

Abstract
Synoviolin, also called HRD1, is an E3 ubiquitin ligase and is implicated in endoplasmic reticulum -associated degradation. In mammals, Synoviolin plays crucial roles in various physiological and pathological processes, including embryogenesis and the pathogenesis of arthropathy. However, little is known about the molecular mechanisms of Synoviolin in these actions. To clarify these issues, we analyzed the profile of protein expression in synoviolin-null cells. Here, we report that Synoviolin targets tumor suppressor gene p53 for ubiquitination. Synoviolin sequestrated and metabolized p53 in the cytoplasm and negatively regulated its cellular level and biological functions, including transcription, cell cycle regulation and apoptosis. Furthermore, these p53 regulatory functions of Synoviolin were irrelevant to other E3 ubiquitin ligases for p53, such as MDM2, Pirh2 and Cop1, which form autoregulatory feedback loops. Our results provide novel insights into p53 signaling mediated by Synoviolin.
Keywords: apoptosis, cell growth, E3 ubiquitin ligase, endoplasmic reticulum-associated degradation, rheumatoid arthritis
Introduction
The ubiquitin–proteasome system (UPS) consists of a small polypeptide ubiquitin, a framework of enzymes that mediates the covalent attachment of ubiquitin to proteolytic substrates and the 26S proteasome that digests the modified proteins into peptides. The formation of ubiquitin conjugates requires the successive action of three classes of enzymes. This process is first activated by an E1 (activating enzyme) in an ATP-dependent manner, forming a high-energy thioester bond between ubiquitin and an E1, and the activated ubiquitin is then transferred to an E2 (conjugating enzyme), forming a similar thioester linkage between ubiquitin and E2, and then E3 ubiquitin ligase transfers ubiquitin to the target proteins. Through repeated reactions of this cycle, a polyubiquitin chain is formed on the target proteins, which is recognized by the 26S proteasome for ultimate degradation (Hershko and Ciechanover, 1998; Pickart, 2001). In the UPS pathway, the E3 ubiquitin ligases play critical roles in the selection of target proteins for degradation, because each distinct E3 ubiquitin ligase usually binds a protein substrate with a degree of selectivity for ubiquitination in a temporally and spatially regulated fashion.
Synoviolin, a representative of endoplasmic reticulum (ER)-resident E3 ubiquitin ligases, is a mammalian homolog of Hrd1p/Der3p that “substrates” misfolded carboxypeptide yscY (CPY*) (Bordallo et al, 1998) and 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGR), a key enzyme of the mevalonate pathway in yeast (Shearer and Hampton, 2004, 2005). We cloned Synoviolin from rheumatoid synovial cells (RSCs) and described that Synoviolin is highly expressed in synoviocytes of patients with rheumatoid arthritis (RA) (Amano et al, 2003). In that report, we demonstrated that overexpression of Synoviolin in transgenic mice leads to advanced arthropathy caused by reduced apoptosis of synoviocytes. On the other hand, synoviolin+/− mice showed resistance to the development of arthritis owing to enhanced apoptosis of synovial cells. These results indicate that Synoviolin is a novel causative factor for arthropathy based on its anti-apoptotic effects. In another study, we reported that all mice fetuses lacking synoviolin (Syno−/−) died in utero around E13.5 (Yagishita et al, 2005), although Hrd1p/Del3p, a yeast ortholog of Synoviolin, was described as non-essential for survival. Syno−/− were anemic owing to enhanced apoptosis of fetal liver cells (Yagishita et al, 2005). It is surprising that an ER-associated degradation (ERAD)-associated E3 ubiquitin ligase, Synoviolin, is involved in cell hyperplasia of dividing cells via its anti-apoptotic effect. In this regard, like RSCs, the anti-apoptotic effect of Synoviolin was observed even for synoviolin expressed ectopically in NIH3T3 cells, which resulted in enhanced cell overgrowth in these cells (Tsuchimochi et al, 2005). These results were confirmed also in the Drosophila fly (Supplementary Figure 1). An important question remains unanswered at this stage. What is the mechanism of Synoviolin-induced cell overgrowth? The present study was designed to identify the substrates for Synoviolin that may be involved in cell growth.
Results
Accumulation of p53 in synoviolin-null cells
To identify target(s) for Synoviolin, we assumed that the lack of Synoviolin results in accumulation of substrate proteins. First, we carried out a two-dimensional polyacrylamide gel electrophoresis (PAGE) using mouse embryonic fibroblasts (MEFs) of Syno−/−. In these experiments, p53 was identified as one of the major targets in the profile by LC-MAS analysis (Supplementary Figure 2). Indeed, the level of p53 was markedly enhanced in Syno−/− MEFs (Figure 1A) and Syno−/− embryos, especially in the posterior part of the body such as somites, brains and maxillary or branchial arches (Supplementary Figure 3), as reported previously (Gottlieb et al, 1997). The accumulated p53 was predominantly localized in the nuclei of Syno−/− MEFs (Figure 1B), although the mRNA level of p53 was not altered in Syno−/− MEFs (Figure 1C). Phosphorylation of p53 was not observed in Syno−/− MEFs (Supplementary Figure 4).
Figure 1.
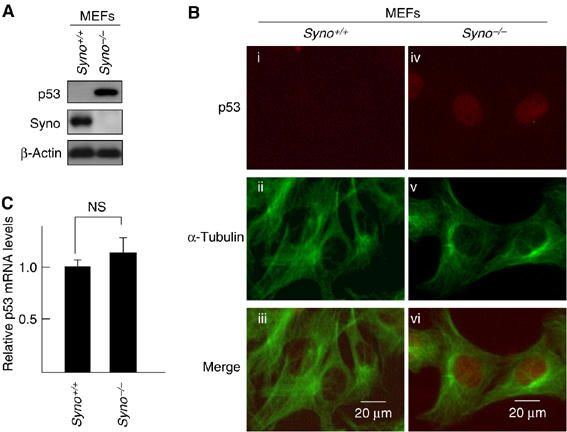
Accumulation of p53 in synoviolin null cells. (A) Accumulation of p53 in Syno−/− MEFs. (B) Nuclear accumulation of p53 in Syno−/− MEFs. p53 in Syno+/+ MEFs (i–iii) and Syno−/− MEFs (iv–vi). Merged images are shown in the bottom panels (iii, vi). (C) Quantification of p53 mRNA. The p53 mRNA level was assessed by real-time PCR and normalized to 18S rRNA. Data are mean±s.e.m. of four experiments. Statistical analysis using Student t-test indicated no significant difference between Syno+/+ and Syno−/− MEFs (NS).
