

Abstract
Background and aims: Recent studies linked cytokine gene polymorphisms to H pylori related gastric cancer development. The current study evaluated the role of cytokine gene polymorphisms for mucosal cytokine expression, the gastric inflammatory response, and bacterial colonisation during H pylori infection.
Patients and methods: In 207 H pylori infected patients with chronic gastritis, polymorphisms at different loci of the interleukin (IL)-10, IL-1B, IL-1 receptor antagonist (IL-1RN), tumour necrosis factor (TNF)-A, and interferon (IFN)-G genes were genotyped by polymerase chain reaction (PCR), restriction fragment length polymorphism (RFLP) analysis, and allelic discriminating TaqMan PCR. Mucosal cytokine mRNA copy numbers were determined by real time quantitative PCR. Presence of bacterial virulence factors was investigated by cagA, vacAs1/2, and babA2 PCR. Biopsies were assessed with regard to the degrees of granulocytic/lymphocytic infiltration and the presence of intestinal metaplasia (IM) and atrophic gastritis (AG).
Results: Proinflammatory IL-1 polymorphisms (IL-1RN*2+/IL-1B−511T/−31C+) were associated with increased IL-1β expression, more severe degrees of inflammation, and an increased prevalence of IM and AG. Carriers of the IL-10−1082G/−819C/−592C alleles (GCC haplotype) had higher mucosal IL-10 mRNA levels than ATA haplotype carriers and were associated with colonisation by more virulent cagA+, vacAs1+, and babA2+ H pylori strains. The TNF-A−307(G/A) and IFN-G+874(A/T) polymorphisms did not influence mucosal cytokine expression or the inflammatory response to H pylori.
Conclusions: Cytokine gene polymorphisms influence mucosal cytokine expression, gastric inflammation, and the long term development of precancerous lesions in H pylori infection. Host polymorphisms are associated with certain bacterial strain types, suggesting host specific colonisation or adaptation. These findings contribute to the understanding of the complex interplay between host and bacterial factors involved in the development of gastric pathology.
Keywords: Helicobacter pylori, cytokines, polymorphisms, cagA, vacA, babA
Infection with Helicobacter pylori leads to persistent colonisation and chronic inflammation of the gastric mucosa, thereby increasing the risk of developing peptic ulceration, distal gastric adenocarcinoma, and gastric lymphoma.1,2 There are high interindividual differences in the extent of gastric inflammation among H pylori infected patients, and clinical consequences develop in only a small subgroup. Bacterial virulence factors, such as the cag pathogenicity island (cagPAI), the vacuolating cytotoxin (VacA), and the blood group antigen binding adhesin (BabA) are associated with enhanced inflammation and cancer development.3–9 However, despite the well defined role of virulence factors, it is unclear why a considerable proportion of patients infected with the cagA+, vacAs1+, or babA2+ H pylori strains do not develop severe pathologies throughout life.
In addition to bacterial factors, mostly unknown host factors seem to influence the inflammatory response and the development of a more severe pathology. H pylori induced inflammation is implicated in the development of mucosal damage and is characterised by strong granulocytic and lymphocytic infiltration.10,11 The T helper cell response towards H pylori is generally considered to be of the Th1 phenotype,10,11 leading to a cell mediated immune response. There is increasing evidence that the H pylori induced Th1 response contributes to cancer development.12 Downregulation of the Th1 response in mice by concurrent enteric helminth infection or p53 mutation was shown to protect against the development of atrophy, intestinal metaplasia, and invasive gastric carcinoma.13,14 However, factors influencing the extent of the H pylori induced Th1 response are currently unknown. Important cytokines characterising Th1 mediated immune responses are interferon γ (IFN-γ), tumour necrosis factor α (TNF-α), and interleukin-1β (IL-1β), all being upregulated during chronic H pylori infection.15–17 IL-10, which is also highly expressed in the H pylori infected stomach,18 is one of the most important regulatory cytokines, inhibiting cell mediated immune responses.
Genes encoding cytokines and related molecules harbour polymorphic regions, which are considered to alter gene transcription and thereby influence inflammatory processes in response to infectious diseases.19,20 Polymorphisms in the human IL-10, IL-1B, TNF-A, IFN-G, and IL-1 receptor antagonist (IL-1RN) genes have been reported to influence cytokine expression. In the IL-10 promoter, single nucleotide polymorphisms (SNPs) at positions −1082 (G/A), −819 (C/T), and −592 (C/A) from the transcription start site have been reported to produce mainly three haplotypes: GCC, ACC, and ATA.21 Although there are stimulus or cell type dependent variations, haplotype GCC seems to be associated with high and ATA with low IL-10 production capacity from in vitro stimulated peripheral blood mononuclear cells (PBMC).22,23 Several polymorphisms have been reported in the TNF-A promoter, most of which are functionally silent.24 The majority of studies focused on the G/A polymorphism at position −307, which was originally misnumbered and is therefore better known as TNF-A−308.24 While in some in vitro studies allele A of this SNP was associated with heightened TNF-α secretion, other studies did not find such a correlation.24,25 In the IFN-G gene, a T/A polymorphism at position +874 has been reported.26 Allele T of this SNP shows an absolute correlation with allele 2 of a penta-allelic CA microsatellite polymorphism, which has been associated with high IFN-γ production from PBMC in one study.27 In the promoter region of the IL-1B gene, IL-1B−511T, which is in complete linkage disequilibrium with IL-1B−31C,28 was previously associated with slightly, but not significantly, increased IL-1β secretion from stimulated PBMC.20 The IL-1RN gene has a penta-allelic 86 bp variable number of tandem repeat region (VNTR) in intron 2, of which allele 2 (IL-1RN*2) was previously associated with enhanced IL-1β secretion.20,29,30
In the context of H pylori infection, the genetic basis for the high interindividual differences in cytokine responses is poorly understood. However, recent reports linked cytokine gene polymorphisms to gastric cancer.28,31,32 In the current study, we investigated a large H pylori infected patient group with chronic gastritis and determined host cytokine gene polymorphisms, corresponding mucosal cytokine expression, histopathological features of gastritis, and the infecting bacterial strain type. The aim of the study was to define the influence of different polymorphisms on the H pylori induced cytokine response and to investigate the resulting consequences for gastric inflammation and bacterial colonisation.
MATERIALS AND METHODS
Patients and biopsies
Five antral and five corpus biopsies were collected from each of 742 patients. A total of 207 patients (106 male and 101 female) with chronic gastritis were H pylori infected. Patients taking non-steroidal anti-inflammatory drugs or receiving antisecretory therapy, as well as those with ulcer disease or gastric carcinoma were excluded. Of the infected patients, 88.9% were of German nationality while the remainder were from different countries in southern Europe. Mean age was 62.5 years (range 32–92). Two antral and corpus sections were stained with haematoxylin-eosin. Histopathological evaluation was performed according to the Sydney classification system with regard to the presence of intestinal metaplasia (IM), atrophic gastritis (AG), and the degree of granulocytic infiltration (G1 mild, G2 moderate, G3 severe) and lymphocytic infiltration (L1–L3). The remaining biopsies were stored in liquid nitrogen and homogenised before DNA/RNA isolation, as described previously.17
Polymerase chain reaction (PCR) for H pylori genotyping
PCR amplification of the H pylori gene loci was performed for the cagA gene, the vacAs mosaics vacAs1/2, and babA2, as published previously.7,17,33
Genotyping of cytokine gene polymorphisms
Cytokine gene polymorphisms were genotyped by PCR, restriction fragment length polymorphism (RFLP) analysis, or 5′ nuclease PCR assay (allelic discriminating TaqMan PCR), as described briefly below. Additional methodological details are available from the authors on request.
