

Abstract
Glucocorticoids (GCs) are potent anti-inflammatory agents that block cytokine production. We investigated whether GCs also block cytokine signaling via the Janus kinase (Jak)-signal transducer and activator of transcription (STAT) pathway. Dexamethasone inhibited IL-2-induced DNA binding, tyrosine phosphorylation, and nuclear translocation of Stat5 in primary T cells. Inhibition of Stat5 correlated with inhibition of expression of IL-2-inducible genes and T cell proliferation. The mechanism of inhibition involved suppression of IL-2 receptor and Jak3 expression. Signaling by IL-4, IL-7, and IL-15, which use IL-2 receptor components, also was inhibited, indicating a block in T cell responses similar to that seen in immunodeficient patients lacking the IL-2 receptor gamma chain or Jak3. IL-2 signaling also was blocked in patients after treatment with GCs, suggesting that inhibition of cytokine signaling contributes to the clinical efficacy of these agents. These results identify inhibition of Jak-STAT signaling by IL-2 and related cytokines as a novel mechanism of GC action and suggest that inhibition of both cytokine production and signaling contribute to their therapeutic potency.
Adrenal glucocorticoids (GCs) are steroid hormones that bind to cytoplasmic receptors and trigger a conformation change leading to nuclear translocation and subsequent modulation of gene transcription. GC receptors can stimulate transcription by binding to specific GC response elements (GREs) in gene promoters, or can repress transcription by binding to negative GREs (1). GCs act on multiple cells and tissues and are important regulators of whole body physiology. Within the immune system, GC synthesis is induced through stimulation of the hypothalamic-pituitary-adrenal axis by inflammatory cytokines such as IL-6, and GCs are thought to play a physiologic role in feedback inhibition of immune/inflammatory responses and in homeostasis (reviewed in ref. 2). GCs accomplish this role through inhibition of effector function of immune cells, inhibition of migration of cells into inflammatory sites, and suppression of proliferation of lymphocytes. GCs are among the most potent and effective immunosuppressive agents and are used in treatment of numerous autoimmune and inflammatory diseases (2).
The molecular mechanisms of GC immunosuppression have been the subject of extensive investigation (1). Two important mechanisms of action are inhibition of cytokine, chemokine, and adhesion molecule production, and antagonism of the action of inflammatory cytokines such as IL-1 and tumor necrosis factor (TNF). It has become clear that inhibition of the activation protein-1 (AP-1) and NF-κB families of transcription factors underlies both of these phenomena. AP-1 and NF-κB are widely expressed and are activated by a variety of immune and inflammatory stimuli, including cytokines such as IL-1 and TNF, as well as crosslinking of antigen receptors and costimulatory molecules. GCs inhibit AP-1 and NF-κB proteins by a variety of mechanisms, the relative roles of which appear to vary according to cell type. One important mechanism is direct physical interaction with AP-1 and NF-κB proteins, to inhibit their transcriptional activity (3–7). This action of GCs is independent of their ability to activate or repress transcription. Another important mechanism is the induction of expression of I-κB, an inhibitory molecule that tethers NF-κB subunits in the cytoplasm (8, 9). Whereas inhibition of AP-1 and NF-κB is an attractive mechanism of GC action, this inhibition is often quite partial (8, 9) and out of proportion to the overall anti-inflammatory potency of GCs, suggesting that additional molecular mechanisms likely contribute to the anti-inflammatory effects of GCs.
IL-2 is a T cell-derived cytokine important in the regulation of lymphocyte proliferation and immune responses (reviewed in ref. 10). IL-2 signaling is mediated by a multichain receptor complex consisting of an α, β, and a common γ chain (γc), the latter used by other cytokine receptors including the IL-4, IL-7, IL-9, and IL-15 receptors (reviewed in ref. 10). Signaling by IL-2 occurs through high or intermediate affinity receptors containing α/β/γc, or β/γc chains, respectively. The IL-2 receptor (IL-2R) α subunit primarily increases the affinity of ligand binding and is not known to contain a signaling domain, whereas the β and γc subunits participate in both ligand binding and signal transduction. The protein tyrosine kinases Jak1 and Jak3 (Janus kinases 1 and 3), which are associated with the IL-2R β and γc subunits, respectively, are activated after binding of IL-2 to its receptor. Subsequently, specific tyrosine residues in the cytoplasmic domains of the IL-2R β and γc subunits become phosphorylated. The β chain phosphotyrosine motifs provide docking sites that are recognized by the Src homology 2 domain of Stat5, and, to a lesser extent, Stat3 (10). The roles of Jak1 and Jak3 in IL-2-mediated proliferation are well established, and the critical role of Stat5 in IL-2 responses and proliferation has been recently demonstrated (11, 12).
Jak-STAT (signal transducer and activator of transcription) signaling by IL-2, IL-4, IL-7, IL-9, and IL-15 is critically important in the generation of immune and inflammatory responses and in T cell expansion and differentiation. Lack of signaling by these cytokines, as occurs in genetic deficiency of γc or Jak3, results in severe immunodeficiency (10). Therefore, suppression of signaling by IL-2 and related cytokines would be predicted to have strong suppressive effects on T cells. We investigated whether GCs, which suppress IL-2 production (13), also might act downstream of cytokine production and inhibit T cells by blocking signaling by IL-2 and related cytokines via the Jak-STAT pathway.
