

Abstract
We have isolated an RGS-GAIP interacting protein that links RGS proteins to protein degradation. GIPN (GAIP interacting protein N terminus) is a 38-kDa protein with an N-terminal leucine-rich region, a central RING finger-like domain, and a putative C-terminal transmembrane domain. GIPN binds exclusively to RGS proteins of subfamily A, RGS-GAIP, RGSZ1, and RGSZ2. The N-terminal leucine-rich region of GIPN interacts with the cysteine-rich motif of RGS-GAIP. GIPN mRNA is ubiquitously expressed, and GIPN is found on the plasma membrane of transfected HEK293 cells. Endogenous GIPN is concentrated along the basolateral plasma membrane of proximal and distal tubules in rat kidney, where many G protein-coupled receptors and some G proteins are also located. Two immunoreactive species are found in rat kidney, a 38-kDa cytosolic form and an ≈94-kDa membrane form. GIPN shows Zn2+- and E1/E2-dependent autoubiquitination in vitro, suggesting that it has E3 ubiquitin ligase activity. Overexpression of GIPN stimulates proteasome-dependent reduction of endogenous Gαi3 in HEK293 cells and reduces the half-life of overexpressed Gαi3-YFP. Thus, our findings suggest that GIPN is involved in the degradation of Gαi3 subunits via the proteasome pathway. RGS-GAIP functions as a bifunctional adaptor that binds to Gα subunits through its RGS domain and to GIPN through its cysteine string motif.
Cells respond to a variety of extracellular stimuli through receptors that are linked to heterotrimeric G proteins. The levels of individual G protein signaling molecules are tightly regulated by modifying their levels of expression or by proteasome-dependent degradation. Many components of G protein signaling pathways have been shown to be ubiquitinated and degraded in proteasomes, including G protein-coupled receptors (GPCRs) (1), kinases (2), and RGS proteins (3). Specific degradation of G alpha subunits (Gα) by the ubiquitin/ proteasome-dependent pathway was previously shown for the yeast Gpa1, a Gα equivalent (4, 5), and more recently for Gαo (6) and Gβγ subunits (7).
RGS proteins are known to bind to Gα (8–10) and to negatively regulate G protein signaling through their GTPase-activating activity by stabilizing the GTP to GDP hydrolysis transition state (11). RGS-GAIP is a member of subfamily A of the RGS proteins and acts as a GTPase-activating protein for Gαi subunits. The N terminus of RGS-GAIP contains a cysteine-rich motif, which is most likely the site of reversible palmitoylation responsible for anchoring of RGS-GAIP to clathrin-coated pits (12). Here, we describe an RGS-GAIP interacting protein, GIPN, which binds to the N terminus of GAIP, has E3 ubiquitin ligase activity, and promotes proteasome-dependent degradation of Gαi3. This interaction provides a functional link between a Gα subunit and the ubiquitin-dependent protein degradation system through an RGS protein intermediate.
Materials and Methods
Antibodies. Monoclonal antibodies against ubiquitin were purchased from Chemicon, anti-GFP (Jl-8) was purchased from Clontech, and anti-mouse IgG was purchased from Bio-Rad. Anti-ezrin antibody was kindly provided by H. Furthmayr (Stanford University, Stanford, CA), and affinity-purified, anti-Gαi3 (EC) IgG was provided by A. Spiegel (National Institute of Diabetes and Digestive and Kidney Diseases). Anti-GIPN antibodies were raised in rabbits against His-6-GIPN (full length); an IgG fraction was prepared by using the caprylic acid method and affinity-purified on either the CNBr immobilized immunogen or a mouse kidney lysate immobilized on poly(vinylidene difluoride) membranes (13).
Cell Culture. HEK293 cells were grown in DMEM-high glucose with 10% (vol/vol) FBS and 100 units/ml penicillin G, 100 units/ml streptomycin sulfate, and 0.3 mg/ml glutamine (Invitrogen).
Sf21 cells (obtained from A. Newton, University of California at San Diego) were grown as monolayers in Hink's TNM (JRH Biosciences, Lenexa, KS) with 10% (vol/vol) FBS and 100 units/ml penicillin G/100 units/ml streptomycin sulfate and transfected in serum-free EX-CELL 400 (JRH Biosciences).
Isolation of Mammalian GIPN cDNAs. Interaction screening was performed with rat RGS-GAIP (amino acids 23–216) in a rat pituitary GC cell library as described (14). Online BLAST searches were performed by using the National Center for Biotechnology Information web site (15). Homologous human and mouse EST clones were purchased from InCyte Pharmaceuticals (Palo Alto, CA). All vectors used contained the rat GIPN sequence. Protein alignments were carried out with the CLUSTALW program (16), PSORT II for protein topology predictions, and PROSITE for motifs. DNA sequencing was performed by the Molecular Pathology Shared Resource of the University of California at San Diego Cancer Center.
In Vitro Interactions. GIPN was cloned into pcDNA3 (Invitrogen) with an N-terminal FLAG-tag and prepared by using the TnT reticulocyte lysate kit (Promega) in the presence of [35S]methionine. Five micrograms of His6-GAIP (17) and AGS3 (residues 424–650; ref. 18) were immobilized on Ni2+-agarose beads (Qiagen, Valencia, CA) and incubated with 35S-labeled GIPN for 1 h at 4°C in 300 μl of buffer A (20 mM Hepes, pH 7.4/150 mM NaCl/0.5 mM EDTA/0.1% Triton X-100/0.1 μg/μl BSA) with gentle rocking. Beads were washed (three times) and boiled in 25 μl of Laemmli buffer; proteins were separated by SDS/12% PAGE and prepared for autoradiography.
