

Abstract
The yeast S. cerevisiae is a central model organism in eukaryotic cell studies and a major component in many food and biotechnological industrial processes. However, the wide knowledge regarding genetics and molecular biology of S. cerevisiae is based on an extremely narrow range of strains. Studies of natural populations of S. cerevisiae, not associated with human activities or industrial fermentation environments, are very few. We isolated a panel of S. cerevisiae strains from a natural microsite, “Evolution Canyon” at Mount Carmel, Israel, and studied their genomic biodiversity. Analysis of 19 microsatellite loci revealed high allelic diversity and variation in ploidy level across the panel, from diploids to tetraploids, confirmed by flow cytometry. No significant differences were found in the level of microsatellite variation between strains derived from the major localities or microniches, whereas strains of different ploidy showed low similarity in allele content. Maximum genetic diversity was observed among diploids and minimum among triploids. Phylogenetic analysis revealed clonal, rather than sexual, structure of the triploid and tetraploid subpopulations. Viability tests in tetrad analysis also suggest that clonal reproduction may predominate in the polyploid subpopulations.
THE budding yeast Saccharomyces cerevisiae is one of the central model organisms of eukaryotic cell studies (Dickinson 2000). The wide knowledge about the genetic and molecular biology of the yeast S. cerevisiae has accumulated on an extremely narrow range of genotypes selected due to their specific technological features or suitability to laboratory conditions and, hence, hardly representing the species (Liu et al. 1996; Mortimer 2000).
The yeast S. cerevisiae is also a central component of many important industrial processes, including baking, brewing, distilling, and wine making. Once again, in this domain, most studies have included selected, commercially available yeast strains removed from the natural adaptation and evolution processes. Relatively limited genetic work has been done on commercial baking, wine, and brewing strains. In the last decade, some studies dealt with isolates from nature, wineries, and grapes (Sangorrin et al. 2001; van der Aa et al. 2001; Fay and Benavides 2005), as well as from contaminants of different lager breweries (van der Aa and Jespersen 1998; Jespersen et al. 2000). In fact, cells of S. cerevisiae are rarely isolated from natural grape surfaces except damaged grapes (Vaughan-Martini and Martini 1995; Martini et al. 1996; Mortimer and Polsinelli 1999), suggesting that insects such as bees or Drosophila are vectors for spreading of this microorganism (Stevic 1962; Snowdon and Cliver 1996; Mortimer and Polsinelli 1999).
Mortimer (2000) reported that attempts to find S. cerevisiae in regions remote from human activities have been unsuccessful. A model proposed by Naumov (1996) states that S. cerevisiae strains in European soil originate from human activity, i.e., are found only in association with human civilization, especially in winery environments. On the other hand, S. cerevisiae was also found in exudates from North American oaks (Naumov et al. 1998). The foregoing evidence calls for extended population-genetic (Zeyl 2000) and molecular-genetic studies of yeast in nature (Liti and Louis 2005).
The yeast S. cerevisiae can exist in a vegetative mode in haploid, diploid, and higher ploidy states. Haploid cells of S. cerevisiae exhibit one of two phenotypes: mating types a or α. Correspondingly, diploid and polyploid cells can exhibit one of three mating phenotypes: a, α, or a/α. When cells of opposite mating types meet they participate in a mating process that results in cell and nuclear fusion to create an a/α-zygote. a/α-cells can either reproduce by mitosis or undergo meiosis and sporulation, with a further possibility of a × α gamete fusion.
Mating type of haploid cells can be stable (heterothallic yeast) or unstable (homothallic yeast). In heterothallic yeasts, haploid culture derived from a single ascospore remains haploid until brought into contact with another haploid culture of the opposite mating type. In contrast, in homothallic culture, derived from a single ascospore, there is an alternation of haploid and diploid generations due to mating-type interconversion followed by mating (for review see Cox 1995). These aspects can be considered as regulation of the mode of reproduction in yeast, from full amphymixis (sex) to automixis. Zeyl and Bell (1997) show that sex increases mean fitness in an environment to which the populations were well adapted, but not in an environment to which new adaptation occurred, supporting the hypothesis that the advantage of sexuality lay in the removal of deleterious mutations.
Zeyl et al. (2003) tested rates of adaptation using asexual diploid and haploid yeast populations. They showed that diploidy may slow down adaptation, but by large populations only. In mutagenesis experiments (Mable and Otto 2001) haploids displayed a more pronounced decrease in apparent growth rate than diploids. Tetraploids did not show increased benefits of masking deleterious mutations compared with diploids. However, all treated tetraploid strains decreased in ploidy level whereas some of the treated haploid lines increased in ploidy level. These findings shed a new light on ploidy level as an adaptive evolutionary trait.
A panel of S. cerevisiae strains, among 25 different yeast species, belonging to 14 genera (Nagornaya et al. 2003), was isolated from a well-studied natural microsite called “Evolution Canyon,” which is located in Lower Nahal Oren in Mount Carmel National Park, Haifa, Israel (32° 24′ N, 34° 58′ E) (Nevo 1995, 1997, 2001). The opposing slopes of this canyon, the “African” south-facing slope (SFS) and the “European” north-facing slope (NFS), are separated by only 100 m at the bottom and 400 m at the top. Despite sharing the same geology and macroclimate, the two slopes differ sharply in microclimatic conditions, because of the higher (up to 200–800%) solar radiation on the SFS as compared with the NFS (Pavlicek et al. 2003), causing also substantial interslope biotic contrasts. The SFS is warmer, drier, microclimatically more variable, and less predictable than the NFS. Parallel genotypic and phenotypic diversity patterns across phylogeny of bacteria, fungi, plants, and animals on the opposing slopes of Evolution Canyon suggest that natural selection plays a major role in biodiversity evolution (Nevo 1995; Andreyuk et al. 1999; Kalendar et al. 2000).
Higher genetic polymorphisms on the SFS are naturally selected as an adaptive strategy to cope with its higher ecological heterogeneity and stress (Lamb et al. 1998; Krugman et al. 2001; Nevo 2001; Saleem et al. 2001; Satish et al. 2001). Indeed, genetic diversity was higher on the more heterogeneous and stressful SFS in 11 of 14 model organisms tested at this site, including wild barley, fruit flies, beetles, and cyanobacteria (Gutterman and Nevo 1994; Rankevich et al. 1996; Lamb et al. 1998; Andreyuk et al. 1999; Krugman et al. 2001; Michalak et al. 2001; Nevo 2001; Saleem et al. 2001; Satish et al. 2001). This could highlight the importance of ecological stress in evolution and its remarkable effect on the genetic system (Grishkan et al. 2003). For instance, strong interslope differentiation for a complex of adaptive traits of Drosophila was found, including physiological and behavioral traits as well as different rates of mutation and recombination (Nevo et al. 1998; Korol et al. 2000; Iliadi et al. 2001; Michalak et al. 2001; Lupu et al. 2004; Singh et al. 2005; Zamorzaeva et al. 2005; Rashkovetsky et al. 2006). This remarkable differentiation has evolved in spite of the small interslope distance. Thus, the obtained evidence leads to a conclusion that strong microclimatic natural selection can override migration and random drift and generate slope-specific multitrait adaptive gene complexes that contribute to fitness at a microsite.
Here we use microsatellite loci to study S. cerevisiae biodiversity in isolates from Evolution Canyon. Microsatellites, or simple sequence repeats (SSRs), represent a class of DNA sequences consisting of tandemly organized, reiterated motifs. SSRs are abundant across eukaryotic genomes including yeasts that show high levels of polymorphism in motif copy number. SSR diversity may result from both external (ecological) and internal (genetic) effects (Li et al. 2002, 2004). In addition to their use as DNA markers, microsatellites have been described as molecular switches controlling gene expression in microbial and eukaryotic species (Trifonov 1989; Kashi et al. 1997; King et al. 1997; van Belkum et al. 1998; Li et al. 2004; Trifonov 2004). Functional significance of at least some SSRs has been demonstrated in critical experiments in many biological phenomena (reviews: Kashi et al. 1997; Kashi and Soller 1999; Li et al. 2002, 2004). SSRs might be a major source of genetic diversity and evolutionary adaptation to environmental stress. Indeed, evidence for microclimatic selection acting on SSR loci has been already reported (Li et al. 2000).
In this article we present results demonstrating high SSR polymorphism, ploidy variation, and clonal rather than sexual population structure, in the natural population of S. cerevisiae in Evolution Canyon.
MATERIALS AND METHODS
Strains:
Sixty-eight S. cerevisiae yeast strains were isolated from Evolution Canyon at seven collection sites, at three altitudes on each slope (lower, middle, and upper) and at valley bottom (VB) (see supplemental figure at http://www.genetics.org/supplemental/) as described in Nagornaya et al. (2003). In short, soil and plant samples were collected in sunny and shady places (including adjacent sites), hereafter sunny, shady, and leaf, respectively. Isolation of S. cerevisiae was conducted by dilution technique and the enrichment method (Beech and Davenport 1971). For identification, morphological, physiological, and biochemical criteria for Saccharomyces were employed (Kurtzman and Fell 1998). For reference, laboratory strains were used: Y422 (Sherman et al. 1993), Y102 and Y103 (MATa and MATα testers, respectively) (all kindly provided by Y. Kassir), and S288C (Mortimer and Johnston 1986) (kindly provided by R. K. Mortimer).
Tetrad dissection:
A micromanipulator equipped with a glass needle was used to isolate spores from asci (kindly made available by D. Kornitzer). The ascus walls were removed using 0.25 mg/ml Zymolase T100 (Sigma, St. Louis) in 1 m sorbitol.
Media:
Yeast cells were usually grown on YPD complete medium (prepared as in Sherman 1991) at 30°. Sporulation was induced on sporulation medium (SPO) (described in Kassir and Simchen 1991). Prototrophy was tested on synthetic dextrose minimal medium (Kaiser et al. 1994).
Mating-type tests:
Mating-type tests were performed as described in Sherman (1991). The tested strain was mixed, in parallel with the MATa and the MATα tester strains on a YPD plate and incubated at 30° overnight. These cultures were then replica plated on a SPO plate, along with the original strain. Sporulation was tested under a microscope after 24 and 48 hr of incubation at room temperature.
