

Abstract
Background and aims: One of the key components of inflammation is changes in vascular structure and function. This suggests that the microcirculation may be a key target of Helicobacter pylori released factors. It has previously been shown in vivo that pooled H pylori extracts from duodenal ulcer/gastritis patients induce platelet aggregation but no leucocyte activation within rat gastric mucosal microcirculation (GMMC). However, infection with strains associated with ulcer disease as compared with gastritis may exert greater effects on the microcirculation. This study used fluorescent in vivo microscopy to determine the acute effects of extracts of genotypically different H pylori strains on the GMMC.
Methods: Three H pylori extracts, with different cagA and VacA toxigenic status, were individually administered to the gastric mucosa of anaesthetised Wistar rats. The mucosal surface was visualised via an incision made in the exteriorised stomach. Fluoroscein isothiocyanate conjugated to bovine serum albumin (FITC-BSA) or acridine orange was used to quantify macromolecular leak (MML) and leucocyte/platelet activity respectively for 120 minutes. Changes in capillary and post-capillary venule (PCV) diameters were also monitored.
Results: The cagA+ VacA toxigenic strain 60190 induced significant and sustained MML by five minutes (p<0.01). Transient and less leakage was observed with its isogenic VacA− mutant and other non-toxigenic strains regardless of cagA status. Significant increases in leucocyte adhesion (p<0.05), platelet aggregation (p<0.05), and PCV vasoconstriction (p<0.05) were only observed with the cag A+ and toxigenic strain.
Conclusion: Extracts of H pylori are capable of inducing marked disturbances within the rat GMMC. These disturbances seem to be dependent on the production of an active vacuolating cytotoxin. Varying effects on the GMMC may explain the clinically diverse outcomes associated with genotypically different strains.
Keywords: Helicobacter pylori, microcirculation, leucocytes, platelets, cagA, VacA−
The pathophysiology of Helicobacter pylori associated gastritis and peptic ulceration, in particular the mechanism by which this non-invasive bacterium induces mucosal inflammation is poorly understood. One of the major components of gastrointestinal inflammation is changes in vascular structure and function. Some of the endoscopic and histological findings related to H pylori infection include erythema, oedema, and vasodilatation as well as inflammatory cell infiltration.1 This suggests that vascular damage induced directly by bacterial chemotactic products or as a result of the inflammatory response could be an important underlying mechanism of injury and a key target of H pylori released factors.2 A variety of microcirculatory variables can be changed including blood flow, vessel diameters, shear rates, leucocyte and platelet activity, and also changes in the endothelial cells.
Many in vitro studies have demonstrated that the surface proteins of the bacterium, extracted into water by centrifugation and vortexing methods, are chemotactic for neutrophils and monocytes.3–6 Limited studies in vivo on the rat mesentery have also demonstrated that aqueous extracts of H pylori cause leucocyte adherence in and emigration from, the microcirculation.7,8 These observations suggest that H pylori recruits inflammatory cells by actively releasing or passively shedding cellular components that, after absorption into the mucosa, serve as chemoattractants for such cells.9 H pylori infection, however, is commonly localised to the stomach and therefore the findings of leucocyte involvement within the mesentery may not be relevant or mirrored in the gastric mucosa.
It is now well recognised that H pylori strains are highly diverse with different strains associated with the presence or absence of gastric mucosal inflammation and peptic ulcers.12,13 Much of this diversity has been attributed to the existence of different genotypes within the bacterium. The most extensively studied and characterised are the vacuolating cytotoxin gene A (vacA) and cytotoxin associated gene A (cagA).12–14 The vacA gene is found in all H pylori strains and encodes for the vacuolating cytotoxin (VacA). This protein has been demonstrated to induce cytoplasmic vacuolation in eukaryotic cells.14 Although the gene is found in all strains, only about 40% of strains exhibit vacuolating cytotoxin activity. This is because vacA nucleotide sequence varies between strains. The region of vacA encoding the VacA signal sequence can be of two main types, s1 or s2.15 The mid-region of the gene can be type m1 or m2. The final structure is a mosaic, and all combinations of signal sequence and mid-region are found.15 The vacA structure determines VacA in vitro cytotoxin activity. Type s2/m2 vacA encodes a non-vacuolating form of VacA.15,16 Type s1/m2 vacA encodes a form of the toxin that induces vacuolation in a narrower range of epithelial cell lines than type s1/m1 vacA.17 Thus, type s1/m1 vacA encodes the most toxic form of VacA.
The second of the highly characterised genotypes found in H pylori is cagA, which encodes a high molecular weight protein, the cytotoxin associated gene product A or CagA. This gene is present in 60%–70% of H pylori strains and is a marker for the presence of the cag pathogenicity island (cag PaI), which comprises about 30 genes.18 The production of cytokines in vitro, has been demonstrated to be dependent on an intact cag PaI.12,13,18 In vivo, infection with CagA positive strains results in chronic gastritis, peptic ulcer development, and also gastric cancer.