Materials and Methods
Cell Culture.
Mononuclear cells were obtained by Ficoll gradient density centrifugation of disease-free donors' peripheral blood, activated with OKT3 for 3 days in RPMI medium 1640 (GIBCO/BRL) containing 10% FCS (HyClone), and activated T cells (>90% CD3+) were washed, rested for 24 h, cultured with dexamethasone (dex), and subsequently treated with IL-2 (Boehringer Mannheim). Several different culture systems, including cultures in which T cells were not rested before addition of dex, were used, and comparable results were obtained.
Cell and Nuclear Extract Preparation, Electrophoretic Mobility Shift Assays (EMSAs), and Immunoblotting (IB).
Whole-cell extracts were prepared as described (14), and nuclear extracts were prepared as described (15), with modifications. A total of 107 cells were resuspended in 200 μl of buffer A (10 mM Hepes, pH 7.9/1.5 mM MgCl2/10 mM KCl/0.1 mM EDTA/0.1 mM EGTA/1 mM DTT/0.5 mM PMSF) and allowed to swell on ice for 15 min. NP-40 was added to a final concentration of 0.2% for 2.5 min, nuclei were obtained by centrifugation, and nuclear proteins were extracted in 50 μl of buffer containing 20 mM Hepes (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 300 mM NaCl, 0.5 mM DTT, 0.2 mM PMSF, and 20% glycerol. EMSAs and IB were performed as described (16).
Flow Cytometry and IL-2R Binding.
Flow cytometry was performed as described (16). The binding of IL-2 to cell surface IL-2Rs was measured by using a Fluorokine kit according to the instructions of the manufacturer (R & D Systems).
Cell Proliferation.
T cells were plated at a concentration of 1 × 106/ml, 0.1 ml/well, in 96-well plates. Cells were cultured in IL-2 for 24 h, 3H-thymidine was added during the last 8 h of culture, and 3H-thymidine incorporation was measured as described (16). Cell viability was monitored in parallel cultures.
Analysis of mRNA Levels.
Total cellular RNA was isolated by using Trizol (GIBCO/BRL) according to the instructions of the manufacturer. For reverse transcriptase (RT)-PCR, RNA was treated with RNase-free DNase, and cDNA was obtained by using Moloney murine leukemia virus RT (GIBCO/BRL). Some 2.5% of each cDNA was subjected to 22–25 cycles of PCR using conditions that result in a single specific amplification product of the correct size, as described (16, 17): 30 sec denaturation at 94°C, 1 min annealing at 55°C, and 30 sec extension at 72°C in a GeneAmp 9600 thermal cycler (Perkin–Elmer). dNTPs were used at 100 μM and 1 μCi of 32P-α-dATP was added to each reaction. No amplification products were obtained when RT was omitted, indicating the absence of contaminating genomic DNA. Amplification was empirically determined to be in the linear range. Oligonucleotide primers used are as follows: glyceraldehyde-3-phosphate dehydrogenase, GTG AAG GTC GGA GTC AAC and TGG AAT TTG CCA TGG GTG; IL-2Rβ, CCG GGC CAT GGC TGA AGA AGG and CCC CGG CCA CAC CCT CAT CAG; CXCR3, AAC GCC ACC CAC TGC CAA TAC and GCT CCC GGA ACT TGA CCC CTA; cytokine inducible Src homology 2-containing protein, CTC CGG CTG GCC CCT TCT GTA and TGA AAG CGG CCG GCC TGA AAG; and FasL, GGA TTG GGC CTG GGG ATG TTT and GTG GCT CAG GGG CAG GTT GTT.
Results
Dex Inhibits IL-2 Stat5 Activation in a Dose- and Time-Dependent Manner.
The effect of the synthetic GC dex on IL-2-induced Stat5 DNA binding was determined in primary human T cells. T cell blasts were obtained and rested as described (18), such that there was no active signaling through the T cell antigen receptor (TcR) (18), and thus effects of GCs on TcR signaling would not confound the analysis of IL-2 signaling. IL-2 activated a DNA-binding complex that corresponded to Stat5 in a rapid and transient fashion (Fig. 1A Upper), as expected (10). Specificity of binding and presence of Stat5 in the DNA-binding complex were confirmed as described (16) (data not shown). Culture with dex (10-6 M) inhibited the activation of Stat5 DNA binding (Fig. 1A, lanes 4, 6, 8, 10, and 12). The similar levels of inhibition of Stat5 at all time points tested shows that dex truly inhibited IL-2 signaling and did not merely alter the kinetics of Stat5 activation. Dex inhibited IL-2 activation of Stat5 DNA binding in a dose-dependent fashion, including concentrations of dex (10-8 M–10-7 M) that correspond to physiological corticosteroid levels (Fig. 1B Upper). Dex did not affect Stat5 protein levels (Fig. 1 A and B Lower), indicating that the decrease in DNA binding cannot be explained on the basis of decreased levels of Stat5 protein. Dex did not affect recovery of viable T cells in this system (data not shown), similar to previously reported results showing that mature human T cells are not susceptible to GC-induced apoptosis (19, 20). Inhibition of Stat5 DNA binding was reproducibly detected in more than 50 independent experiments using different blood donors and several different culture systems (data not shown).