DNA extraction:
DNA was extracted by the yeast DNA miniprep method described in Kaiser et al. (1994).
Sequence polymorphism:
Microsatellite analysis:
Nineteen SSR (or microsatellite) loci were used to characterize the panel. The published genome of S. cerevisiae (http://www.yeastgenome.org/) was scanned by a computer program developed in our laboratory (ftp://ftp.technion.ac.il/pub/supported/biotech/) to detect SSR loci. Unique PCR primers were designed to amplify selected SSR loci according to the published genome of S. cerevisiae. In addition, some published microsatellite markers were used as well (Field and Wills 1998). The employed SSRs included di- and trinucleotide repeats dispersed over nine chromosomes (Table 1).
TABLE 1.
List of SSR loci selected for the analysis of genomic diversity within S. cerevisiae isolates from Evolution Canyon
| Name of SSR locus | Chromosome no. | Coordinates (bp) | Core motif | No. of alleles found |
|---|---|---|---|---|
| ChII-ATP I | II | 370450 | (AT)13 | 6 |
| ChIV-1038.6 | IV | 1038641 | (TCA)10 | 3 |
| ChVI-FAB1 | VI | 186212 | (ATA)19 | 6 |
| ChIX-105.7 | IX | 105769 | (TAA)19 | 7 |
| ChX-188.7 | X | 188731 | (TCT)6 | 4 |
| ChX-403.7 | X | 403763 | (CAG)8 | 4 |
| ChX-423.8 | X | 423865 | (AT)12 | 5 |
| ChX-469.8 | X | 469823 | (TAA)16 | 6 |
| ChX-518.90 | X | 518948 | (CA)20 | 9 |
| ChX-GRR | X | 593880 | (TTG)10 | 3 |
| ChX-639.6 | X | 639684 | (AC)19 | 6 |
| ChXI-126.1 | XI | 126106 | (GAA)10 | 9 |
| ChXI-184.5 | XI | 184504 | (TAA)8 | 3 |
| ChXI-576.1 | XI | 576129 | (TCG)13 | 7 |
| ChXII-511.5 | XII | 511525 | (CAG)10 | 5 |
| ChXII-823.4 | XII | 823442 | (GA)32 | 6 |
| ChXIII-CMP II | XIII | 159854 | (AT)12 | 5 |
| ChXIII-ORF4 | XIII | 209872 | (TGA)11 | 4 |
| ChXIII-388.6 | XIII | 388685 | (AT)14 | 4 |
The number of alleles found in SSR loci analyzed in the panel is shown. The name of the locus, its chromosomal location, the SSR motif, and the number of repeats according to the published genome are given.
Size of PCR products was determined using an automated sequencer, ALFexpress (Pharmacia, Uppsala, Sweden). Sequencing gel-running conditions were: 1800 V, 50 mA, and 50 W at 50° for 5 hr. Product sizes were calculated by the ALFwin fragment analysis software using an ALF size marker (50–500 bp) as an external marker. For validation of SSR marker analysis, direct sequencing was conducted for some of the amplified fragments.
Taxonomic analysis:
To validate the allocation of the natural isolates to S. cerevisiae, sequence analyses of two loci were performed in a part of our isolates (see Kurtzman and Robnett 2003):
ITS1-5.8S-ITS2 rDNA: primers (for amplification and sequencing) were TCCTCCGCTTATTGATAT(f) and GGAAGTAAAAGTCGTAACAAGG(r). The annealing temperature for PCR was 50°.
Translation EF-1 αA gene: primers (for amplification and sequencing) were TTCTTCGACTATGCTGGAGG(f), and TAAGGTTACCAAGGCTGCTC(r). The annealing temperature for PCR was 58°.
All sequences were aligned with reference sequences of all Saccharomyces sensu stricto species using ClustalW. GenBank accession numbers for the reference sequences are AY046146–AY046152 and AY130303–AY130313 for the ITS1-5.8S-ITS2 locus and AF402004–AF402009, AF402010–AF402016, AY130808, and AY130810–AY130813 for the translation EF-1 αA locus. Phylogenetic analysis was conducted for the ITS1-5.8S-ITS2 locus using the Fitch–Margoliash algorithm (Fitch and Margoliash 1967) in the PHYLIP software package. The minimum evolution distance matrix method was used to obtain the best tree (not shown).
Mating-type and HO analysis:
Mating type was tested by PCR as described in Bradbury et al. (2006) with minor changes: each PCR reaction included only two primers, flanking either the a- or the α-mating type. Amplification products were loaded on a 1.5% agarose gel to examine which primer set produced amplification. The HO gene encodes an endonuclease responsible for initiating mating-type switching, a gene-conversion process where MATa cells change to MATα cells or vice versa through the generation of a double-strand DNA break (Russell et al. 1986). This locus was amplified using the primers TTGAGAAAGGCTGAAGTTGG(f) and TGTTGAAGCATGATGAAGCG(r). The annealing temperature for PCR was 56°. Primers for sequencing were: ACACTCTGGTCCTTTAAC, GACATTGGACTTTTCTTCC, CTGGCTCTTTTGTTGTAC, and TCACCTTCAAAAGCTCTG, along with the forward PCR primer. The resulting sequences were aligned with the wild-type sequence, GenBank accession no. M14678.
Assessment of ploidy level:
The results of SSR analysis indicated that the collected strains varied in ploidy level, from haploid up to tetraploid. Independent scoring of the ploidy level was conducted using flow cytometry analysis (FACS). Protocol was adopted for this scoring from Foiani et al. (1994), using a Becton–Dickinson FACScan analyzer. In particular, yeast cultures were grown to logarithmic phase, fixated in ethanol, treated by pepsin and ribonuclease A, and stained with propidium iodide. Each tested strain was scored a few times, in each experiment with two replicates. Each experiment included at least two reference strains, haploid and diploid. The ploidy level was scored on the basis of the fluorescence intensity compared to the haploid and diploid reference stains.
Statistical analysis:
Cluster analysis and phylogenetic reconstructions based on SSR polymorphism were made using the Fitch–Margoliash algorithm (Fitch and Margoliash 1967). Bootstrap consensus trees were constructed using the PHYLIP software package. The minimum evolution distance matrix method was used to obtain the best tree. We used normalized Euclidian distance 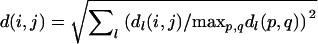 between strains, where dl is the contribution of the lth locus. Distance (dl) between two tetraploid strains was calculated in the coordinate system of ordered allele sizes:
between strains, where dl is the contribution of the lth locus. Distance (dl) between two tetraploid strains was calculated in the coordinate system of ordered allele sizes: 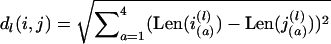 , where
, where 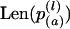 is the length of the ath allele at locus l of strain p. Our way of genetic distance calculation takes into account the size of alleles, but is different from the standard one (reviewed in Takezaki and Nei 1996). In contrast to standard models, we suppose that population is asexual and mutations are rare and cause only small changes in allele length. Actually, we consider mutation process like a four-dimensional Brownian motion in the space of allele lengths transforming one tetraploid genotype to another. According to our definition, genetic distance between two tetraploid genotypes is equal to the length of the shortest (and most probable) mutation way. This value is proportional to mean time required to reach the observed level of genetic difference between the compared genotypes (Feller 1957).
is the length of the ath allele at locus l of strain p. Our way of genetic distance calculation takes into account the size of alleles, but is different from the standard one (reviewed in Takezaki and Nei 1996). In contrast to standard models, we suppose that population is asexual and mutations are rare and cause only small changes in allele length. Actually, we consider mutation process like a four-dimensional Brownian motion in the space of allele lengths transforming one tetraploid genotype to another. According to our definition, genetic distance between two tetraploid genotypes is equal to the length of the shortest (and most probable) mutation way. This value is proportional to mean time required to reach the observed level of genetic difference between the compared genotypes (Feller 1957).
Distance between a tetraploid strain and a strain with another ploidy level was calculated by “redefining” the non-tetra-strain as a virtual “tetraploid.” This was done (separately for each locus) using the following rules, which obey the triangle inequality for the defined distance measure: haploids, tetraploids were constructed by taking four times the alleles of the haploid at every locus; diploids, tetraploids were constructed by taking two times the alleles of the diploid at every locus; triploids, tetraploids were constructed using the three alleles of the triploid and an addition of a fourth allele, with a length equal to that of the second-in-size allele of the triploid. The proposed distance definition has close physical sense in the case of strains with the same ploidy level. Also, it obeys the triangle inequality. By creating a virtual tetraploid from the triploid via a duplicating median allele, we get a genotype that can be produced from this triploid without additional mutations (mutations are considered as small and rare).
In the case of disconcordance between the ploidy and the number of observed alleles maximum-likelihood estimations were used. A χ2-test was employed to assess the significance of population differentiation into ecological groups (e.g., between SFS and NFS). Phylogenetic trees were drawn using the program TreeView (Page 1996). Bootstrap support for phylogenetic trees was determined from 1000 replications.
Expected homozygosity he was calculated as the sum of squares of allele frequencies. Allele diversity He and allele combination diversity D were calculated as 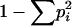 , where pi is the frequency of allele i or frequency of genotype i, correspondingly (Weir 1990). This characteristic of allele diversity (and allele-combination diversity) is close to zero in the case when almost all alleles (or allele combinations) are the same at the considered locus across strains and is close to 1 when all of them are different. The value He = 1 − he can characterize the mean expected heterozygote proportion in the diploid case under panmixia.
, where pi is the frequency of allele i or frequency of genotype i, correspondingly (Weir 1990). This characteristic of allele diversity (and allele-combination diversity) is close to zero in the case when almost all alleles (or allele combinations) are the same at the considered locus across strains and is close to 1 when all of them are different. The value He = 1 − he can characterize the mean expected heterozygote proportion in the diploid case under panmixia.