The different clinical outcomes in infected patients could be dependent upon pathogenic strains exerting greater disturbances within the gastric mucosal microcirculation. This study therefore aimed to determine the acute effects of extracts of genotypically different H pylori strains (cagA and VacA toxigenicity present or absent) on the rat gastric mucosal microcirculation in vivo. Specifically, macromolecular leakage of labelled albumin indicating disturbances in vascular integrity, platelet and leucocyte activation, and mucosal vessel diameters were assessed.
METHODS
Preparation of H pylori water extracts
Three different strains of H pylori were studied: (a) 60190 (ATCC 49503), a cagA+ strongly toxigenic vacA s1/m1 strain,19 (b) 60190 vacA::km, its VacA-, non-toxigenic isogenic mutant, but still cagA+. This was created as previously described except that natural transformation was used to transform 60190 with the pCTB8:km construct.20 Immunoblots confirmed that no VacA or VacA fragments were expressed and (c) J154, a cagA-, vacA s2/m2 strain producing a non-toxigenic form of the VacA protein.
Strains were cultured separately on blood agar plates for four to five days under microaerobic conditions after which they were harvested into distilled water, with approximately 109-1010 bacteria in 1 ml of solution. The cell suspension was sonicated five times on ice for five seconds with 30 seconds between each sonication and then centrifuged at 12 000 rpm for 15 minutes at 4°C. The initial supernatant was further centrifuged at 18 000 rpm for 30 minutes and the pellet discarded. The protein extract was obtained by filtration through a sterile 0.2 μm micropore filter. The final supernatant was stored in aliquots of 0.5 ml at −20°C until use. The observer was not blinded to the nature of the H pylori strain for any of the experiments described.
Animals
Experiments were carried out on 48 adult male Wistar rats weighing between 120 g–150 g obtained from the University of Sheffield Field Laboratories. Animals were divided into eight groups of six and fasted overnight before experimentation, housed individually in wire bottomed cages to prevent coprophagy. All experimental procedures were carried out in accordance with Home Office approval, (Project Licence Number PIL 50/01135).
Surgical procedure
The method for preparing animals for fluorescent in vivo microscopy and quantification of results have been detailed previously.10,11 Briefly, animals were anaesthetised with a subcutaneous injection of diazepam (5 mg/ml, Dumex Ltd) and hypnorm (fentanyl citrate 0.315 mg/ml and fluanisone 10 mg/ml, Janssen Pharmaceutical Ltd) in the ratio of 1:1 at a volume of 0.1 ml/100g body weight with further anaesthetic administered as required. A tracheotomy was performed and an oesophageal thermistor probe (Fluke Ltd, RS Components, UK) was inserted to maintain body temperature between at 36°C–38°C. A cannula connected to a pressure transducer (MicroMed, USA) and placed in the left carotid artery monitored mean arterial blood pressure, heart rate and also provided access for administration of fluorochromes.
After laparotomy a 1.0 cm incision was made with an electric microcautery in the anterior wall of the exteriorised stomach parallel to the “limiting ridge”, (separates the squamous cell lined forestomach from the more distal glandular stomach). Observations were made from the glandular portion of the stomach. Care was taken to avoid burning through blood vessels. A glass microscope slide was mounted on perspex pegs of a specially designed animal board and heating mat, and the stomach was gently extended onto it and held in place by a stay suture. The incision in the anterior wall was opened using microclamps to allow visualisation of the posterior mucosal surface and covered with Saran wrap, an impermeable membrane.
Fluorescent in vivo microscopy
The animal, warming pad, and perspex board were transferred to the stage of a Nikon Optiphot-2 microscope (Nikon, UK) equipped with a mercury arc lamp for epi-illumination fluorescent light microscopy. Images of the preparation were monitored using a cooled coupled device camera (CCD, Hitachi; UK) displayed on a high resolution monitor (Sony PVM-1443; UK) and recorded on video (Sony SLV-373-UB; UK) tape for later offline analysis. After a 30 minute stabilisation period, either Acridine Red, (0.1 ml/100 g bodyweight; Sigma UK; n=6 in each group) or fluoroscein isothiocyanate conjugated to bovine serum albumin, (FITC-BSA; Sigma, UK; 0.2 ml/100 g bodyweight; n=6 in each group) was injected via the carotid cannula. Acridine red labels leucocytes and platelets and FITC-BSA labels plasma, which permits blood flow to be visualised.21,22 Under circumstances resulting in increased microvascular permeability to macromolecules, FITC-BSA can be observed to leak from the vasculature appearing as a flare in the interstitium. Interstitial fluorescent intensity is proportional to the degree of FITC-BSA leakage from the vessels.22
The microscopy technique does not permit the entire mucosa to be kept under surveillance simultaneously, therefore three areas were pre-selected in each animal, well removed from each other, for detailed observation. These pre-selected areas included the characteristic honeycomb-like network of mucosal capillaries and at least one post-capillary venule (PCV). Capillaries had a typical diameter of 10 μm–12 μm and drained into PCVs with diameters of 20 μm–25 μm. The location of these three areas was marked on the impermeable membrane. Changes in macromolecular leakage and vessel diameter were quantified and mucosal blood flow changes were assessed qualitatively as being as rapid as in a control situation, visibly reduced, sluggish, or static. For the acridine red experiments only two areas of interest were identified as it was difficult to assess three areas simultaneously.