Increment of functional p53 in synoviolin-null cells
Next, we tested whether impairment of Synoviolin influences the functions of p53 in the cell. Knockdown of Synoviolin by small interfering RNA (siRNA) for synoviolin (Syno siRNA) in RKO cells, a human colon cancer cell line known to express wild-type (WT) p53 (Smith et al, 1995), resulted in almost complete disappearance of Synoviolin expression (Figure 2A). Synoviolin knockdown was associated with increased p53 protein level and nuclear accumulation of p53 (Figure 2A and B), but no change in p53 mRNA levels (Figure 2C). No changes were noted in the expression levels of other ubiquitin ligases for p53 such as MDM2, Arf-BP (data not shown) and Parc (see Figure 4B), in synoviolin-null RKO cells (Brooks and Gu, 2006). On the other hand, the expression levels of unfolded protein response (UPR) markers such as HERP and PERK (Wu and Kaufman, 2006) were increased, which suggests that accumulation of unfolded proteins in synoviolin-knockdown RKO cells caused ER stress, followed by UPR (data not shown). These results were confirmed in other cell lines (HEK293 cells and HeLa cells, data not shown). In another experiment, a marked increase was noted in the binding of p53 to its consensus sequences such as GADD45, MDM2 and p21 promoter in synoviolin-knockdown cells compared with GFP-knockdown cells (Figure 2D). Furthermore, further additions of the respective competitor abrogated the binding capacity dose-dependently, confirming the specific interactions of p53 on electrophoretic mobility shift assay (EMSA) (Figure 2D). We also noted three times increment of luciferase activities on GADD45-MLP-Luciferase reporter plasmid in synoviolin-deficient RKO cells compared with GFP-siRNA-treated RKO cells (Figure 2E). Moreover, in Syno siRNA-treated RKO cells, we detected enhanced expression of p21, one of the target genes of p53 (Figure 2F), and the accumulation of cells in G1 phase and decreased cells in S phase (Figure 2G). Taken together, the above results indicate that Synoviolin deficiency is not only associated with increased levels of p53, but also with functional activation of p53.
Figure 2.
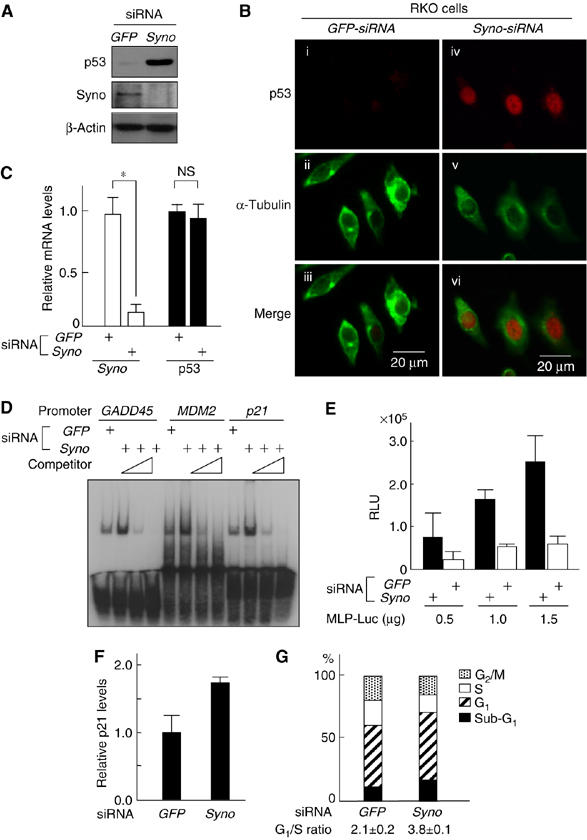
Functional evaluation of increased p53 in synoviolin-deficient RKO cells. (A) Increment of endogenous p53 by depletion of synoviolin. (B) Depletion of synoviolin causes nuclear accumulation of p53. Merged images are shown in the bottom panels (iii, vi). (C) synoviolin depletion does not affect mRNA levels of p53. Real-time PCR was performed as in Figure 1C. *P<0.01. (D) DNA-binding activity of p53 for promoters of the indicated genes increases by depletion of synoviolin. (E) Transactivation activity of p53 is increased upon depletion of Synoviolin. Relative transactivation activity was determined by normalizing luciferase to an internal control, β-Gal activity from RSV-β-gal plasmid. RLU, relative light units. (F) siRNA depletion of synoviolin causes activation of p21 expression. (G) siRNA-induced depletion of synoviolin induces G1 arrest. The cell-cycle profile was determined by propidium iodide staining and FACS. The results represent the average of triplicate experiments. Data in (C), (E) and (F) are mean±s.e.m. of four experiments.
Figure 4.
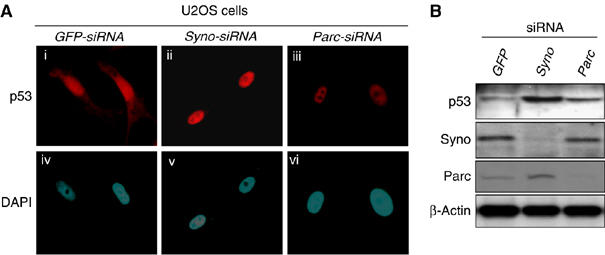
p53-related functional differences between Synoviolin and Parc. (A) Nuclear accumulation of p53 by knockdown of synoviolin or Parc. (B) Different levels of p53 following knockdown of synoviolin compared with Parc.
Synoviolin sequestrates p53 in the cytoplasm
To understand the molecular mechanism of Synoviolin-induced control of p53, we investigated the interaction between Synoviolin and p53 in vitro. As shown in Figure 3A, GST-SynoΔTM interacted directly with p53 (lane 8). A series of N-terminus Synoviolin-TM deletion mutants showed that the amino-acid sequence 236–270 of Synoviolin is responsible for binding with p53 (lanes 1–6) (this binding domain was termed provisionally as ‘53BD'). Furthermore, a synthetic 53BD peptide inhibited Synoviolin-p53 interaction in a dose-dependent manner, whereas a peptide representing amino acids 322–332 of Synoviolin, used as a negative control, did not show any inhibitory activity (Supplementary Figure 5). We also confirmed in vivo, using co-immunoprecipitation assay, the interaction of transiently expressed exogenous Synoviolin WT-FLAG and p53, and the necessity of 53BD was also apparent (Figure 3B). The interaction of these two molecules was independent of ubiquitin ligase activity of Synoviolin, because Synoviolin C307S-FLAG lacking E3 activity bound to p53, as its WT (Figure 3B). Furthermore, endogenous interaction of p53 and Synoviolin was also confirmed in HEK293 cells (Figure 3C).
Figure 3.