Two-Hybrid Interactions. For analysis of the interaction between GIPN and RGS-GAIP, we used deletion mutants for hGAIP (1–79 and 80–217) in pGBT9 vector (14). RGS-GAIP1–38 was made by introducing a stop codon at Cys-39 in the 1–79 pGBT9 construct with the QuikChange mutagenesis kit (Stratagene). RET-RGS1 and RGS4 constructs in pGBT9 were obtained as described (14). Human RGSZ1, amino acid 1–217 (19), and human RGSZ2, amino acid 1–212 (20), were used as template cDNAs in PCR for subcloning into pGBKT7 vector (Clontech). GIPN1–110 was made by introducing a stop codon after the indicated residue in full-length GIPN (pACT2 vector) as above. All constructs were verified by automated DNA sequencing. For one-to-one interactions, we used a colony lift assay in Y187 yeast cells as described (9).
Northern Blot Analysis. Multiple tissue blots of human (Clontech) and rat and mouse (Seegene, Seoul, Korea) poly(A+) RNA were hybridized to random-primed, 32P-labeled, species-specific probes (Bio-Rad, ≈1,000 bp long, specific activity 109 cpm/μg). ExpressHyb solution (Clontech) was used under high stringency conditions for hybridization (68°C), and high stringency washes were performed in 0.1× SSC (150 mM NaCl/15 mM sodium citrate, pH 7) plus 0.1% SDS at 55°C. Blots were exposed for autoradiography for 18 and 72 h at -70°C with intensifying screens.
Preparation of Membrane and Cytosolic Fractions from Mouse Kidney. Kidneys were excised from Swiss–Webster mice and placed in PBS containing a protease inhibitor mixture (Sigma) at 4°C. The tissue was minced and homogenized, and membrane (100,000 × g pellet) and cytosolic (100,000 × g supernatant) fractions were prepared as described (13). Proteins were separated by SDS/ 10% PAGE and immunoblotted with anti-GIPN (0.4 μg/ml).
Immunofluorescence. GIPN was cloned into pcDNA 3.1(+) zeo (Invitrogen). HEK293 cells were plated on coverslips and transfected with GIPN using FuGENE 6 (Roche Molecular Biochemicals); 22-h posttransfection cells were fixed for 30 min with 2% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) permeabilized with 0.1% Triton X-100 in PBS, and incubated with affinity-purified anti-GIPN (6.7 μg/ml), followed by Alexa Fluor-488 goat anti-rabbit F(ab′)2 (Molecular Probes; 8 μg/ml). Rat kidneys were perfused with DMEM followed by 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) for 15 min, excised, and immersion fixed in 4% paraformaldehyde for an additional 4 h. Samples were cryoprotected as described (12), and semithin cryosections (0.5–1.0 μm) were cut and incubated as above. Images were captured by using a Zeiss Axiophot microscope equipped with a Hamamatsu Orca ER CCD camera run by OPENLAB software (Improvision, Lexington, MA).
Immunogold Labeling of Rat Kidney. For immunoelectron microscopy, ultrathin cryosections (70–80 nm) were prepared as described (12) and incubated with affinity-purified anti-GIPN for 2 h at 25°C, followed by goat anti-rabbit IgG conjugated to 10 nm gold (Amersham Biosciences). Sections were stained with 2% neutral uranyl acetate, and adsorption stained with 0.2% uranyl acetate in 1.8% methyl cellulose on ice and analyzed in a JEOL 1200EX II electron microscope.
In Vitro Ubiquitination Assays. GIPN was cloned into pGEX-KG (Pharmacia), and GST-fusion protein was expressed in Escherichia coli and purified as described (21). The in vitro ubiquitination assay was from Matsuda et al. (22). Briefly, 2 μg of GST or GST-GIPN were immobilized on glutathione-agarose beads in 400 μl of buffer B (40 mM Tris·HCl, pH 7.5/5 mM MgCl2/2 mM ATP/2 mM DTT/300 ng/μl ubiquitin/25 μM MG-132/20 μl of TnT reticulocyte lysate) and incubated for 2 h at 25°C. To demonstrate Zn2+ dependence, the reaction mixture was preincubated in tetrakis (2-pyridylmethyl) ethylenediamine (TPEN, Sigma; 2 mM final) for 1 h at 25°C, followed by addition of ZnCl2 (20 mM final). After washes, beads were boiled in Laemmli buffer, and proteins were separated by SDS/10% PAGE and transferred onto nitrocellulose membranes (0.45 μm, Transblot, Bio-Rad), which were autoclaved and incubated (1 h, 25°C) with anti-ubiquitin (0.5 μg/ml in TBS, 0.45% Tween 20/2% calf serum) followed by horseradish peroxidase-conjugated goat anti-mouse IgG (0.3 μg/ml) and ECL detection using Super-Signal (Pierce).
For assays with purified proteins, His6-GIPN was expressed in Sf21 insect cells infected with a recombinant baculovirus following the instructions from the Bac to Bac manual (Invitrogen). His6-GIPN virion underwent two rounds of amplification, and 900 μl were used to infect three T75 flasks of Sf21 cells for 48 h. Sf21 cells were lysed, and His6-GIPN was purified on Ni-NTA (Qiagen) agarose beads. The ubiquitination assay was carried out in the laboratory of Randy Hampton (University of California at San Diego) as described (23). In brief, 0.5 μg Hrd1 were incubated in the presence of 1 μg of purified His6-E1 and 1 μg His6-Ubc4, ubiquitin, and ATP for 2 h at 25°C. Proteins were separated by SDS/3–8% PAGE and transferred onto nitrocellulose before immunoblotting as above.