IL-1B−511/−31 polymorphisms and the 86 bp VNTR in the IL-1RN gene were genotyped by allelic discrimination TaqMan PCR, RFLP analysis, and PCR, as described previously.28
IL-10 SNPs were genotyped by RFLP analysis. Primer sequences for PCR are shown in table 1 ▶. For genotyping of the −1082 polymorphism, PCR products were digested with 10 units of MnlI (New England Biolabs, Frankfurt, Germany) at 37°C for three hours. This gave products of 98 bp+46 bp (−1082G) and 144 bp (−1082A). An additional MnlI restriction site at position −1059 was eliminated from the sequence by introducing a point mutation into the amplified PCR product through a base exchange in the antisense primer (C to G, underlined in the primer sequence). To analyse the IL-10−819 polymorphism, PCR products were digested with 1 unit of RsaI (Roche, Mannheim, Germany) at 37°C for two hours. Digestion yielded five bands (−819A: 240, 11, 85, 42, 8 bp) or four bands (−819C: 351, 85, 42, 8 bp). Typing of the IL-10−592 SNP was performed by digestion of the PCR products with MaeIII (New England Biolabs) at 55°C, which gave products of 217, 144, and 125 bp (−592C) or of 361 and 125 bp (−592T).
Table 1.
Primer sequences and restriction enzymes used for restriction fragment length polymorphism analysis of tumour necrosis factor A (TNF-A) and interleukin 10 (IL-10) polymorphisms. Typing of the remaining single nucleotide polymorphisms was performed by allelic discriminating TaqMan PCR, as described in materials and methods
| Target sequence | Sequences of primers and probes | Restriction enzyme | |
| TNF-A-307 | FP | 5′- GAG GCA ATA GGT TTT GAG GGC CAT-3′ | NcoI |
| RP | 5′- GGG ACA CAC AAG CAT CAA G-3′ | ||
| IL-10-592 | FP | 5′-ATC CAA GAC AAC ACT ACT AA-3′ | MaeIII |
| RP | 5′-TAA ATA TCC TCA AAG TTC C-3′ | ||
| IL-10-1082 | FP | 5′- TCG CTG CAA CCC AAC TGG C-3′ | MnlI |
| RP | 5′- GGT CCC TTA CTT TGC TCT TAC C-3′ | ||
| IL-10-819 | FP | 5′-GAC TCC AGC CAC AGA AGC TTA C-3′ | RsaI |
| RP | 5′-AGG TCT CTG GGC CTT AGT-3′ | ||
FP, forward primer; RP, reverse primer.
The IFN-G+874 SNP was genotyped by allelic discriminating TaqMan PCR, using the following primers and probes: forward primer 5′-ATT CAG ACA TTC ACA ATT GAT TTT ATT CTT AC-3′ and reverse primer 5′-ACT GTG CCT TCC TGT AGG GTA TTA TT-3′; probe 1: 5′-FAM-AAT CAA ATC TCA CAC ACA C-TAMRA-3′ and probe 2: 5′-VIC-ATC AAA TCA CAC ACA CAC-TAMRA-3′. PCR and end point analysis was performed in a volume of 25 μl on a ABI PRISM 7700 Sequence Detection System (Perkin-Elmer, Weiterstadt, Germany).
Typing of the TNF-A−307 SNP was performed by RFLP analysis. Primer sequences are shown in table 1 ▶. PCR products were digested with 10 units of NcoI (New England Biolabs) at 37°C for three hours, giving fragments of 126+21 bp (−307G). The −307A allele was not cut.
Real time quantitative reverse transcription-PCR
Quantitative cytokine mRNA determination was performed by TaqMan PCR, as described previously.17 In order to obtain absolute cytokine mRNA copy numbers, standard curves were generated using plasmid dilution series containing the corresponding target sequence. Cytokine copy numbers were normalised to GAPDH copies. TaqMan primers (MWG, Ebersberg, Germany) and probes (Perkin-Elmer) were designed to span exon junctions or to lie in different exons to prevent amplification of genomic DNA. Sequences of GAPDH, IFN-γ, and TNF-α primers and probes have been published previously.17 For IL-10 and IL-1β quantification, the following primers and probes were used: IL-10 forward primer 5′-CAT CGA TTT CTT CCC TGT GAA-3′; IL-10 reverse primer 5′-TCT TGG AGC TTA TTA AAG GCA TTC-3′; IL-10 probe 5′-FAM-ACA AGA GCA AGG CCG TGG AGC A-TAMRA-3′; IL-1β forward primer 5′-CTG ATG GCC CTA AAC AGA TGA AG-3′; IL-1β reverse primer 5′-GGT CGG AGA TTC GTA GCT GGA T-3′; IL-1β probe 5′-FAM-TTC CAG GAC CTG GAC CTC TGC CCT C-TAMRA-3′;
Statistical analysis
Statistical analysis was performed using the Mann-Whitney rank sum test and the χ2 test, depending on the data set. The test applied is indicated in the figures and tables. A p value of <0.05 was considered significant. Odds ratios (OR) and corresponding 95% confidence intervals (CI) were calculated by StatXact.4.0.1 software. Hardy-Weinberg equilibrium of alleles at individual loci was assessed by χ2 statistics.
RESULTS
Patient population, polymorphism frequencies, and H pylori strain characteristics
Strain characteristics of H pylori were investigated in 207 infected patients by PCR. The vacAs1 genotype was found in 78.3% (162/207), cagA in 70.5% (146/207), and babA2 in 35.3% (73/207). Almost all cagA+ strains were simultaneously vacAs1+ (143/146) and almost all babA2+ strains were cagA+ (67/73). The frequencies of different alleles in the IL-1B and IL-1RN polymorphisms have been published previously.33 Allele frequencies of IL-10, TNF-A, and IFN-G polymorphisms and of different IL-10 haplotypes are shown in fig 1 ▶. For all control populations (G0/G1; L1; non-IM; non-AG), alleles at the different loci were in Hardy-Weinberg equilibrium, with non-significant χ2 values. As the development of severe histological changes is influenced by age and sex, these parameters were investigated in different patient groups. There were no significant differences in age or sex between carriers of different cytokine gene polymorphisms or patients infected with different strain types (data not shown).
Figure 1.