Figure 1.
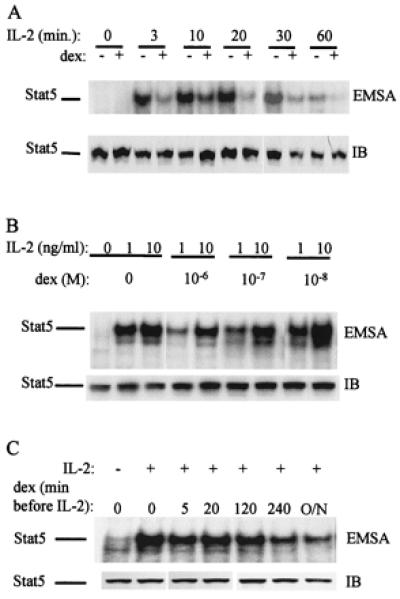
Dex inhibits IL-2 induction of Stat5 DNA binding in a dose- and time-dependent manner. Mononuclear cells were obtained by Ficoll gradient density centrifugation of disease-free donors' peripheral blood, activated with OKT3 for 3 days, and then T cells (>90% CD3+) were washed, rested for 24 h, cultured with dex, and subsequently treated with IL-2. (A) Effect of dex on IL-2-induced-Stat5 DNA binding. T cells were cultured for 16 h with dex (10-6 M), and subsequently treated with IL-2 (1 ng/ml) for the times indicated. Whole-cell extracts were assayed for Stat5 DNA binding using the IFN responsive factor oligonucleotide as described (16) (Upper). Stat5 protein levels in the same extracts were determined by using IB (Lower). (B) Dose dependency of the effect of dex on IL-2-induced Stat5 DNA binding. T cells were cultured for 16 h with dex at the doses indicated, then treated with IL-2 (1 or 10 ng/ml) for 10 min. (C) Time dependency of the effect of dex on IL-2-induced Stat5 DNA binding. T cells were cultured with dex (10-6 M) for the times indicated, and subsequently treated with a dose of 1 ng/ml of IL-2.
The time of culture with dex that was required to achieve inhibition of Stat5 activation was investigated. The level of inhibition of Stat5 increased in a time-dependent manner over 16 h of culture with dex and was consistently detected after 4 h (Fig. 1C; a representative experiment of five performed is shown). The requirement for preincubation may explain why, although some effects of dex on cytokine action have been described, inhibition of Jak-STAT signaling by dex has not been previously appreciated as an important mechanism of GC action (21–25). The length of time required for dex to exert its inhibitory effect suggests that dex treatment does not directly modify IL-2 signaling components, but instead acts by inducing the synthesis of inhibitory proteins or inhibiting the synthesis of components of the IL-2-Jak-STAT signaling pathway.
Inhibition of Stat5 Occurs at the Level of Tyrosine Phosphorylation and Nuclear Translocation.
Dex could potentially block Stat5 DNA binding by inhibiting the binding activity of tyrosine-phosphorylated Stat5 dimers, or by blocking tyrosine phosphorylation. The effect of dex on Stat5 tyrosine phosphorylation was investigated. Dex suppressed Stat5 tyrosine phosphorylation in a dose-dependent fashion (Fig. 2A). Nuclear translocation of Stat5 depends on prior tyrosine phosphorylation, and the effect of dex on nuclear translocation was determined. Dex treatment effectively blocked Stat5 nuclear translocation at every time point tested (Fig. 2B). Levels of SP1 DNA binding activity were comparable in nuclear extracts from control cells or cells treated with dex (Fig. 2B) and SP1 protein levels were not changed by dex treatment (data not shown), indicating that extraction of nuclear proteins was comparable, and that dex did not nonspecifically block DNA binding of all transcription factors. These results, together with the DNA binding studies (Fig. 1), suggest that Stat5 activation is blocked at a proximal step in signal transduction.
Figure 2.
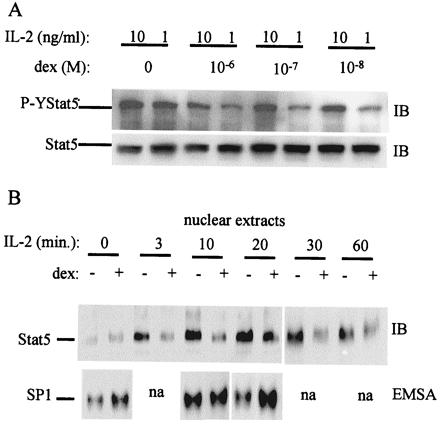
Dex inhibits IL-2 induction of Stat5 tyrosine phosphorylation and nuclear translocation. (A) Preactivated T cells were cultured for 16 h with dex at the doses indicated, then treated with IL-2 (1 or 10 ng/ml), and levels of tyrosine-phosphorylated and total Stat5 were measured by sequential IB of the same membrane. (B) T cells cultured with dex for 16 h were stimulated with IL-2 (1 ng/ml) for the times indicated. Nuclear extracts were prepared as described in Materials and Methods, and Stat5 was detected by IB (Upper). Nuclear levels of Sp1 DNA binding were measured as a control (Lower).