Immunoblotting. Cells were scraped into ice-cold PBS, passed (ten times) through a 30.5-gauge needle and centrifuged at 600 × g for 10 min at 4°C. Proteins in the postnuclear supernatant (25 μg) were separated by SDS/10% PAGE, transferred to poly(vinylidene difluoride) membranes (Millipore), and immunoblotted with anti-ezrin (1:10,000) or anti-Gαi3 (EC, 1:1,000) and horseradish peroxidase-conjugated goat anti-rabbit IgG (0.3 μg/ml). Detection was by ECL. Protein concentrations were determined by using the BCA assay (Pierce).
Metabolic Labeling and Turnover Studies. Cells were plated in 6-cm dishes and transfected with GIPN and Gαi3-YFP (24) by using Fugene 6. Twenty-four hours thereafter, cells were pulse labeled for 20 min with 100 μCi/ml Easytag (NEN Life Sciences) in Cys/Met-free DMEM-high glucose, 10% (vol/vol) FBS, 100 units/ml penicillin G, 100 units/ml streptomycin sulfate, 0.3 mg/ml glutamine (Invitrogen), and 20 mM Hepes and chased for 0, 1, and 2 h with labeling medium containing 6 mM Met and Cys (25). Some cells were treated with MG-132 (5 μm for 2 h) before labeling. Cells were then washed and lysed in 500 μl of lysis buffer (1% Nonidet P-40/0.5% Na deoxycholate/50 mM Tris, pH 7.4/150 mM NaCl/2.5 mM MgCl2/1 mM EDTA/1 mM DTT) containing protease inhibitors. Lysates were cleared by centrifugation (10,000 × g for 10 min), and incorporated radio-activity was determined as trichloroacetic acid precipitable counts (26). Equal amounts of radioactivity were immunoprecipitated with 1.5 μg of anti-GFP, and immune complexes were collected on protein A-Sepharose (Amersham Biosciences), separated by SDS/10% PAGE, exposed for autoradiography, and quantified by using SCAN ANALYSIS software (Biosoft, Ferguson, MD).
Results
GIPN Contains a RING Finger-Like Motif and a Putative C-Terminal Transmembrane Domain. Using the two-hybrid system with RGS-GAIP as a bait and a rat pituitary cell library, we isolated a previously undocumented sequence, which we named GIPN. The rat cDNA we isolated contained the full ORF encoding a 338-aa protein (Fig. 1A). Bioinformatics analysis (Kyte-Doolittle, PSORT) predicts GIPN is an integral membrane protein with a large cytoplasmic domain and a short (≈30 aa) hydrophilic, C-terminal ectodomain (Fig. 1 B and C). Prosite analysis and detailed protein sequence inspection suggested the presence of a RING finger motif. To follow up this possibility, we aligned the putative RING finger domain with the RING fingers of Rbx1 and Mdm2 (27) (Fig. 1B). The characteristic feature of RING fingers is the presence of regularly spaced C/H pairs involved in Zn2+ binding. The main divergence between the consensus sequence of RING fingers and GIPN resides in (i) a longer loop between the second and third pair of C/H (27) and (ii) a CXXXC sequence (instead of CXXC) at the level of the fourth pair (28). Variations from the consensus could be found in Mdm2 between the second and third pair of C/H (27) and in Rbx1 at the level of the fourth C/C pair (28). We believe that GIPN can be best classified as a RING finger-like protein.
Fig. 1.

(A) Alignment of rat (R), mouse (M), and human (H) GIPN proteins. Identical residues are shaded in black, and conserved residues are shaded in gray; cysteine and histidine pairs are boxed. (B) The RING finger-like domain of GIPN (amino acids 175–283) was aligned with the RING finger of Rbx1(amino acids 42–97) and Mdm2 (amino acids 438–478). (C) GIPN is predicted to be an integral membrane protein with a large RING finger-like cytoplasmic domain, a putative transmembrane domain (amino acids 287–309), and a short (30-aa) C terminus. The N terminus of GIPN (amino acids 1–110) contains several dileucine motifs (LL-rich region).
GIPN Is a Unique Gene. BLAST searches with the rat cDNA sequence revealed significant mouse and human EST sequences and a human HSPC019 cDNA sequence (GenBank accession no. XM004529). We sequenced a mouse EST (GenBank accession no. BF161283) that contained an in-frame stop codon upstream from the putative start codon and a human EST (GenBank accession no. BE276148) containing a full ORF. We found the corresponding human gene on a BAC clone (GenBank accession no. Z98200). The human GIPN gene is located on chromosome 6(q16–21) and contains six coding exons interspaced by five introns over a distance of 35 kb. The rat and mouse ORFs translate into a 338-aa protein and the human ORF translates into a 334-aa protein (Fig. 1 A). We also found an ortholog of GIPN in Drosophila (GH26007) but none in Caenorhabditis elegans or Saccharomyces cerevisiae. Further searches in mammalian databases provided no DNA or protein sequences with lower homologies, suggesting that GIPN is not part of a larger protein family.
GIPN Interacts with RGS-GAIP in Vitro. When in vitro labeled GIPN was incubated with His6-GAIP or His6-AGS3 (amino acids 424–650) on Ni2+-agarose beads, it bound to GAIP but not to beads alone (Fig. 2A). These results confirm that binding of GIPN to GAIP is specific.
Fig. 2.