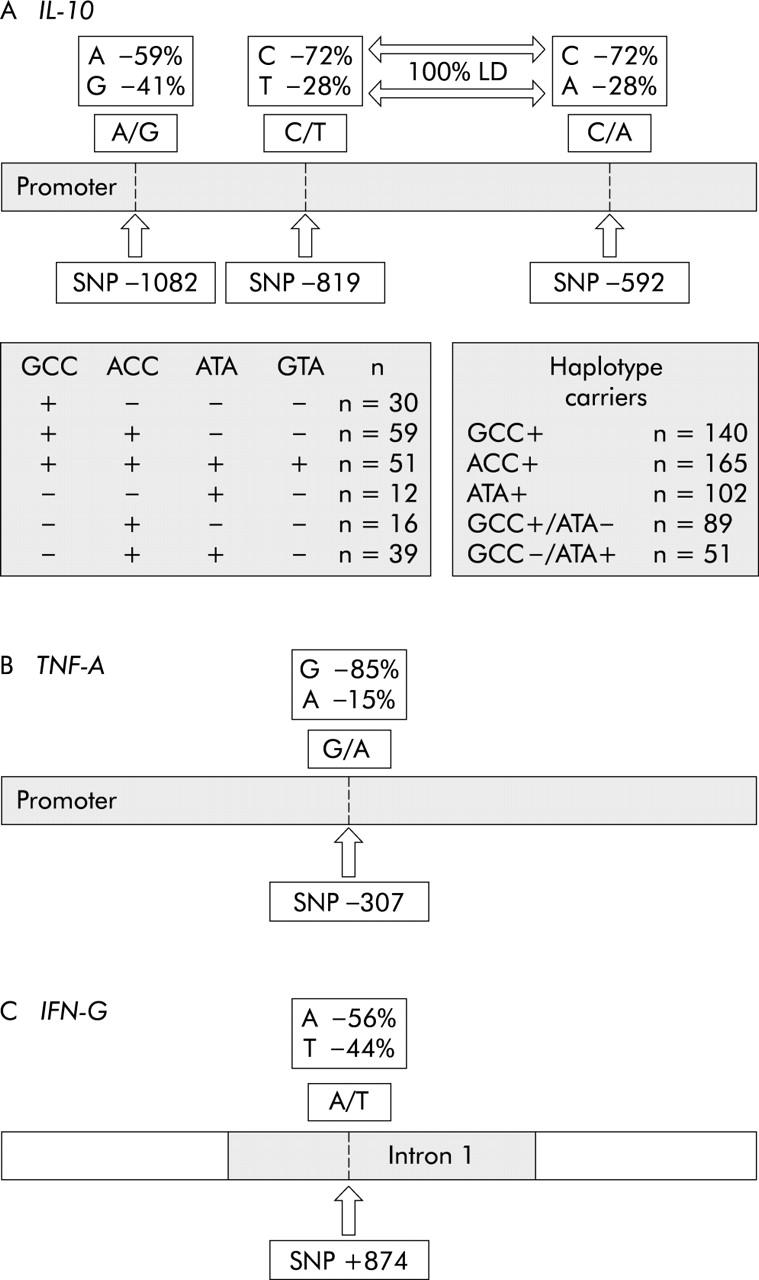
Allele/haplotype frequencies of interleukin 10 (IL-10), tumour necrosis factor A (TNF-A), and interferon G (IFN-G) polymorphisms. Single nucleotide polymorphisms (SNPs) at positions −819 and −592 in the IL-10 promoter were in complete linkage disequilibrium (LD). Four IL-10 haplotypes were observed and the occurrence of each haplotype and of haplotype combinations are listed in the shaded boxes.
Mucosal IL-1β expression in response to H pylori infection is influenced by IL-1 polymorphisms
We have previously shown that proinflammatory IL-1B and IL-1RN polymorphisms are associated with heightened degrees of inflammation and the presence of IM/AG during H pylori infection.33 To investigate the influence of IL-1 polymorphisms on mucosal IL-1β expression, IL-1β mRNA amounts were determined in the gastric mucosa of infected patients by quantitative TaqMan PCR. As illustrated in fig 2 ▶, IL-1β mRNA levels tended to be higher in patients with the IL-1B–511T allele compared with −511CC carriers. However, differences between the groups were low and not significant.
Figure 2.

Interleukin 1β (IL-1β) mRNA amounts in Helicobacter pylori infected patients harbouring different interleukin 1B (IL-1B) and interleukin 1receptor antagonist (IL-1RN) alleles, as well as the proinflammatory IL-1 polymorphism combination IL-1B−511T+/IL-1RN2+. As IL-1B−511T is in almost complete linkage disequilibrium with IL-1B−31C, data are not shown for the latter single nucleotide polymorphism. Bars within the plots represent median values and numbers above indicate mean IL-1β mRNA amounts for each group. p values were calculated using the Mann-Whitney U test. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
In contrast, IL-1β mRNA amounts were significantly higher in patients with at least one IL-1RN*2 allele compared with IL-1RN1/1 carriers (fig 2 ▶). The highest cytokine mRNA levels were found in patients who harboured simultaneously at least one proinflammatory allele of each polymorphism (simultaneous IL-1B−511T and IL-1RN*2 carriers).
IL-10 polymorphisms influence mucosal IL-10 expression during chronic H pylori infection
Correlating mucosal IL-10 mRNA amounts with host IL-10 polymorphisms, –1082AA was associated with low IL-10 expression, −1082GA with intermediate, and –1082GG with high mucosal IL-10 mRNA levels (fig 3A ▶). Similarly, the IL-10–592C and −819C alleles, which are in complete linkage disequilibrium, were associated with higher IL-10 mRNA levels. As shown in fig 3A ▶, −592CC carriers had almost twofold higher mRNA levels than −592AA carriers. We then explored the influence of IL-10 haplotypes and haplotype combinations on IL-10 expression. Figure 3B ▶ shows that the GCC haplotype (−1082G, −819C, −592C) was associated with significantly higher IL-10 mRNA levels than the ATA haplotype. ACC haplotype carriers had intermediate IL-10 mRNA levels. Furthermore, the highest IL-10 mRNA levels were observed in patients with the haplotype combination GCC+/ATA− whereas GCC−/ATA+ carriers were associated with the lowest IL-10 mRNA levels.
Figure 3.
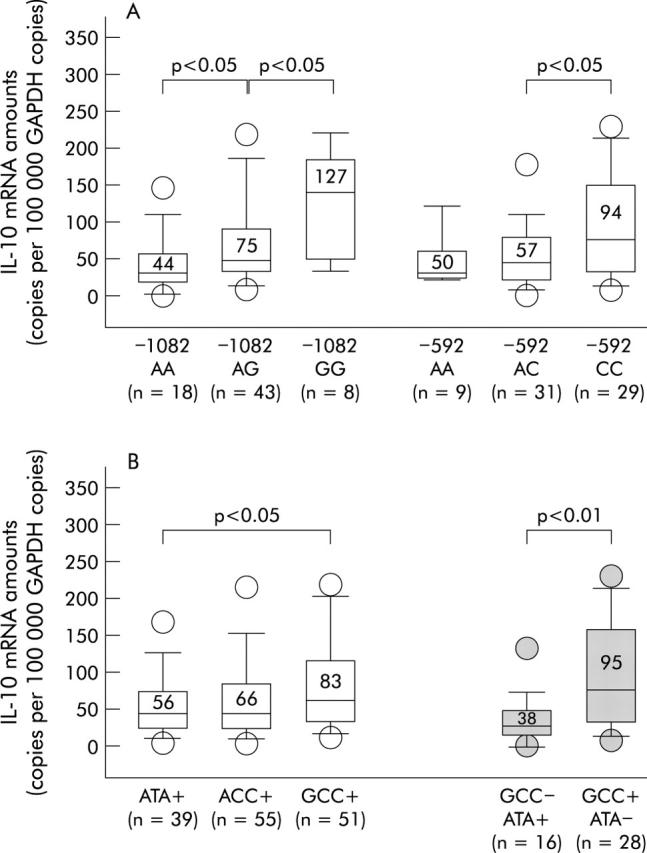
Interleukin 10 (IL-10) mRNA amounts in Helicobacter pylori infected patients harbouring different IL-10 single nucleotide polymorphism (SNP) alleles (A) and different IL-10 SNP haplotypes/haplotype combinations (B). As the IL-10-592C allele is in complete linkage disequilibrium with IL-10-819C, data are not shown for the latter. Bars within the plots represent median values and numbers above indicate mean IL-10 mRNA amounts for each group. p values were calculated using the Mann-Whitney U test. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
High IL-10 secreting haplotypes are associated with infection by virulent cagA+ and vacAs1+ strains
Next, the infecting strain type was correlated to the different IL-10 haplotypes. Interestingly, the prevalence of cagA+ strains was higher among GCC carriers than among ATA carriers (104/140 (74.3%) v 64/102 (62.8%); p = 0.05, χ2 test). Patients with the ACC haplotype had an intermediate frequency of cagA+ strains (115/165 (69.7%)). Similarly, the frequency of vacAs1+ and babA2+ strains was higher among GCC carriers than among ATA carriers (vacAs1+ strains: 115/140 (82.1%) v 72/102 (70.6%); p<0.05, χ2 test; babA2+ strains: 59/140 (42.1%) v 31/102 (32.4%); p = 0.06, χ2 test).