Segregation analysis:
To test whether segregations of SSR loci in considered tetraploid yeasts are random we compared observed results of segregations with expected ones. Denote by k = k(l) the number of different possible sets of four diploid genotypes at locus l that can be obtained in segregation of the tetraploid strain. If k = 1 (for example, for tetraploid genotype AAAa) then locus l is not informative for segregation analysis. If k > 1 then denote by p1,…,pk the probabilities to obtain a corresponding set of diploid genotypes. Denote by m = m(l) the number of different possible sets of diploid genotypes at locus l, which can be obtained in segregation of a tetraploid strain when probably not all of four diploid genotypes are observed. Denote by pij (i = 1,…,k; j = 1,…,m) the probability to obtain a set of genotypes j in the case of segregation producing a set of four diploid genotypes i (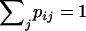 ).
).
If segregations are random and all diploid products have the same probability to be observed, then values pi and pij (i = 1,…,k; j = 1,…,m) can be calculated using combinatorial formulas. Let Nj (j = 1,…,m) be the number of segregation cases where a set of diploid genotypes j was observed (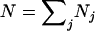 is the total number of successful segregations). The number of cases where segregation produces a set of four diploid products i can be estimated by
is the total number of successful segregations). The number of cases where segregation produces a set of four diploid products i can be estimated by 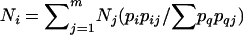 (i = 1,…,k). If segregation is random, then we expect that the value
(i = 1,…,k). If segregation is random, then we expect that the value 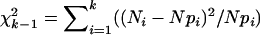 will have standard
will have standard 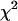 -distribution with d.f. = k − 1.
-distribution with d.f. = k − 1.
RESULTS
All 68 isolates were found to be prototrophic and sporulated on SPO media. After tetrad dissection of representative strains the spores either were successfully mated with haploid testers MATa or MATα or underwent additional sporulation that showed the polyploid nature of the parental strain. A very low sporulation rate was obtained in the second sporulation test (see below).
Microsatellite variation:
Nineteen primer pairs designed for SSR analysis on the basis of the S. cerevisiae genome sequence proved efficient in amplifying fragments in the expected length range (see Table 2). To validate the relevance of these PCR products, parts of the amplimeres were sequenced. The resulting sequences were in agreement with the published genome and the predicted number of repeats from sizing analysis (data not shown). The scored microsatellite loci showed high polymorphism of allele size, with 2–9 alleles per locus and an average of 5.1 alleles per locus (Table 1). No significant differences were found in the level of SSR variation in any locus between the three major localities (NFS, SFS, and VB): the maximum of 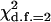 among the loci in the likelihood test for microsite heterogeneity was 7.3 (found for locus X-188.70), P = 0.05. Likewise, between the three microniches (habitats) tested (sunny, shady, and leaf), the maximum of
among the loci in the likelihood test for microsite heterogeneity was 7.3 (found for locus X-188.70), P = 0.05. Likewise, between the three microniches (habitats) tested (sunny, shady, and leaf), the maximum of 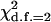 among the loci in the likelihood test was 8.9 (found for locus XIII-388.60), P = 0.03. Keeping in mind the multiple-comparison nature of our analysis, these values cannot be considered as significant because the probability to reach a minimal P-value = 0.03 in 19 tests when H0 (no real difference between the locations) is true is ∼0.6 (Benjamini and Hochberg 1995).
among the loci in the likelihood test was 8.9 (found for locus XIII-388.60), P = 0.03. Keeping in mind the multiple-comparison nature of our analysis, these values cannot be considered as significant because the probability to reach a minimal P-value = 0.03 in 19 tests when H0 (no real difference between the locations) is true is ∼0.6 (Benjamini and Hochberg 1995).
TABLE 2.
PCR primers for amplification of the selected SSR loci
| Locus | Forward | Reverse | Temperature |
|---|---|---|---|
| ChII-ATP I | tgccatccgtgtacgctagg | gcgaacagagccgtttaccg | 57° |
| Ch IV-1038.6 | aattgctgtcattggatctat | attattcctacgtatgaagtg | 52° |
| ChVI-FAB1 | ctacaattccaaaggtccttcgc | cgtgccattgtcgtttgaggg | 53° |
| ChIX-105.7 | gcacttgctgaacataagc | aggtagtttaggaagtgaggc | 51° |
| ChX-188.7 | cagaggaggaccaccagtttg | aaaggaaccacagcagcagg | 53° |
| ChX-403.7 | cacaaataggttagagacacag | ctaaatcgtcctcccattg | 55° |
| ChX-423.8 | gctggctctatatctcctctcg | actgtgtggcgggtaatgc | 55° |
| ChX-469.8 | caatgctaaaggacaccaag | ggcgaagagaagaagcatctg | 55° |
| ChX-518.90 | cgccgatattagacgtgtg | gggctttcactccactttac | 53° |
| ChX-GRR | cgttgcatccctaacctcactt | gctgcacccacctgatatacatcc | 53° |
| ChX-639.6 | gtagcataacagcagcgtag | cttcaaactcagtagtcgtcc | 53° |
| ChXI-126.1 | tgaatctggcgacgatag | acttttggccaatttctcaagat | 55° |
| ChXI-184.5 | aagcgtcctaacatactatccacc | atttcaattggctatatatcctta | 52° |
| ChXI-576.1 | agatacagaagataagaacgaaaa | ttattgatgcttatctattatacc | 55° |
| ChXII-511.5 | cttaaacaacagctcccaaa | atgaatcagcgcatcagaaat | 53° |
| ChXII-823.4 | ctggaatgaaattaaacaaaagc | tcttccttttctactatcttctc | 51° |
| ChXIII-CMP II | cggactctcgtcctactattg | ggggacaatgttggcgctag | 60° |
| ChXIII-ORF4 | Gctcgcagggagaaatctgcttcc | cttcatcggtatccgttccactagg | 53° |
| ChXIII-388.6 | Atgcactcaaacagtcgatcctt | cctatccatcgcttatagaacaa | 57° |
Ploidy variation:
High heterozygosity at the scored SSR loci indicated a variation in ploidy level, with one to four alleles per locus per strain. Flow cytometry analysis (FACS) was conducted for a sample of strains, representing the four putative levels of ploidy revealed from SSR analysis, and for a part of their progeny obtained by tetrad dissection. Our diploid isolates showed the same level of fluorescence per cell as the laboratory diploid control strain, whereas presumably tetraploid strains (according to SSR analysis) showed a doubled level of fluorescence per cell. High consistence was found between these sources of ploidy assessment (see Figure 1).
Figure 1.—

Analysis of DNA content by flow cytometry. Yeast cells were stained with propidium iodide for cell cycle analysis. Cell counts and relative DNA content are shown. (a) Diploid, triploid, and tetraploid natural isolates. (b) A tetraploid natural isolate and one of its diploid offspring.
Consequently, the numbers of di-, tri-, and tetraploid strains in our panel were found to be 21, 7, and 40 (31, 10, and 59%), correspondingly. No significant differences were found between the three microsite subpopulations (NFS, SFS, and VB) or the three microniches (sunny, shady, and leaf) with respect to variation in ploidy level (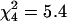 and
and 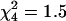 , correspondingly, P > 0.1).
, correspondingly, P > 0.1).
At all three ploidy levels (di-, tri-, and tetraploid) the sporulation test was positive for all strains. As expected, our trials for tetrad analysis of triploid strains gave an extremely low level of viable spores. An interesting result was obtained by tetrad analysis of tetraploid strains: they showed high sporulation level and high viability of spores. Unlike the tetraploids themselves, a part of the offspring derived from the tetraploids displayed low sporulation level (<1%), with no viable spores, and therefore were considered a/α-diploid. Other diploid offspring, derived from meiosis of tetraploid strains, did not sporulate at all. These strains were successfully mated with an α-tester strain and the resulting triploids were able to undergo sporulation. The mating type deduced from these tests was confirmed by PCR. Almost all haploids derived from diploids as well as diploids derived from tetraploids displayed stability of mating type (being therefore heterothallic). It is noteworthy that homothallism is believed to be characteristic of most yeast in nature (e.g., Mortimer 2000). In light of the foregoing results obtained in our mating experiments, it was desirable to check the sequence of the HO locus that controls mating-type switch (Russell et al. 1986). Such tests were conducted on a few haploids derived from diploids. Sequence analysis followed by alignment to the standard HO sequence revealed several point mutations, including deletions and SNPs. The first substitution, causing a nonsense mutation, appeared at position 235 (after the ATG codon). This mutation causes the formation of a short mutant polypeptide of only 79 amino acids, while the wild-type protein is 586 amino acids long (Russell et al. 1986).
SSR allele diversity varied across ploidy levels (Table 3). Two tests shown below were conducted to estimate the significance of this variation.
TABLE 3.
Variation in allele diversity across ploidy levels and in entire population of S. cerevisiae isolates from Evolution Canyon
| Allele diversity (He)
|
P-value (for test of allele diversity equivalence)
|
||||||
|---|---|---|---|---|---|---|---|
| Locus | Diploids | Triploids | Tetraploids | Entire population | Di-triploids | Di-tetraploids | Tri-tetraploids |
| ChII-ATP I | 0.62 | 0.00 | 0.38 | 0.44 | <0.001 | <0.001 | <0.001 |
| Ch IV-1038.6 | 0.61 | 0.09 | 0.37 | 0.57 | <0.001 | <0.001 | <0.001 |
| ChVI-FAB1 | 0.67 | 0.67 | 0.63 | 0.76 | NS | 0.01 | NS |
| ChIX-105.7 | 0.73 | 0.67 | 0.70 | 0.75 | 0.01 | 0.02 | NS |
| ChX-188.7 | 0.49 | 0.09 | 0.38 | 0.55 | <0.001 | <0.001 | <0.001 |
| ChX-403.7 | 0.39 | 0.44 | 0.38 | 0.47 | 0.02 | NS | 0.02 |
| ChX-423.8 | 0.69 | 0.44 | 0.63 | 0.67 | <0.001 | 0.002 | <0.001 |
| ChX-469.8 | 0.69 | 0.67 | 0.63 | 0.69 | NS | 0.002 | NS |
| ChX-518.90 | 0.80 | 0.67 | 0.75 | 0.82 | <0.001 | 0.005 | 0.005 |
| ChX-GRR | 0.41 | 0.44 | 0.50 | 0.50 | NS | <0.001 | 0.01 |
| ChX-639.6 | 0.74 | 0.44 | 0.50 | 0.60 | <0.001 | <0.001 | 0.01 |
| ChXI-126.1 | 0.75 | 0.44 | 0.65 | 0.75 | <0.001 | <0.001 | <0.001 |
| ChXI-184.5 | 0.56 | 0.00 | 0.38 | 0.46 | <0.001 | <0.001 | <0.001 |
| ChXI-576.1 | 0.83 | 0.00 | 0.38 | 0.52 | <0.001 | <0.001 | <0.001 |
| ChXII-511.5 | 0.52 | 0.00 | 0.38 | 0.47 | <0.001 | <0.001 | <0.001 |
| ChXII-823.4 | 0.77 | 0.44 | 0.39 | 0.57 | <0.001 | <0.001 | 0.02 |
| ChXIII-CMP II | 0.70 | 0.00 | 0.50 | 0.61 | <0.001 | <0.001 | <0.001 |
| ChXIII-ORF4 | 0.60 | 0.44 | 0.62 | 0.67 | <0.001 | NS | <0.001 |
| ChXIII-388.6 | 0.35 | 0.00 | 0.02 | 0.09 | <0.001 | <0.001 | NS |
NS, not significant. Allele diversity He was calculated as 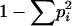 , where pi is the frequency of allele i (as described in Statistical analysis in materials and methods). Zero allele diversity means monomorphism at the locus. Results on significance of differences in allele diversity between different ploidy levels are presented in the right side. P-values are calculated using a maximum-likelihood test (see text).