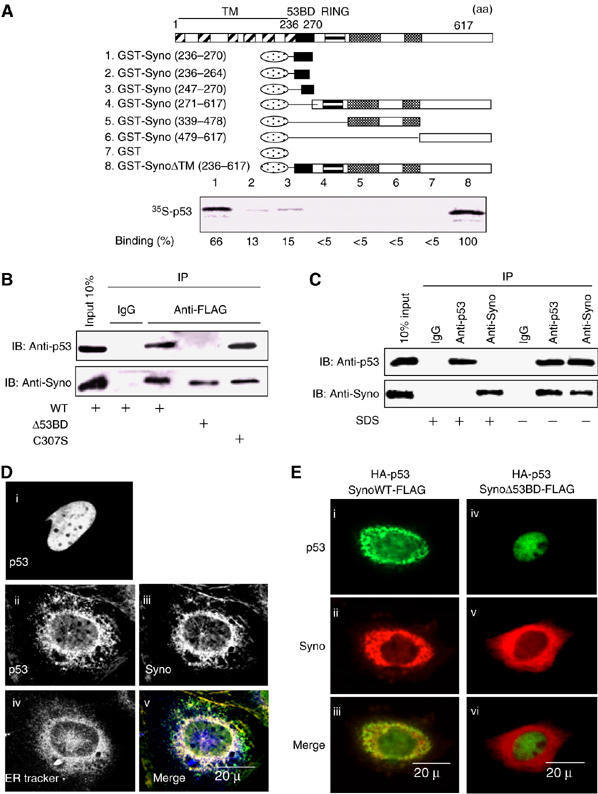
Synoviolin sequestrates p53 in the ER through its 53BD-dependent interaction with p53. (A) Identification of p53-binding domain of Synoviolin in vitro. Black box: p53-binding domain (53BD), gray box: proline-rich domain, oval box: GST. Relative binding ability is denoted as percentage (100%=SynoΔTM, lane 8). (B) 53BD-dependent in vivo binding of Synoviolin with p53 in HEK293 cell. (C) Interaction between endogenous Synoviolin and p53 in HEK293 cells. Cell lysates were immunoprecipitated in the presence or absence of SDS by using anti-p53 antibodies, anti-Synoviolin antibodies or control IgG. Inputs and immunoprecipitates were analyzed by Western blot by using anti-p53 or anti-Synoviolin antibodies. (D) P53 is anchored around ER in a Synoviolin-dependent manner. Saos-2 cells were transfected with HA-p53 (i) or co-transfected with HA-p53 and Synoviolin-FLAG (ii–v). Panel v shows a merged image with p53 (green), Synoviolin (red) and ER-Tracker stain (blue). (E) Binding of p53 with Synoviolin is required for p53 anchoring in the ER. Saos-2 cells were co-transfected with HA-p53 and Synoviolin WT-FLAG (I–iii) or SynoviolinΔ53BD-FLAG (iv–vi). Merged images are shown in the bottom panels (iii, vi).
Considering the interplay between the ER-resident Synoviolin and the nuclear p53, we next investigated their cellular localization in Saos-2 cells, a human osteosarcoma cell line that lacks the endogenous p53 gene (Fogh et al, 1977), under conditions of transient expression of exogenous HA-p53 with Synoviolin WT-FLAG or SynoviolinΔ53BD-FLAG (Figure 3D and E). By overexpression of HA-p53 alone in these cells, HA-p53 was localized in the nucleus (Figure 3Di), as reported previously (Shaulsky et al, 1990). On the other hand, when HA-p53 was coexpressed with Synoviolin WT-FLAG, HA-p53 was predominantly colocalized with Synoviolin WT-FLAG in the perinuclear regions, but not in the nucleus (Figure 3Dii, iii and v). The perinuclear regions were confirmed to be the ER, by counterstaining with ER-Tracker Blue-White DPX (Figure 3Div and v). In addition, ectopically expressed SynoviolinΔ53BD-FLAG did not affect the translocation of HA-p53 into the nucleus (Figure 3E). These results clearly indicate that Synoviolin entraps p53 around ER, and that 53BD is required for this sequestration in vivo. In this regard, a previous study reported that another RING finger protein, Parc (Nikolaev et al, 2003), also acts as a cytoplasmic anchor for p53. To compare the characteristics of Synoviolin and Parc, we investigated p53 localization in U2OS cells, a human osteosarcoma cell line known to express WT p53 (Ponten and Saksela, 1967), after depletion of synoviolin or Parc (Nikolaev et al, 2003). Treatment with either Syno siRNA (Figure 4Aii and v) or Parc siRNA (Figure 4Aiii and vi) resulted in accumulation of p53 in the nucleus with diffused and lesser staining in the cytoplasm, different from treatment with GFP siRNA. Whereas the nuclear translocation of p53 was comparable in both Syno siRNA and Parc siRNA cells, a higher expression of p53 was observed in synoviolin-deficient U2OS cells (Figure 4Aii and iii). Western blotting analysis also revealed increased level of p53 in Synoviolin-knockdown but not in Parc-knockdown cells (Figure 4B). These findings indicate that Synoviolin regulates both localization and quantity of p53, whereas Parc does not affect the amount of p53, as reported previously (Nikolaev et al, 2003).
Synoviolin functions as a novel E3 ubiquitin ligase for p53 degradation
Considering that Synoviolin interacts with p53 in vitro and in vivo, we next examined whether Synoviolin ubiquitinates p53. As shown in Figure 5A, polyubiquitinated GST-p53 was detected only in the presence of ATP, PK-His-HA-Ub, E1, E2 (UbcH5c) and SynoviolinΔTM (SynoΔTM). This activity was not observed when we used Synoviolin with mutation in the RING finger domain (Figure 5B), and the deletion of 53BD also did not show any ubiquitination activity on p53 (Figure 5B), but this mutant by itself still preserved the auto-ubiquitination activity (Supplementary Figure 6). In addition, the 53BD peptide also inhibited polyubiquitination of p53 compared with a control peptide (amino acids 322–332), although the 53BD peptide did not influence the auto-ubiquitination activity of Synoviolin (Supplementary Figure 7). Moreover, ubiquitinated FLAG-p53 was observed when HA-tagged ubiquitin and Synoviolin WT were coexpressed in HEK293 cells because of its easy transfection, but Synoviolin C307S did not (Figure 5C). As a positive control, p53 was ubiquitinated by MDM2 in an in vivo ubiquitination assay (Figure 5C) (Haupt et al, 1997; Kubbutat et al, 1997).
Figure 5.