After a 30 minute equilibration period, either FITC-BSA or acridine red was administered intra-arterially. Recordings were made from pre-selected areas five minutes after the administration of the fluorochromes. After the recording of the pre-treatment images, 0.5 ml of the H pylori aqueous extract of one of the strains or distilled water was applied to the exposed gastric mucosa. This was left in continuous contact with the gastric mucosa. After treatment recordings were made in all groups after five minutes and then every 15 minutes for a period of two hours.
Data collection and quantification
FITC-BSA leakage
To measure FITC-BSA leakage from the vessels, an area of interest adjacent to three segments of the PCV and surrounding three separate capillaries was mapped out on the television screen. Computerised image analysis (Image Pro Plus, Media Cybernetics, USA) was used to measure interstitial fluorescence at each time point during the experiment. The image analysis programme stored the recorded image as a palette of 255 grey levels. A pre-treatment grey level was taken and used as the baseline value. After H pylori treatment, the grey level was again measured at each time point. As an increase in fluorescence represents a proportional increase in leak, values for percentage increase in leak could be calculated.
Vessel diameters and blood flow
Diameters of the PCVs and capillaries in the pre-selected areas were also measured using computerised image analysis calibrated to produce values in micrometres. Mucosal vessel blood flow was assessed qualitatively. This was based simply on blood flow being rapid as in a control situation, visibly reduced, sluggish, or static.
Leucocyte adhesion and platelet activity
The sum of adherent leucocytes in both PCVs and capillaries within a single field of view was determined offline during playback of videotaped images. A leucocyte was considered adherent to the vessel endothelium if it remained stationary for ≥30 seconds. Adherent leucocytes were expressed as the number per field of view observed on the monitor during a period of one minute at each time point. Leucocytes adherent within the blood vessel were often difficult to distinguish from those that had emigrated to the interstitium and were still in close proximity to the capillaries. As a consequence both types of stationary leucocytes were regarded as adherent in this study. The difficulty in distinguishing these leucocyte populations primarily resulted from the images obtained from thick tissue, the small diameter of the visualised capillaries, and the presence of the mucosal surface in different focal planes. Any leucocytes that were adherent at the onset of the experiment were subtracted from the total number that accumulated subsequently. Adherent platelet thrombi were difficult to quantitate as they were often of varying sizes. However, platelet emboli were easily identified circulating within the PCVs and the total counted were expressed as the number per field of view during a period of one minute at each time point.
Statistics
Macromolecular leakage before and after H pylori application or distilled water was tested for within group variations using the Wilcoxon test for non-parametric data. Macromolecular leakage in the experimental and control groups was compared using the Kruskall-Wallis test, followed by the Mann-Whitney U test for significance between group variations for unpaired, two tailed non-parametric data. Changes in vessel diameters, leucocyte activity, and platelet activity were compared using a one way analysis of variance followed by a two tailed Student’s t test for significance on normally distributed data. Results were considered statistically significant at p≤0.05.
RESULTS
Macromolecular leakage
Vascular integrity was maintained in control animals exposed to distilled water as evidenced by no significant increases in interstitial fluorescence surrounding the PCVs or capillaries during the experimental period (fig 1 and 5A ▶ ▶). Significant increases in MML at all time points were only observed after exposure of the gastric mucosa to the cagA+ and toxigenic H pylori strain (60190). Interstitial fluorescence increased significantly from a grey level of 41.4 (5.2) (mean (SD)) at five minutes to 80.1 (20.1) at 90 minutes ((p<0.01; figs 1 and 5B ▶ ▶). Transient, increases in MML were also observed with the non-toxigenic strains, including 60190 vacA::km, reaching significance (p<0.05) at 5 or 15 minutes only. However, this leakage was of a significantly lesser magnitude than that induced by strain 60190. A similar pattern of events was observed from the mucosal capillaries (data not shown).
Figure 1.

The effects of topical administration of genotypically different strains of H pylori on macromolecular leakage from mucosal PCVs. Only strain 60190 induced significant leakage from PCVs at all time points. Strains 60190 vacA::km and J154 only induced significant leakage at five minutes. Distilled water had no significant effect at any time point. Each point represents the means of 18 areas. * p<0.05, **p<0.01 when compared with controls using the Mann-Whitney U test. Results are presented as means (SD).
Figure 5.