More pronounced were the differences between carriers of combined pro- and anti-inflammatory haplotypes. Figure 4 ▶ shows that the prevalence of cagA+, vacAs1+, and babA2+ strains was lower among GCC−/ATA+ patients (cagA+ strains: 29/51 (56.9%); vacAs1+ strains: 35/51 (68.6%); babA2+ strains: 9/51 (17.6%)) than among GCC+/ATA− patients (cagA+ strains: 69/89 (77.5%); vacAs1+ strains: 78/89 (87.6%); babA2+ strains: 37/89 (41.6%)). The most obvious difference was observed for babA2+ strains: the prevalence of babA2+ strains was 2.4 times higher among patients with the high IL-10 secreting haplotype combination compared with the low IL-10 secreting haplotype combination. Interestingly, polymorphisms in other cytokine genes investigated in this study were not associated with certain bacterial strain types (data not shown).
Figure 4.

Prevalence of infection with cagA+ and cagA− strains (A), vacAs1+ and vacAs1− strains (B), and babA2− and babA2+ strains in carriers of different interleukin 10 (IL-10) haplotype combinations (GCC−/ATA+, low IL-10 secreting haplotype combination; GCC+/ATA−, high IL-10 secreting haplotype combination). p values were calculated using the χ2 test.
The role of IL-10 polymorphisms for the gastric inflammatory response and the development of IM/AG
To investigate the role of IL-10 polymorphisms on the development of gastric inflammation and IM/AG, the frequency of IL-10 haplotypes was compared in different patient groups. Interestingly, although IL-10 polymorphisms influenced IL-10 expression, the frequency of the IL-10 haplotypes was similar in patients with high or low degrees of gastritis and in patients with or without IM/AG (data not shown). Table 2 ▶ shows that even the frequency of high or low IL-10 secreting haplotype combinations did not differ significantly among groups. Also, the frequency of the GCC+/ATA− haplotype combination tended to be higher among patients with IM/AG (OR 1.2–4.2); differences compared with the GCC−/ATA+ group were not significant in most cases.
Table 2.
Frequencies of the interleukin 10 (IL-10) haplotype combinations in patients with different degrees of granulocytic (G) and lymphocytic (L) infiltration, as well as the presence or absence of intestinal metaplasia (IM) and atrophic gastritis (AG)
| Antrum | G2/G3 (n = 152) | G0/G1 (n = 55) | OR (95% CI) | L2/L3 (n = 162) | L1 (n = 45) | OR (95% CI) | IM (n = 64) | Non-IM (n = 143) | OR (95% CI) | AG (n = 49) | Non-AG (n = 158) | OR (95% CI) |
| GCC−ATA+ | 35 | 16 | 1.0 | 44 | 7 | 1.0 | 11 | 40 | 1.0 | 7 | 44 | 1.0 |
| 23% | 29% | Ref | 27% | 16% | Ref | 17% | 30% | Ref | 14% | 28% | Ref | |
| GCC+ATA− | 65 | 24 | 1.2 | 69 | 20 | 0.6 | 34 | 55 | 2.3 | 24 | 65 | 2.3 |
| 43% | 44% | (0.5–1.8) | 43% | 44% | (0.2–1.5) | 53% | 39% | (1.0–5.5) | 49% | 41% | (0.9–6.9) |
| G2/G3 | G0/G1 | OR | L2/L3 | L1 | OR | IM | Non-IM | OR | AG | Non-AG | OR | |
| Corpus | (n = 84) | (n = 123) | (95% CI) | (n = 62) | (n = 145) | (95% CI) | (n = 12) | (n = 195) | (95% CI) | (n = 12) | (n = 195) | (95% CI) |
| Ref, reference. | ||||||||||||
| G0, absent; G1, mild; G2, moderate; G3, severe granulocytic infiltration. | ||||||||||||
| L1, mild; L2, moderate; L3, severe lymphocytic infiltration. | ||||||||||||
| GCC−/ATA+, low IL-10 secreting haplotype combination; GCC+/ATA−, high IL-10 secreting haplotype combination. | ||||||||||||
| Odds ratios (OR) with exact 95% confidence intervals (CI) were calculated using StatXact-4.0.1 software. | ||||||||||||
| GCC−ATA+ | 15 | 36 | 1.0 | 13 | 38 | 1.0 | 3 | 48 | 1.0 | 1 | 50 | 1.0 |
| 18% | 29% | Ref | 21% | 26% | Ref | 25% | 25% | Ref | 8% | 26% | Ref | |
| GCC+ATA− | 38 | 51 | 1.8 | 32 | 57 | 1.6 | 6 | 83 | 1.2 | 7 | 82 | 4.2 |
| 45% | 42% | (0.8–4.0) | 52% | 39% | (0.7–3.9) | 50% | 43% | (0.2–7.5) | 58% | 42% | (0.5–196.1) | |
The role of TNF-A-307 and IFN-G+874 polymorphisms for mucosal cytokine expression and the gastric inflammatory response
The influence of the TNF-A−307 and IFN-G+874 polymorphisms on mucosal cytokine expression was assessed. Figure 5 ▶ shows that there were no significant differences in IFN-γ and TNF-α levels between different allele carriers. This suggests that these polymorphisms do not influence cytokine expression in the course of chronic H pylori infection. Accordingly, there were no significant associations of these polymorphisms with gastric inflammation or the presence of IM or AG (tables 3 ▶, 4 ▶). Although the frequency of the TNF-A−307A allele tended to be higher among patients with IM/AG, differences compared with the TNF-A–307TT group did not reach statistically significant levels.
Figure 5.

Tumour necrosis factor α (TNF-α) and interferon γ (IFN-γ) mRNA amounts in Helicobacter pylori infected patients harbouring different TNF-A−307 and IFN-G−874 alleles. Bars within the plots represent median values and numbers above indicate mean cytokine mRNA amounts for each group. p values were calculated using the Mann-Whitney U test but there were no significant differences between the groups.