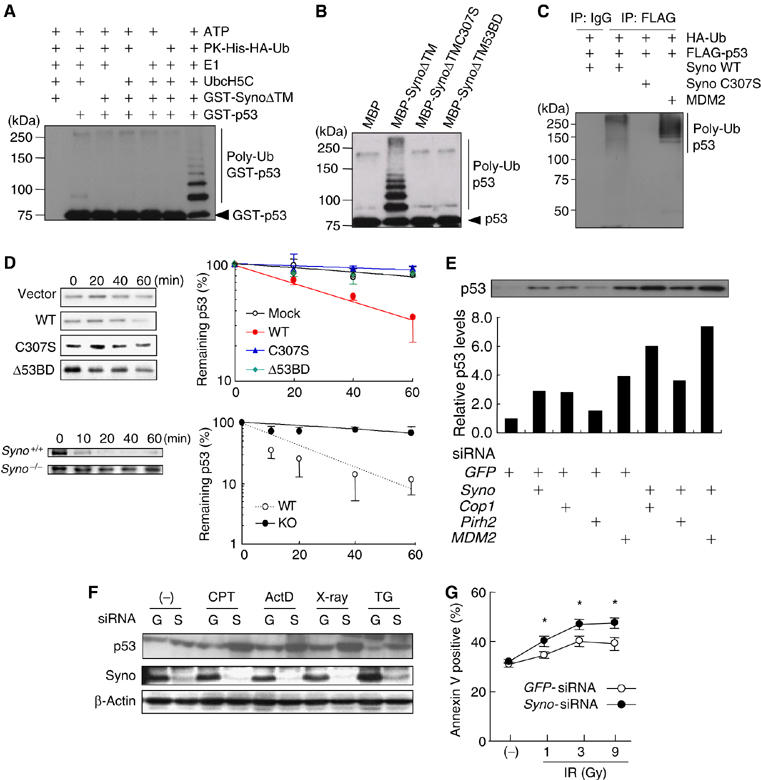
Synoviolin up-regulates p53 level in cells under normal and genotoxic stress conditions. (A) Synoviolin ubiquitinates p53 in vitro. (B) The ubiquitination of p53 is dependent on an intact RING finger domain. (C) WT Synoviolin, but not RING finger mutant, ubiquitinates p53 in vivo. (D) Synoviolin over-expression in HEK293 cells increases degradation of p53 (top). At 24 h after the transfection with empty vector, wild type Synoviolin (WT), mutants in RING finger domain (C307S) or p53 binding domain deletion mutant (Δ53BD), p53 expression was examined at indicated time of cycloheximide treatment. Degradation of p53 is inhibited in synoviolin knockout MEFs (Syno−/−) compared with wild type MEFs (Syno+/+). The p53 expression was examined after the indicated time of cycloheximide treatment. The remaining p53 expressions were normalized to β-actin expression and plotted against time (minutes). (E) Effects of knockdown of Synoviolin and/or other known E3 ubiquitin ligases for p53 on the level of p53 in RKO cells. Quantified p53 level is expressed as relative levels (1.0=GFP-siRNA treated RKO cells). (F) Genotoxic stress induces p53 accumulation in the absence of Synoviolin. GFP- siRNA (G). syno siRNA (S). At 48 h after transfection, the RKO cells were treated with vehicle, camptothecine (CPT, 0.5 μM), actinomycin D (ActD, 5 nM), X-Ray (9 Gray) or thapsigargin (TG, 1 μM) for 6 h. (G) Synoviolin knockdown sensitizes cells to genotoxic stress. At 48 h after transfection, cells were exposed to the indicated doses of ionizing-radiation for 24 h, followed by FACS analysis to detect annexin V-positive cells. Data in (D) and (G) are mean±s.e.m. of n⩾3. *P<0.05.
In the next step, we tested the implication of ubiquitination of p53 by Synoviolin in the degradation of p53 in vivo. In HEK293 cells, overexpressed Synoviolin WT significantly shortened the half-life of endogenous p53, whereas Synoviolin C307S and SynoviolinΔ53BD did not increase the degradation rate of p53 (Figure 5D top, mock: 125.5±18.2 min, Synoviolin WT: 44.8±3.8 min, Synoviolin C307S: 177.3±26.8 min and SynoviolinΔ53BD: 161.0±41.4 min). These results indicate that Synoviolin is responsible for the turnover of p53 as its E3 ubiquitin ligase in vivo. Consistent with these data, the half-life of p53 was significantly prolonged in Syno−/− MEFs (Figure 5D (bottom), Syno+/+ MEFs: 26.1±1.6 min; and Syno−/− MEFs: 120.0±30.3 min. P<0.05) as well as RKO cells treated with synoviolin siRNA (Supplementary Figure 8). In this regard, several ubiquitin ligases, such as Cop1 (Dornan et al, 2004), Pirh2 (Leng et al, 2003) and MDM2 (Haupt et al, 1997; Kubbutat et al, 1997), are already reported to negatively regulate p53 (Bode and Dong, 2004). To ascertain the significance of Synoviolin relative to these ligases, we compared the effects of depletion of synoviolin and/or Cop1, Pirh2 or MDM2 on the expression level of p53 in RKO cells. The amount of p53 by synoviolin ablation was less than that by MDM2 ablation, but equivalent to that by Cop1 ablation. Depletion of synoviolin in cells treated with siRNA for Cop1, Pirh2 or MDM2 non-redundantly increased p53 levels (Figure 5E). Therefore, Synoviolin functionally targets p53 independent of other ubiquitin ligase pathways. Then, does Synoviolin regulate p53 activation process? To address this question, we applied genotoxic stress as a stimulus for p53 activation (Kastan et al, 1991; Vogelstein et al, 2000). Syno siRNA and GFP siRNA-transfected RKO cells were treated with or without genotoxic stresses such as camptothecin, actinomycin D and γ-irradiation. As expected, increased level of p53 by Synoviolin knockdown was cooperatively enhanced by treatment with genotoxic stresses in these cells (Figure 5F). Thapsigargin induced Synoviolin expression, as reported previously (Yagishita et al, 2005), which was abolished in Syno siRNA-transfected RKO cells (Figure 5F). Consistent with the findings of a previous report (Qu et al, 2004), thapsigargin, an ER stress inducer, destabilized p53 expression compared with vehicle treatment in GFP siRNA-treated RKO cells. Interestingly, the thapsigargin-induced inhibition of p53 expression was reversed, at least in part, by the ablation of Synoviolin (Figure 5F). In addition, the sensitivity of syno siRNA-treated RKO cells to γ-irradiation was significantly increased compared with GFP siRNA-treated RKO cells (Figure 5G). Considered together, these results indicate that Synoviolin also participates in genotoxic stress response through the mechanism identified here.
Synoviolin regulates p53-dependent apoptotic pathway in vivo
The above experiments provided several pieces of evidence that Synoviolin is a novel class of E3 ubiquitin ligase for p53. However, the majority of the results obtained from cultured cells may not fully reflect the physiological function of p53 in the context of the whole organism. Therefore, we used Drosophila to confirm the association between Synoviolin and p53. Among Drosophila clones, CG1937 was identified by BLASTP (protein–protein blast analysis using flybase—http://flybase.bio.indiana.edu/blast/) as the gene with 63% homology to mammalian Synoviolin, and the RING domain of CG1937 is highly conserved (82%) and in vitro ubiquitination assay evidently indicated that Drosophila Synoviolin (dSyno) ubiquitinates Drosophila p53 (Dmp53) (Figure 6A) (Ollmann et al, 2000). To investigate the role of Synoviolin in p53 regulation in the whole organism, we generated transgenic flies in which Dmp53 or dSyno was overexpressed by tissue-specific Gal4 driver (Harrison et al, 1995). By crossing each transgenic fly, we generated e16E-Gal4/UAS-Dmp53;UAS-dSyno/+ flies, in which both Dmp53 and dSyno could be overexpressed in the posterior halves of wings by e16E-Gal4 driver. The expression level of Dmp53 in the wing discs was significantly decreased in e16E-Gal4/UAS-Dmp53;UAS-dSyno/+ discs compared with e16E-Gal4/ Dmp53;+/+ discs (Figure 6B). Moreover, acridine orange staining of apoptotic cells in these discs demonstrated that the level of apoptosis induced by overexpression of Dmp53 was diminished by dSyno overexpression (Figure 6C). These results indicate that dSyno affects Dmp53 protein levels in the fly system, similar to the results of the cell culture system.
Figure 6.