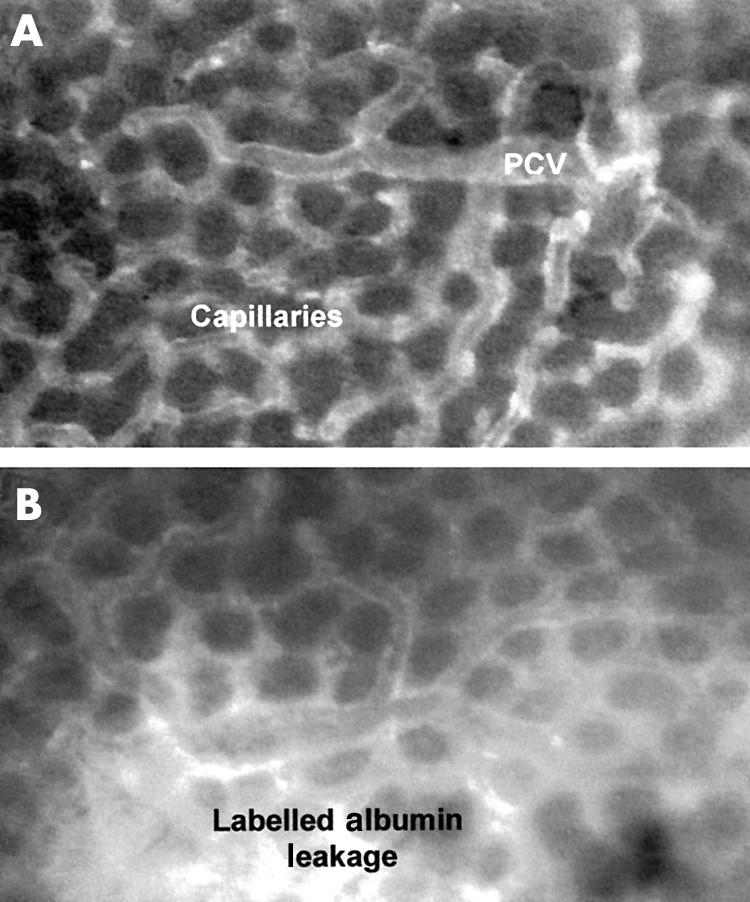
(A) Control rat gastric mucosal microcirculation exposed to distilled water with fluorescently labelled albumin (FITC-BSA) contained with the vessels, which appear white against a relatively black interstitial background. Images for the isogenic 60190 vacA::km would be identical to the distilled water control images. PCV=post-capillary venule. (B) Gastric mucosal microcirculation after topical administration of 60190, a cagA+ and VacA toxigenic H pylori strain. FITC-BSA leakage appears as a flare in the interstitium surrounding the vessels, possibly indicative of vascular injury.
Blood flow
Gastric mucosal blood flow was rapid and stable throughout the experimental period in control and all H pylori treated animals, with homogenous blood flow observed in all areas of interest. No vascular stasis or sluggish flow was observed in any experimental group at any time point.
Vessel diameters
Rapid and significant vasoconstriction of PCVs (p<0.05) was observed within five minutes with the cagA+/toxigenic strain, 60190, only (fig 2 ▶). This vasoconstriction was a transient event with no differences in PCV diameter observed at any other time point. No changes in capillary diameter were induced with any strain (fig 2 ▶).
Figure 2.

The effects of topical administration of genotypically different strains of H pylori on gastric mucosal vessel diameters. Only strain 60190 induced vasoconstriction of PCVs when compared with distilled water, which had no effect. Strains 60190 vacA::km and J154 also had no significant effect on vessel diameters and therefore are not included on the figure. Each point represents the means of 18 areas. *p<0.05 when compared with control using unpaired Student’s t test on normal data. Results are presented as means (SD).
Platelet aggregation
Circulating platelet emboli were only observed within the PCVs after exposure to the cagA+ / toxigenic strain, 60190, and reached statistical significance after 30 minutes (p<0.05; figs 3 and 6A ▶ ▶). Scanning of the whole gastric mucosa occasionally revealed adherent, but non-occlusive platelet thrombi within the capillaries. Distilled water and the other three H pylori strains had no effect on platelet aggregation.
Figure 3.

The effects of topical administration of genotypically different strains of H pylori on platelet aggregates within gastric mucosal vessels. Only strain 60190 induced significant platelet aggregation when compared with distilled water, which had no effect. Strains 60190 vacA::km and J154 also had no significant effect and therefore are not included on the figure. Each point represents the means of 12 areas. *p<0.05 when compared with controls using unpaired Student’s t test on normal data. Results are presented as means (SD).
Figure 6.