Table 3.
Frequencies of the tumour necrosis factor (TNF)-A−307 alleles in patients with different degrees of granulocytic (G) and lymphocytic (L) infiltration, as well as the presence or absence of intestinal metaplasia (IM) and atrophic gastritis (AG)
| Antrum | G2/G3 (n = 152) | G0/G1 (n = 55) | OR (95% CI) | L2/L3 (n = 162) | L1 (n = 45) | OR (95% CI) | IM (n = 64) | Non-IM (n = 143) | OR (95% CI) | AG (n = 49) | Non-AG (n = 158) | OR (95% CI) |
| GG | 113 | 34 | 1.0 | 115 | 32 | 1.0 | 40 | 107 | 1.0 | 34 | 113 | 1.0 |
| 74% | 62% | Ref | 71% | 71% | Ref | 62% | 75% | Ref | 69% | 72% | Ref | |
| A carrier | 39 | 21 | 0.6 | 47 | 13 | 1.0 | 24 | 36 | 1.8 | 15 | 45 | 1.1 |
| 26% | 38% | (0.3–1.1) | 29% | 29% | (0.5–2.3) | 38% | 25% | (0.9–3.5) | 31% | 28% | (0.5–2.3) |
| G2/G3 | G0/G1 | OR | L2/L3 | L1 | OR | IM | Non-IM | OR | AG | Non-AG | OR | |
| Corpus | (n = 84) | (n = 123) | (95% CI) | (n = 62) | (n = 145) | (95% CI) | (n = 12) | (n = 195) | (95% CI) | (n = 12) | (n = 195) | (95% CI) |
| Ref, reference. | ||||||||||||
| G0, absent; G1, mild; G2, moderate; G3, severe granulocytic infiltration. | ||||||||||||
| L1, mild; L2, moderate; L3 severe lymphocytic infiltration. | ||||||||||||
| Odds ratios (OR) with exact 95% confidence intervals (CI) were calculated using StatXact-4.0.1 software. | ||||||||||||
| GG | 59 | 88 | 1.0 | 43 | 104 | 1.0 | 6 | 141 | 1.0 | 6 | 141 | 1.0 |
| 70% | 72% | Ref | 69% | 72% | Ref | 50% | 72% | Ref | 50% | 72% | Ref | |
| A carrier | 25 | 35 | 1.1 | 19 | 41 | 1.1 | 6 | 54 | 2.6 | 6 | 54 | 2.6 |
| 30% | 28% | (0.6–2.0) | 31% | 28% | (0.6–2.2) | 50% | 28% | (0.7–10.1) | 50% | 28% | (0.7–10.1) | |
Table 4.
Frequencies of the interferon (IFN)-G+874 alleles in patients with different degrees of granulocytic (G) and lymphocytic (L) infiltration, as well as the presence or absence of intestinal metaplasia (IM) and atrophic gastritis (AG)
| Antrum | G2/G3 (n = 152) | G0/G1 (n = 55) | OR (95% CI) | L2/L3 (n = 162) | L1 (n = 45) | OR (95% CI) | IM (n = 64) | Non-IM (n = 143) | OR (95% CI) | AG (n = 49) | Non-AG (n = 158) | OR (95% CI) |
| AA | 47 | 18 | 1.0 | 54 | 11 | 1.0 | 20 | 45 | 1.0 | 17 | 48 | 1.0 |
| 31% | 33% | Ref | 33% | 24% | Ref | 31% | 31% | Ref | 35% | 30% | Ref | |
| T carrier | 105 | 37 | 1.1 | 108 | 34 | 0.7 | 44 | 98 | 1.0 | 32 | 110 | 0.8 |
| 69% | 67% | (0.5–2.2) | 67% | 76% | (0.3–1.4) | 69% | 69% | 0.5–2.0 | 65% | 70% | 0.4–1.7 |
| G2/G3 | G0/G1 | OR | L2/L3 | L1 | OR | IM | Non-IM | OR | AG | Non-AG | OR | |
| Corpus | (n = 84) | (n = 123) | (95% CI) | (n = 62) | (n = 145) | (95% CI) | (n = 12) | (n = 195) | (95% CI) | (n = 12) | (n = 195) | (95% CI) |
| Ref, reference. | ||||||||||||
| G0, absent; G1, mild; G2, moderate; G3, severe granulocytic infiltration. | ||||||||||||
| L1, mild; L2, moderate; L3 severe lymphocytic infiltration. | ||||||||||||
| Odds ratios (OR) with exact 95% confidence intervals (CI) were calculated using StatXact-4.0.1 software. | ||||||||||||
| AA | 28 | 37 | 1.0 | 25 | 40 | 1.0 | 4 | 61 | 1.0 | 4 | 61 | 1.0 |
| 33% | 30% | Ref | 40% | 28% | Ref | 33% | 31% | Ref | 33% | 31% | Ref | |
| T carrier | 56 | 86 | 0.9 | 37 | 105 | 0.6 | 8 | 134 | 0.9 | 8 | 134 | 0.9 |
| 67% | 70% | (0.5–1.6) | 60% | 72% | (0.3–1.1) | 67% | 69% | (0.2–4.3) | 67% | 69% | (0.2–4.3) | |
DISCUSSION
Allelic variants in cytokine genes have been shown to influence gene expression and subsequently the susceptibility to and severity of various infectious diseases.19,20 While inflammation is advantageous for a host when microbes can be eliminated, it may be deleterious when infection cannot be eradicated as it leads to continuous impairment of tissue structure and function. An interesting question is therefore how genetic variants in candidate genes influence the H pylori induced inflammatory response and the accompanying development of gastric pathology.
Studies investigating the role of genetic polymorphisms on cytokine secretion are controversial and the experimental approaches used (mainly in vitro) have several disadvantages.19,24 For example, experiments with stimulated PBMC from different polymorphism carriers are often not generally applicable as the cytokine response may differ depending on the stimulus (pathogen).24 Furthermore, the stimulus dose in vitro may not reflect the situation in vivo. To overcome these difficulties, we determined actual cytokine mRNA levels in vivo in the infected mucosa.
In the current study, we found that IL-10 SNPs markedly influenced mucosal IL-10 expression in the course of chronic H pylori infection: GCC haplotype carriers (IL-10 −1082G; −819C; −592C) were associated with high and ATA carriers with low IL-10 expression. These results show the functional relevance of IL-10 SNPs in vivo.
Interestingly, IL-10 polymorphisms were associated with infection by different bacterial strain types. H pylori strains have co-evolved to colonise specific human host populations via the selection of certain bacterial properties34,35 but it is not known which evolutionary forces influence strain selection. Ethnic differences in the frequency of host genetic polymorphisms may account for the diversity of H pylori strain distribution in human populations. In the current study, the prevalence of virulent cagA+/vacAs1+/babA2+ H pylori strains was higher among GCC than ATA carriers. There are two possible explanations for this association: either (i) initial infection with cagA+ strains may be more successful in GCC carriers, suggesting a host specific colonisation or (ii) host specific adaptation of the bacterium may occur in the course of chronic infection.