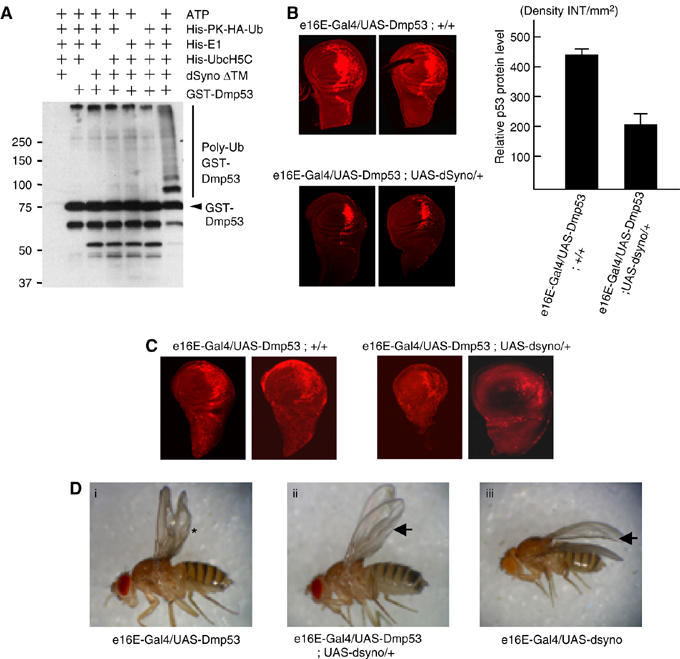
Synoviolin directly regulates p53-dependent apoptotic pathway in Drosophila fly. (A) Fly homolog of Synoviolin ubiquitinates fly homolog of p53 in vitro. GST-fusion Drosophila melanogaster p53 (Dmp53) was incubated with or without ATP, His-PK-HA-Ub, His-E1 (human), His-UbcH5C (human) and Drosophila Synoviolin (dSyno)ΔTM. Ubiquitinated proteins were probed with anti-HA antibody. (B) P53 protein level of wing discs was determined by immunostaining using anti-Dmp53 antibody (left). 2 representative pictures of each fly are shown. The fluorescence intensity of each 15 fly disc was quantified, and the net density level (Density INT/mm2) was determined by subtracting the density level of the background area (anterior half of disc) from the measured level of the target area (posterior half of disc) (right). Data are mean±s.e.m. of n=15. (C) Apoptosis was examined by Acridine orange staining of wing disc. Overexpression of dSyno in the posterior half of the discs reduced Dmp53 overexpression-induced apoptosis. (D) Overexpression of dSyno suppressed the Dmp53-induced bubbled wing phenotype. The extent of wing bubble (*) in e16E-Gal4/UAS-Dmp53 flies varied with age, but the penetrance of bubbled wing phenotype was close to 100% (Supplementary Table 1). Overexpression of dSyno suppressed the Dmp53-induced bubbled wing phenotype, but dSyno-induced wrinkled phenotype at the posterior edge of wing was still observed (arrow).
In addition to decreased Dmp53 protein level by dSyno in the wing discs of adult flies, dSyno altered the wing phenotype. Namely, overexpression of Dmp53 by e16E-Gal4 driver caused bubbled wing phenotype at the posterior half of wings (Figure 6Di). This phenotype was completely suppressed by dSyno overexpression (Figure 6Dii and Supplementary Table 1). Overexpression of dSyno alone by e16E-Gal4 driver also produced wing phenotype (weak wrinkling of the posterior edge of the wing) (Figure 6Diii). This wrinkled phenotype induced by dSyno overexpression was not affected by Dmp53 in the double overexpressing flies (Figure 6Dii and iii, arrow). These results confirm that dSyno regulates Dmp53 protein level in vivo and such regulation might be accomplished through ubiquitination of dDmp53.
To determine whether this phenotypic suppression of Dmp53 by dSyno is specific to Dmp53, we investigated the interaction between dSyno and the upstream activators of p53 such as dATM, CHK2, using the same strategy. None of these activators showed interaction with dSyno (data not shown), suggesting that dSyno regulates Dmp53 protein level directly in vivo.
Discussion
We provide concrete evidence for the first time of the functional relationship between Synoviolin and p53. As a target for Synoviolin, p53 is evidently a non-ERAD substrate. In this regard, Doa10p, the RING finger E3 ubiquitin ligase, is known not only to be involved exclusively in removing ER proteins in the ERAD, but also to eliminate cytoplasmic targets, especially the soluble transcriptional factor Matα2, which translocates into the nucleus similar to p53 (Swanson et al, 2001; Laney and Hochstrasser, 2003). Thus, our finding can be viewed within the same framework of yeast though in higher eukaryotes. In the meantime, it was proposed that the ERAD in yeast is composed of two distinct surveillance mechanisms, that is, the folded state of luminal domains and the cytosolic domains are monitored by ERAD-luminal (ERAD-L) and ERAD-cytosolic (ERAD-C) pathways, respectively (Vashist and Ng, 2004; Nishikawa et al, 2005). Hrd1p is recognized as an ERAD-L ligase; however, this classification is not applicable to Synoviolin as a human homolog of yeast Hrd1, because Synoviolin can target both ERAD-L substrate and cytoplasmic p53 (ERAD-C substrate). Therefore, we propose the novel regulatory system of Synoviolin as a different classification of the ERAD-L/C pathway.
Maintenance of homeostasis is an important cellular function, and cells are equipped with various processes to maintain their conditions. Transcriptional alteration mediated by p53 results in a variety of cell fate changes, including growth arrest and apoptosis (Vousden and Lu, 2002; Meek, 2004). Normally, the cell maintains low levels of p53 through rapid protein degradation via the UPS by the function of ubiquitin ligases. In contrast, under genotoxic stress conditions, stabilization of p53 is promoted and the diffusely distributed p53 translocates to the nucleus owing to growth inhibition and apoptosis by its transcriptional activity. Thus, adjusting the level and nuclear localization of p53 are two essential processes for cells in order to maintain the physiological state. Although p53 mutations have been documented in more than half of all human tumors (Hollstein et al, 1999), it is also known that tumor cells retain WT p53. In this regard, functional inactivation of WT p53 by abnormal cytoplasmic sequestration is frequently observed in many tumor types (Moll et al, 1992, 1996; Schlamp et al, 1997). The RING finger protein Parc is considered to act as a cytoplasmic anchoring molecule of p53, but this clone does not have a p53 ubiquitination activity (Nikolaev et al, 2003). On the other hand, our present findings demonstrated that Synoviolin not only anchors p53 in the cytoplasm, but also ubiquitinates it, and thus differs from Parc (Figure 4). Moreover, Synoviolin diverges from other ligases for p53; each of the three ubiquitin ligases for p53 (MDM2, Pirh2 and Cop1) forms an autoregulatory negative feedback loop, resulting in lower p53 activity upon its expression, but these three ligases are target for the p53 transcriptional pathway (Dornan et al, 2004; Leng et al, 2003), whereas the expression of Synoviolin is not regulated by p53 (Figure 5F). Indeed, the synoviolin promoter region does not have a p53 target sequence, whereas it contains the ER stress responsive element (Tsuchimochi et al, 2005) and responds to the stress (Figure 5F). The reason for the multiple post-translational steps for p53 is the enormous importance of this molecule in maintaining cellular homeostasis. p53 is negatively regulated by various ubiquitin ligases, such as MDM2, MdmX, HAUSP, ARF, COP1, Pirh2 and ARF-BP1 (Brooks and Gu, 2006), and it is assumed that each molecule has its specific roles in p53 control. Among them, Synoviolin is also a unique regulator of p53 because of its independency from other ligases and transcriptional regulation by p53, ER localization and canonical function in ERAD.