, where pi is the frequency of allele i (as described in Statistical analysis in materials and methods). Zero allele diversity means monomorphism at the locus. Results on significance of differences in allele diversity between different ploidy levels are presented in the right side. P-values are calculated using a maximum-likelihood test (see text).
Likelihood test for equivalence of allele diversity for different ploidy levels:
This test employs maximum-likelihood estimation of allele frequencies, under two conditions for allele diversity equivalence. We compared likelihood function for observed data under hypotheses H1 and H0: “allele distribution was different (H1) vs. the same (H0) for different ploidy levels.” Supposing multinomial distribution of the allele numbers in the sampled strains, we calculated log-likelihood as
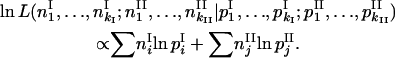 |
Here 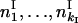 and
and 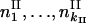 are the observed numbers of alleles for two ploidy levels I and II;
are the observed numbers of alleles for two ploidy levels I and II; 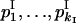 and
and 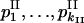 are expected allele frequencies (they are positive and fit the trivial condition
are expected allele frequencies (they are positive and fit the trivial condition 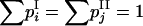 ). The symbol “∝” denotes that, in the equality, the term independent of
). The symbol “∝” denotes that, in the equality, the term independent of 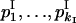 and
and 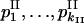 is omitted. For the compared hypotheses H1 and H0,
is omitted. For the compared hypotheses H1 and H0,
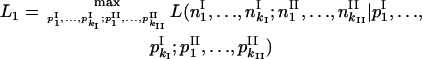 |
and
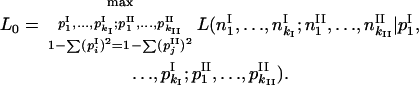 |
If H0 is true then we expect that the statistic 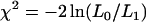 is asymptotically distributed as
is asymptotically distributed as 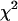 with d.f. = 1.
with d.f. = 1.
Rank test of diversity values comparison:
For diversity scores at ploidy levels I and II, the expected number of signs that “level I is greater than level II” in 19 pairs (corresponding to 19 marker loci) of diversity comparison is 9.5. In the case of diversity equivalence, the value 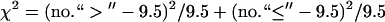 has
has 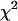 -distribution with d.f. = 1. Allele diversity was significantly higher in diploids than in tetraploids (P-value 0.005) and in tetraploids than in triploids (P-value 0.015). Allele-combination diversity significantly differed from the expected values in tetraploids (Table 4).
-distribution with d.f. = 1. Allele diversity was significantly higher in diploids than in tetraploids (P-value 0.005) and in tetraploids than in triploids (P-value 0.015). Allele-combination diversity significantly differed from the expected values in tetraploids (Table 4).
TABLE 4.
Allele combination diversity for the three ploidy levels of S. cerevisiae isolates from Evolution Canyon
| Allele combination diversity (D)
|
||||||
|---|---|---|---|---|---|---|
| Observed
|
Expecteda
|
|||||
| Locus | Diploids | Triploids | Tetraploids | Diploids | Triploids | Tetraploids |
| ChII-ATP I | 0.66 | 0.00 | 0.05 | 0.78 | 0.00 | 0.68 |
| Ch IV-1038.6 | 0.74 | 0.24 | 0.05 | 0.80 | 0.24 | 0.67 |
| ChVI-FAB1 | 0.72 | 0.00 | 0.14 | 0.85 | 0.87 | 0.89 |
| ChIX-105.7 | 0.63 | 0.00 | 0.42 | 0.89 | 0.87 | 0.95 |
| ChX-188.7 | 0.65 | 0.24 | 0.00 | 0.64 | 0.24 | 0.68 |
| ChX-403.7 | 0.54 | 0.00 | 0.00 | 0.55 | 0.66 | 0.68 |
| ChX-423.8 | 0.73 | 0.00 | 0.00 | 0.85 | 0.66 | 0.89 |
| ChX-469.8 | 0.69 | 0.00 | 0.00 | 0.84 | 0.87 | 0.89 |
| ChX-518.90 | 0.83 | 0.00 | 0.00 | 0.93 | 0.87 | 0.96 |
| ChX-GRR | 0.49 | 0.00 | 0.00 | 0.57 | 0.66 | 0.73 |
| ChX-639.6 | 0.72 | 0.00 | 0.00 | 0.89 | 0.66 | 0.73 |
| ChXI-126.1 | 0.76 | 0.00 | 0.42 | 0.90 | 0.66 | 0.90 |
| ChXI-184.5 | 0.74 | 0.00 | 0.05 | 0.74 | 0.00 | 0.68 |
| ChXI-576.1 | 0.77 | 0.00 | 0.00 | 0.95 | 0.00 | 0.68 |
| ChXII-511.5 | 0.65 | 0.00 | 0.05 | 0.72 | 0.00 | 0.68 |
| ChXII-823.4 | 0.74 | 0.00 | 0.05 | 0.91 | 0.66 | 0.68 |
| ChXIII-CMP II | 0.70 | 0.00 | 0.05 | 0.85 | 0.00 | 0.73 |
| ChXIII-ORF4 | 0.65 | 0.00 | 0.05 | 0.76 | 0.66 | 0.89 |
| ChXIII-388.6 | 0.53 | 0.00 | 0.05 | 0.53 | 0.00 | 0.09 |
Allele-combination diversity D was calculated for each ploidy level as 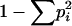 , where pi is the frequency of genotype i (as described in Statistical analysis in materials and methods). Zero allele-combination diversity means that all strains of the ploidy level have the same genotype.
, where pi is the frequency of genotype i (as described in Statistical analysis in materials and methods). Zero allele-combination diversity means that all strains of the ploidy level have the same genotype.
Expected allele combination diversity was calculated on the basis of observed allele frequencies for the ploidy level. The difference between observed and expected allele-combination diversity was not significant for diploids, but it was highly significant (P < 0.0001) for tetraploids for almost all loci, except XIII-388.60.
Sexual vs. clonal reproduction:
An exceptional feature of the inspected population is the seemingly clonal structure of its higher ploidy parts. Namely, at both tri- and tetraploid levels, we observed a very limited number of different multilocus genotypes (clones), representing the majority of strains (Figure 2, Table 4).
Figure 2.—

Bootstrap consensus phylogenetic tree, demonstrating the relationship between S. cerevisiae isolates from Evolution Canyon and their relationship with laboratory strains. The tree was constructed on the basis of 19 SSR loci with distance matrices calculated as described in Statistical analysis in materials and methods. The numbers above the branches are the percentages of bootstrap trees containing each bipartition (only values >50% are given).
We think that the tight clustering shown in Figure 2 reflects clonality. Even with recent origin, one would expect that just one to two cycles of sexual reproduction of tetraploids would generate various segregants per each heterozygous locus (see also Table 4) and recombinant genotypes, which we do see in diploids, but not in tri- and tetraploids.
Low similarity in allele content was found between strains of different ploidy levels, except diploid strain nos. 03, 05, 07, and 23 that share their alleles with the cluster of tetraploids and might be either offspring or parental strains of the tetraploid strains. As indicated above, an interesting effect was found in tetrad analysis: all tested tetraploid strains produced diploid progeny, but all efforts to obtain haploids from this progeny failed. By contrast, haploid progeny was easily obtained from diploid strains.
In light of the foregoing findings, it is interesting to test whether the proportion of heterozygotes for SSR markers will show any pattern compatible with the assumed tendency of clonality. A simple test for deviation from Hardy–Weinberg (HW) proportions was, therefore conducted on diploids, the least clonal part of the population (Table 5). The observed number of homozygotes was found to exceed significantly the HW expectations, albeit the discrepancy varied among chromosomes and even within chromosomes. The majority of chromosomes displayed an excess of homozygotes despite the high polymorphism of the diploid part of the population. An exception was chromosome X with only one marker of seven that showed deviation from HW proportions. Presumably, this may reflect selection against heterozygotes for loci of this chromosome or complex genetic architecture of modifiers of mating preferences (hence proportions of homozygotes). The fact that significant paucity of heterozygotes compared to HW expectation is found for loci distantly located from the centromere and near-centromeric loci indicates that mitotic recombination alone cannot be the only cause, and selfing should be a playing factor here. The results of the HO sequence test show that at least some of the strains are strictly heterothallic, whereas the HW test indicates that at least part should be or should have been homothallic.
TABLE 5.