Dex Suppresses Expression of IL-2Rs and Jak3.
The mechanism of inhibition was further explored by analyzing the effect of dex on expression of proteins in the IL-2-Jak-STAT signaling pathway. Levels of cell surface expression of IL-2R α, β, and γc subunits were measured by using flow cytometry. The exposure of T cells to dex for 16 h did not alter the expression of cell surface IL-2R α subunit at any of the concentrations tested (Fig. 3A), whereas β and γc subunit expression was suppressed (Fig. 3A). Expression of IL-2Rα was not affected by dex in more than 20 independent experiments with different donors. Suppression of the β and γc subunits was consistently detected, the extent of suppression varied among donors, and a representative experiment is shown. These results indicating inhibition of cell surface IL-2R expression were confirmed by measuring the binding of IL-2 to functional IL-2Rs on T cells exposed for 4 or 16 h to dex. Dex suppressed the number of IL-2 binding sites in a time-dependent fashion (Table 1). Because a quantitative relationship between IL-2R levels and signal transduction is well established (26), these results indicate that modulation of IL-2R expression represents one mechanism of inhibition of IL-2 signal transduction by dex. Dex-mediated suppression of β and γc expression suggested that dex would block signaling by other cytokines that use these signaling proteins (10), and this was confirmed for IL-4, IL-7, and IL-15 (Fig. 3B). A block in IL-2 signaling upstream of Stat5 activation would be predicted to also suppress the activation of parallel signaling pathways, such as activation of the Akt kinase that is important for IL-2-induced proliferation. Activation of Akt by IL-2 was indeed blocked by dex treatment (Fig. 3C), strengthening the evidence for a proximal block in the IL-2 signaling pathway.
Figure 3.
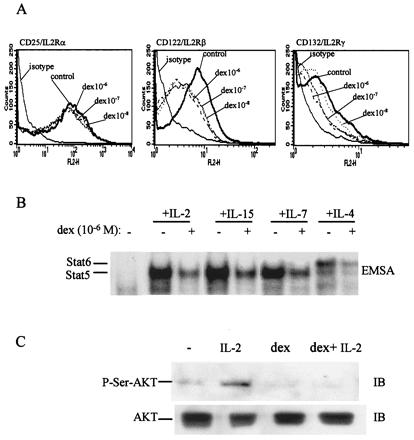
Dex inhibits surface expression of IL-2R β and γc subunits and activation of Stat5 by other cytokines that use γc. (A) T cells were cultured with dex for 16 h at the doses indicated and the surface expression of IL-2R α (CD25), β (CD122), and γc (CD132) was assessed by flow cytometry as described (16). (B) T cells cultured with dex for 16 h were stimulated for 10 min with IL-2 (1 ng/ml), IL-15 (10 ng/ml), IL-7 (10 ng/ml), or IL-4 (10 ng/ml). (C) Akt kinase activation is blocked in dex-treated cells. Cell extracts were analyzed by sequential IB of the same membrane.
Table 1.
Dex inhibits the binding of IL-2 to its receptor
| Time of culture, hr | IL-2R density | Negative control | Specificity control |
|---|---|---|---|
| 4 | 584 | 56 | 31 |
| 4+dex | 289 | 31 | 33 |
| 16 | 410 | 38 | 16 |
| 16+dex | 146 | 27 | 9 |
Measured using biotinylated IL-2 as recommended by the manufacturer (R&D Systems). Soybean trypsin inhibitor, biotinylated to the same degree as IL-2, was used as negative control for staining. Specificity of the IL-2 biotin reaction was tested by measuring the binding after preincubation with αhIL-2 Ab.
These results do not exclude the possibility that dex also may accelerate deactivation of signaling, possibly through the induction of a tyrosine phosphatase or of degradation of signaling proteins by proteosomes (27–29). The effects of dex on the rate of decay of Stat5 activity were investigated. When IL-2 signaling was terminated by removing cytokine as described (28), the rate of inactivation of Stat5 was similar in control and dex-treated cells (Fig. 4A), suggesting that dex does not act by accelerating Stat5 inactivation. A potential role for tyrosine phosphatases or proteosomes in dex-mediated inhibition was investigated by using vanadate, a potent inhibitor of protein tyrosine phosphatases, and MG132, a proteosome inhibitor. Vanadate increased the level of IL-2-induced Stat5 activity in both control and dex-treated cells to a similar extent (Fig. 4B). Importantly, the level of Stat5 activity in dex-cultured cells exposed to vanadate (Fig. 4B, lane 8) was substantially lower than Stat5 activity in control cells exposed to vanadate (lane 4), indicating that dex did not act solely by inducing tyrosine phosphatase activity. MG132 did not significantly affect inhibition of IL-2 signaling by dex (Fig. 4C), excluding an important role for proteosome-dependent degradation. Taken together, the results shown in Figs. 3 and 4 indicate that dex primarily inhibits IL-2 signaling upstream of the activation of Stat5.
Figure 4.