In the present study, we demonstrated that Synoviolin participates in genotoxic stress-mediated p53 signaling, and its to participation in the ER stress-induced apoptosis is also well known (Bordallo et al, 1998; Kaneko et al, 2002; Kikkert et al, 2004; Yagishita et al, 2005). Therefore, Synoviolin seems to regulate two distinct apoptotic pathways and the ubiquitination of p53 by Synoviolin may be another target for crosstalk between them. Another linkage between ER stress and p53 pathway is also implicated by our finding that UPR markers are increased in cells with synoviolin knockdown (data not shown). Two reports described a crosstalk of p53- and ER stress- induced apoptosis pathways, that is, ER stress antagonizes p53-mediated apoptosis through the cytoplasmic localization of p53 due to phosphorylation by glycogen synthase kinase-3β (GSK-3β) (Qu et al, 2004), and p53 destabilization utilized the cooperative action of MDM2 and GSK-3β in ER-stressed cells (Pluquet et al, 2005). In this regard, it is important to note that UPR activation upon Synoviolin knockdown in RKO cells may be related to ER stress with impaired ERAD system. Since Synoviolin null cells show upregulation of p53, it is possible that the effect of p53 stabilization by Synoviolin knockdown exceeds the p53 destabilization effect of UPR induced by Synoviolin knockdown. This hypothesis may be supported by the finding that synoviolin siRNA treatment seemed to restore the expression of p53 at least in part, which was suppressed by ER stress (Figure 5F). The regulatory action of Synoviolin on p53 under ER stress is obviously more complex, because ER stress also induces Synoviolin expression. Further studies are necessary to determine the physiological regulatory role of Synoviolin in p53 expression under ER stress conditions.
The function of p53 in patients with RA is still controversial (Firestein et al, 1997; Reme et al, 1998; Inazuka et al, 2000; Muller-Ladner and Nishioka, 2000; Sun and Cheung, 2002). Mice lacking p53 do not develop spontaneous arthropathy but have severe collagen-induced arthritis (CIA) (Yamanishi et al, 2002; Simelyte et al, 2005). As we reported previously, overexpression of Synoviolin resulted in spontaneous arthropathy and its deficiency resulted in resistance to CIA in mice (Amano et al, 2003). Therefore, we assumed that the severity of arthritis could be determined by the Synoviolin–p53 control pathway and that the onset of spontaneous arthropathy may be caused by p53-independent pathway in these models. The influence of these relationships on arthritis is currently being examined in our laboratories, using synoviolin and p53 double null mutant mice. We hope that our research could uncover new pathogenic mechanisms of RA. Furthermore, since p53 is an important tumor suppressor gene, we believe that Synoviolin could be a useful therapeutic target for not only RA but also cancer based on its cytological and biochemical features, i.e., cytoplasmic localization and enzymatic activity (Hopkins and Groom, 2002).
In conclusion, we demonstrated that Synoviolin acts as an ERAD E3 ubiquitin ligase that controls cellular p53 and thus opens, a new concept for proliferative disorders such as RA and cancer.
Materials and methods
Plasmids
pcDNA3/Synoviolin WT or C307S-FLAG, pcDNA3/HA- and FLAG-p53, pcDNA3/MDM2 and pcDNA/HA-Ub plasmids have been described previously (Amano et al, 2003; Matsushita et al, 2005). Deletion of 53BD, MBP- and GST-fusions in Synoviolin deletion mutants was performed by PCR-based method in this study. To clone a cDNA encoding Drosophila homolog of human Synoviolin (dSyno), 2282 bp of CG1937 was cut out from EST GH11117 with EcoRI/XhoI, and subcloned into pUAST vector (Brand and Perrimon, 1993). The sequences of all plasmids generated by PCR were confirmed by ABI auto-sequencer.
Cells and transfections
RKO and HEK293 cells were cultured in Minimum Essential Medium (Sigma) and U2OS and Saos-2 in Dulbecco's modified Eagle's medium (Sigma). The sense sequences of siRNA oligonucleotides to synoviolin are (1) GGUGUUCUUUGGGCAACUG, (2) GCUGUGACAGAUGCCAUCA, (3) GGUUCUGCUGUACAUGGCC. Changes in p53 protein in RKO cells were determined by all these siRNAs. The sense sequence of siRNA oligonucleotides to GFP is GGCUACGUCCAGGAGCGC.
GST pull-down assay
GST-fusion proteins were expressed in Escherichia coli strain BL21 (Invitrogen) and purified by using glutathione-Sepharose beads (Amersham Biosciences). In vitro-translated 35S-labeled p53 was pre-cleaned with 10 μg GST protein for 1 h at 4°C, followed by incubation with 10 μg of each GST-fusion protein in binding buffer (20 mM N-2-hydroxyethylpiperazine-N′-ethanesulfonic acid (HEPES), pH 7.9, 150 mM NaCl and 0.2% TritonX-100) for 1 h at 4°C. After washing, bound proteins were separated by SDS–PAGE and detected by BAS.
Immunoprecipitation assay
For co-immunoprecipitation assay between exogenous Synoviolin and exogenous p53, HEK293 cells were co-transfected with HA-p53 and pcDNA3-Synoviolin WT-FLAG, pcDNA3-SynoviolinΔ53BD-FLAG or pcDNA3-Synoviolin C307S-FLAG plasmids. Cell extracts were prepared with high-salt buffer (20 mM HEPES pH 7.2, 420 mM NaCl, 10% glycerol, 0.5% NP-40, 0.5 mM dithiothreitol (DTT), and 1 mM phenylmethylsulfonyl fluoride (PMSF)) and diluted at three-fold with 0.5 mM DTT and a protease inhibitor solution, followed by incubation with mouse IgG or anti-FLAG antibody. Precipitated proteins were detected by anti-HA or anti-FLAG antibodies.
To detect the interaction between endogenous Synoviolin and p53, HEK293 cells were lysed in 100 mM Tris–HCl, 80 mM NaCl, 1 mM EDTA, 5 mM EGTA, 5% glycerol, 2%(w/v) digitonin, 0.1% Brij 35, protease inhibitor cocktail and 20 μM of MG132. Immunoprecipitation was carried out in the presence or absence of SDS by using anti-p53 antibodies, anti-Synoviolin antibodies or control IgG. The immunoprecipitated samples were analyzed by western blot by using anti-p53 or anti-Synoviolin antibodies.
In vitro and in vivo ubiquitination assays
The in vitro ubiquitination assay was conducted as described previously (Amano et al, 2003). For the peptide inhibition assay, reaction solutions lacking no MBP-SynoviolinΔTM-6xHis and ATP were incubated with 53BD or control peptides (50, 100, and 200 μM) for 30 min at 4°C. Reactions were started by addition of MBP-SynoviolinΔTM-6xHis and ATP and incubating at 37°C.