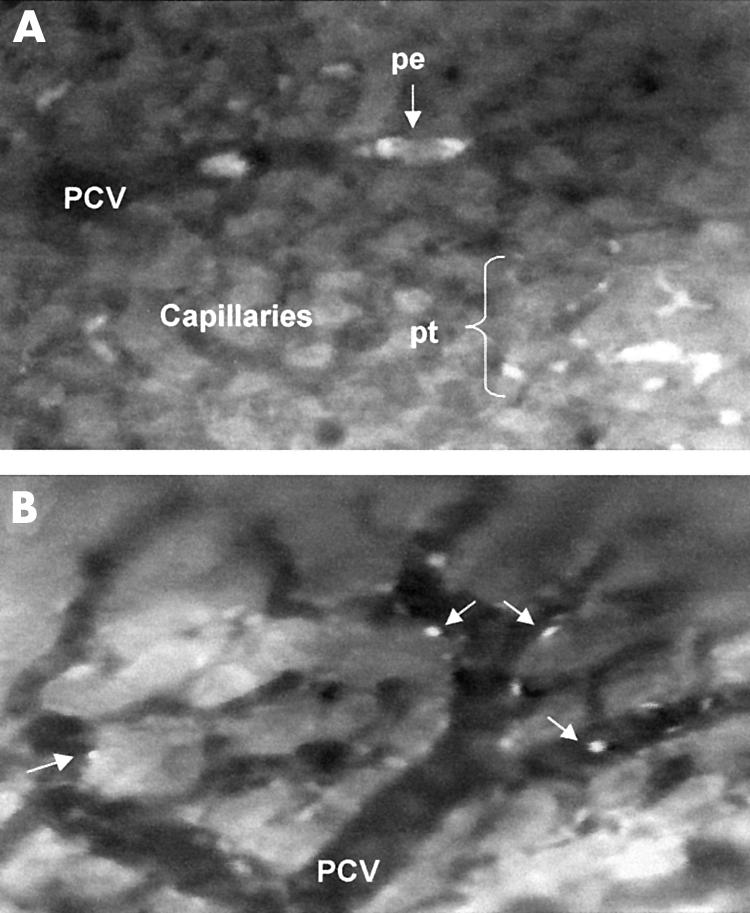
Gastric mucosal microcirculation after topical administration of a cagA+/vacA+ strain of H pylori. (A) Platelets appear as adherent thrombi (pt) or circulating emboli (pe) within the capillaries and post-capillary venules (PCV). (b) Increased leucocytes (arrows) are observed rolling or adherent within the microvessels. These images were not obtained for strains 60190 vacA::km and J154.
Leucocyte adherence
Only occasional leucocytes were adherent within the capillaries of control animals throughout the experimental period (fig 4 ▶). The cagA+/toxigenic strain, 60190, induced a rapid, sustained and significant increase in leucocyte adherence within mucosal capillaries (p<0.01; figs 4 and 6B ▶ ▶). Although leucocyte adhesion was also observed with the other strains, it was of a significantly lower magnitude than that observed in 60190 treated animals (fig 4 ▶). Fast circulating leucocytes or “flyers” were seldom observed in control animals because of their high velocity making them difficult to visualise. However, scanning of the whole gastric mucosa revealed flyers within the mucosal vessels in animals exposed to the cagA+/toxigenic strain, 60190. Rolling leucocytes were also observed within the PCVs in these animals.
Figure 4.

The effects of topical administration of genotypically different strains of H pylori on the numbers of adherent leucocytes within gastric mucosal vessels. Only strain 60190 induced significant leucocyte adhesion at all time points. Strains 60190 vacA::km, J154, and distilled water had no significant effect at any time point (the latter has not been included on the figure). Each point represents the means of 12 areas. **p<0.01 when compared with controls using unpaired Student’s t test on normal data. Results are presented as means (SD).
The physiological parameters, heart rate, and mean arterial blood pressure, remained constant throughout the course of the experiment for all study groups.
DISCUSSION
In acute gastritis caused by H pylori infection, the initial inflammatory response comprises of neutrophil activation. This study shows that acute administration of aqueous H pylori extracts can result in the rapid activation of these potent inflammatory cells within the gastric mucosal microcirculation. However, this effect was observed to be of a greater extent in those animals topically exposed to the virulent strain of H pylori, that was cagA+ and secreted the toxigenic form of the VacA protein, encoded by the vacA gene. Macromolecular leakage, indicative of a disturbance in the vascular wall integrity, was also of a greater magnitude with platelet activation and PCV constriction uniquely associated with this strain. This study therefore presents novel data demonstrating an active vacuolating cytotoxin is essential for inducing microcirculatory disturbances. Collectively, these observations suggest that the microcirculation could be a key target for H pylori released factors.
Neutrophil chemotaxis and activation can be induced by the synthesis of chemokines from the gastric epithelial cells in response to bacterial infection.23,24 H pylori strains positive for cagA have been associated with increased synthesis of chemokines in the gastric epithelium, in particular interleukin 8 (IL8), both clinically and experimentally.25,26 The consequences of the local release of potent inflammatory cytokines from endothelial cells directly into the microcirculation are numerous, including adhesion molecule up-regulation, leucocyte recruitment, and synthesis of other inflammatory and vasoactive substances that would further amplify the inflammatory cascade.2,27 Interestingly, in this study, even though strain 60190 vacA::km was identical to its parent in possessing cagA and the cag PaI, this strain was virtually inactive with respect to its effects on the GMMC. This observation shows that in our model the cag PaI is insufficient for leucocyte adherence in the microcirculation.