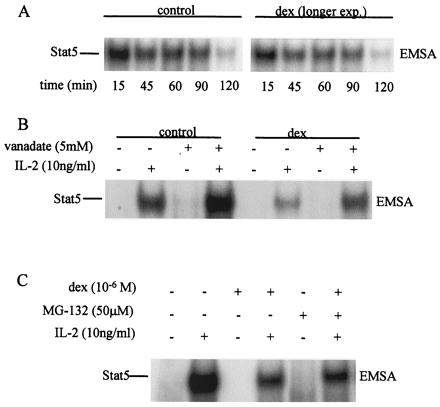
Dex does not accelerate deactivation of Stat5 by phosphatases or proteosomes. (A) Kinetics of Stat5 deactivation. IL-2 was removed and the decay of Stat5 activity was analyzed by using EMSA. A representative experiment of four performed is shown. (B) Inhibition of tyrosine phosphatases does not reverse dex inhibitory activity. Vanadate was added 2 min before adding IL-2. (C) MG132 was added 5 min before adding IL-2.
The mechanisms by which dex inhibited expression of IL-2R components were further examined. Interestingly, dex inhibited IL-2R β, but not γc mRNA expression (Fig. 5A, and data not shown). Cyclohexemide did not affect inhibition of IL-2Rβ mRNA expression, indicating that the dex effect is independent of de novo protein synthesis (Fig. 5A). Further analysis of the regulation of IL-2Rβ and γc expression at the translational level was hampered by the inability of currently available reagents to detect the low levels of intracellular protein in primary T cells. However, Jak3 protein levels were readily detectable, and Jak3 expression was suppressed by dex (Fig. 5B Middle), whereas Jak1 expression was not affected (Fig. 5B Bottom). The effect of RU486, a competitive antagonist of dex, on Jak3 expression and Stat5 DNA binding was determined. RU486 reversed dex-mediated inhibition of both Jak3 expression and Stat5 DNA binding (Fig. 5B). This finding indicates that inhibition by dex is mediated by the GC receptor and demonstrates a correlation between Jak3 expression and IL-2 signaling.
Figure 5.
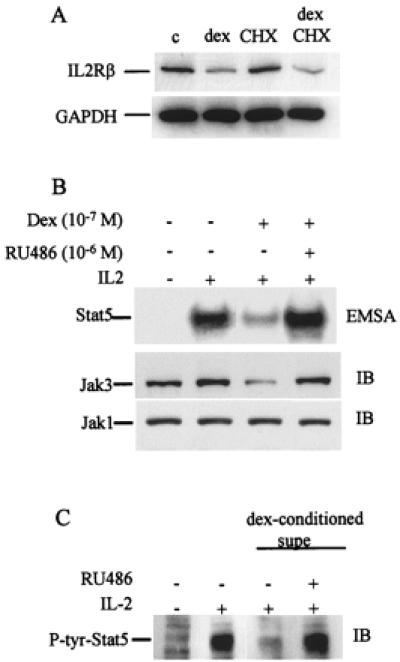
(A) Dex inhibits IL-2R β mRNA expression independent of de novo protein synthesis. T cells were cultured for 8 h with dex (10-6 M), in the presence or absence of cyclohexemide (CHX) (40 μg/ml), and mRNA levels were measured by using semiquantitative RT-PCR as described (16, 17). GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (B) Dex suppresses JAK3 protein levels, and suppression is reversed by RU486. T cells cultured for 16 h with dex (10-7 M) in the presence or absence of RU486 (10-6 M). (C) RU486 blocks the inhibitory activity present in supernatants of cells cultured with dex. Supernatants from cells cultured with 10-6 M dex for 16 h were added to new cultures (50% final concentration) for 16 h in the presence or absence of RU486.
Dex-mediated suppression of IL-2Rβ mRNA levels in the absence of de novo protein synthesis (Fig. 5A) suggested that dex may act, at least in part, directly. This possibility was further explored by testing whether culture supernatants from dex-treated cells contained any inhibitory activity (in addition to dex) toward IL-2 signaling. Supernatants from dex-treated cells did inhibit IL-2 signaling (Fig. 5C, lane 3), but this activity was reversed when the dex present in these supernatants was blocked by using RU486 (Fig. 5C, lane 4), suggesting that the supernatants did not contain newly synthesized inhibitory molecules. In addition, neutralization of IL-10 and transforming growth factor β, the two cytokines known to inhibit IL-2 signaling that might have been induced by dex, had no effect (data not shown). Taken together, these results indicate that one component of inhibition of IL-2 signaling by dex is direct. These results do not exclude the possibility that dex also may induce the expression of negative factors that contribute to inhibition.
Inhibition of IL-2 Signaling by Dex Results in Suppression of IL-2-Induced Gene Activation and Proliferation.