For the in vivo ubiquitination assay, HEK293 cells were transfected with pcDNA3/HA-Ubiquitin, pcDNA3/FLAG-p53, and pcDNA3/Synoviolin WT, C307S or pcDNA3/MDM2. At 24 h post-transfection, cells were treated with MG132 (10 μM) for 1 h, then the cells were lysed in SDS containing buffer (50 mM Tris, pH 7.5, 0.5 mM EDTA, 1% SDS, and 1 mM DTT) and boiled for 5 min to denature the proteins. The denatured samples were diluted with immunoprecipitation buffer (50 mM Tris, pH 7.5, 2 mM EDTA, 150 mM NaCl, and 0.1% NP-40 and protease inhibitor cocktail) and the p53 protein was immunopurified by using anti-p53 antibody. Ubiquitinated p53 was detected by western blotting by using anti-HA antibody.
Immunostaining of fly wing discs
Fly wing discs were dissected in PBS, fixed in a buffer containing 50 mM Tris–HCl, pH 6.8, 1 mM EGTA, 1% Triton × -100, 2 mM MgSO4, 150 mM NaCl, and 2.2% formaldehyde for 15 min, and blocked using a blocking buffer (50 mM Tris–HCl, pH 6.8, 150 mM NaCl, 0.5% NP-40 and 5 mg/ml BSA). The fixed wing discs were incubated overnight at 4°C in a 1:200 dilution of anti-Dmp53 (d-200) antibody. After washing in a wash buffer (50 mM Tris–HCl, pH 6.8, 150 mM NaCl, 0.5% NP-40 and 1 mg/ml BSA), they were incubated for 3 h at 4°C in donkey anti-rabbit FITC at 1:200 dilution, washed with the wash buffer and then mounted in a mounting solution (50 mM Tris–HCl, pH 6.8, 30% glycerol, 150 mM NaCl, and 5 mg/ml phenylethylendiamine). The fluorescence intensity of each disc was quantified with Quantity One software (Bio-Rad Laboratories). Acridine orange staining was performed as reported previously (Brodsky et al, 2000).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
Supplementary Figure 8
Supplementary Table 1
Supplementary methods
Acknowledgments
We are grateful to MR Montminy, G Verdine, R Nagata, H Shimizu, I Hishinuma, H Yokohama, H Kato, S Kitamura, K Yoshimatsu, Yuichiro ITAKURA OFFICE and ES Takagi, for advice and encouragement, and to H Takahashi, M Sato, S Otani, A Sugamiya, N Takagi, S Shinkawa, Y Nakagawa, Y Sato, M Yamanashi and members of Toshi's Laboratory for the excellent technical assistance. This study was supported in part by LocomoGene Inc., Eisai Co., Ltd, National Institute of Biomedical Innovation, the Japanese Ministry of Education, Culture, Sports, Science and Technology, the Japanese Ministry of Health, Labour and Welfare, the Kato Memorial Trust for Nanbyo Research, the Japan Medical Association, Nagao Memorial Fund, Kanae Foundation for Life & Socio-medical Science, Japan Research Foundation for Clinical Pharmacology, Kanagawa Nanbyo Foundation, Kanagawa Academy of Science and Technology Research Grants, Japan College of Rheumatology, the Nakajima Foundation, Japan Society for Promotion of Science, New Energy and Industrial Technology Development Organization, Mochida Pharmaceutical Co. Ltd, Kanto Bureau of Economy, Trade and Industry, and the Uehara Memorial Foundation. HF is supported by Japan Society for the Promotion of Science.
References
- Amano T, Yamasaki S, Yagishita N, Tsuchimochi K, Shin H, Kawahara K, Aratani S, Fujita H, Zhang L, Ikeda R, Fujii R, Miura N, Komiya S, Nishioka K, Maruyama I, Fukamizu A, Nakajima T (2003) Synoviolin/Hrd1, an E3 ubiquitin ligase, as a novel pathogenic factor for arthropathy. Genes Dev 17: 2436–2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode AM, Dong Z (2004) Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer 4: 793–805 [DOI] [PubMed] [Google Scholar]
- Bordallo J, Plemper RK, Finger A, Wolf DH (1998) Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol Biol Cell 9: 209–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118: 401–415 [DOI] [PubMed] [Google Scholar]
- Brodsky MH, Nordstrom W, Tsang G, Kwan E, Rubin GM, Abrams JM (2000) Drosophila p53 binds a damage response element at the reaper locus. Cell 101: 103–113 [DOI] [PubMed] [Google Scholar]
- Brooks CL, Gu W. (2006) p53 ubiquitination: Mdm2 and beyond. Mol Cell 21: 307–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dornan D, Wertz I, Shimizu H, Arnott D, Frantz GD, Dowd P, O'Rourke K, Koeppen H, Dixit VM (2004) The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 429: 86–92 [DOI] [PubMed] [Google Scholar]
- Firestein GS, Echeverri F, Yeo M, Zvaifler NJ, Green DR (1997) Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proc Natl Acad Sci USA 94: 10895–10900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogh J, Wright WC, Loveless JD (1977) Absence of HeLa cell contamination in 169 cell lines derived from human tumors. J Natl Cancer Inst 58: 209–214 [DOI] [PubMed] [Google Scholar]
- Gottlieb E, Haffner R, King A, Asher G, Gruss P, Lonai P, Oren M (1997) Transgenic mouse model for studying the transcriptional activity of the p53 protein: age- and tissue-dependent changes in radiation-induced activation during embryogenesis. EMBO J 16: 1381–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DA, Binari R, Nahreini TS, Gilman M, Perrimon N (1995) Activation of a Drosophila Janus kinase (JAK) causes hematopoietic neoplasia and developmental defects. EMBO J 14: 2857–2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M (1997) Mdm2 promotes the rapid degradation of p53. Nature 387: 296–299 [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A (1998) The ubiquitin system. Annu Rev Biochem 67: 425–479 [DOI] [PubMed] [Google Scholar]
- Hollstein M, Hergenhahn M, Yang Q, Bartsch H, Wang ZQ, Hainaut P (1999) New approaches to understanding p53 gene tumor mutation spectra. Mutat Res 431: 199–209 [DOI] [PubMed] [Google Scholar]
- Hopkins AL, Groom CR (2002) The druggable genome. Nat Rev Drug Discov 1: 727–730 [DOI] [PubMed] [Google Scholar]
- Inazuka M, Tahira T, Horiuchi T, Harashima S, Sawabe T, Kondo M, Miyahara H, Hayashi K (2000) Analysis of p53 tumour suppressor gene somatic mutations in rheumatoid arthritis synovium. Rheumatology 39: 262–266 [DOI] [PubMed] [Google Scholar]
- Kaneko M, Ishiguro M, Niinuma Y, Uesugi M, Nomura Y (2002) Human HRD1 protects against ER stress-induced apoptosis through ER-associated degradation. FEBS Lett 532: 147–152 [DOI] [PubMed] [Google Scholar]
- Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW (1991) Participation of p53 protein in the cellular response to DNA damage. Cancer Res 51: 6304–6311 [PubMed] [Google Scholar]