Regarding the first hypothesis (host specific colonisation), there is evidence from animal models that individuals differ in their susceptibility to H pylori strains during the initial stages of colonisation. For example, after challenge of rhesus monkeys with a mixture of seven different H pylori strains, different animals were colonised by different strains.36 In addition, recently Philpott et al have shown in the mouse model that cagPAI−H pylori strains have a selective advantage for colonisation.37 As IL-10 lowers gastric inflammation,38cagPAI+ strains (which usually induce a strong inflammatory reaction) may be more successful in the initial colonisation of hosts with the high IL-10 producing haplotype GCC.
In relation to the second hypothesis (host specific bacterial adaptation during chronic infection), it has been shown that H pylori exists within its ecological niche as a quasispecies, a bacterial population in a continuous state of genetic flux, which allows optimal adaptation to changing conditions.39–41 Exaggerated inflammation may be deleterious for the bacterium as it leads to impairment of tissue structure/function and finally to loss of its niche.3 Therefore, in patients with low IL-10 production (proinflammatory ATA haplotype carriers), it may be beneficial for H pylori to loose its cagPAI (which is an unstable locus8,42) to avoid an exaggerated immune response. Alternatively, in cases of mixed infections, cagPAI− strains may have a selective advantage in such hosts.
Correlating IL-10 SNPs with gastric histopathology, we could not find significant associations with gastric inflammation or atrophy and intestinal metaplasia. In a mouse model, IL-10 has been shown to protect from the development of severe gastritis and mucosal damage through downregulation of the H pylori induced Th1 response.38,43 In humans, two recent large studies associated IL-10 polymorphims with gastric cancer, leading to conflicting results. Whereas one report found a higher prevalence of gastric cancer in patients with the proinflammatory (low IL-10 secreting) haplotype ATA,32 the other study reported an association between carcinoma and the anti-inflammatory (high IL-10 secreting) haplotype GCC.31 The apparent paradoxical observation in the latter study may be explained by our finding that cagA+ strains were more prevalent among GCC carriers. Further studies are needed to clarify the role of IL-10 polymorphisms in H pylori infection.
The TNF-A−307 and IFN-G+874 SNPs were not associated with changes in mucosal cytokine expression and the inflammatory response to H pylori. Several polymorphisms have been reported in the TNF-A promoter, almost all being functionally silent.24 The G/A polymorphism at position −307 is considered to be related to differences in TNF-α secretion by some authors (reviewed by Allen24). However, the results of the available studies are conflicting: two studies found 20–40% increased TNF-α production from lipopolysaccharide stimulated PBMC of allele A carriers44,45 whereas the majority of reports found no significant differences.25,46–48 Our data are in agreement with the latter reports and indicate that the –307 polymorphism is either generally functionally silent or does not play a functional role for H pylori induced TNF-α expression. An alternative explanation for associations of cytokine gene polymorphisms with heightened inflammatory responses could be linkage of these loci with other immunologically important genes (such as association of TNF-A–307A with the HLA-DR3 allele). However, in this study, we did not find that the TNF-A-307 polymorphism influenced the severity of gastric inflammation.
There is only one study which has examined the relationship between IFN-G polymorphisms and IFN-γ transcription, reporting an association of allele 2 of a polymorphic microsatellite marker (which shows an absolute correlation with the IFN-G+874T allele26) with heightened IFN-γ secretion.27 However, in the H pylori infected stomach, the +874A/T SNP seems to have no influence on H pylori induced IFN-γ expression and the subsequent inflammatory response.
Recent studies have linked proinflammatory IL-1 polymorphisms to low acid secretion, gastric inflammation, and gastric carcinoma.28,30,33,49–51 We found an association between IL-1 polymorphisms and high mucosal IL-1β expression. Although the association was significant only for the IL-1RN*2 allele and not for IL-1B−511T/-31C, the highest IL-1β mRNA levels were found in individuals who were simultaneous carriers of both proinflammatory alleles (IL-1B−511T/−31C and IL-1RN*2). These results on mRNA levels are in accordance with a recent study30 which investigated the role of IL-1 polymorphisms on IL-1β protein secretion in the H pylori infected mucosa. In that study, IL-1B−511T/T and IL-1RN*2 carriers had higher mucosal IL-1β levels than non-carriers, and patients who simultaneously harboured both genotypes had the highest IL-1β protein levels.30 Accordingly, we and others found that carriers of the IL-1RN*2 and IL-1B−511T/-31C alleles were associated with more severe gastric inflammation than non-carriers.30,33,50,51 The effect of IL-1 polymorphisms may be due to the antisecretory and proinflammatory capacity of IL-1β.52–56 Several studies have shown that high acid output and antrum predominant gastritis are associated with duodenal ulceration whereas low acid output and corpus gastritis lead to an increased risk for the development of non-cardia gastric cancer.57–59 Excessive acid secretion has a growth inhibitory effect on H pylori.60 Therefore, IL-1β induced hypochlorhydria probably promotes colonisation of the acid secreting corpus region thus leading to an increased cancer risk.61 On the other hand, IL-1β induced hypochlorhydria may also ameliorate growth conditions of the antrum, explaining the association of IL-1 polymorphisms with more severe antral gastritis, as observed in the current study. However, despite this association with antral gastritis, proinflammatory IL-1 polymorphisms seem not to be linked to duodenal ulceration.62 The explanation for this observation may be that high acid secretion, which is a major feature of duodenal ulceration,61 is not likely to develop in patients with high IL-1β secretion.
In summary, the current study shows how genetic polymorphisms influence cytokine expression, gastric inflammation, and bacterial strain selection in H pylori infection. In the future, genotyping of host polymorphisms may allow identification of patients with an increased risk for the development of gastric pathology. The association of polymorphisms with certain bacterial strain types indicates that host specific colonisation or adaptation occurs, which may explain the heterogenity of H pylori strains in different populations.2 Taken together, these findings contribute to the understanding of the complex host-pathogen interaction and support the model of multifactorial development of gastric pathology.