Although gastric epithelial cell vacuolation has been observed in vitro after exposure to the Vac A toxin, a clear role for the vacA gene or its encoded protein Vac A has not been defined in vivo. Attempts have been made to correlate vacA to gastric inflammation and injury.28,29 However, these studies did not support a role for vacA in relation to inflammatory cell infiltrate. The current in vivo study presents novel data suggesting that the toxigenic VacA may also be involved in leucocyte activation. In this study not only were significant numbers of adherent leucocytes observed, but circulating leucocytes or “flyers” could also be detected within the microvessels. These have not previously been observed, even under control conditions, within the gastric mucosal microcirculation using in vivo microscopy. This has been attributed to their high flow velocity within the gastric mucosa, making observation and therefore quantification of circulating leucocytes difficult. The ability to identify flyers after exposure to the cagA+ and toxigenic strain of H pylori suggest that perhaps this extract reduced either the blood or leucocyte velocity allowing leucocyte visualisation to be possible.
A number of studies have provided evidence that H pylori can indeed lead to decreases in gastric mucosal blood flow. Atuma and colleagues demonstrated significant decreases in rat gastric blood flow by H pylori extracts, but interestingly, regardless of cagA and vacA status.30,31 In separate studies, exposure to water extracts of cagA+/toxigenic VacA or cagA-/non-toxigenic VacA strains and also infection with these strains decreased gastric blood flow at the margin of acid induced ulcers.32–34 In a clinical study, increased antral blood flow was observed after eradication of H pylori, but not if H pylori persisted.35 Although gastritis in its own right has been observed to reduce gastric mucosal blood flow, H pylori probably releases factors that further reduce the blood flow.
Platelet aggregation has been a common feature of the in vivo microcirculatory studies.7,8,10,11 The categorisation of platelets as potent vascular inflammatory cells, such as the neutrophils, monocytes, and mast cells, is under debate. However, they do act as a storage site for a wide range of vasoactive substances, including histamine, 5HT, platelet activating factor, thromboxane A2 and leukotrienes.36 Recent evidence suggests that platelets can also generate cytotoxic superoxide and hydroxyl radicals, which may be another mechanism by which aggregated platelets could induce microcirculatory disturbances.37 Besides a direct involvement in vascular inflammation, platelets may participate in neutrophil recruitment through their adhesion to endothelium. It has been demonstrated that activated platelets, expressing P-selectin on their surface, not only adhere to monocytes and neutrophils, but also prime these cells for subsequent responses that augment inflammation.38 Interestingly, platelet surface expression of P-selectin is also increased in other gastrointestinal inflammatory conditions including Crohn’s disease and ulcerative colitis as well as after exposure to H pylori.39,40
In this study, the cagA+/toxigenic strain also induced a very rapid and significant constriction of the PCVs. The extract was only in contact with the gastric mucosa for a duration of two hours. However, it would be of concern if this effect reflects that occurring in humans where the H pylori infection is of a longer duration. Blood vessel constriction, especially of small diameter vessels, is associated with a number of deleterious events, including decreased flow, increased resistance to flow, increased platelet and entrapment of leucocytes. It is also possible that focal areas of vasoconstriction in addition to infarction resulting from platelet aggregation may lead to ischaemia. This may provide an explanation for the focal nature of ulcer disease despite widespread bacterial infection.
There are still many areas of research that remain unexplored with regards to the effects of H pylori on the gastric microcirculation in vivo, including the identification of major cytokines contributing to neutrophil capture and activation within the microvessels. Although a pivotal role for the chemokine IL8 has been implicated, its importance in vivo on leucocyte recruitment has not been identified. This study has shown varying degrees of microcirculatory disturbances dependent on the secretion of toxigenic VacA. However, the contribution of additional newly identified virulence factors remains unknown. Elizalde and colleagues, using fluorescent in vivo microscopy, investigated the effects of chronic infection in mice with fresh isolates of H pylori that also differed with respect to cagA and toxigenic strains.41 Again, increased platelet aggregates and leucocyte rolling was observed but the degree of microcirculatory involvement observed was similar regardless of strain, suggesting a possible role for other virulence genotypes or factors. An important role for an H pylori protein possessing neutrophil activating activity (HP-NAP) has also been identified. HP-NAP not only served as a chemoattractant for human leucocytes but could also activate the cells to produce reactive oxygen intermediates.42 Vaccination of mice with HP-NAP also conferred significant protection against subsequent H pylori challenge. The in vivo effects of this protein on the microcirculation have yet to be elucidated.
Along with previous in vivo studies, these results further confirm the view that H pylori may exert some of its pathogenic effects by acting on the gastric microcirculation. However, this study provides novel and additional data demonstrating that these microcirculatory disturbances are not all identical but vary depending on the virulence of the strain tested. These varying effects may provide a possible explanation for the different clinical outcomes associated with H pylori infection. The mucosal microcirculation is one of the most important gastric defence mechanisms. Recognition that abnormal microvascular function frequently accompanies gastrointestinal dysfunction and disease has led to a renewed interest in defining the contribution of the microcirculation to tissue injury, especially after H pylori infection.