Discrepancy between the observed and expected (assuming panmixia) proportions of homozygotes among the diploid S. cerevisiae isolates from Evolution Canyon
| Locus | h observed | h expected | P-value |
|---|---|---|---|
| ChII-ATP I | 0.90 | 0.38 | <0.001 |
| Ch IV-1038.6 | 0.67 | 0.38 | 0.03 |
| ChVI-FAB1 | 0.48 | 0.33 | NS |
| ChIX-105.7 | 0.48 | 0.28 | NS |
| ChX-188.7 | 0.57 | 0.51 | NS |
| ChX-403.7 | 0.62 | 0.64 | NS |
| ChX-423.8 | 0.19 | 0.30 | NS |
| ChX-469.8 | 0.29 | 0.30 | NS |
| ChX-518.90 | 0.43 | 0.20 | 0.02 |
| ChX-GRR | 0.43 | 0.59 | NS |
| ChX-639.6 | 0.33 | 0.25 | NS |
| ChXI-126.1 | 0.71 | 0.25 | <0.001 |
| ChXI-184.5 | 0.62 | 0.42 | NS |
| ChXI-576.1 | 0.48 | 0.17 | <0.001 |
| ChXII-511.5 | 0.57 | 0.48 | NS |
| ChXII-823.4 | 0.62 | 0.23 | <0.001 |
| ChXIII-CMP II | 0.81 | 0.30 | <0.001 |
| ChXIII-ORF4 | 0.81 | 0.40 | 0.003 |
| ChXIII-388.6 | 0.57 | 0.65 | NS |
NS, not significant. Expected homozygosity was estimated as the sum of squares of allele frequencies. The P-value was calculated using a 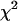 -test with d.f. = 1.
-test with d.f. = 1.
Segregation analysis:
We considered 27 segregation cases to test whether segregations of the tetraploid strain are random. Analysis was made only for 9 loci from the 19, because the others could produce only a k = 1 possible set of four diploid genotypes. It was found that only two loci CMP2 (XIII-159.9) and ORF4 (XIII-209.8) demonstrate significant deviation from random segregation (P-values 0.07 and 0.005 correspondingly, see Table 6). It can be noted that both of them are situated on chromosome XIII and the locus that demonstrated high significance is closer to the centromere.
TABLE 6.
Results of test on random segregation
| Locus | χ2 | d.f. | P-value |
|---|---|---|---|
| IX-105.7 | 1.79 | 2 | NS |
| X-423.8 | 3.37 | 2 | NS |
| X-469.8 | 0.17 | 2 | NS |
| X-518.9 | 1.25 | 4 | NS |
| GRR (X-593.9) | 2.89 | 2 | NS |
| X-639.6 | 0.82 | 2 | NS |
| XI-126.1 | 2.8 | 2 | NS |
| CMP2 (XIII-159.9) | 5.37 | 2 | 0.07 |
| ORF4 (XIII-209.8) | 10.67 | 2 | 0.005 |
The P-value was calculated using a χ2-test (see Segregation analysis in materials and methods). Only markers from chromosome XIII [both CMP2 (XIII-159.9) and ORF4 (XIII-209.8)] demonstrate significant deviation from random segregation. The locus that demonstrated higher significance is closer to the centromere (with coordinate 268.1 kb in the physical map).
Taxonomic consideration:
Very high amplification efficiency of primer pairs designed for SSR loci on the basis of the published genome sequence of S. cerevisiae was achieved, while for S. paradoxus, a close relative of S. cerevisiae, it was reported recently that only 3 of 20 primer pairs, designed on the basis of S. cerevisiae genome sequence, resulted in amplimeres (Johnson et al. 2004). This is a strong indication that the collected isolates are indeed S. cerevisiae. However, no simple explanation seems to fit the observed complicated pattern of ploidy variation, SSR distribution among and within the stains, and the outcomes of tetrad analysis of diploid isolates compared to the diploid progeny of tetraploid strains. This motivated us to conduct in-depth specific taxonomic tests on the basis of sequence comparison at two selected loci: ITS1-5.8S-ITS2 rDNA and translation EF-1 αA. Multiple sequence alignment for the representative isolates from Evolution Canyon with all available Saccharomyces sensu stricto sequences from GeneBank (http://www.ncbi.nlm.nih.gov) were conducted. For the translation EF-1 αA locus, the tested isolates showed high sequence similarity to S. cerevisiae. In fact, we found only one SNP out of 1127 bp.
For the ITS1-5.8S-ITS2 locus, sequencing of tetraploids, diploids, and their offspring yielded overlap of different sequences that could not be obviously merged to a single sequence. Only triploid strains tested formed a single sequence that could be analyzed. Most of the variation found between these triploid isolates and the members of the Saccharomyces sensu stricto group was due to mononucleotide repeats (MNRs). Our natural isolates tend to show a larger number of repeats compared to those in published sequences of three MNR tracts [all poly(T/A)] in the locus. Still, these results indicate that these isolates have closest homology to S. cerevisiae.
Microscale (microclimatic) differentiation:
The differences in number of alleles, allele diversity, and allele-combination diversity were tested for association with microclimatic contrasts: (a) major localities SFS, NFS, and VB and (b) variation among niches/habitats sun vs. shade vs. leaf surface. Only tetraploid strains displayed significant interslope differentiation in their allele combinations that involved two loci, IX-107.70 and XI-126.10 (χ2 = 11.1, P < 0.001). No intraslope (i.e., interstation) variation was detected. A slight tendency toward between-niche variation was found for diploids (for shade-derived strains vs. remainder strains χ2 = 2.7, P < 0.1; and for leaf vs. remainder χ2 = 3.9, P < 0.05). Additional tests, especially using candidate stress-related genes (for tolerance to high temperature, desiccation, UV, etc.), are needed to check whether the foregoing diversity reflects ecological adaptation to microclimatic variation.
DISCUSSION
The yeast S. cerevisiae is one of the most important organisms for biotechnology and the food industry. It is also the most studied eukaryote and an important model for cell biology. Thus, despite the huge amount of genetic and molecular studies on S. cerevisiae, only a narrow range of strains, which hardly represent the species, were characterized. Consequently, there is no understanding of the adaptive structure of yeast natural populations and evolutionary strategies (Liti and Louis 2005).
We assessed the genetic diversity in a natural population of S. cerevisiae from a well-studied natural model, called Evolution Canyon at Mount Carmel, where dozens of other species are analyzed at the phenotypic, genetic, and molecular-genetic levels. The collected S. cerevisiae strains were characterized for 19 SSR markers. Several findings are described in this article, including high SSR polymorphism, ploidy variation from diploid up to tetraploid, and a tendency toward a clonal rather than a sexual population structure. Our results seem to differ markedly from those reported in other studies of wild yeasts. In a population study of the yeast S. paradoxus, a close relative of S. cerevisiae, strains were isolated from the bark of oak trees in southern England (Johnson et al. 2004). The isolation procedure in that study allowed getting S. cerevisiae yeast, but no strain of this species was found among the isolates. All S. paradoxus strains were found to be homothallic diploid, with a very low level of heterozygosity. These differences between the two studies can be explained by the environmental characteristics of extremely sharp spatial microclimatic contrasts at Evolution Canyon and relatively mild variation in Silwood Park and Windsor Great Park. In the studies of natural isolates of S. cerevisiae derived from noninoculated wine fermentations, most isolates were found to be homothallic diploids. The isolates displayed tremendous diversity, and 35% of isolates were completely homozygous to the assessed systems (Mortimer 2000).
Proving the taxonomic affiliation:
The employed criteria for yeast isolation and characterization of the isolates were suitable for S. cerevisiae (Beech and Davenport 1971; Barnett et al. 2000). All SSRs were successfully amplified using specific primers designed on the basis of the published genome of S. cerevisiae, resulting in fragments of the expected size range for S. cerevisiae. For further validation, samples of the amplified products were sequenced and proved to coincide very well with the published sequence of the S. cerevisiae genome. One of the most popular genetic tools for differentiation among yeast species is sequence variation at the ribosomal DNA ITS1-5.8S-ITS2 locus (Kurtzman and Robnett 2003). Sequences of the ITS1-5.8S-ITS2 locus were successfully obtained only from triploid isolates. All other tested strains (tetraploids, diploids, and offspring of both) showed multiple sequences at this locus, presumably due to the large number of rDNA repeats in the yeast genome (100–200 repeats) (Johnston et al. 1997). Sequence variation in these ribosomal DNA regions arising from array duplication events is a known phenomenon (see detailed review by Alvarez and Wendel 2003). We hypothesize that triploids reproduce only clonally, and therefore all copies of the rDNA could have undergone a complete homogenization process (Gangloff et al. 1996). Multiple alignment of this sequence was conducted and a phylogenetic tree was generated for a triploid isolate and all Saccharomyces sensu stricto sequences from GenBank (not shown). The tested isolate showed the closest homology to S. cerevisiae. In addition, multiple alignment of the translation EF-1 αA locus was conducted, and all tested isolates showed good sequence similarity to S. cerevisiae. All these results give us high confidence that the studied material belongs to the S. cerevisiae species.
The ploidy challenge:
The yeast S. cerevisiae can exist in haploid, diploid, or polyploid states. There are variations both within and between laboratory, commercial, and clinical strains and natural isolates (e.g., our results). In recent decades, evolution of ploidy level became an important subject within the problems of genome molecular evolution and sex evolution. In particular, yeast may be a very relevant model organism for such studies (e.g., Wolfe and Shields 1997; Mable and Otto 1998; Korona 1999; Mable and Otto 2001; Wolfe 2001; Zeyl 2004). It is noteworthy that the recent renaissance of the polyploidy studies is, to a large extent, due to the interest in the (presumably existing) mechanisms of (very) fast evolution of polyploid genomes toward diploidization and novel tools allowing us to address this “old” problem (Belyayev et al. 2000; Ozkan et al. 2001; Feldman and Levy 2005). The relevance of this problem to our project is caused both by the paleopolyploid nature of the yeast genome (Wong et al. 2002) and by the results presented here.
Our SSR analysis and subsequent test of the tetrads show that the isolated strains differ in ploidy level. Flow cytometry analysis confirmed the assessed ploidy levels based on the SSR analysis (diploids, triploids, and tetraploids) (see Figure 1). This result presumably reflects the transitory nature of the haploid stage in yeast in nature. We assume that these results indicate higher adaptive potential of heterozygous polyploids combined with higher flexibility of a population harboring genotypes with different ploidy levels. Indeed, it is known that a certain proportion of genes may be either repressed or induced in response to increased ploidy even if most genes will not be affected by variation in ploidy (Galitski et al. 1999).