The physiologic significance of dex-induced inhibition of IL-2 signaling was investigated by examining the effect of dex on expression of IL-2-inducible genes and on T cell proliferation. Steady-state mRNA levels of IL-2-inducible genes were determined by using semiquantitative RT-PCR, as described (16, 17). Treatment with IL-2 induced expression of the CIS, IFN-γ, CXC chemokine receptor 3 (CXCR3), and Fas ligand (FasL) genes (Fig. 6A). These genes have been previously shown to be IL-2-inducible, and CIS expression is regulated primarily by Stat5. Pretreatment with dex suppressed IL-2 activation of CIS, IFN-γ, and CXCR3 expression (Fig. 6A, lane 4). Induction of FasL mRNA levels was not suppressed by dex (Fig. 6A). This indicates that dex did not nonspecifically inhibit all transcription, and that dex did not block the induction of all IL-2-inducible genes (some of which may depend less on Stat5). The effect of dex on IL-2-triggered T cell proliferation was determined. Dex suppressed proliferation in a dose-dependent fashion, as assessed by 3H-thymidine incorporation (Fig. 6B). The suppression of proliferation by dex was detected at dex concentrations as low as 10-8 M (data not shown) and was reproducibly in the range of 75% to 90% inhibition of proliferation at a dose of 10-6 M in experiments with multiple independent donors. A dex inhibitory effect on T cell proliferation was detected at high doses of IL-2 (50 ng/ml) and was not secondary to cell death or apoptosis, as parallel wells treated with or without dex contained equal numbers of viable cells (data not shown). These results demonstrate that inhibition of IL-2 signaling by dex leads to inhibition of T cell function. Activation of Stat5 in response to IL-2 was evaluated in seven patients with autoimmune diseases before and after i.v. treatment with the GC methylprednisolone. IL-2-induced activation of Stat5 was suppressed in 5 of 7 patients (P = 0.021 by Fisher's exact test); a representative experiment is shown in Fig. 6C. These results demonstrate that the inhibition of Stat5 activation that occurred in vitro also occurred in vivo in patients treated with steroids and suggest that suppression of Jak-STAT signaling plays a role in the anti-inflammatory effects of GC therapy.
Figure 6.
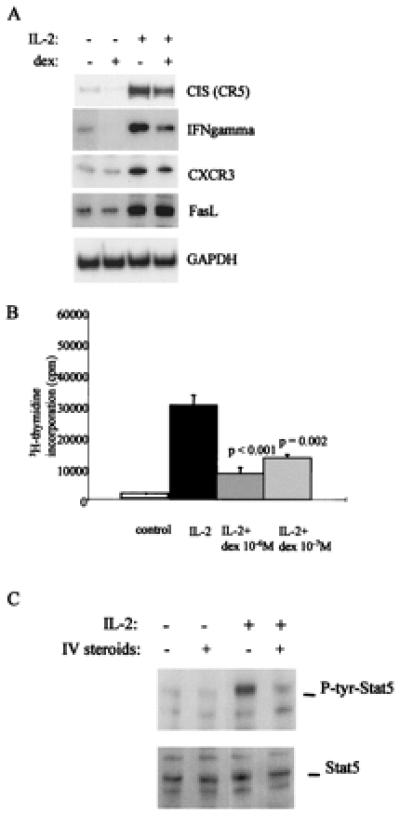
Dex inhibits expression of IL-2-inducible genes and T cell proliferation. (A) T cells were cultured for 4 h in presence of dex (10-6 M), then stimulated with IL-2 (1 ng/ml) and mRNA levels were measured by using semiquantitative RT-PCR, as described in Materials and Methods. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (B) After 16 h of culture with dex at the doses indicated, IL-2-dependent T cell proliferation was measured by using 3H-thymidine incorporation. (C) Dex suppresses IL-2 activation of Stat5 in vivo. Mononuclear cells were isolated from patients immediately before and 24 h after treatment with 1 g of i.v. methylprednisolone, and immediately stimulated with 1 ng/ml of IL-2 (−, before steroids and +, 24 h after steroid treatment).
Discussion
The major finding reported herein is that GCs block signaling by IL-2, and by related cytokines that share the γc receptor subunit, in T cells. This identifies a level of GC action that works downstream of the previously reported GC-mediated inhibition of T cell antigen receptor signaling and of production of multiple cytokines, including IL-2. IL-2 and related cytokines are particularly important for the development, expansion, and survival of T cells, and lack of signaling by these cytokines results in severe immunodeficiency (10). Thus, inhibition of both cytokine production and cytokine action in T cells will contribute to the potent suppressive effect of GCs on these cells.
Inhibition of IL-2 activation of Stat5 occurred at the level of tyrosine phosphorylation (Fig. 2). Lower levels of Stat5 tyrosine phosphorylation may be secondary either to more rapid dephosphorylation or degradation of Stat5 (28, 29) or to a block in upstream signaling. Several lines of evidence suggest that an important component of dex-mediated inhibition occurs upstream of Stat5 in the IL-2 signaling pathway: (i) A quantitative relationship exists between IL-2 receptor levels and signal transduction over a broad range of IL-2R expression (26), and thus the lower expression of IL-2Rs (Fig. 3 and Table 1) contributes to lower levels of signaling. (ii) Expression of an additional component of the IL-2R, Jak3, was suppressed by GCs, and reversal of inhibition of Jak3 expression by RU486 correlated with reversal of inhibition of Stat5 DNA binding (Fig. 5). Because Jak3 is expressed in limiting quantities (30), suppression of Jak3 expression also contributes to lower Stat5 activation. (iii) The kinetics of Stat5 deactivation were not altered by dex, and experiments using vanadate and MG132 suggested that induction of tyrosine phosphatases or of degradation by the proteosome did not play an important role in dex-mediated inhibition. Taken together, these results indicate that GC-mediated inhibition of IL-2 signaling occurs, at least in part, upstream of Stat5 activation, and the mechanism likely corresponds to inhibition of expression of IL-2 signaling components. Inhibition of cytokine receptor and Jak3 expression represents a novel mechanism of GC action, as GCs have been previously reported not to change, or to paradoxically increase, expression of cytokine receptors (31).