Acknowledgments
This research was supported by the Bardhan Research and Education Trust. Registered charity number 328452.
Abbreviations
vacA, vacuolating cytotoxin gene A
cagA, cytotoxin associated gene A
GMMC, gastric mucosal microcirculation
FITC-BSA, fluorscein isothiocyanate-bovine serum albumin
MML, macromolecular leak
PCV, post-capillary venule
REFERENCES
- 1.Wyatt JI. Histopathology of gastroduodenal inflammation: the impact of Helicobacter pylori. Histopathology 1995;26:1–15. [DOI] [PubMed] [Google Scholar]
- 2.Granger DN, Grisham MB, Kvietys PR. Mechanisms of microvascular injury. In: Johnson LR, ed. Physiology of the gastrointestinal tract. 3rd edn. New York: Raven Press, 1994:1693–722.
- 3.Neilson H, Anderson LP. Chemotactic activity of Helicobacter pylori sonicate for human polymorphonuclear leukocytes and monocytes. Gut 1992;33:738–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Craig PM, Territo MC, Karnes WE, et al. Helicobacter pylori secretes a chemotactic factor for monocytes and neutrophils. Gut 1992;33:1020–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mai UE, Perez-Perez GI, Wahl LM, et al. Soluble surface proteins from Helicobacter pylori activate monocytes/macrophages by lipopolysaccharide-independent mechanism. J Clin Invest 1991;87:894–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reymunde A, Deren J, Nachamkin I, et al. Production of chemoattractant by Helicobacter pylori. Dig Dis Sci 1993;38:1697–701. [DOI] [PubMed] [Google Scholar]
- 7.Yoshida N, Granger DN, Evans DJ, et al. Mechanisms involved in Helicobacter pylori-induced inflammation. Gastroenterology 1993;105:1431–40. [DOI] [PubMed] [Google Scholar]
- 8.Kurose I, Granger DN, Evans DJ, et al. Helicobacter pylori-induced microvascular protein leakage in rats: role of neutrophils, mast cells and platelets. Gastroenterology 1994;107:70–9. [DOI] [PubMed] [Google Scholar]
- 9.Mai UE, Perez-Perez GI, Allen JB, et al. Surface proteins from Helicobacter pylori exhibit chemotactic activity for human leukocytes and are present within the gastric mucosa. J Exp Med 1992;175:517–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalia N, Brown NJ, Jacob S, et al. Studies on the gastric mucosal microcirculation. 2. Platelet aggregation induced by Helicobacter pylori water soluble extracts in vivo. Gut 1997;41:748–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalia N, Bardhan KD, Reed MWR, et al. Mechanisms of Helicobacter pylori-induced rat gastric mucosal microcirculation in vivo. Dig Dis Sci 2000;45:763–72 [DOI] [PubMed] [Google Scholar]
- 12.Atherton JC, Peek RM, Tham KT, et al. Clinical and pathological importance of heterogeneity in vac A, the vacuolating cytotoxin gene of Helicobacter pylori. Gastroenterology 1997;112:92–9. [DOI] [PubMed] [Google Scholar]
- 13.Atherton JC. The clinical relevance of strain types in Helicobacter pylori. Gut 1997;40:710–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cover TL. The vacuolating cytotoxin of Helicobacter pylori. Mol Microbiol 1996;20:241–6. [DOI] [PubMed] [Google Scholar]
- 15.Atherton JC, Cao P, Peek RM Jr, et al. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem 1995;270:17771–7 [DOI] [PubMed] [Google Scholar]
- 16.Letley DP, Atherton JC. Natural diversity in the N terminus of the mature vacuolating cytotoxin of Helicobacter pylori determines cytotoxin activity. J Bacteriol 2000;182:3278–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pagliaccia C, de Bernard M, Lupetti, P, et al. The m2 form of the Helicobacter pylori cytotoxin has cell type-specific vacuolating activity. Proc Natl Acad Sci USA 1998;95:10212–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Censini S, Lange C, Xiang Z, et al. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci USA 1996; 93:14648–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leunk RD, Johnson PT, David BC, et al. Cytotoxic activity in broth-culture filtrates of Campylobacter pylori. J Med Microbiol 1988;26:93–9. [DOI] [PubMed] [Google Scholar]
- 20.Cover TL, Tummuru MK, Cao P, et al. Divergence of genetic sequences for the vacuolating cytotoxin among Helicobacter pylori strains. J Biol Chem 1994; 269:10566–73. [PubMed] [Google Scholar]
- 21.Tangelder GJ, Slaaf DW, Reneman RS. Fluorescent labeling of blood platelets in vivo. Thromb Res 1982; 28:803–20. [DOI] [PubMed] [Google Scholar]