Acknowledgments
The authors wish to thank Dr Stefan Wagenpfeil for statistical advice, Dr Vera Pravica for kindly providing us with control DNA, and Dr El-Omar for providing us with primer and probe sequences for IL-1B polymorphisms. Part of this work was performed by Roland Rad as an MD thesis at the TUM. Sponsored by DFG Pr 411/7-1 (Heisenberg Program to CP); a grant from Astra Hässle, Wedel, Germany; and Kuratorium für Klinische Forschung der TUM 8733153 and 8733156. Roland Rad is a recipient of a grant from the “Bund der Freunde der TU München”. Sponsored by Dr Else Kröner Fresenius Stiftung, Germany
Abbreviations
cagPAI, cag pathogenicity island
VacA, vacuolating cytotoxin
BabA, blood group antigen binding adhesin
IFN-γ, interferon γ
TNF-α, tumour necrosis factor α
IL, interleukin
IL-1RN, IL-1 receptor antagonist
SNP, single nucleotide polymorphism
PBMC, peripheral blood mononuclear cells
VNTR, variable number of tandem repeat region
IM, intestinal metaplasia
AG, atrophic gastritis
PCR, polymerase chain reaction
RFLP, restriction fragment length polymorphism
OR, odds ratio
GAPDH, glyceraldehyde-3-phosphate dehydrogenase
REFERENCES
- 1.Suerbaum S , Michetti P. Helicobacter pylori infection. N Engl J Med 2002;347:1175–86. [DOI] [PubMed] [Google Scholar]
- 2.Covacci A , Telford JL, Del GG, et al. Helicobacter pylori virulence and genetic geography. Science 1999;284:1328–33. [DOI] [PubMed] [Google Scholar]
- 3.Blaser MJ, Berg DE. Helicobacter pylori genetic diversity and risk of human disease. J Clin Invest 2001;107:767–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prinz C , Schoniger M, Rad R, et al. Key importance of the Helicobacter pylori adherence factor blood group antigen binding adhesin during chronic gastric inflammation. Cancer Res 2001;61:1903–9. [PubMed] [Google Scholar]
- 5.Gerhard M , Lehn N, Neumayer N, et al. Clinical relevance of the Helicobacter pylori gene for blood-group antigen-binding adhesin. Proc Natl Acad Sci U S A 1999;96:12778–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blaser MJ, Perez PG, Kleanthous H, et al. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res 1995;55:2111–15. [PubMed] [Google Scholar]
- 7.Atherton JC, Cao P, Peek-RM J, et al. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem 1995;270:17771–7. [DOI] [PubMed] [Google Scholar]
- 8.Censini S , Lange C, Xiang Z, et al. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci U S A 1996;93:14648–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crabtree JE, Taylor JD, Wyatt JI, et al. Mucosal IgA recognition of Helicobacter pylori 120 kDa protein, peptic ulceration, and gastric pathology. Lancet 1991;338:332–5. [DOI] [PubMed] [Google Scholar]
- 10.Ernst PB, Gold BD. The disease spectrum of Helicobacter pylori: the immunopathogenesis of gastroduodenal ulcer and gastric cancer. Annu Rev Microbiol 2000;54:615–40. [DOI] [PubMed] [Google Scholar]
- 11.Shimoyama T , Crabtree JE. Bacterial factors and immune pathogenesis in Helicobacter pylori infection. Gut 1998;43 (suppl 1) :S2–5. [PMC free article] [PubMed] [Google Scholar]
- 12.Ernst P . Review article: the role of inflammation in the pathogenesis of gastric cancer. Aliment Pharmacol Ther 1999;13 (suppl 1) :13–18. [DOI] [PubMed] [Google Scholar]
- 13.Fox JG, Beck P, Dangler CA, et al. Concurrent enteric helminth infection modulates inflammation and gastric immune responses and reduces Helicobacter-induced gastric atrophy. Nat Med 2000;6:536–42. [DOI] [PubMed] [Google Scholar]
- 14.Fox JG, Sheppard BJ, Dangler CA, et al. Germ-line p53-targeted disruption inhibits Helicobacter-induced premalignant lesions and invasive gastric carcinoma through down-regulation of Th1 proinflammatory responses. Cancer Res 2002;62:696–702. [PubMed] [Google Scholar]
- 15.Bodger K , Crabtree JE. Helicobacter pylori and gastric inflammation. Br Med Bull 1998;54:139–50. [DOI] [PubMed] [Google Scholar]
- 16.Yamaoka Y , Kita M, Kodama T, et al. Induction of various cytokines and development of severe mucosal inflammation by cagA gene positive Helicobacter pylori strains. Gut 1997;41:442–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rad R , Gerhard M, Lang R, et al. The Helicobacter pylori blood group antigen-binding adhesin facilitates bacterial colonization and augments a nonspecific immune response. J Immunol 2002;168:3033–41. [DOI] [PubMed] [Google Scholar]
- 18.Karttunen RA, Karttunen TJ, Yousfi MM, et al. Expression of mRNA for interferon-gamma, interleukin-10, and interleukin-12 (p40) in normal gastric mucosa and in mucosa infected with Helicobacter pylori. Scand J Gastroenterol 1997;32:22–7. [DOI] [PubMed] [Google Scholar]
- 19.Bidwell J , Keen L, Gallagher G, et al. Cytokine gene polymorphism in human disease: on-line databases. Genes Immun 1999;1:3–19. [DOI] [PubMed] [Google Scholar]
- 20.Hurme M , Lahdenpohja N, Santtila S. Gene polymorphisms of interleukins 1 and 10 in infectious and autoimmune diseases. Ann Med 1998;30:469–73. [DOI] [PubMed] [Google Scholar]
- 21.Eskdale J , Keijsers V, Huizinga T, et al. Microsatellite alleles and single nucleotide polymorphisms (SNP) combine to form four major haplotype families at the human interleukin-10 (IL-10) locus. Genes Immun 1999;1:151–5. [DOI] [PubMed] [Google Scholar]
- 22.Turner DM, Williams DM, Sankaran D, et al. An investigation of polymorphism in the interleukin-10 gene promoter. Eur J Immunogenet 1997;24:1–8. [DOI] [PubMed] [Google Scholar]
- 23.Crawley E , Kay R, Sillibourne J, et al. Polymorphic haplotypes of the interleukin-10 5′ flanking region determine variable interleukin-10 transcription and are associated with particular phenotypes of juvenile rheumatoid arthritis. Arthritis Rheum 1999;42:1101–8. [DOI] [PubMed] [Google Scholar]
- 24.Allen RD. Polymorphism of the human TNF-alpha promoter—random variation or functional diversity? Mol Immunol 1999;36:1017–27. [DOI] [PubMed] [Google Scholar]
- 25.de Jong BA, Westendorp RG, Bakker AM, et al. Polymorphisms in or near tumour necrosis factor (TNF)-gene do not determine levels of endotoxin-induced TNF production. Genes Immun 2002;3:25–9. [DOI] [PubMed] [Google Scholar]
- 26.Pravica V , Perrey C, Stevens A, et al. A single nucleotide polymorphism in the first intron of the human IFN-gamma gene: absolute correlation with a polymorphic CA microsatellite marker of high IFN-gamma production. Hum Immunol 2000;61:863–6. [DOI] [PubMed] [Google Scholar]
- 27.Pravica V , Asderakis A, Perrey C, et al. In vitro production of IFN-gamma correlates with CA repeat polymorphism in the human IFN-gamma gene. Eur J Immunogenet 1999;26:1–3. [DOI] [PubMed] [Google Scholar]
- 28.El Omar EM, Carrington M, Chow WH, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 2000;404:398–402. [DOI] [PubMed] [Google Scholar]
- 29.Santtila S , Savinainen K, Hurme M. Presence of the IL-1RA allele 2 (IL1RN*2) is associated with enhanced IL-1beta production in vitro. Scand J Immunol 1998;47:195–8. [DOI] [PubMed] [Google Scholar]