In vitro translated GIPN interacts specifically with GAIP. His6-GAIP and His6-AGS3 (424–650) bound to Ni2+-agarose beads were incubated in the presence of 35S-labeled, in vitro translated GIPN. (A) GIPN (arrow) bound specifically to GAIP but not to beads alone and marginally to AGS3. The fourth lane represents 20% of the total [35S]GIPN input. (B) Coomassie blue staining of the gel (Left) showing His6-GAIP and His6-AGS3 bound to the beads.
GIPN Interacts with Subfamily A RGS Proteins. By two-hybrid analysis, GIPN interacted with RGS-GAIP, RGSZ1, and RGSZ2, members of subfamily A (29), but not with RGS4, a member of subfamily B (Fig. 3A). Interestingly, GIPN did not interact with RET-RGS1, an alternatively spliced form of RGSZ1 with a 156-aa, N-terminal extension (30, 31).
Fig. 3.

One-on-one two-hybrid interactions. (A) GIPN interacts with RGS-GAIP (row 2), RGSZ1 (row 4), and RGSZ2 (row 5) but not with RGS4 (row 3) or RET-RGS1 (row 6). The interaction between RGS-GAIP and Gαi3 (row 1) serves as positive control. (B) The N terminus of GIPN (amino acids 1–110) interacts with RGS-GAIP (row 7), RGSZ1 (row 8), and RGSZ2 (row 9). GIPN interacts with RGS-GAIP1–79 (row 10) but not with RGS-GAIP80–206 (row 11). RGS-GAIP1–38 lacking the cysteine string motif does not interact with GIPN (row 12). RGS-GAIP1–79 interacts with the N terminus of GIPN (amino acids 1–110) (row 13). Yeast were cotransformed with the indicated constructs. For each interaction pair, three separate colonies were streaked on a filter and scored for β-galactosidase activity.
The Cysteine String Region of RGS-GAIP Interacts with the N Terminus of GIPN. To determine the sites of interaction between GIPN and GAIP, we used deletion mutants in a two-hybrid assay (Fig. 3B). Neither GAIP80–217 nor GAIP1–38 interacted with GIPN (Fig. 3B). We conclude that the region between amino acids 39 and 79, N-terminal to the RGS domain containing the cysteine string, is the domain in GAIP that interacts with GIPN. This domain is also the signature region in the N termini of subfamily A RGS proteins, in keeping with the specificity of GIPN for members of this subfamily. In GIPN, the first 110 residues are sufficient for interaction with GAIP, RGSZ1, and RGSZ2 (Fig. 3B).
GIPN mRNA Is Ubiquitously Expressed. Northern blot analysis was performed on multiple tissues of adult human, rat, and mouse. In humans (Fig. 4A), two mRNAs of different sizes, ≈4.4 kb (minor) and 3.2 kb (major), were expressed in all tissues examined (highest in brain followed by lung). In rat and mouse (not shown), we found a single ≈4.4-kb mRNA present in all tissues examined.
Fig. 4.

(A) Human multiple tissue Northern blot. GIPN mRNA is expressed in all tissues tested. h, heart; br, brain; pl, placenta; lg, lung; li, liver; sm, skeletal muscle; k, kidney; p, pancreas. Two different mRNAs (4.4 and 3.2 kb) are expressed in all human tissues, whereas in rat and mouse (not shown) only a single band (4.4 kb) could be detected. (B) Immunoblot for endogenous GIPN in mouse kidney. Membrane (100,000 × g pellet, P) and cytosolic (100,000 × g supernatant, S) fractions were prepared from a postnuclear supernatant. Proteins (50 μg) were separated by SDS/10% PAGE and immunoblotted with affinity-purified anti-GIPN (0.4 μg/ml). Two bands, 94 and 38 kDa, are found in postnuclear supernatant; the 38-kDa band is present exclusively in soluble (S, cytosolic) fractions, and the 94-kDa band is present exclusively in the pellet (P, membrane) fractions.
GIPN Is Found in Both Cytosolic and Membrane Fractions. When cytosolic and membrane fractions prepared from mouse kidney were immunoblotted for GIPN, we found a 38-kDa immunoreactive band present in the cytosolic fraction and an ≈94-kDa band in the membrane fraction (Fig. 4B). Anti-GIPN IgG affinity-purified on the 38-kDa band recognized the 94-kDa band; similarly, IgG affinity-purified on the 94-kDa band recognized the 38-kDa protein, suggesting that the two bands represent different forms of GIPN.
GIPN Is Concentrated at the Cell Membrane in Transfected HEK293 Cells. When GIPN was expressed in HEK293 cells, staining for GIPN was detected along the plasma membrane by immunofluorescence (Fig. 5A). When semithin cryosections of rat kidney were similarly labeled, there was a strong signal at the base of distal tubules and a similar but weaker staining in proximal tubules (Fig. 5B). By immunogold labeling at the EM level, it was apparent that in these cells GIPN is present on the characteristic infoldings of the basolateral plasma membrane (Fig. 6).
Fig. 5.