At least 4–16 h of preincubation with dex were required for inhibition of IL-2 signaling to become established (Fig. 1), suggesting that dex may, at least in part, act indirectly via induction of expression of inhibitory proteins. Reversal of inhibition of IL-2 signaling and Jak3 expression by RU486, an antagonist GC receptor-dependent transcription, also suggests a role for newly expressed gene products in inhibition. The identity of these proteins remains unknown, but a role for suppressor of cytokine signaling (SOCS) proteins, IL-10, and transforming growth factor β has been excluded (data not shown). However, the evidence also supports a direct mechanism of dex action: suppression of IL-2Rβ mRNA levels occurred in the presence of cycloheximide (Fig. 5A), and the inhibitory activity of supernatants of dex-treated cells was reversed by RU486 (Fig. 5C). The most likely mechanism of direct action of dex would be inhibition of the expression of signaling proteins such as IL-2Rβ, possibly through inhibition of transcription mediated by a negative GC response element. These results, taken together, indicate that GCs act by both direct and indirect mechanisms, and target multiple components of the IL-2 signaling machinery at the level of expression. This complex regulation is similar to GC inhibition of NF-κB, where direct mechanisms at the level of GC receptor/NF-κB protein–protein interactions, and indirect mechanisms mediated by induction of I-κB expression, or inhibition of kinases, both play important roles (3–9, 32).
Interactions between GCs and cytokine Jak-STAT signaling pathways have not been widely appreciated, but represent an emerging area of research. Previous reports show that simultaneous addition of GCs and the cytokine prolactin (that activates Stat5) leads to synergistic activation of genes important in lactation (25), an effect that is mediated by direct interactions between the GC receptor and Stat5. In contrast, the results presented herein reveal an unexpected GC-mediated inhibition of signaling of IL-2 and related cytokines. This inhibitory mechanism differs from most previously described mechanisms of Jak-STAT positive and negative regulation, which depend on posttranslational modification of pre-existing signaling components or induction of Jak inhibitors (14, 33). Inhibition of signaling by IL-2 had clear-cut physiologic consequences in terms of suppressing T cell proliferation and expression of IL-2-inducible genes. In addition, IL-2 signaling was suppressed in patients that were treated with steroids (Fig. 6C). Thus, inhibition of IL-2 signaling likely contributes to the immunosuppressive effects of GCs and to the efficacy of GCs in the treatment of inflammatory diseases.
Acknowledgments
We thank John O'Shea and Kendall Smith for helpful discussions and Simi Ahmed and Pietro Ghezzi for reviewing the manuscript. This work was supported by a grant from the National Institutes of Health.
Abbreviations
- GC
glucocorticoid
- STAT
signal transducer and activator of transcription
- Jak
Janus kinase
- EMSA
electrophoretic mobility shift assay
- AP-1
activation protein-1
- γc
common γ chain
- dex
dexamethasone
- IL-2R
IL-2 receptor
- IB
immunoblotting
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.160099797.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.160099797
References
- 1.Karin M. Cell. 1998;93:487–490. doi: 10.1016/s0092-8674(00)81177-0. [DOI] [PubMed] [Google Scholar]
- 2.Boumpas D T, Chrousos G P, Wilder R L, Cupps T R, Balow J E. Ann Intern Med. 1993;119:1198–1208. doi: 10.7326/0003-4819-119-12-199312150-00007. [DOI] [PubMed] [Google Scholar]
- 3.Diamond M I, Miner J N, Yoshinaga S K, Yamamoto K R. Science. 1990;249:1266–1272. doi: 10.1126/science.2119054. [DOI] [PubMed] [Google Scholar]
- 4.De Bosscher K, Schmitz M L, Vanden Berghe W, Plaisance S, Fiers W, Haegeman G. Proc Natl Acad Sci USA. 1997;94:13504–13509. doi: 10.1073/pnas.94.25.13504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jonat C, Rahmsdorf H J, Park K K, Cato A C, Gebel S, Ponta H, Herrlich P. Cell. 1990;62:1189–1204. doi: 10.1016/0092-8674(90)90395-u. [DOI] [PubMed] [Google Scholar]