- 30.Hwang IR, Kodama T, Kikuchi S, et al. Effect of interleukin 1 polymorphisms on gastric mucosal interleukin 1beta production in Helicobacter pylori infection. Gastroenterology 2002;123:1793–803. [DOI] [PubMed] [Google Scholar]
- 31.Wu MS, Wu CY, Chen CJ, et al. Interleukin-10 genotypes associate with the risk of gastric carcinoma in Taiwanese Chinese. Int J Cancer 2003;104:617–23. [DOI] [PubMed] [Google Scholar]
- 32.El Omar EM, Rabkin CS, Gammon MD, et al. Increased risk of noncardia gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology 2003;124:1193–201. [DOI] [PubMed] [Google Scholar]
- 33.Rad R , Prinz C, Neu B, et al. Synergistic effect of Helicobacter pylori virulence factors and interleukin-1 polymorphisms for the development of severe histological changes in the gastric mucosa. J Infect Dis 2003;188:272–81. [DOI] [PubMed] [Google Scholar]
- 34.Covacci A , Rappuoli R. Helicobacter pylori: molecular evolution of a bacterial quasi-species. Curr Opin Microbiol 1998;1:96–102. [DOI] [PubMed] [Google Scholar]
- 35.Kirschner DE, Blaser MJ. The dynamics of Helicobacter pylori infection of the human stomach. J Theor Biol 1995;176:281–90. [DOI] [PubMed] [Google Scholar]
- 36.Dubois A , Berg DE, Incecik ET, et al. Host specificity of Helicobacter pylori strains and host responses in experimentally challenged nonhuman primates. Gastroenterology 1999;116:90–6. [DOI] [PubMed] [Google Scholar]
- 37.Philpott DJ, Belaid D, Troubadour P, et al. Reduced activation of inflammatory responses in host cells by mouse-adapted Helicobacter pylory isolates. Cell Microbiol 2002;4:285–96. [DOI] [PubMed] [Google Scholar]
- 38.Sutton P , Kolesnikow T, Danon S, et al. Dominant nonresponsiveness to Helicobacter pylori infection is associated with production of interleukin 10 but not gamma interferon. Infect Immun 2000;68:4802–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuipers EJ, Israel DA, Kusters JG, et al. Quasispecies development of Helicobacter pylori observed in paired isolates obtained years apart from the same host. J Infect Dis 2000;181:273–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Israel DA, Salama N, Krishna U, et al. Helicobacter pylori genetic diversity within the gastric niche of a single human host. Proc Natl Acad Sci U S A 2001;98:14625–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Webb GF, Blaser MJ. Dynamics of bacterial phenotype selection in a colonized host. Proc Natl Acad Sci U S A 2002;99:3135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kersulyte D , Chalkauskas H, Berg DE. Emergence of recombinant strains of Helicobacter pylori during human infection. Mol Microbiol 1999;31:31–43. [DOI] [PubMed] [Google Scholar]
- 43.Eaton KA, Mefford M, Thevenot T. The role of T cell subsets and cytokines in the pathogenesis of Helicobacter pylori gastritis in mice. J Immunol 2001;166:7456–61. [DOI] [PubMed] [Google Scholar]
- 44.Bouma G , Crusius JB, Oudkerk PM, et al. Secretion of tumour necrosis factor alpha and lymphotoxin alpha in relation to polymorphisms in the TNF genes and HLA-DR alleles. Relevance for inflammatory bowel disease. Scand J Immunol 1996;43:456–63. [DOI] [PubMed] [Google Scholar]
- 45.Louis E , Franchimont D, Piron A, et al. Tumour necrosis factor (TNF) gene polymorphism influences TNF-alpha production in lipopolysaccharide (LPS)-stimulated whole blood cell culture in healthy humans. Clin Exp Immunol 1998;113:401–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huizinga TW, Westendorp RG, Bollen EL, et al. TNF-alpha promoter polymorphisms, production and susceptibility to multiple sclerosis in different groups of patients. J Neuroimmunol 1997;72:149–53. [DOI] [PubMed] [Google Scholar]
- 47.Pociot F , Briant L, Jongeneel CV, et al. Association of tumor necrosis factor (TNF) and class II major histocompatibility complex alleles with the secretion of TNF-alpha and TNF-beta by human mononuclear cells: a possible link to insulin-dependent diabetes mellitus. Eur J Immunol 1993;23:224–31. [DOI] [PubMed] [Google Scholar]
- 48.Mycko M , Kowalski W, Kwinkowski M, et al. Multiple sclerosis: the frequency of allelic forms of tumor necrosis factor and lymphotoxin-alpha. J Neuroimmunol 1998;84:198–206. [DOI] [PubMed] [Google Scholar]
- 49.Machado JC, Pharoah P, Sousa S, et al. Interleukin 1B and interleukin 1RN polymorphisms are associated with increased risk of gastric carcinoma. Gastroenterology 2001;121:823–9. [DOI] [PubMed] [Google Scholar]
- 50.Furuta T , El Omar EM, Xiao F, et al. Interleukin 1beta polymorphisms increase risk of hypochlorhydria and atrophic gastritis and reduce risk of duodenal ulcer recurrence in Japan. Gastroenterology 2002;123:92–105. [DOI] [PubMed] [Google Scholar]
- 51.Figueiredo C , Machado JC, Pharoah P, et al. Helicobacter pylori and interleukin 1 genotyping: an opportunity to identify high-risk individuals for gastric carcinoma. J Natl Cancer Inst 2002;94:1680–7. [DOI] [PubMed] [Google Scholar]
- 52.Schepp W , Dehne K, Herrmuth H, et al. Identification and functional importance of IL-1 receptors on rat parietal cells. Am J Physiol 1998;275:G1094–105. [DOI] [PubMed] [Google Scholar]
- 53.Beales IP, Calam J. Interleukin 1 beta and tumour necrosis factor alpha inhibit acid secretion in cultured rabbit parietal cells by multiple pathways. Gut 1998;42:227–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prinz C , Neumayer N, Mahr S, et al. Functional impairment of rat enterochromaffin-like cells by interleukin 1 beta. Gastroenterology 1997;112:364–75. [DOI] [PubMed] [Google Scholar]
- 55.Mahr S , Neumayer N, Gerhard M, et al. IL-1beta-induced apoptosis in rat gastric enterochromaffin-like cells is mediated by iNOS, NF-kappaB, and Bax protein. Gastroenterology 2000;118:515–24. [DOI] [PubMed] [Google Scholar]
- 56.Takashima M , Furuta T, Hanai H, et al. Effects of Helicobacter pylori infection on gastric acid secretion and serum gastrin levels in Mongolian gerbils. Gut 2001;48:765–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gillen D , El Omar EM, Wirz AA, et al. The acid response to gastrin distinguishes duodenal ulcer patients from Helicobacter pylori-infected healthy subjects. Gastroenterology 1998;114:50–7. [DOI] [PubMed] [Google Scholar]
- 58.El Omar EM, Penman ID, Ardill JE, et al. Helicobacter pylori infection and abnormalities of acid secretion in patients with duodenal ulcer disease. Gastroenterology 1995;109:681–91. [DOI] [PubMed] [Google Scholar]
- 59.El Omar EM, Oien K, El Nujumi A, et al. Helicobacter pylori infection and chronic gastric acid hyposecretion. Gastroenterology 1997;113:15–24. [DOI] [PubMed] [Google Scholar]
- 60.Sachs G , Weeks DL, Melchers K, et al. The gastric biology of Helicobacter pylori. Annu Rev Physiol 2003;65:349–69. [DOI] [PubMed] [Google Scholar]
- 61.El Omar EM. The importance of interleukin 1beta in Helicobacter pylori associated disease. Gut 2001;48:743–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Garcia-Gonzalez MA, Lanas A, Santolaria S, et al. The polymorphic IL-1B and IL-1RN genes in the aetiopathogenesis of peptic ulcer. Clin Exp Immunol 2001;125:368–75. [DOI] [PMC free article] [PubMed] [Google Scholar]