- 22.Miller FN, Joshua IG, Anderson GL. Quantitation of vasodilator induced macromolecular leakage by in vivo fluorescent microscopy. Microvasc Res 1982;24:56–67. [DOI] [PubMed] [Google Scholar]
- 23.Peek RM, Miller GG, Tham KT, et al. Heightened inflammatory response and cytokine expression in vivo to cagA+ Helicobacter pylori strains. Lab Invest 1995;73:760–70. [PubMed] [Google Scholar]
- 24.Shimada T, Terano A. Chemokine expression in Helicobacter pylori-infected gastric mucosa. J Gastroenterology 1998;33:613–17. [DOI] [PubMed] [Google Scholar]
- 25.Li SD, Kersulyte D, Lindley IJ, et al. Multiple genes in the left half of the cag pathogenicity island of Helicobacter pylori are required for tyrosine kinase-dependent transcription of interleukin-8 in gastric epithelial cells. Infect Immun 1999;67:3893–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ando T, Kusugami K, Ohsuga M. Interleukin-8 activity correlates with histological severity in Helicobacter pylori-associated antral gastritis. Am J Gastroenterol 1996;91:1150–6. [PubMed] [Google Scholar]
- 27.Ding SZ, Cho CH, Lam SK. Helicobacter pylori induces interleukin-8 expression in endothelial cells and the signal pathway is protein tyrosine kinase dependent. Biochem Biophys Res Commun 1997;240:561–5. [DOI] [PubMed] [Google Scholar]
- 28.Yamaoka Y, Kodama T, Kita M, et al. Relationship of vacA genotypes of Helicobacter pylori to cagA status, cytotoxin production, and clinical outcome. Helicobacter 1998;3:241–53. [DOI] [PubMed] [Google Scholar]
- 29.Ghiara P, Marchetti M, Blaser MJ, et al. Role of the Helicobacter pylori virulence factors vacuolating cytotoxin, CagA, and urease in a mouse model of disease. Infect Immun 1995;63:4154–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Atuma C, Engstrand L, Holm L. Extracts of Helicobacter pylori reduce gastric mucosal blood flow through a Vac A - and Cag A – independent pathway in rats. Scand J Gastroenterol 1998;33:1256–61. [DOI] [PubMed] [Google Scholar]
- 31.Atuma C, Engstrand L, Holm L. Helicobacter pylori extracts reduce gastric mucosal blood flow by a nitric oxide-independent but mast cell and platelet activating factor receptor-dependent pathway in rats. Scand J Gastroenterol 1999;34:1183–9. [DOI] [PubMed] [Google Scholar]
- 32.Brzozowski T, Konturek PC, Konturek SJ, et al. Water extracts of Helicobacter pylori delay healing of chronic gastric ulcers in rats: role of cytokines and gastrin-somatostatin link. Digestion 1999;60:22–33. [DOI] [PubMed] [Google Scholar]
- 33.Brzozowski T, Konturek PC, Konturek SJ, et al. Gastric secretion and ulcer healing in mouse stomach infected with cytotoxin expressing strain of Helicobacter pylori. J Physiol Pharmacol 1998;49:387–403. [PubMed] [Google Scholar]
- 34.Konturek PC, Brzozowski T, Konturek SJ, et al. Mouse model of Helicobacter pylori infection: studies of gastric function and ulcer healing. Aliment Pharmacol Ther 1999;13:333–46. [DOI] [PubMed] [Google Scholar]
- 35.Wu DC, Wang WM, Lu CYL, et al. The hemodynamic changes of gastroduodenal regional blood flow after Helicobacter pylori eradication in patients with duodenal ulcer scar. Kaohsiung J Med Sci 1999;15:19–25. [PubMed] [Google Scholar]
- 36.Page CP. Platelets as inflammatory cells. Immunopharmacology 1989;17:51–9. [DOI] [PubMed] [Google Scholar]
- 37.Caccese D, Pratico D, Ghiselli A, et al. Superoxide anion and hydroxyl radical release by collagen-induced platelet aggregation-role of arachidonic acid metabolism. Thromb Haemost 2000;83:485–90. [PubMed] [Google Scholar]
- 38.Mannaioni PF, Di Bello MG, Masini E. Review. Platelets and inflammation: role of platelet derived growth factor, adhesion molecules and histamine. Inflamm Res 1997;46:4–18. [DOI] [PubMed] [Google Scholar]
- 39.Collins CE, Rampton DS. Platelet dysfunction: a new dimension in inflammatory bowel disease. Gut 1995;36:5–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Collins CE, Cahill MR, Newland AC, et al. Platelets circulate in an activated state in Inflammatory Bowel Disease. Gastroenterology 1994;106:840–5. [DOI] [PubMed] [Google Scholar]
- 41.Elizalde JI, Gomez J, Lozano M, et al. Platelet activation in mice and human Helicobacter pylori infection. J Clin Invest 1997;100:996–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Satin B, Del Giudice G, Della Bianca V, et al. The neutrophil-activating protein (HP-NAP) of Helicobacter pylori is a protective antigen and a major virulence factor. J Exp Med 2000;191:1467–76. [DOI] [PMC free article] [PubMed] [Google Scholar]