An interesting pattern was revealed in our tetrad analysis. We managed to obtain diploid a/α progeny from tetraploid strains, but had no success in obtaining viable progeny of these diploids, despite the fact that a similar test on natural diploids did give viable haploid progeny. Possible explanations for this result can be the accumulation of a variety of epistatic relations between alleles in tetraploids strains or aneuploid “diploid” offspring that segregated to unbalanced haploids. Another possibility is an assumption of some hybridization event(s) that could have occurred in the past between cells of S. cerevisiae and a sibling yeast species, resulting in a fertile allotetraploid. Hybridizations between different species of Saccharomyces are known to occur and are a subject to a number of studies (e.g., Naumov et al. 2000; de Barros et al. 2002; Greig et al. 2002a,b). Such an event may promote a fast intragenome evolution toward further perfection of diploid-like behavior (Shaked et al. 2001). Offspring of the evolved amphidiploids would not be able to undergo one more cycle of meiosis giving haploid products (Loidl 1995). Another consequence of such a hypothetical process would be a tendency for directed segregation.
To test the last hypothesis we conducted an analysis of the segregations of tetraploids to diploids. Only 9 of 19 SSR loci, located in chromosomes IX, X, XI, and XIII, could be tested. Our results (see Table 6) demonstrate that the segregation of chromosome XIII was nonrandom. For locus OFR4, which is close to the centromere, significance was higher than for locus CMP2, situated more distantly from the centromere, fitting the expectation that direction of segregation is determined by the centromere and correlation with the centromere decreases by recombination toward the telomeres. It is possible that different chromosomes behave unlike each other due to a different level of sequence similarity. The nonsignificant results of other chromosomes can be also partially explained by different power of analysis, depending on tetraploid genotype and haplotypes. A larger sample size and more suitable genomic composition of targeted tetraploids could clarify whether or not segregations in other chromosomes are random.
Microsatellite polymorphism:
Nineteen SSR loci were amplified using specific primer pairs, to give amplimeres in the expected length range. The scored microsatellite loci showed two to nine alleles per locus (Table 1). However, no significant differences were found in the level of SSR variation in any ecological niche/habitat. Allele diversity varied with ploidy level (see Table 3). In particular, maximum diversity was observed among diploids and minimum among triploids. We found no correlation between the chromosomal position and the inter- and intrastrain diversity of the scored SSR, as one would expect for a sexual population with a recombination gradient within the chromosome caused by a centromeric effect on recombination (Lambie and Roeder 1988). This expectation derives from the mechanisms of selective sweep (caused by spreading of new favorable mutations) and background selection (which considers the consequences of elimination of deleterious mutations) (Maynard Smith and Haigh 1974; Charlesworth et al. 1993). Under these models, sequence polymorphism at the assayed loci is interpreted mainly as a result of their linkage to other negatively or positively selected loci; i.e., the assayed sequences themselves are considered as neutral ones. Recombination reduces the dependence of the majority of neutral sequence variation on the minority of sporadically appearing new variants at the selected loci. Hence, lower diversity is expected in genomic regions with a low recombination rate. However, multilocus selection can cause opposite, either positive or negative, effects of recombination on sequence polymorphism (Kirzhner et al. 2003). Therefore, the absence of any positional (relative to centromere) pattern seemingly corroborates the hypothesis about a tendency toward asexual reproduction of yeast in Evolution Canyon.
Another interesting possibility of employing “chromosomal gradients” is to look for similar patterns in the excess of homozygosity (h) observed for some loci in diploid strains (see Table 5). Namely, it would be instructive to learn whether h increases with deviation from the centromere. Such a tendency would indicate simultaneously the shaping role of mitotic recombination and asexual reproduction. To address this question, we calculated rank correlation between homozygosity and the chromosomal position of the markers relative to the centromere. Because of the difference in allele diversity at different loci, we normalized observed homozygosity as 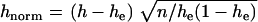 , where n = 21 is the number of diploid strains. Rank of marker chromosomal position was calculated according to the percentage of physical distance to the centromere from the length of the corresponding chromosomal arm. Rank order correlation was +0.42 (P = 0.07). A similar trend was obtained for nonnormalized tests (not shown). This result indicates that the observed excess in homozygosity might be a consequence of mitotic recombination upon asexual reproduction. Indeed, even with a high rate of mitotic recombination and a corresponding increase of homozygosity (compared to Hardy–Weinberg expectations) with distance from the centromere, just one cycle of outbreeding would be sufficient to recover the expected proportions.
, where n = 21 is the number of diploid strains. Rank of marker chromosomal position was calculated according to the percentage of physical distance to the centromere from the length of the corresponding chromosomal arm. Rank order correlation was +0.42 (P = 0.07). A similar trend was obtained for nonnormalized tests (not shown). This result indicates that the observed excess in homozygosity might be a consequence of mitotic recombination upon asexual reproduction. Indeed, even with a high rate of mitotic recombination and a corresponding increase of homozygosity (compared to Hardy–Weinberg expectations) with distance from the centromere, just one cycle of outbreeding would be sufficient to recover the expected proportions.
Population structure:
The inspected population shows a clonal structure of its higher ploidy part. Both the tri- and the tetraploid subgroups are composed of a very limited number of different multilocus genotypes (clones), representing the majority of strains. In other words, the polyploid part of the population can be characterized as a set of a few clusters with high proximity of strains within clusters and relatively high genetic distance between the clusters (see Figures 2 and 3). No correlation was found between polymorphism level and chromosomal position of loci relative to the centromere. This may also point to predominance of asexual reproduction in the studied population. An additional argument for this assumption may be very low allele-combination diversity (relative to the expected level) displayed in triploids and tetraploids (Table 4). Analysis of SSR loci across the genome showed that even for the diploid part of the population, a significant excess of homozygotes is characteristic despite a high level of polymorphism. For asexual reproduction, there is no expectation of changing homozygote:heterozygote ratios. In other words, the homozygosity test (deviations from HW) asks actually how frequent the meiosis events are followed by selfing vs. outcrossing. The fact that a significant paucity of heterozygotes compared to the HW expectation is found for loci located both distantly from the centromere and near the centromere indicates that mitotic recombination alone cannot be the only explanation and selfing should be a playing factor here. Seemingly, this fact may reflect the homothallic nature of the collected natural strains, but this explanation does not fit our tests that showed high mating-type stability of the derived a- and α-strains in the lab and our HO sequence results. Apparently, these results indicate that at least some of the strains are strictly heterothallic, whereas at least part should be or should have been homothallic.
Figure 3.—

Phylogenetic analysis of tetraploid S. cerevisiae isolates from Evolution Canyon. The tree is based on three SSR loci differentiating between tetraploids [FAB1 (VI-186.2), IX-105.70, and XI-126.10]. The numbers above the branches are the percentages of bootstrap trees containing each bipartition (only values >50% are given). Excluding locus FAB1 from the analysis leads to the increasing clade credibility of main bipartition up to 100%.
Low similarity in allele content was found between strains of different ploidy levels, except diploid strain nos. 03, 05, 07, and 23 that share their alleles with the tetraploid cluster. In tetrad analysis all tested tetraploid strains produced diploid progeny, but all efforts to obtain haploids from this progeny failed. By contrast, haploid progeny was easily obtained from diploid strains. These results allow us to speculate that clonal reproduction tends to predominate in the polyploid part of the tested population of S. cerevisiae.
Unlike the results obtained on other species studied in Evolution Canyon (Krugman et al. 2001; Michalak et al. 2001; Singh et al. 2005; Zamorzaeva et al. 2005; Rashkovetsky et al. 2006), no interslope divergence was found in this study, except in two loci in tetraploids. It is possible that analysis of adaptive traits may be a much better source of evidence for interslope differential selection than genetic distances estimated using molecular markers. Differentiation for adaptively valuable gene complexes can better withstand destruction by migration and recombination due to the protective effect of differential selection. However, such adaptive differentiation would not necessarily be sufficient to preserve allelic combinations of selectively neutral markers, unless the latter are in linkage disequilibrium with selected loci. This last condition can also persist despite migration, but only under very tight linkage and strong selection. On the other hand, it is possible that from the yeast's perspective our classification of the different habitats by location and niches was too broad.
Conclusions:
S. cerevisiae yeasts were isolated from soil and leaf samples in Evolution Canyon at Mount Carmel, Israel. Analysis of 19 SSR loci showed one to four alleles per strain, indicating different ploidy levels, diploids, triploids, and tetraploids, confirmed by flow cytometry tests. Maximum genetic diversity was observed among diploids and minimum among triploids. Clonal structure of triploid and tetraploid subpopulations was revealed using SSR data. In tetrad analysis, all tested tetraploids gave viable diploid progeny. No viable haploids were obtained from this progeny in a second round of tetrad dissection, while natural diploids gave high viability of haploid offspring. These results suggest that clonal reproduction tends to predominate in the polyploid subpopulation of the studied natural population of S. cerevisiae.
Acknowledgments
The authors are grateful to S. S. Nagornaya for collecting and classifying the strains employed in this study, to R. K. Mortimer and Y. Kassir for providing laboratory strains, and to D. Kornitzer and Y. Kassir for access to instruments and helpful advice. Constructive criticism and important comments from anonymous reviewers are acknowledged with thanks.
References
- Alvarez, I., and J. F. Wendel, 2003. Ribosomal ITS sequences and plant phylogenetic inference. Mol. Phylogenet. Evol. 29: 417–434. [DOI] [PubMed] [Google Scholar]
- Andreyuk, E. I., A. F. Antipchuk, G. A. Iutinskaja, T. Fahima and E. Nevo, 1999. Species characteristics of the soil bacterial communities at “Evolution Canyon”, Nahal Oren, Mount Carmel Natural Reserve, Israel. Mikrobiol. Z. 61: 3–9.10330872 [Google Scholar]
- Barnett, J. A., R. W. Payne and D. Yarrow, 2000. Yeasts, Characteristics and Identification. Cambridge University Press, New York.
- Beech, F. W., and R. R. Davenport, 1971. Isolation, purification and maintenance of yeasts, pp. 153–182 in Methods in Microbiology, edited by J. R. Morris and D. W. Ribbons. Academic Press, New York.
