.5.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Farese R V, Jr, Veniant M M, Cham C M, Flynn L M, Pierotti V, Loring J F, Traber M, Ruland S, Stokowski R S, Huszar D, et al. Proc Natl Acad Sci USA. 1996;93:6393–6398. doi: 10.1073/pnas.93.13.6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brown M L, Ramprasad M P, Li R, Umeda P K, Gianturco S H, Bradley W A. FASEB J. 1998;12:A1383. [Google Scholar]
- 35.Brown M L, Ramprasad M P, Umeda P K, Tanaka A, Kobayashi Y, Watanabe T, Shimoyamada H, Bradley W A, Gianturco S H. Circulation. 1999;100 Suppl. I:1728. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}