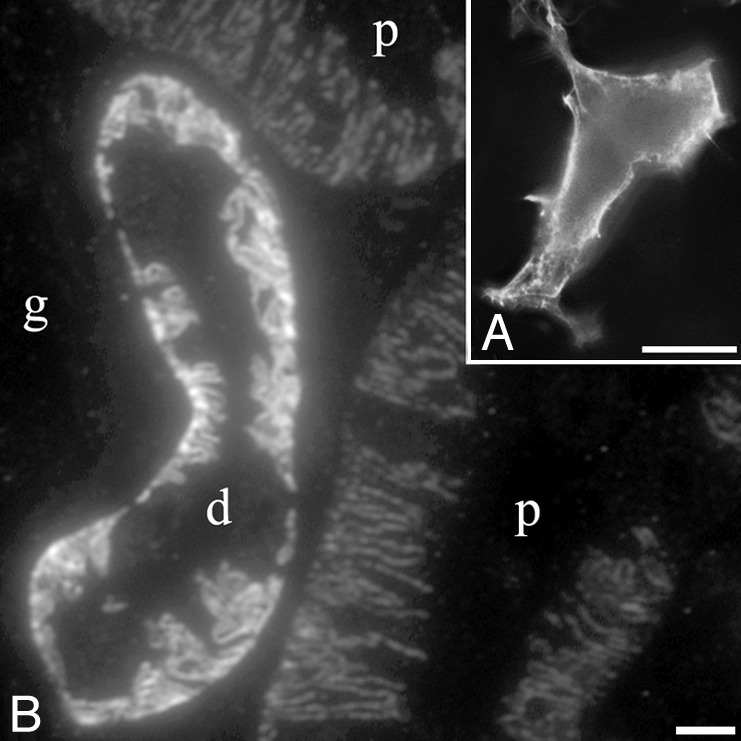
Localization of GIPN. (A) GIPN is found mainly along the plasma membrane when expressed in HEK293 cells. Immunofluorescence was performed on fixed and permeabilized cells with affinity-purified anti-GIPN followed by Alexa 488-conjugated goat anti-rabbit F(ab)′2. (B) Endogenous GIPN is concentrated on basal infoldings of the cell membranes of distal (d) and proximal (p) tubules in rat kidney. Semithin cryosection of rat kidney cortex incubated as in A. g, glomerulus. (Scale bars = 5 μm.)
Fig. 6.

Immunoelectron microscopic localization of GIPN in a distal kidney tubule showing gold particles concentrated along the infoldings of the basal cell membrane (arrows). Shown is an ultrathin cryosection of rat kidney cortex incubated with affinity-purified anti-GIPN followed by goat anti-rabbit IgG conjugated to 5 nm gold. bm, basement membrane; m, mitochondria; *, intercellular spaces. (Scale bar = 100 nm.)
GIPN Shows Zn-Dependent, Autoubiquitination Activity in Vitro. Our alignment analysis suggested that GIPN has a RING finger-like motif, suggesting that it could have E3 ubiquitin ligase activity (32). RING fingers need to coordinate two Zn2+ ions to maintain their conformation (33). To test for the presence of such activity, we determined whether GIPN is capable of Zn2+-dependent and E1/E2-dependent autoubiquitination. Initially, we used reticulocyte lysate as a source for E1 ubiquitin-activating enzyme and E2 ubiquitin-conjugating enzyme. In the presence of GST-GIPN, ATP, lysate, and the proteasome inhibitor MG-132, polyubiquitination was observed as visualized by the typical broad band (Fig. 7A). Ubiquitination of GIPN is Zn2+-dependent because addition of the divalent cation chelator, TPEN, inhibited autoubiquitination of GIPN (Fig. 7A). Addition of ZnCl2 to a mixture preincubated with TPEN restored autoubiquitination of GIPN, thereby demonstrating the Zn2+ dependency of the reaction.
Fig. 7.

GIPN has in vitro Zn2+-sensitive ubiquitination activity. (A) Autopolyubiquitination of GIPN is Zn2+-dependent. GST-GIPN bound to glutathione-agarose beads was incubated with ubiquitin and MG-132 in the presence or absence of ATP and lysate. Proteins were immunoblotted with anti-ubiquitin. The broad band (lane 2) indicates polyubiquitination. When lysates are preincubated with the Zn2+ chelator TPEN, polyubiquitination is significantly diminished (lane 3). When a 10-fold concentration of ZnCl2 (relative to TPEN) is added, polyubiquitination is greatly enhanced (lane 4). No polyubiquitination is detected in the absence of ATP and lysate (lane 1). (B) GIPN undergoes autoubiquitination in the presence of E1 and E2. Purified recombinant E1, E2 (Ubc4), and Hrd1 or GIPN were incubated with ubiquitin and ATP. Proteins were immunoblotted with anti-ubiquitin. GIPN is able to autoubiquitinate itself in a concentration-dependent manner (lanes 3 and 4). In the absence of E3 (lane 2), no signal can be detected. Hrd1 (lane 1) was used as a positive control.
Next, we investigated whether GIPN can be autoubiquitinated in a reconstituted system. As a positive control, E1-activating enzyme (UBA1), E2-conjugating enzyme (UBC4), and the E3 ligase Hrd1 were incubated together, which resulted in autoubiquitination of Hrd1 (Fig. 7B). Replacing Hrd1 with GIPN resulted in autoubiquitination of GIPN, although ubiquitination was significantly weaker than when equivalent amounts of Hrd1 protein were added. We conclude from these experiments that GIPN has E3 ubiquitin ligase activity.
Overexpression of GIPN Down-Regulates Gαi3 Expression. Our findings that GIPN binds to GAIP, which in turn binds to Gαi subunits, together with the fact that G proteins are known to be down-regulated via the ubiquitin/proteasome pathway, led us to explore whether overexpression of GIPN might affect turnover of Gαi. To find out whether this is the case, we determined the total endogenous Gαi3 in cells expressing GIPN vs. mock-transfected controls by immunoblotting. We found that GIPN-transfected cells contained significantly less (≈30%) endogenous Gαi3 protein, whereas the levels of the cytoskeletal protein ezrin used as a control were the same (Fig. 8A).