- Belyayev, A., O. Raskina, A. Korol and E. Nevo, 2000. Coevolution of A and B genomes in allotetraploid Triticum dicoccoides. Genome 43: 1021–1026. [PubMed] [Google Scholar]
- Benjamini, Y., and Y. Hochberg, 1995. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J. R. Stat. Soc. B Met. 57: 289–300. [Google Scholar]
- Charlesworth, B., M. T. Morgan and D. Charlesworth, 1993. The effect of deleterious mutations on neutral molecular variation. Genetics 134: 1289–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury, J. E., K. D. Richards, H. A. Niederer, S. A. Lee, D. P. Rod et al., 2006. A homozygous diploid subset of commercial wine yeast strains. Antonie Leeuwenhoek 89: 27–37. [DOI] [PubMed] [Google Scholar]
- Cox, B. S., 1995. Genetic analysis in Saccharomyces cerevisiae and Schizosaccharomyces pombe, pp. 7–67 in The Yeasts, edited by A. H. Rose and J. S. Harrison. Academic Press, London.
- de Barros, L. M., J. R. Bellon, N. J. Shirley and P. F. Ganter, 2002. Evidence for multiple interspecific hybridization in Saccharomyces sensu stricto species. FEMS Yeast Res. 1: 323–331. [DOI] [PubMed] [Google Scholar]
- Dickinson, J. R., 2000. Yeasts: providing questions and answers for modern biology. Sci. Prog. 83: 173–192. [PubMed] [Google Scholar]
- Fay, J. C., and J. A. Benavides, 2005. Evidence for domesticated and wild populations of Saccharomyces cerevisiae. PLoS Genet. 1: 66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman, M., and A. A. Levy, 2005. Allopolyploidy—a shaping force in the evolution of wheat genomes. Cytogenet. Genome Res. 109: 250–258. [DOI] [PubMed] [Google Scholar]
- Feller, W., 1957. An Introduction to Probability Theory and Its Applications, Vol. 1, Ed. 3. John Wiley & Sons, New York.
- Field, D., and C. Wills, 1998. Abundant microsatellite polymorphism in Saccharomyces cerevisiae, and the different distributions of microsatellites in eight prokaryotes and S. cerevisiae, result from strong mutation pressures and a variety of selective forces. Proc. Natl. Acad. Sci. USA 95: 1647–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch, W. M., and E. Margoliash, 1967. Construction of phylogenetic trees. Science 155: 279–284. [DOI] [PubMed] [Google Scholar]
- Foiani, M., F. Marini, D. Gamba, G. Lucchini and P. Plevani, 1994. The B subunit of the DNA polymerase alpha-primase complex in Saccharomyces cerevisiae executes an essential function at the initial stage of DNA replication. Mol. Cell. Biol. 14: 923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galitski, T., A. J. Saldanha, C. A. Styles, E. S. Lander and G. R. Fink, 1999. Ploidy regulation of gene expression. Science 285: 251–254. [DOI] [PubMed] [Google Scholar]
- Gangloff, S., H. Zou and R. Rothstein, 1996. Gene conversion plays the major role in controlling the stability of large tandem repeats in yeast. EMBO J. 15: 1715–1725. [PMC free article] [PubMed] [Google Scholar]
- Greig, D., R. H. Borts, E. J. Louis and M. Travisano, 2002. a Epistasis and hybrid sterility in Saccharomyces. Proc. Biol. Sci. 269: 1167–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig, D., E. J. Louis, R. H. Borts and M. Travisano, 2002. b Hybrid speciation in experimental populations of yeast. Science 298: 1773–1775. [DOI] [PubMed] [Google Scholar]
- Grishkan, I., A. B. Korol, E. Nevo and S. P. Wasser, 2003. Ecological stress and sex evolution in soil microfungi. Proc. Biol. Sci. 270: 13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutterman, Y., and E. Nevo, 1994. Temperatures and ecological genetic differentiation affecting the germination of Hordeum-Spontaneum caryopses harvested from 3 populations—the Negev Desert and opposing slopes on Mediterranean Mount Carmel. Isr. J. Plant Sci. 42: 183–195. [Google Scholar]
- Iliadi, K., N. Iliadi, E. Rashkovetsky, I. Minkov, E. Nevo et al., 2001. Sexual and reproductive behaviour of Drosophila melanogaster from a microclimatically interslope differentiated population of “Evolution Canyon” (Mount Carmel, Israel). Proc. R. Soc. Lond. B Biol. Sci. 268: 2365–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jespersen, L., K. A. van der Aa and K. M. Petersen, 2000. Phenotypic and genetic diversity of Saccharomyces contaminants isolated from lager breweries and their phylogenetic relationship with brewing yeasts. Int. J. Food Microbiol. 60: 43–53. [DOI] [PubMed] [Google Scholar]
- Johnson, L. J., V. Koufopanou, M. R. Goddard, R. Hetherington, S. M. Schafer et al., 2004. Population genetics of the wild yeast Saccharomyces paradoxus. Genetics 166: 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, M., L. Hillier, L. Riles, K. Albermann, B. Andre et al., 1997. The nucleotide sequence of Saccharomyces cerevisiae chromosome XII. Nature 387: 87–90. [PMC free article] [PubMed] [Google Scholar]
- Kaiser, C., S. Michaelis and A. Mitchell, 1994. Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Kalendar, R., J. Tanskanen, S. Immonen, E. Nevo and A. H. Schulman, 2000. Genome evolution of wild barley (Hordeum spontaneum) by BARE-1 retrotransposon dynamics in response to sharp microclimatic divergence. Proc. Natl. Acad. Sci. USA 97: 6603–6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashi, Y., and M. Soller, 1999. Functional roles of microsatellites and minisatellites, pp. 10–23 in Microsatellites, Evolution and Applications, edited by D. B. Goldstein and C. Schlotterer. Oxford University Press, Oxford.
- Kashi, Y., D. King and M. Soller, 1997. Simple sequence repeats as a source of quantitative genetic variation. Trends Genet. 13: 74–78. [DOI] [PubMed] [Google Scholar]
- Kassir, Y., and G. Simchen, 1991. Monitoring meiosis and sporulation in Saccharomyces cerevisiae. Methods Enzymol. 194: 94–110. [DOI] [PubMed] [Google Scholar]
- King, D. G., M. Soller and Y. Kashi, 1997. Evolutionary tuning knobs. Endeavour 21: 36–40. [Google Scholar]
- Kirzhner, V. M., A. V. Ryndin, E. Nevo and A. B. Korol, 2003. Balancing selection, recombination rate and polymorphism: a commentary. Preprint No. 1334. The Erwin Schräodinger International Institute for Mathematical Physics, Vienna.
- Korol, A., E. Rashkovetsky, K. Iliadi, P. Michalak, Y. Ronin et al., 2000. Nonrandom mating in Drosophila melanogaster laboratory populations derived from closely adjacent ecologically contrasting slopes at “Evolution Canyon”. Proc. Natl. Acad. Sci. USA 97: 12637–12642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korona, R., 1999. Unpredictable fitness transitions between haploid and diploid strains of the genetically loaded yeast Saccharomyces cerevisiae. Genetics 151: 77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krugman, T., N. Satish, O. N. Vinogradova, A. Beharav, Y. Kashi et al., 2001. Genome diversity in the cyanobacterium Nostoc linckia at ‘Evolution Canyon’, Israel, revealed by inter-HIP1 size polymorphisms. Evol. Ecol. Res. 3: 899–915. [Google Scholar]
- Kurtzman, C. P., and J. W. Fell, 1998. The Yeasts. A Taxonomic Study. Elsevier, Amsterdam.