- 6.Schule R, Rangarajan P, Kliewer S, Ransone L J, Bolado J, Yang N, Verma I M, Evans R M. Cell. 1990;62:1217–1226. doi: 10.1016/0092-8674(90)90397-w. [DOI] [PubMed] [Google Scholar]
- 7.Yang-Yen H F, Chambard J C, Sun Y L, Smeal T, Schmidt T J, Drouin J, Karin M. Cell. 1990;62:1205–1215. doi: 10.1016/0092-8674(90)90396-v. [DOI] [PubMed] [Google Scholar]
- 8.Scheinman R I, Cogswell P C, Lofquist A K, Baldwin A S., Jr Science. 1995;270:283–286. doi: 10.1126/science.270.5234.283. [DOI] [PubMed] [Google Scholar]
- 9.Auphan N, DiDonato J A, Rosette C, Helmberg A, Karin M. Science. 1995;270:286–290. doi: 10.1126/science.270.5234.286. [DOI] [PubMed] [Google Scholar]
- 10.Gaffen S L, Goldsmith M A, Greene W C. Interleukin-2 and the Interleukin-2 Receptor. San Diego: Academic; 1998. [Google Scholar]
- 11.Teglund S, McKay C, Schuetz E, van Deursen J M, Stravopodis D, Wang D, Brown M, Bodner S, Grosveld G, Ihle J N. Cell. 1998;93:841–850. doi: 10.1016/s0092-8674(00)81444-0. [DOI] [PubMed] [Google Scholar]
- 12.Moriggl R, Sexl V, Piekorz R, Topham D, Ihle J N. Immunity. 1999;11:225–230. doi: 10.1016/s1074-7613(00)80097-7. [DOI] [PubMed] [Google Scholar]
- 13.Papanicolaou D A, Wilder R L, Manolagas S C, Chrousos G P. Ann Intern Med. 1998;128:127–137. doi: 10.7326/0003-4819-128-2-199801150-00009. [DOI] [PubMed] [Google Scholar]
- 14.Sengupta T K, Talbot E S, Scherle P A, Ivashkiv L B. Proc Natl Acad Sci USA. 1998;95:11107–11112. doi: 10.1073/pnas.95.19.11107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schreiber E, Matthias P, Muller M M, Schaffner W. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Castro A, Sengupta T K, Ruiz D C, Yang E, Ivashkiv L B. J Immunol. 1999;162:1261–1269. [PubMed] [Google Scholar]
- 17.Lee I H, Li W P, Hisert K B, Ivashkiv L B. J Exp Med. 1999;190:1263–1274. doi: 10.1084/jem.190.9.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cantrell D A, Smith K A. J Exp Med. 1983;158:1895–1911. doi: 10.1084/jem.158.6.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brunetti M, Martelli N, Colasante A, Piantelli M, Musiani P, Aiello F B. Blood. 1995;86:4199–4205. [PubMed] [Google Scholar]
- 20.Tuosto L, Cundari E, Montani M S, Piccolella E. Eur J Immunol. 1994;24:1061–1065. doi: 10.1002/eji.1830240508. [DOI] [PubMed] [Google Scholar]
- 21.Gillis S, Crabtree G R, Smith K A. J Immunol. 1979;123:1624–1631. [PubMed] [Google Scholar]
- 22.Franchimont D, Galon J, Gadina M, Visconti R, Zhou Y, Aringer M, Frucht D M, Chrousos G P, O'Shea J J. J Immunol. 2000;164:1768–1774. doi: 10.4049/jimmunol.164.4.1768. [DOI] [PubMed] [Google Scholar]
- 23.Louahed J, Renauld J C, Demoulin J B, Baughman G, Bourgeois S, Sugamura K, Van Snick J. J Immunol. 1996;156:3704–3710. [PubMed] [Google Scholar]
- 24.Paliogianni F, Ahuja S S, Balow J P, Balow J E, Boumpas D T. J Immunol. 1993;151:4081–4089. [PubMed] [Google Scholar]
- 25.Stocklin E, Wissler M, Gouilleux F, Groner B. Nature (London) 1996;383:726–728. doi: 10.1038/383726a0. [DOI] [PubMed] [Google Scholar]
- 26.Smith K A. Ann NY Acad Sci. 1995;766:263–271. doi: 10.1111/j.1749-6632.1995.tb26674.x. [DOI] [PubMed] [Google Scholar]
- 27.Haspel R L, Darnell J E., Jr Proc Natl Acad Sci USA. 1999;96:10188–10193. doi: 10.1073/pnas.96.18.10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang D, Moriggl R, Stravopodis D, Carpino N, Marine J C, Teglund S, Feng J, Ihle J N. EMBO J. 2000;19:392–399. doi: 10.1093/emboj/19.3.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu C L, Jin Y J, Burakoff S J. J Biol Chem. 2000;275:599–604. doi: 10.1074/jbc.275.1.599. [DOI] [PubMed] [Google Scholar]
- 30.Kolenko V, Rayman P, Roy B, Cathcart M K, J, O S, Tubbs R, Rybicki L, Bukowski R, Finke J. Blood. 1999;93:2308–2318. [PubMed] [Google Scholar]
- 31.Almawi W Y, Beyhum H N, Rahme A A, Rieder M J. J Leukocyte Biol. 1996;60:563–572. doi: 10.1002/jlb.60.5.563. [DOI] [PubMed] [Google Scholar]
- 32.Caelles C, Gonzalez-Sancho J M, Munoz A. Genes Dev. 1997;11:3351–3364. doi: 10.1101/gad.11.24.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hilton D J. Cell Mol Life Sci. 1999;55:1568–1577. doi: 10.1007/s000180050396. [DOI] [PMC free article] [PubMed] [Google Scholar]