Fig. 8.

(A) Overexpression of GIPN reduces endogenous Gαi3. HEK293 cells were transfected with FLAG-GIPN or FLAG mock. Proteins (25 μg) were immunoblotted with anti-Gαi3 (EC) or anti-ezrin and detected by ECL. In cells transfected with GIPN, total endogenous Gαi3 is reduced compared with mock-transfected cells. The level of ezrin remains unchanged. (B) GIPN shortens the half-life (t1/2) of Gαi3. HEK 293 cells were transfected with Gαi3-YFP and either FLAG-GIPN or FLAG-mock. Cells were pulse-labeled for 20 min and chased for 0, 1, or 2 h, and Gαi3-YFP was immunoprecipitated with anti-GFP followed by autoradiography. Some cells were treated with the proteasome inhibitor MG-132 before labeling. The amount of Gαi3 remaining at a 2-h chase is considerably reduced in cells expressing GIPN. In the presence of GIPN, the t1/2 of Gαi3 is shorter (1.4 h ± 0.1 vs. 2.0 h ± 0.2, n = 5). The increased degradation of Gαi3 is proteasome-dependent as it is reversed (t1/2 = 2.9 ± 0.6, n = 3) by pretreatment with MG-132.
We also carried out pulse–chase experiments on cells transfected with Gαi3-YFP and determined the half-life (t1/2) of Gαi3-YFP (Fig. 8B). The t1/2 of Gαi3 in mock-transfected cells was found to be 2 h (±0.2) vs. 1.4 h (±0.1) in GIPN-transfected cells. This effect is proteasome-dependent because when the cells were preincubated with the proteasome inhibitor MG-132, the effect of GIPN was abolished. We conclude from these experiments that GIPN promotes proteasome-dependent degradation of Gαi3.
Discussion
We have isolated a protein, GIPN, that interacts with subfamily A RGS proteins (RGS-GAIP, RGSZ1, and RGSZ2; ref. 29), which share a common cysteine string motif. GIPN has a leucine-rich N terminus that binds to RGS-GAIP, a putative C-terminal transmembrane domain, and a central RING finger-like domain. GIPN is highly conserved from flies to humans, and its mRNA is ubiquitously expressed. We further demonstrated that GIPN has E3 ubiquitin ligase activity and that overexpression of GIPN promotes turnover (degradation) of Gαi3 via the proteasome pathway.
Ubiquitination is a posttranslational modification that conjugates ubiquitin, a 76-aa peptide, to the side chains of specific lysine residues in protein targets. Polyubiquitination leads to the degradation of specific protein substrates by the proteasome (27, 34). Monoubiquitination or limited ubiquitination (<n = 4) triggers internalization of some receptors and targeting of other proteins to lysosomes for degradation (35, 36). Ubiquitination is performed by the sequential action of an E1 ubiquitin-activating enzyme that activates ubiquitin, which is transesterified onto an E2-conjugating enzyme, which in turn couples to an E3 ubiquitin ligase (37). There are two main classes of E3s: RING finger and HECT E3s (33). Each RING finger E3 binds to a specific E2 via the RING finger domain, which is a prerequisite for ubiquitination of the substrate protein. Recently, several proteins have been described to have divergent motifs that are structurally and functionally similar to RING fingers and have E3 ligase activity (38, 39).
A key question to be answered is: what is the substrate ubiquitinated by GIPN? Our finding that GIPN can interact with RGS-GAIP, RGSZ1, and RGSZ2, members of the same subfamily of RGS proteins, suggested that these specific RGS proteins might represent the substrates. E3 ligases use protein–protein interaction domains outside the catalytic domain to bind their substrate (33). This also holds for GIPN, which uses its N-terminal leucine-rich domain to bind RGS-GAIP. We found no evidence for enhanced degradation of RGS-GAIP or for its polyubiquitination by simply overexpressing GIPN in cells. However, we found a clear decrease in the t1/2 of Gαi3 upon overexpression of GIPN, with a concomitant decrease in Gαi3 protein expression levels. The effects we observed on Gαi3 are modest but reproducible, and we propose two possible, reasonable explanations: (i) overexpressing GIPN might not be sufficient for effective Gαi3 degradation, and (ii) GIPN-dependent Gαi3 degradation might be part of a medium to long-term process requiring longer times for expression of GIPN. Because RGS-GAIP can also interact with other members of the Gαi subfamily, we cannot exclude that other Gαi members might be better substrates for GIPN.
Most RGS proteins specifically interact with Gα in its GTP to GDP hydrolysis transition state. RGS-GAIP is also able to interact with the GTP-bound (activated) form of Gαi3 (40). We speculate that this might increase the interaction period of RGS-GAIP with Gαi3 and represent a mechanism for selectively degrading the active form of Gα. This assumption is in keeping with reports that the t1/2 of G proteins is shortened by activation (41, 42).
We found GIPN concentrated at the cell membrane when expressed in HEK293 cells and endogenous GIPN to be located on the basolateral plasma membrane in the epithelia of proximal and distal kidney tubules. Interestingly, this is the site where the majority of GPCRs and many G proteins (43, 44) are located in these cells, suggesting that GIPN could be part of a GPCR/Gα/ RGS-GAIP signaling complex. The presence of two different cross-immunoreactive species in cytosolic (38 kDa) and membrane (>94 kDa) fractions from mouse kidney raises the question of the relationship between the two forms. By Northern blot analysis, we detected two mRNA species with a difference of 1,200 bp for GIPN, suggesting the existence of an alternatively spliced form. Further studies are needed to resolve this issue.