- Kikkert M, Doolman R, Dai M, Avner R, Hassink G, vanVoorden S, Thanedar S, Roitelman J, Chau V, Wiertz E (2004) Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J Biol Chem 279: 3525–3534 [DOI] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Vousden KH (1997) Regulation of p53 stability by Mdm2. Nature 387: 299–303 [DOI] [PubMed] [Google Scholar]
- Laney JD, Hochstrasser M (2003) Ubiquitin-dependent degradation of the yeast Mat(alpha)2 repressor enables a switch in developmental state. Genes Dev 17: 2259–2270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng RP, Lin Y, Ma W, Wu H, Lemmers B, Chung S, Parant JM, Lozano G, Hakem R, Benchimol S (2003) Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell 112: 779–791 [DOI] [PubMed] [Google Scholar]
- Matsushita N, Kitao H, Ishiai M, Nagashima N, Hirano S, Okawa K, Ohta T, Yu DS, McHugh PJ, Hickson ID, Venkitaraman AR, Kurumizaka H, Takata M (2005) A FancD2-Monoubiquitin Fusion Reveals Hidden Functions of Fanconi Anemia Core Complex in DNA Repair. Mol Cell 19: 841–847 [DOI] [PubMed] [Google Scholar]
- Meek DW (2004) The p53 response to DNA damage. DNA Repair 3: 1049–1056 [DOI] [PubMed] [Google Scholar]
- Moll UM, Ostermeyer AG, Haladay R, Winkfield B, Frazier M, Zambetti G (1996) Cytoplasmic sequestration of wild-type p53 protein impairs the G1 checkpoint after DNA damage. Mol Cell Biol 16: 1126–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll UM, Riou G, Levine AJ (1992) Two distinct mechanisms alter p53 in breast cancer: mutation and nuclear exclusion. Proc Natl Acad Sci USA 89: 7262–7266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Ladner U, Nishioka K (2000) p53 in rheumatoid arthritis: friend or foe? Arthritis Res 2: 175–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev AY, Li M, Puskas N, Qin J, Gu W (2003) Parc: a cytoplasmic anchor for p53. Cell 112: 29–40 [DOI] [PubMed] [Google Scholar]
- Nishikawa S, Brodsky JL, Nakatsukasa K (2005) Roles of molecular chaperones in endoplasmic reticulum (ER) quality control and ER-associated degradation (ERAD). J Biochem 137: 551–555 [DOI] [PubMed] [Google Scholar]
- Ollmann M, Young LM, Di Como CJ, Karim F, Belvin M, Robertson S, Whittaker K, Demsky M, Fisher WW, Buchman A, Duyk G, Friedman L, Prives C, Kopczynski C (2000) Drosophila p53 is a structural and functional homolog of the tumor suppressor p53. Cell 101: 91–101 [DOI] [PubMed] [Google Scholar]
- Pickart CM (2001) Mechanisms underlying ubiquitination. Annu Rev Biochem 70: 503–533 [DOI] [PubMed] [Google Scholar]
- Pluquet O, Qu L, Baltzis D, Koromilas AE (2005) Endoplasmic reticulum stress accelerates p53 degradation by the cooperative actions of Hdm2 and Glycogen synthase kinase 3β. Mol Cell Biol 25: 9392–9405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponten J, Saksela E (1967) Two established in vitro cell lines from human mesenchymal tumours. Int J Cancer 2: 434–447 [DOI] [PubMed] [Google Scholar]
- Qu L, Huang S, Baltzis D, Rivas-Estilla AM, Pluquet O, Hatzoglou M, Koumenis C, Taya Y, Yoshimura A, Koromilas AE (2004) Endoplasmic reticulum stress induces p53 cytoplasmic localization and prevents p53-dependent apoptosis by a pathway involving glycogen synthase kinase-3beta. Genes Dev 18: 261–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reme T, Travaglio A, Gueydon E, Adla L, Jorgensen C, Sany J (1998) Mutations of the p53 tumour suppressor gene in erosive rheumatoid synovial tissue. Clin Exp Immunol 111: 353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlamp CL, Poulsen GL, Nork TM, Nickells RW (1997) Nuclear exclusion of wild-type p53 in immortalized human retinoblastoma cells. J Natl Cancer Inst 89: 1530–1536 [DOI] [PubMed] [Google Scholar]
- Shaulsky G, Goldfinger N, Ben-Ze'ev A, Rotter V (1990) Nuclear accumulation of p53 protein is mediated by several nuclear localization signals and plays a role in tumorigenesis. Mol Cell Biol 10: 6565–6577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer AG, Hampton RY (2004) Structural control of endoplasmic reticulum-associated degradation: effect of chemical chaperones on 3-hydroxy-3-methylglutaryl-CoA reductase. J Biol Chem 279: 188–196 [DOI] [PubMed] [Google Scholar]
- Shearer AG, Hampton RY (2005) Lipid-mediated, reversible misfolding of a sterol-sensing domain protein. EMBO J 24: 149–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simelyte E, Rosengren S, Boyle DL, Corr M, Green DR, Firestein GS (2005) Regulation of arthritis by p53: Critical role of adaptive immunity. Arthritis Rheum 52: 1876–1884 [DOI] [PubMed] [Google Scholar]
- Smith ML, Chen IT, Zhan Q, O'Connor PM, Fornace AJ Jr (1995) Involvement of the p53 tumor suppressor in repair of u.v.-type DNA damage. Oncogene 10: 1053–1059 [PubMed] [Google Scholar]
- Sun Y, Cheung HS (2002) p53, proto-oncogene and rheumatoid arthritis. Semin Arthritis Rheum 31: 299–310 [DOI] [PubMed] [Google Scholar]
- Swanson R, Locher M, Hochstrasser M (2001) A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matalpha2 repressor degradation. Genes Dev 15: 2660–2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchimochi K, Yagishita N, Yamasaki S, Amano T, Kato Y, Kawahara K, Aratani S, Fujita H, Ji F, Sugiura A, Izumi T, Sugamiya A, Maruyama I, Fukamizu A, Komiya S, Nishioka K, Nakajima T (2005) Identification of a crucial site for synoviolin expression. Mol Cell Biol 25: 7344–7356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashist S, Ng DT (2004) Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J Cell Biol 165: 41–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ (2000) Surfing the p53 network. Nature 408: 307–310 [DOI] [PubMed] [Google Scholar]
- Vousden KH, Lu X (2002) Live or let die: the cell's response to p53. Nat Rev Cancer 2: 594–604 [DOI] [PubMed] [Google Scholar]
- Wu J, Kaufman RJ (2006) From acute ER stress to physiological roles of the Unfolded Protein Response. Cell Death Differ 13: 374–384 [DOI] [PubMed] [Google Scholar]
- Yagishita N, Ohneda K, Amano T, Yamasaki S, Sugiura A, Tsuchimochi K, Shin H, Kawahara K, Ohneda O, Ohta T, Tanaka S, Yamamoto M, Maruyama I, Nishioka K, Fukamizu A, Nakajima T (2005) Essential role of synoviolin in embryogenesis. J Biol Chem 280: 7909–7916 [DOI] [PubMed] [Google Scholar]
- Yamanishi Y, Boyle DL, Pinkoski MJ, Mahboubi A, Lin T, Han Z, Zvaifler NJ, Green DR, Firestein GS (2002) Regulation of joint destruction and inflammation by p53 in collagen-induced arthritis. Am J Pathol 160: 123–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
Supplementary Figure 8
Supplementary Table 1
Supplementary methods