- Kurtzman, C. P., and C. J. Robnett, 2003. Phylogenetic relationships among yeasts of the ‘Saccharomyces complex’ determined from multigene sequence analyses. FEMS Yeast Res. 3: 417–432. [DOI] [PubMed] [Google Scholar]
- Lamb, B. C., M. Saleem, W. Scott, N. Thapa and E. Nevo, 1998. Inherited and environmentally induced differences in mutation frequencies between wild strains of Sordaria fimicola from “Evolution Canyon.” Genetics 149: 87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambie, E. J., and G. S. Roeder, 1988. A yeast centromere acts in cis to inhibit meiotic gene conversion of adjacent sequences. Cell 52: 863–873. [DOI] [PubMed] [Google Scholar]
- Li, Y., T. Fahima, A. B. Korol, J. Peng, M. S. Roder et al., 2000. Microsatellite diversity correlated with ecological-edaphic and genetic factors in three microsites of wild emmer wheat in North Israel. Mol. Biol. Evol. 17: 851–862. [DOI] [PubMed] [Google Scholar]
- Li, Y. C., A. B. Korol, T. Fahima, A. Beiles and E. Nevo, 2002. Microsatellites: genomic distribution, putative functions and mutational mechanisms: a review. Mol. Ecol. 11: 2453–2465. [DOI] [PubMed] [Google Scholar]
- Li, Y. C., A. B. Korol, T. Fahima and E. Nevo, 2004. Microsatellites within genes: structure, function, and evolution. Mol. Biol. Evol. 21: 991–1007. [DOI] [PubMed] [Google Scholar]
- Liti, G., and E. J. Louis, 2005. Yeast evolution and comparative genomics. Annu. Rev. Microbiol. 59: 135–153. [DOI] [PubMed] [Google Scholar]
- Liu, H., C. A. Styles and G. R. Fink, 1996. Saccharomyces cerevisiae S288C has a mutation in FLO8, a gene required for filamentous growth. Genetics 144: 967–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loidl, J., 1995. Meiotic chromosome pairing in triploid and tetraploid Saccharomyces cerevisiae. Genetics 139: 1511–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupu, A., A. Pechkovskaya, E. Rashkovetsky, E. Nevo and A. Korol, 2004. DNA repair efficiency and thermotolerance in Drosophila melanogaster from “Evolution Canyon”. Mutagenesis 19: 383–390. [DOI] [PubMed] [Google Scholar]
- Mable, B. K., and S. P. Otto, 1998. The evolution of life cycles with haploid and diploid phases. BioEssays 20: 453–462. [Google Scholar]
- Mable, B. K., and S. P. Otto, 2001. Masking and purging mutations following EMS treatment in haploid, diploid and tetraploid yeast (Saccharomyces cerevisiae). Genet. Res. 77: 9–26. [DOI] [PubMed] [Google Scholar]
- Martini, A., M. Ciani and G. Scorzetti, 1996. Direct enumeration and isolation of wine yeasts from grape surfaces. Am. J. Enol. Viticult. 47: 435–440. [Google Scholar]
- Maynard Smith, J., and J. Haigh, 1974. The hitch-hiking effect of a favourable gene. Genet. Res. 23: 23–35. [PubMed] [Google Scholar]
- Michalak, P., I. Minkov, A. Helin, D. N. Lerman, B. R. Bettencourt et al., 2001. Genetic evidence for adaptation-driven incipient speciation of Drosophila melanogaster along a microclimatic contrast in “Evolution Canyon,” Israel. Proc. Natl. Acad. Sci. USA 98: 13195–13200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer, R., and M. Polsinelli, 1999. On the origins of wine yeast. Res. Microbiol. 150: 199–204. [DOI] [PubMed] [Google Scholar]
- Mortimer, R. K., 2000. Evolution and variation of the yeast (Saccharomyces) genome. Genome Res. 10: 403–409. [DOI] [PubMed] [Google Scholar]
- Mortimer, R. K., and J. R. Johnston, 1986. Genealogy of principal strains of the yeast genetic stock center. Genetics 113: 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagornaya, S. S., T. V. Babich, V. S. Podgorsky, A. Beharav, E. Nevo et al., 2003. Yeast interslope divergence in soils and plants of “Evolution canyon”, Lower Nahal Oren, Mount Carmel, Israel. Isr. J. Plant Sci. 51: 55–57. [Google Scholar]
- Naumov, G. I., 1996. Genetic identification of biological species in the Saccharomyces sensu stricto complex. J. Ind. Microbiol. 17: 295–302. [Google Scholar]
- Naumov, G. I., E. S. Naumova and P. D. Sniegowski, 1998. Saccharomyces paradoxus and Saccharomyces cerevisiae are associated with exudates of North American oaks. Can. J. Microbiol. 44: 1045–1050. [PubMed] [Google Scholar]
- Naumov, G. I., E. S. Naumova, I. Masneuf, M. Aigle, V. I. Kondratieva et al., 2000. Natural polyploidization of some cultured yeast Saccharomyces sensu stricto: auto- and allotetraploidy. Syst. Appl. Microbiol. 23: 442–449. [DOI] [PubMed] [Google Scholar]
- Nevo, E., 1995. Asian, African and European biota meet at Evolution-Canyon Israel—local tests of global biodiversity and genetic diversity patterns. Proc. R. Soc. Lond. Ser. B Biol. Sci. 262: 149–155. [Google Scholar]
- Nevo, E., 1997. Evolution in action across phylogeny caused by microclimatic stresses at “Evolution Canyon”. Theor. Popul. Biol. 52: 231–243. [DOI] [PubMed] [Google Scholar]
- Nevo, E., 2001. Evolution of genome-phenome diversity under environmental stress. Proc. Natl. Acad. Sci. USA 98: 6233–6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevo, E., E. Rashkovetsky, T. Pavlicek and A. Korol, 1998. A complex adaptive syndrome in Drosophila caused by microclimatic contrasts. Heredity 80: 9–16. [DOI] [PubMed] [Google Scholar]
- Ozkan, H., A. A. Levy and M. Feldman, 2001. Allopolyploidy-induced rapid genome evolution in the wheat (Aegilops-Triticum) group. Plant Cell 13: 1735–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page, R. D. M., 1996. TreeView: an application to display phylogenetic trees on personal computers. Comput. Appl. Biosci. 12: 357–358. [DOI] [PubMed] [Google Scholar]
- Pavlicek, T., D. Sharon, H. Kravchenko, H. Saaroni and E. Nevo, 2003. Microclimatic interslope differences underlying biodiversity contrasts in “Evolution Canyon”, Mt. Carmel, Israel. Isr. J. Earth Sci. 52: 1–9. [Google Scholar]
- Rankevich, D., B. Lavie, E. Nevo, A. Beiles and Z. Arad, 1996. Genetic and physiological adaptations of the prosobranch landsnail Pomatias olivieri to microclimatic stresses on Mount Carmel, Israel. Isr. J. Zool. 42: 425–441. [Google Scholar]
- Rashkovetsky, E., K. Iliadi, P. Michalak, A. Lupu, E. Nevo et al., 2006. Stable adaptive differentiation of thermotolerance in Drosophila along a microclimatic gradient. Heredity 96: 353–359. [DOI] [PubMed] [Google Scholar]
- Russell, D. W., R. Jensen, M. J. Zoller, J. Burke, B. Errede et al., 1986. Structure of the Saccharomyces cerevisiae HO gene and analysis of its upstream regulatory region. Mol. Cell. Biol. 6: 4281–4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleem, M., B. C. Lamb and E. Nevo, 2001. Inherited differences in crossing over and gene conversion frequencies between wild strains of Sordaria fimicola from “Evolution Canyon.” Genetics 159: 1573–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangorrin, M. P., I. E. Zajonskovsky, C. A. Lopes, M. E. Rodriguez, M. R. Giraudo de van Broock et al., 2001. Killer behaviour in wild wine yeasts associated with Merlot and Malbec type musts spontaneously fermented from northwestern Patagonia (Argentina). J. Basic Microbiol. 41: 105–113. [DOI] [PubMed] [Google Scholar]
- Satish, N., T. Krugman, O. N. Vinogradova, E. Nevo and Y. Kashi, 2001. Genome evolution of the cyanobacterium Nostoc linckia under sharp microclimatic divergence at “Evolution Canyon,” Israel. Microb. Ecol. 42: 306–316. [DOI] [PubMed] [Google Scholar]
- Shaked, H., K. Kashkush, H. Ozkan, M. Feldman and A. A. Levy, 2001. Sequence elimination and cytosine methylation are rapid and reproducible responses of the genome to wide hybridization and allopolyploidy in wheat. Plant Cell 13: 1749–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman, A., M. Shefer, S. Sagee and Y. Kassir, 1993. Post-transcriptional regulation of IME1 determines initiation of meiosis in Saccharomyces cerevisiae. Mol. Gen. Genet. 237: 375–384. [DOI] [PubMed] [Google Scholar]
- Sherman, F., 1991. Getting started with yeast. Methods Enzymol. 194: 3–21. [DOI] [PubMed] [Google Scholar]
- Singh, S. R., E. Rashkovetsky, K. Iliadi, E. Nevo and A. Korol, 2005. Assortative mating in Drosophila adapted to a microsite ecological gradient. Behav. Genet. 35: 753–764. [DOI] [PubMed] [Google Scholar]
- Snowdon, J. A., and D. O. Cliver, 1996. Microorganisms in honey. Int. J. Food Microbiol. 31: 1–26. [DOI] [PubMed] [Google Scholar]
- Stevic, S., 1962. The significance of bees (Apis sp.) and wasps (Vespa sp.) as carriers of yeast for the micoflora of grapes and the quality of wine. Arh. Poljopr. Nauke 50: 80–92. [Google Scholar]
- Takezaki, N., and M. Nei, 1996. Genetic distances and reconstruction of phylogenetic trees from microsatellite DNA. Genetics 144: 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifonov, E. N., 1989. The multiple codes of nucleotide sequences. Bull. Math. Biol. 51: 417–432. [DOI] [PubMed] [Google Scholar]
- Trifonov, E. N., 2004. The tuning function of the tandemly repeating sequences: molecular device for fast adaptation, pp. 115–138 in Evolutionary Theory and Processes: Modern Horizons, edited by S. P. Wasser. Kluwer Academic Publishers, Dordrecht, The Netherlands/Norwell, MA.
- van Belkum, A., S. Scherer, L. van Alphen and H. Verbrugh, 1998. Short-sequence DNA repeats in prokaryotic genomes. Microbiol. Mol. Biol. Rev. 62: 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Aa, K., and L. Jespersen, 1998. Detection and identification of wild yeasts in lager breweries. Int. J. Food Microbiol. 43: 205–213. [DOI] [PubMed] [Google Scholar]
- van der Aa, K. A., L. Jesperen, R. L. Glover, B. Diawara and M. Jakobsen, 2001. Identification and characterization of Saccharomyces cerevisiae strains isolated from West African sorghum beer. Yeast 18: 1069–1079. [DOI] [PubMed] [Google Scholar]
- Vaughan-Martini, A., and A. Martini, 1995. Facts, myths and legends on the prime industrial microorganism. J. Ind. Microbiol. 14: 514–522. [DOI] [PubMed] [Google Scholar]
- Weir, B. S., 1990. Genetic Data Analysis. Sinauer, Sunderland, MA.
- Wolfe, K. H., 2001. Yesterday's polyploids and the mystery of diploidization. Nat. Rev. Genet. 2: 333–341. [DOI] [PubMed] [Google Scholar]
- Wolfe, K. H., and D. C. Shields, 1997. Molecular evidence for an ancient duplication of the entire yeast genome. Nature 387: 708–713. [DOI] [PubMed] [Google Scholar]
- Wong, S., G. Butler and K. H. Wolfe, 2002. Gene order evolution and paleopolyploidy in hemiascomycete yeasts. Proc. Natl. Acad. Sci. USA 99: 9272–9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamorzaeva, E., E. Rashkovetsky, E. Nevo and A. Korol, 2005. Sequence polymorphism of candidate behavioural genes in Drosophila melanogaster flies from ‘Evolution Canyon’. Mol. Ecol. 14: 3235–3245. [DOI] [PubMed] [Google Scholar]
- Zeyl, C., 2000. Budding yeast as a model organism for population genetics. Yeast 16: 773–784. [DOI] [PubMed] [Google Scholar]
- Zeyl, C., 2004. Experimental studies of ploidy evolution in yeast. FEMS Microbiol. Lett. 233: 187–192. [DOI] [PubMed] [Google Scholar]
- Zeyl, C., and G. Bell, 1997. The advantage of sex in evolving yeast populations. Nature 388: 465–468. [DOI] [PubMed] [Google Scholar]
- Zeyl, C., T. Vanderford and M. Carter, 2003. An evolutionary advantage of haploidy in large yeast populations. Science 299: 555–558. [DOI] [PubMed] [Google Scholar]