GIPN interacts with all three RGS subfamily A members, i.e., RGS-GAIP, RGSZ1, and RGSZ2, but not with RET-RGS1. RET-RGS1 is a retina-specific, alternatively spliced form of RGSZ1 with a 156-aa N-terminal extension (30) that most likely masks interaction with GIPN. We found GIPN interacts specifically with the cysteine string region, the signature domain of subfamily A.
There are now numerous examples of ubiquitination as a regulatory event in GPCR signaling and trafficking. Ubiquitination of the β2-adrenergic receptor and of β-arrestin by the E3 ubiquitin ligase Mdm2 has been shown to regulate trafficking of the receptor (1). G protein-coupled receptor kinase 2 (GRK2) is ubiquitinated and degraded by the proteasome after β2-adrenergic receptor activation (2). CXCR4 and DOR, both GPCRs, are ubiquitinated and sorted to lysosomes after ligand-induced internalization (45, 46). In yeast, Ste7 (mitogen-activated protein kinase kinase) is also ubiquitinated when a G protein pathway is activated by pheromone (47). Degradation of the yeast G protein Gpa1 (4, 5), Go (6), and Gβγ (7) subunits has also been documented. Moreover, RGS4 and RGS16 were recently shown to be subject to degradation by the N-end rule pathway in vitro (3), implying degradation by the ubiquitin-proteasome system. Direct binding of RGS proteins to elements of the ubiquitin degradation and targeting system has not previously been described.
Our results suggest that RGS-GAIP, like many other RGS proteins (14), is more than just a GTPase-activating protein for Gαi3. They also suggest that RGS-GAIP could act as a bifunctional adaptor that uses the RGS domain as a Gα binding domain and its cysteine string motif for membrane anchoring as well as to bind to GIPN, which degrades the bound Gα.
Acknowledgments
We thank Dr. R. Hampton, R. Garza, O. Bazirgan, and N. Bays (Division of Biology, University of California at San Diego) for helpful advice and assistance with the ubiquitin assays. This work was supported by National Institutes of Health Research Grants CA58689, DK17780, and CA100768 (to M.G.F.).
Abbreviations: Gα, G alpha subunits; GPCR, G protein-coupled receptors; TPEN, tetrakis (2-pyridylmethyl) ethylenediamine.
References
- 1.Shenoy, S. K., McDonald, P. H., Kohout, T. A. & Lefkowitz, R. J. (2001) Science 294 1307-1313. [DOI] [PubMed] [Google Scholar]
- 2.Penela, P., Ruiz-Gomez, A., Castano, J. G. & Mayor, F., Jr. (1998) J. Biol. Chem. 273 35238-35244. [DOI] [PubMed] [Google Scholar]
- 3.Davydov, I. V. & Varshavsky, A. (2000) J. Biol. Chem. 275 22931-22941. [DOI] [PubMed] [Google Scholar]
- 4.Madura, K. & Varshavsky, A. (1994) Science 265 1454-1458. [DOI] [PubMed] [Google Scholar]
- 5.Marotti, L. A., Jr., Newitt, R., Wang, Y., Aebersold, R. & Dohlman, H. G. (2002) Biochemistry 41 5067-5074. [DOI] [PubMed] [Google Scholar]
- 6.Busconi, L., Guan, J. & Denker, B. M. (2000) J. Biol. Chem. 275 1565-1569. [DOI] [PubMed] [Google Scholar]
- 7.Obin, M., Lee, B. Y., Meinke, G., Bohm, A., Lee, R. H., Gaudet, R., Hopp, J. A., Arshavsky, V. Y., Willardson, B. M. & Taylor, A. (2002) J. Biol. Chem. 277 44566-44575. [DOI] [PubMed] [Google Scholar]
- 8.De Vries, L., Zheng, B., Fischer, T., Elenko, E. & Farquhar, M. G. (2000) Annu. Rev. Pharmacol. Toxicol. 40 235-271. [DOI] [PubMed] [Google Scholar]
- 9.De Vries, L. & Farquhar, M. G. (2002) Methods Enzymol. 344 657-673. [DOI] [PubMed] [Google Scholar]
- 10.Ross, E. M. & Wilkie, T. M. (2000) Annu. Rev. Biochem. 69 795-827. [DOI] [PubMed] [Google Scholar]
- 11.Tesmer, J. J., Berman, D. M., Gilman, A. G. & Sprang, S. R. (1997) Cell 18 251-261. [DOI] [PubMed] [Google Scholar]
- 12.De Vries, L., Elenko, E., McCaffery, J. M., Fischer, T., Hubler, L., McQuistan, T., Watson, N. & Farquhar, M. G. (1998) Mol. Biol. Cell 9 1123-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spector, D. L., Goldman, R. D. & Leinwand, L. A. (1998) in Cells: A Laboratory Manual (Cold Spring Harbor Lab. Press, Plainview, NY), Vol. 1, pp. 73.9-73.12. [Google Scholar]
- 14.De Vries, L., Lou, X., Zhao, G., Zheng, B. & Farquhar, M. G. (1998) Proc. Natl. Acad. Sci. USA 95 12340-12345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Madden, T. L., Tatusov, R. L. & Zhang, J. (1996) Methods Enzymol. 266 131-141. [DOI] [PubMed] [Google Scholar]
- 16.Higgins, D. G., Thompson, J. D. & Gibson, T. J. (1996) Methods Enzymol. 266 383-402. [DOI] [PubMed] [Google Scholar]
- 17.Fischer, T., Elenko, E., McCaffery, J. M., DeVries, L. & Farquhar, M. G. (1999) Proc. Natl. Acad. Sci. USA 96 6722-6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Vries, L., Fischer, T., Tronchère, H., Brothers, G. M., Strockbine, B., Siderovski, D. P. & Farquhar, M. G. (2000) Proc. Natl. Acad. Sci. USA 97 14364-14369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang, J., Ducret, A., Tu, Y., Kozasa, T., Aebersold, R. & Ross, E. M. (1998) J. Biol. Chem. 273 26014-26025. [DOI] [PubMed] [Google Scholar]
- 20.Jordan, J. D., Carey, K. D., Stork, P. J. & Iyengar, R. (1999) J. Biol. Chem. 274 21507-21510. [DOI] [PubMed] [Google Scholar]
- 21.De Vries, L., Mousli, M., Wurmser, A. & Farquhar, M. G. (1995) Proc. Natl. Acad. Sci. USA 92 11916-11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsuda, N., Suzuki, T., Tanaka, K. & Nakano, A. (2001) J. Cell Sci. 114 1949-1957. [DOI] [PubMed] [Google Scholar]
- 23.Bays, N. W., Wilhovsky, S. K., Goradia, A., Hodgkiss-Harlow, K. & Hampton, R. Y. (2001) Mol. Biol. Cell 12 4114-4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weiss, T. S., Chamberlain, C. E., Takeda, T., Lin, P., Hahn, K. M. & Farquhar, M. G. (2001) Proc. Natl. Acad. Sci. USA 98 14961-14966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonifacino, J. S., Dasso, M., Harford, J. B., Lippincott-Schwartz, J. & Yamada, K. M. (1998) in Current Protocols in Cell Biology, ed. Morgan, K. S. (Wiley, New York), Vol. 1, pp. 714-715. [Google Scholar]
- 26.Bence, N. F., Sampat, R. M. & Kopito, R. R. (2001) Science 292 1552-1555. [DOI] [PubMed] [Google Scholar]
- 27.Weissman, A. M. (2001) Nat. Rev. Mol. Cell Biol. 2 169-178. [DOI] [PubMed] [Google Scholar]
- 28.Zheng, N., Schulman, B. A., Song, L., Miller, J. J., Jeffrey, P. D., Wang, P., Chu, C., Koepp, D. M., Elledge, S. J., Pagano, M., et al. (2002) Nature 416 703-709. [DOI] [PubMed] [Google Scholar]
- 29.Zheng, B., De Vries, L. & Farquhar, M. G. (1999) Trends Biochem. Sci. 24 411-414. [DOI] [PubMed] [Google Scholar]
- 30.Barker, S. A., Wang, J., Sierra, D. A. & Ross, E. M. (2001) Genomics 78 223-229. [DOI] [PubMed] [Google Scholar]
- 31.Faurobert, E. & Hurley, J. B. (1997) Proc. Natl. Acad. Sci. USA 94 2945-2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Joazeiro, C. A. & Weissman, A. M. (2000) Cell 102 549-552. [DOI] [PubMed] [Google Scholar]
- 33.Jackson, P. K., Eldridge, A. G., Freed, E., Furstenthal, L., Hsu, J. Y., Kaiser, B. K. & Reimann, J. D. (2000) Trends Cell Biol. 10 429-439. [DOI] [PubMed] [Google Scholar]
- 34.Thrower, J. S., Hoffman, L., Rechsteiner, M. & Pickart, C. M. (2000) EMBO J. 19 94-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Terrell, J., Shih, S., Dunn, R. & Hicke, L. (1998) Mol. Cell 1 193-202. [DOI] [PubMed] [Google Scholar]
- 36.Hicke, L. (1999) Trends Cell Biol. 9 107-112. [DOI] [PubMed] [Google Scholar]
- 37.Bonifacino, J. S. & Weissman, A. M. (1998) Annu. Rev. Cell Dev. Biol. 14 19-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aravind, L. & Koonin, E. V. (2000) Curr. Biol. 10 132-134. [DOI] [PubMed] [Google Scholar]
- 39.Lu, Z., Xu, S., Joazeiro, C., Cobb, M. H. & Hunter, T. (2002) Mol. Cell 9 945-956. [DOI] [PubMed] [Google Scholar]
- 40.De Vries, L., Elenko, E., Hubler, L., Jones, T. L. & Farquhar, M. G. (1996) Proc. Natl. Acad. Sci. USA 93 15203-15208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Levis, M. J. & Bourne, H. R. (1992) J. Cell Biol. 119 1297-1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jewell-Motz, E. A., Donnelly, E. T., Eason, M. G. & Liggett, S. B. (1998) Biochemistry 37 15720-15725. [DOI] [PubMed] [Google Scholar]
- 43.Stow, J. L., Sabolic, I. & Brown, D. (1991) Am. J. Physiol. 261 F831-F840. [DOI] [PubMed] [Google Scholar]
- 44.Brunskill, N., Bastani, B., Hayes, C., Morrissey, J. & Klahr, S. (1991) Kidney Int. 40 997-1006. [DOI] [PubMed] [Google Scholar]
- 45.Marchese, A. & Benovic, J. L. (2001) J. Biol. Chem. 276 45509-45512. [DOI] [PubMed] [Google Scholar]
- 46.Chaturvedi, K., Bandari, P., Chinen, N. & Howells, R. D. (2001) J. Biol. Chem. 276 12345-12355. [DOI] [PubMed] [Google Scholar]
- 47.Wang, Y. & Dohlman, H. G. (2002) J. Biol. Chem. 277 15766-15772. [DOI] [PubMed] [Google Scholar]